
Tuning crystallochromism in diketopyrrolopyrrole-co-thieno[3,2-b]thiophene derivatives by the architecture of their alkyl side chains†
Nicolas
Genevaz
a,
Patricia
Chávez
b,
Viktoriia
Untilova
a,
Alex
Boeglin
c,
Corinne
Bailly
d,
Lydia
Karmazin
 d and
Laure
Biniek
*a
d and
Laure
Biniek
*a
aUniversité de Strasbourg, CNRS, ICS UPR 22, F-67034 Strasbourg, France. E-mail: laure.biniek@ics-cnrs.unistra.fr
bUniversité de Strasbourg, CNRS, ICPEES UMR 7515, F-67034 Strasbourg, France
cUniversité de Strasbourg, CNRS, IPCMS UMR 7504, F-67087 Strasbourg, France
dService de Radiocristallographie, Fédération de Chimie Le Bel, FR2010, 1 rue Blaise Pascal – BP296/R8, 67008 Strasbourg Cedex, France
First published on 13th August 2018
Abstract
Two diketopyrrolopyrrole-co-thieno[3,2-b]thiophene derivatives substituted with either branched ethylhexyl (TTDPP-EH) or linear hexyl side chains (TTDPP-C6) have been synthesized. The impact of the side chain architecture on the structure and optical properties has been evaluated. TTDPP molecules crystallize in triclinic unit cells observed in both single crystals and in thin films. The most striking difference between the two compounds is the packing of the molecules. For TTDPP-EH, pairs of molecules overlap only at their thienothiophene (TT) ring tips leading to a weak excitonic coupling of the J-type character. In contrast, TTDPP-C6 molecules stack in a 1D columnar structure with extended molecular overlapping. A transverse displacement of the molecules along their molecular axis allows a partial overlap of electron-rich TT and electron-poor DPP units. This leads to a stronger excitonic coupling with apparent coexistence of H- and J-like absorption features. Interestingly, both single crystals and oriented thin films change color with light polarization. This sensitivity to light polarization is related to the presence of two different excitonic couplings within TTDPP-C6.
Introduction
Diketopyrrolopyrroles (DPPs) are the most used family of organic pigments in organic electronics applications. Their simple synthesis, excellent stability and tunable optical properties with intense color make them excellent candidates for application in semi-conducting electronic devices (e.g. OLEDs, OFETs, and OPV).1 Numerous studies have been reported on the control of their optical and electronic properties by varying the chemical design. Chemical engineering of DPPs involves the introduction of (hetero)-aromatic units on the main core and/or N-substitution. (a) For instance the nature of the heteroaromatic units (from phenyl, pyridine, thiophene, thiazole or furan units to more complex architectures) added onto the DPP core modulates the molecular conformation and the conjugation length.2 The dihedral angle between the planes of the adjacent unit and the central DPP can vary from a few degrees up to almost 50°. High torsional angles disturb the molecular packing which has a direct impact on all opto-electronic properties. More generally, the nature of the heteroaromatic units has an impact also on the packing of the molecules in the solid state and their intermolecular couplings. For instance, thieno[3,2-b]thiophene units can bring strong intermolecular couplings within the molecular and polymeric system through good packing properties.3 As an example, absorption in solution of non-substituted TTDPP can red-shift from 35 nm up to 60 nm in comparison to bithiophene DPP or biphenyl-DPP derivatives, respectively.4 Yet understanding the structure–property relationships in such derivatives is difficult because no complete structural data are available. (b) The N-substitution of the DPP is another key parameter to be taken into account in the design of new DPP-based materials. By default, the DPP core presents two electron-donating N–H groups and two electron-accepting C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups which generate strong hydrogen bonding. This capability has been used extensively to form self-assembled systems based on DPPs.5 However these strong intermolecular interactions result in low solubility and can therefore hamper the film formation ability needed in the device elaboration process. Moreover it has been shown that N-unsubstituted DPP or mono-substituted DPP can form different polymorphs in the solid state which is problematic for a good control of the properties.6 To limit polymorphism, N-alkylation can be done easily on the DPP units. However, these substituents can also have an important impact on the dihedral angles and the overall electronic properties.7 For instance DPP dyes bearing various alkyl substituents at the amide positions (n-butyl, n-pentyl, n-hexyl, n-heptyl, n-octyl, and 2-ethylhexyl) showed field effect hole mobilities ranging from 0.01 to 0.7 cm2 V−1 s−1.14c For other conjugated oligomers, it has also been demonstrated that the nature of the side chains can have a strong impact on the solid state luminescence properties and on the phenomenon of crystallochromy, i.e. the extreme sensitivity of a crystal's color to the intermolecular packing.8 It is apparent that the impact of the side chains’ nature on the properties of new compounds needs to be evaluated.
O groups which generate strong hydrogen bonding. This capability has been used extensively to form self-assembled systems based on DPPs.5 However these strong intermolecular interactions result in low solubility and can therefore hamper the film formation ability needed in the device elaboration process. Moreover it has been shown that N-unsubstituted DPP or mono-substituted DPP can form different polymorphs in the solid state which is problematic for a good control of the properties.6 To limit polymorphism, N-alkylation can be done easily on the DPP units. However, these substituents can also have an important impact on the dihedral angles and the overall electronic properties.7 For instance DPP dyes bearing various alkyl substituents at the amide positions (n-butyl, n-pentyl, n-hexyl, n-heptyl, n-octyl, and 2-ethylhexyl) showed field effect hole mobilities ranging from 0.01 to 0.7 cm2 V−1 s−1.14c For other conjugated oligomers, it has also been demonstrated that the nature of the side chains can have a strong impact on the solid state luminescence properties and on the phenomenon of crystallochromy, i.e. the extreme sensitivity of a crystal's color to the intermolecular packing.8 It is apparent that the impact of the side chains’ nature on the properties of new compounds needs to be evaluated.
Herein, the synthesis, thermal and optical characterizations and structural order of two diketopyrrolopyrrole-co-thieno[3,2-b]thiophene (TTDPP) derivatives substituted either with branched ethylhexyl side chains (TTDPP-EH) or with linear hexyl side chains (TTDPP-C6) are described. These materials can be considered as model compounds for their parent higher molecular weight materials that are difficult to crystallize. In this work, we highlight the major impact of the molecular structure on the optical properties. Macroscopic single crystals of the two alkylated TTDPP have been produced enabling a complete structural characterization in the solid state. Optical properties are evaluated and correlated with their solid state structure. The nature of the side chains has a strong impact on the intra- and inter-molecular interactions in the solid state and influences strongly the colour of the crystals. A first evidence of polarization dependent colour of DPP molecules with a possible link with the structure is described. The apparent coexistence of H and J aggregates, which leads to a broad absorption in the visible range of the linear derivative, is discussed.
Experimental
Single crystals have been obtained by slow evaporation of a dichloromethane/methanol mixture.Thin film preparation
Glass slides and silicon wafers were cleaned prior to use according to the following process: sonication for 15 min at 45 °C in acetone, ethanol, Hellmanex/water (1/50), and deionized water (three times). Oriented polytetrafluoroethylene (PTFE) substrates were prepared according to the method described elsewhere9 by sliding a PTFE rod at a constant pressure (6 bar) across a clean glass slide held at 300 °C at a rate of 1 mm s−1. Films were prepared by drop-casting or doctor blading a solution in chloroform at 5 mg mL−1 on the substrates. Thermal evaporation has been used to provide better film homogeneity, in particular for the highly crystalline TTDPP-C6 derivatives. Further thermal annealings have been applied using a Linkam temperature controlled microscope stage. Under ambient conditions, under nitrogen, evaporation of the materials occurs at 280 °C for TTDPP-EH and at 300 °C for TTDPP-C6.Spectroscopic characterization
The absorption spectra of solutions and thin films were recorded in the range of 250–800 nm with a spectral resolution of 1 nm, under ambient conditions using an Agilent Cary 5000. Polarized incident light was used for aligned samples on PTFE. The emission spectra of solutions and thin films were recorded using a FluoroMax-4 spectrofluorometer (Horiba Jobin Yvon) under ambient conditions with a slit width of 3 nm.Thermal characterization
To determine if degradation of the products could occur during the thermal evaporation or the annealing process, thermal gravimetric analysis (TGA) was conducted on both samples. The TGA instrument used was a TA Q5000 IR. The measurements were performed in helium. The sample masses used were typically 2–5 mg. The scan rate was 50 °C min−1 and the temperature range was 4–500 °C. The degradation temperature was determined at 5% weight loss. Differential scanning calorimetry (DSC) analysis was performed using a TA Instruments Q1000 instrument, operating at a scanning rate of 10 °C min−1 on heating and on cooling. Only the second cycles are displayed.DFT calculations
The geometry of TTDPP-C6 has been optimized at the B3-LYP/6-31G(d,p) level of accuracy with and without the PCM (polarizable continuum model) option for chloroform using the Gaussian 09 release D01 software.10 TD-DFT calculations have been performed at the 6-311++G(d,p) level of accuracy, again with and without the PCM model for solvation by chloroform in the respective geometries, providing the convergence of 32 excited singlet states.Transmission electron microscopy (TEM) and electron diffraction
The areas of interest were identified for TEM analysis by optical microscopy (Leica DMR-X microscope). The films were coated with a thin amorphous carbon film and removed from the glass substrate by floating on a diluted aqueous HF solution (10 wt%) and subsequently recovered on TEM copper grids. TEM was performed in bright field and diffraction modes using a CM12 Philips microscope equipped with a MVIII (Soft Imaging System) Charge Coupled Device camera. Calibration of the reticular distances in the ED patterns was done using an oriented PTFE film.X-ray crystallography
For TTDPP-C6, X-ray diffraction data collection was carried out using a Bruker APEX II DUO Kappa-CCD diffractometer equipped with an Oxford Cryosystem liquid N2 device, using Cu-Kα radiation (λ = 1.54178 Å). The crystal-detector distance was 40 mm. The cell parameters were determined (APEX2 software)11 from reflections taken from three sets of 20 frames, each at 10 s exposure. The structure was solved by direct methods using the program SHELXS-2013.12 The refinement and all further calculations were carried out using SHELXL-2013.13 The H-atoms were included in calculated positions and treated as riding atoms using SHELXL default parameters. The non-H atoms were refined anisotropically, using weighted full-matrix least-squares on F2. A semi-empirical absorption correction was applied using SADABS in APEX2;11 transmission factors: Tmin/Tmax = 0.5499/0.7528. For TTDPP-EH, X-ray diffraction data collection was carried out using a Bruker APEX II DUO Kappa-CCD diffractometer equipped with an Oxford Cryosystem liquid N2 device, using Mo-Kα radiation (λ = 0.71073 Å). The crystal-detector distance was 38 mm. The cell parameters were determined (APEX2 software)11 from reflections taken from three sets of 6 frames, each at 10 s exposure. The structure was solved by direct methods using the program SHELXS-2013.12 The refinement and all further calculations were carried out using SHELXL-2013.13 The H-atoms were included in calculated positions and treated as riding atoms using SHELXL default parameters. The non-H atoms were refined anisotropically, using weighted full-matrix least-squares on F2. A semi-empirical absorption correction was applied using SADABS in APEX2;11 transmission factors: Tmin/Tmax = 0.6060/0.7456. The atoms C16 and C17 were disordered over two positions with an occupancy ratio of 0.5/0.5.Results
A. Syntheses and thermal characterizations
In order to evaluate the impact of the architecture of the side chains on the packing behavior of bis-thieno[3,2-b]thiophene-DPP, two derivatives have been synthesized. One is functionalized with linear hexyl side chains (hereafter TTDPP-C6) and the other with branched ethyl hexyl side chains (hereafter TTDPP-EH). Their chemical structures are shown in Fig. 1a and f. Both compounds are obtained following a two step procedure described previously (see Scheme S1,7a synthetic details are reported in the ESI†).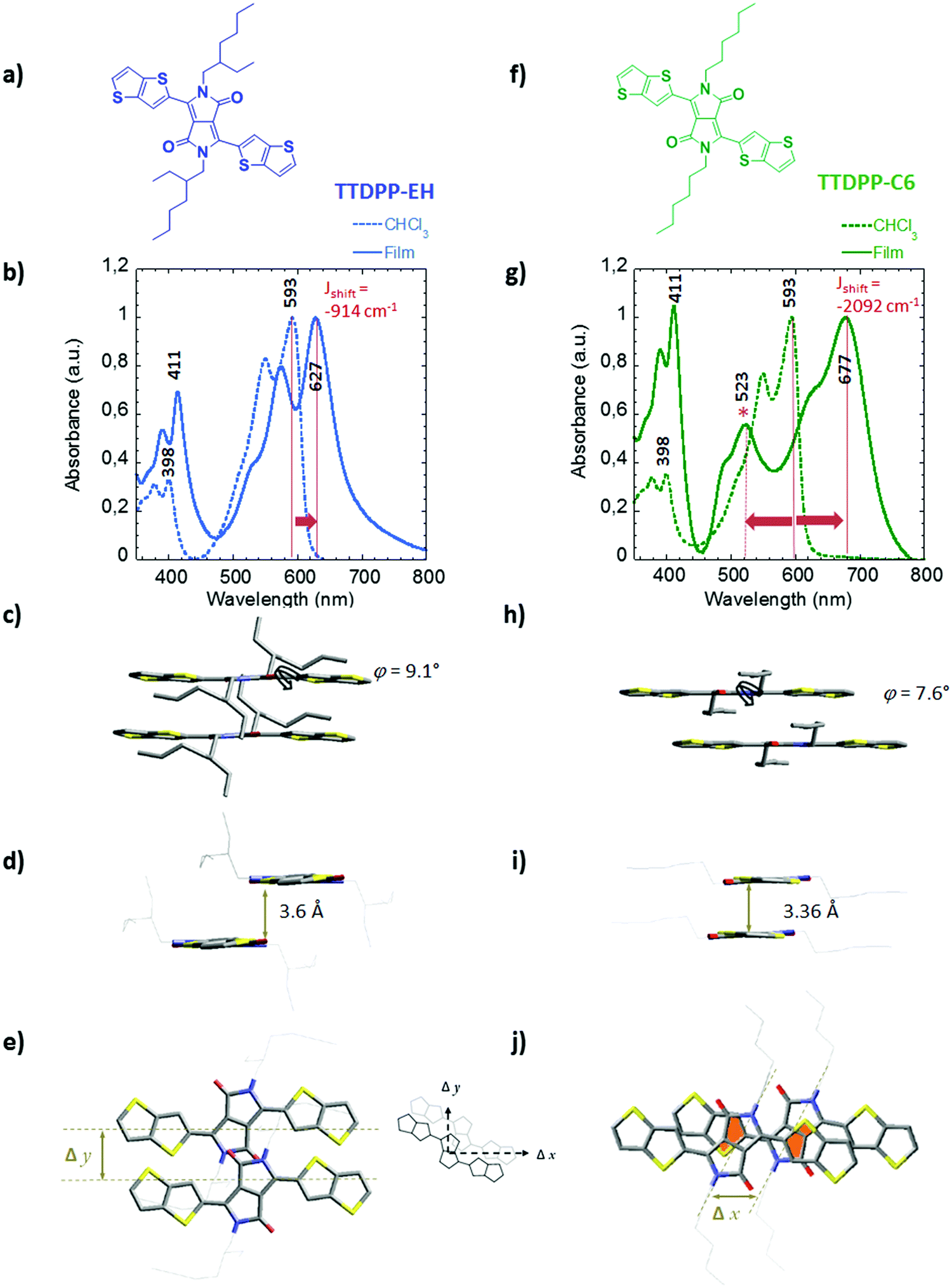 | ||
| Fig. 1 Chemical structures and comparison of the packing behavior in the solid state of TTDPP-EH (a–e) and TTDPP-C6 (f–j). (b and g) Changes in the absorption spectra from the dilute solution (in chloroform, dashed lines) to solid state (drop-cast films, solid lines). The plain lines and arrows highlight the bathochromic shifts observed from the dilute state to the solid state. The bathochromic shift energies are −914 cm−1 and −2092 cm−1 for TTDPP-EH and TTDPP-C6, respectively. The asterisk (*) indicates a characteristic optical transition band observed in the solid state of the C6 linear derivatives (hypsochromic shift energy of 2257 cm−1). (c, d and h, i) Side views onto a stack of two molecules, (e and j) top view onto a stack of two molecules of TTDPP-EH and TTDPP-C6 respectively. A diagram of transverse (Δx) and longitudinal (Δy) offsets is also shown. Partial overlaps of the thiophene and lactam rings are highlighted in orange. Torsional angles φ between the thienothiophene and the lactam rings and the short contact distances between stacks of molecules are also indicated. | ||
The thermal behavior has been studied by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) (see Fig. S1, ESI,† and Table 1). The two compounds have a high thermal stability (they decompose above 320 °C), and are suitable for vacuum deposition techniques. For both materials, single melting and crystallization peaks suggest a single polymorph. In thin films TEM did not provide evidence for polymorphism. TTDPP-EH with branched side chains displays lower melting/crystallization temperatures (Tm = 224 °C and Tc = 190 °C) than TTDPP-C6 (292 °C and 283 °C, respectively). As expected the nature of the side chains has an impact on the crystallization/melting temperatures of small molecular systems, an issue that will be discussed later on. In addition, substitution of the DPP core with two thienothiophene units leads to a ∼100 °C increase of the thermal transitions compared to that with the bithiophene analogues.14 This important shift illustrates the strong aggregation ability of the thienothiophene units.
λ
max![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sol
[nm] sol
[nm] |
λ
maxfilm
[nm] |
E
goptical
[eV] |
T m [°C] | T c [°C] | ΔHmd [J g−1] | ΔHcd [J g−1] | |
|---|---|---|---|---|---|---|---|
| a Maxima of absorption measured in dilute CHCl3 solution. b Maxima of absorption measured in thin films drop-cast from CHCl3 solution. c Optical band gap determined in the solid state. d Melting and crystallization peak temperatures and enthalpy determined by DSC. | |||||||
| TTDPP-C6 | 315, 398, 593 | 315, 405, 523, 677 | 1.84 | 292 | 283 | 54.84 | 53.04 |
| TTDPP-EH | 315, 398, 593 | 315, 412, 627 | 1.97 | 224 | 190 | 50.91 | 50.30 |
B. Optical properties of isolated molecules (in solution)
The absorption spectra of the two TTDPP derivatives in dilute chloroform solutions are shown in Fig. 1b, g and Fig. S2a (ESI†). The absorption properties are similar indicating that side chains have no impact on the spectroscopic properties in solution, a fact that had already been observed in studies on the effect of alkyl chains branched on DPP.14 The absorption spectra display three main absorption bands whose maxima are located at 315 nm, 398 nm and 593 nm. The most intense absorption band at low energy is characterized by a typical vibronic progression (ΔE = 0.16 eV).
| Electronic transition | Orbital assignment (probability) | λ (nm) | Oscillator strength (f) | Debye |
|---|---|---|---|---|
| S0 → S1 | HOMO → LUMO (0.71) | 599 | 0.9531 | 11.01 |
| S0 → S4 | HOMO−2 → LUMO (0.69) | 387 | 0.1728 | 3.77 |
| S0 → S6 | HOMO−4 → LUMO (0.68) | 364 | 0.3176 | 4.95 |
| HOMO → LUMO+2 (−0.15) | ||||
| S0 → S10 | HOMO → LUMO+2 (0.67) | 306 | 0.4081 | 5.15 |
| HOMO−2 → LUMO (0.14) |
The four associated transition dipole moments are located in the plane of the molecule. The main transition, at lowest energy, has its transition dipole moment vector oriented along C4–C4′ of the thienothiophene unit (see μ(1) in Fig. S4a, ESI†). This vector is close to the long molecular axis. A similar analysis can be conducted for the isolated EH derivative molecule since it presents similar experimental optical properties. The optical behavior of the crystals in the visible range will be related to the main S0 → S1 transition.
Whereas the nature of the side chains has no impact on the optical properties in solution, let us now examine how it impacts the properties in the solid state.
C. Structure and optical properties in the solid state
Both solutions are fluorescent pink/violet, but thin films of TTDPP-EH and TTDPP-C6 are deep violet and blue, respectively. The color change is mainly due to the low energy band broadening up to 800 nm and the red-shift of the most intense absorption peak from solution to thin films (see orange arrows). The effect is more pronounced for the linear C6 derivatives than that for the branched EH chain ones (84 versus 34 nm red-shift). The two transitions at high energy are less sensitive to the molecular interaction in the solid state (the shifts are about ∼13 nm). Thus alkyl substitution on the DPP unit derivatives has mainly a strong impact on the S0 → S1 transition in the solid state. This effect has already been observed in DPP-co-bis-thiophene derivatives bearing different alkyl side chains. However such strong red shifts (especially for TTDPP-C6) have never been observed in DPP-based materials of comparable molecular length.14a The change in absorption mainly arises from strong intermolecular interactions. The energies of the shifts are shown in Fig. 1b and g. They are proportional to the excitonic coupling (J0) as the excitonic shift of the k = 0 exciton is given by Jk=0 ≈ 2J0.26b Accordingly TTDPP-EH aggregates belong to a weak excitonic coupling regime (∼450 cm−1) whereas TTDPP-C6 aggregates show a stronger excitonic coupling (∼1050–1150 cm−1) (intermediate regime). According to Kasha's theory, shifts of the S0 → S1 transition towards the lower energy, as compared with the solution spectra (monomer), are the defining characteristics of J-aggregates.18 One would also expect a narrowing of the J band. In our case the J bands of both compounds are very broad. It is likely that the highly crystalline nature of the films (with a lot of grain boundaries, see the TEM image in ESI 8,† leading to high static disorder) is at the origin of some broadening of the band.18d In both compounds, the J bands are characterized by a vibronic progression of ∼1465–1475 cm−1. The vibronic ratios (relative absorption intensity of 0–0/0–1) are 1.25 and 1.47 for TTDPP-EH and TTDPP-C6 respectively. This reinforces the fact that excitonic coupling is stronger in TTDPP-C6 than that in TTDPP-EH as the vibronic peak ratio of the J band increases within the linear derivative.18e,26b The more sterically hindered EH side chains reduce the excitonic coupling. This hindrance results in smaller changes between solution and thin films and a larger optical bandgap (estimated from thin films at 1.97 eV) in comparison to TTDPP-C6 derivatives (optical band gap of 1.84 eV).
Interestingly a new band at 523 nm also appears in the spectrum of TTDPP-C6 (see in Fig. 1g). We have attempted to fit and deconvoluate this band from the J-like band using a Franck–Condon progression built upon two vibrational modes (see the ESI 2†). The new bands at 523 and 489 nm do not belong to the vibronic progression of the S0 → S1 electronic transition (maxima located at 677 nm). The difference in energy between 0–1 and the band located at 523 nm (>3000 cm−1) is much larger than the typical vibrational energy observed for other DPP derivatives (in the range 1300–1500 cm−1) (see Fig. S2b, ESI†).16,17 Because of its hypsochromic shift (∼2257 cm−1) compared to the solution spectrum, the 523/489 nm bands have an H-type character. This band is characterized by a vibronic progression with an absorption ratio intensity of 1.3 (estimated from the fit and peak deconvolution).
In the solid state, the fluorescence is very weak (almost quenched) for both derivatives, and even more for TTDPP-C6 (see Fig. S3c, ESI†). The photoluminescence spectra do not help in identifying the nature of the aggregates. The PL spectra in the solid state show mainly one broad emission band at 650–750 nm and 700–850 nm for TTDPP-EH and TTDPP-C6, respectively (in sharp contrast with the absorption band). The high crystalline state of the films could be at the origin of the disappearance of the vibronic structure.
The structural parameters are shown in Table 3 (and Table S3, ESI†), and the packing of the molecules is illustrated in Fig. 1 and 2 and Fig. S6 (ESI†).19 Both compounds crystallize in fairly similar triclinic unit cells with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. Both unit cells contain only one molecule. However, the C6 derivative has a higher density than the EH one, which is in agreement with the stronger intermolecular couplings observed in the solid state absorption spectra. Finally, the branched EH derivative show some disorder in the side chain positions (see CIF file and structure, ESI†).
space group. Both unit cells contain only one molecule. However, the C6 derivative has a higher density than the EH one, which is in agreement with the stronger intermolecular couplings observed in the solid state absorption spectra. Finally, the branched EH derivative show some disorder in the side chain positions (see CIF file and structure, ESI†).
| a [Å] | b [Å] | c [Å] | α [Å] | β [Å] | γ [Å] | Space group | ρ (g cm−3) | |
|---|---|---|---|---|---|---|---|---|
| TTDPP-C6 | 4.88 | 10.26 | 14.18 | 106.95 | 94.11 | 97.99 |
P |
1.442 |
| TTDPP-EH | 5.72 | 10.46 | 15.05 | 73.2 | 84.94 | 89.14 |
P |
1.231 |
The two thieno[3,2-b]thiophene rings (TT) are in anti-orientation with respect to each other (a more favorable trans–cis orientation has also been observed in other D–A–D systems).20 For TTDPP-C6 and TTDPP-EH, the TT rings make a dihedral angle of 7.5° and 9.5° with respect to the mean plane of the DPP core, respectively. The molecules are almost fully planar along their conjugated backbone. Thus the intramolecular couplings should be very similar in both compounds. Let us consider now in more detail the packing of the molecules in the unit cells. We will first look at a stack of two molecules along the a axis as shown in Fig. 1 and then at a larger view as shown in Fig. 2.
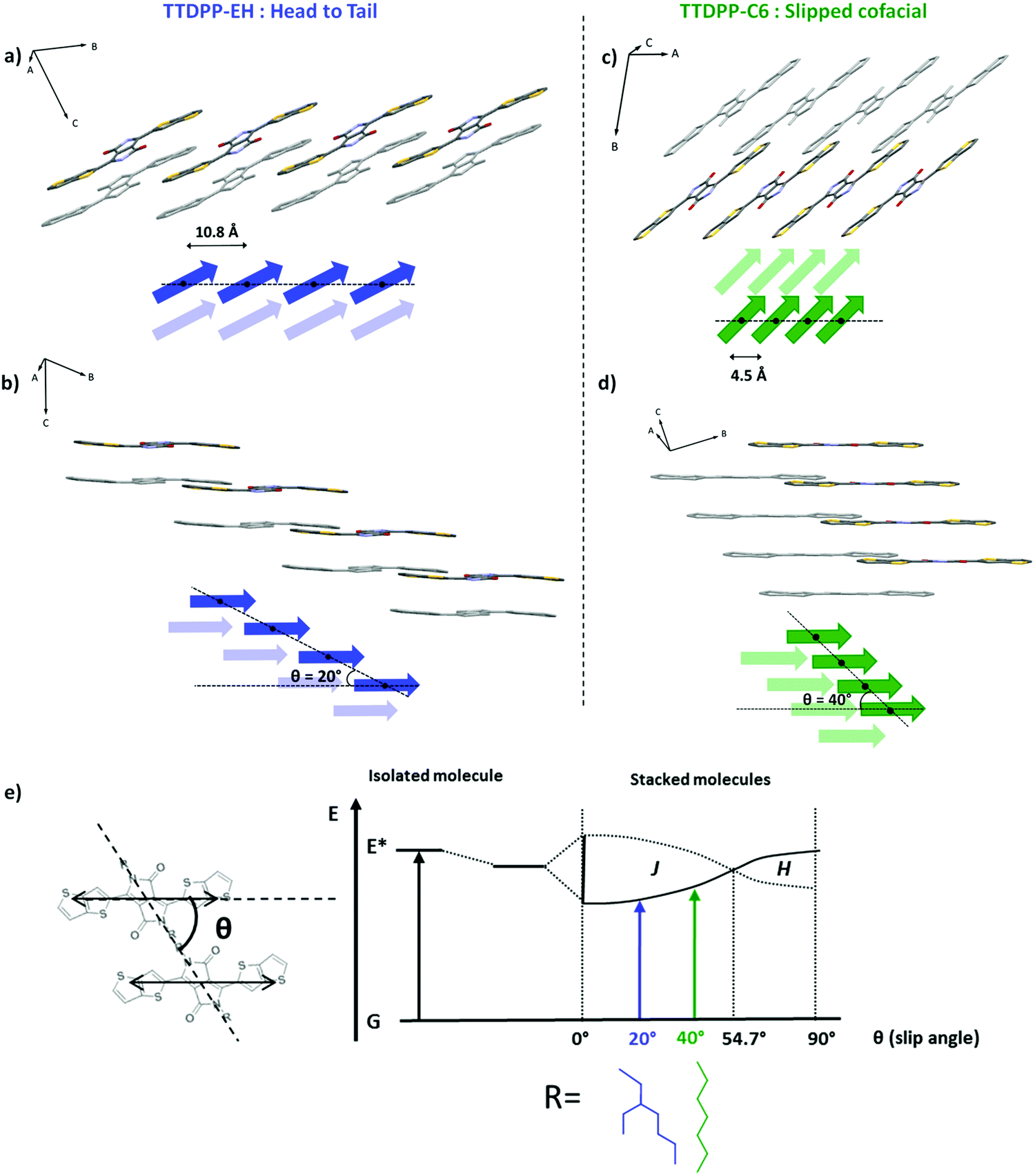 | ||
| Fig. 2 Representation of two stacks of 4 molecules taken from the crystal structures of TTDPP-EH (a and b) and TTDPP-C6 (c and d) and schematic illustrations of phase relation transition dipole moments (side chains and H are omitted for clarity; the next layers of molecules are represented in grey in the structure and pale color in the schematics). The center-to-center distances between neighbor molecules are 10.8 Å and 4.5 Å and the slip angles are 20° and 40° respectively for TTDPP-EH and TTDPP-C6. (b and d) Illustration of the assembly into pairs of molecules for TTDPP-EH and the 1D columnar structure of TTDPP-C6. (e) Exciton band energy diagram for a pair of TTDPP molecules bearing branched or linear side chains. The diagram is represented according to Kasha's model of point dipole approximation for coplanar-induced transition dipoles. It compares both structures regarding their slip angles.18 | ||
The crystal packing of TTDPP-EH (Fig. 1c–e) shows that the two molecules involved in two different layers are separated along the longitudinal y axis (Δy = 4.5 Å) which prevents from any ring overlapping along the a axis. Only the lactam groups (separated by an interplanar distance of 3.6 Å) are slightly overlapping in that direction. However, the TT rings at the tips of the molecules overlap (with a stacking distance of 3.6–3.7 Å) but always between pairs of molecules. Thus no 1D columnar stacks involving the whole molecule are formed. One could measure a slipping distance of Δxb = 10 Å along the long molecular axis direction leading to a clear “head-to-tail” configuration (see Fig. 2).
By contrast, TTDPP-C6 displays an almost cofacial layered structure along the a axis with a strong π–π stacking (a stacking distance of 3.4 Å) (Fig. 1h–j). The two stacked molecules are slipped along their long axis (Δx = 3 Å, θ = 40°), while maintaining a strong intermolecular packing. Note that this transverse displacement leads to a stronger overlap between the electron-rich thienothiophene unit and the electron deficient lactam ring (highlighted in orange in Fig. 1j), and the overlap is not seen in TTDPP-EH. Furthermore, TTDPP-C6 forms a columnar structure along the a axis (a chain of molecules involving the whole backbone) which reinforces the intermolecular interaction at a longer distance (see Fig. 2).
For both derivatives, the molecules of adjacent stacks are also engaged in a supramolecular planar 1D network formed through intermolecular weak bonds between the lactam CO and the thienothiophene S atom (see Fig. S6, ESI†). The bond distances are similar for both compounds (S2⋯O1 = 3.17 Å) despite the bulky side chains of TTDPP-EH. This supramolecular 1D network is different from the one obtained on bithiophene-DPP bearing similar side chains. In this case, the intermolecular interactions take place between the proton of the thiophene units and the CO of the lactam groups as indicated by the small C–H⋯O distances (2.33–2.67 Å).14b,c
To summarize, the nature of the side chains has little influence on the molecular conformation but a strong influence on the intermolecular couplings of the molecules in the crystal. For TTDPP-EH, neighbor molecules show π-overlaps only at the tips of the molecules. Each TT unit is only coupled two by two with an adjacent TT unit. In contrast, for TTDPP-C6, the whole molecule is involved in a strong π-overlap with its neighbor forming a 1D columnar stack at a large distance. The longitudinal slip leads to a mixed stack of D and A units.
According to Kasha's model of point dipole approximation, the slipped cofacial structures of both TTDPP derivatives are of J-type since slip angles θ < 54.7° (θEH = 20° and θC6 = 40°, see Fig. 2). This model suggests that the spectral shift between a pair of molecules depends on their mutual orientation and separation. The extent of the shift is proportional to the slip angle and inversely proportional to the cube of their center-to-center distance from one another.18 This model is consistent with our observation that TTDPP-C6 with the shortest center-to-center distance and a higher slip angle exhibit the largest red shift. However the appearance of the so-called H band in the TTDPP-C6 solid state spectra cannot be analyzed only by the point dipole approximation. Kirkus and co-workers have also observed the coexistence of J/H features for DPP-oligothiophene bearing linear side chains. These authors assign the high energy band to the presence of H aggregates (co-existing with J-aggregates).21 Such optical transitions (at high energy) nearly disappear by introducing branched alkyl chains. A gel phase of a DPP-amide oligomer has also been reported to show complete visible-spectrum coverage due to the simultaneous formation of both H- and J-type aggregates.22 This phenomenon has been assigned to Davydov splitting while it can be seen only for unit cells containing at least two molecules or more. Kirkus et al. considered that the crystal structure of their compounds (dithiophene-DPP derivatives) should be fairly similar to that of the dihexyl-3,6-diphenyl DPP analogue2e whose structure was known at the time of their work.21 However diphenyl DPP contains two molecules per unit cell (P21/c) while dithiophene-DPP14c and dithienothiophene DPP (our work) contain only one molecule per unit cells (P). Their explanation of Davydov splitting being at the origin of the joint presence of J and H is ruled out by the fact that their structures contain only one molecule per unit cell. Davydov splitting cannot be at the origin of the presence of H- and J-like bands in TTDPP-C6. This spectral signature may arise from the specific packing of the molecules and intermolecular couplings. In particular the wave function overlap between neighboring chromophores must play a role. Let us examine if the correlations between optical properties and structure still hold in thin films.
C.4.1. TTDPP-C6 oriented films on PTFE. Optical microscopy of TTDPP-C6 deposited on PTFE illustrates a preferential nucleation of the needle-shape crystallites along the PTFE c axis (Fig. 3b). In addition, a small fraction of the crystals are oriented at ±70° to the PTFE c axis. The crystallites are blue when the light polarization is oriented along the long axis of the needle, and red/purple when at right angle to it, as is the case for single crystals under polarized light (Fig. 3a). As opposed to most aligned and crystalline molecules oriented on PTFE, there was no total extinction of the birefringence when the PTFE friction direction was oriented at 90° to the polarizers. Instead we observe a remarkable change in color. Polarized absorption spectra (shown in Fig. 3c) on these oriented thin films show the same optical features as those of the non oriented films (coexistence of J- and H-like bands characterized by different vibronic progressions). The most important information holds in the polarization difference of the bands. The J aggregate band at 693 nm is mostly polarized parallel to the PTFE chains (dichroic ratio = 2.88 at 693 nm). In strong contrast, the H-like band at 525 nm is weakly polarized along the PTFE (dichroic ratio = 1.34). This highlights the fact that the two bands must have a different electronic origin. In TTDPP-C6 oriented films, the high energy band located at 398 nm is not polarized. It is likely that the blue color is linked with the strong absorption band at 693 nm. In the perpendicular orientation, it is the mixture of different transitions (centered at 525 nm and 693 nm) that makes the absorbing bodies appear red/violet. At the moment it is not evident to rationalize the polarization difference of the J and the H bands but this difference underlines the different electronic origin of the two bands.
 | ||
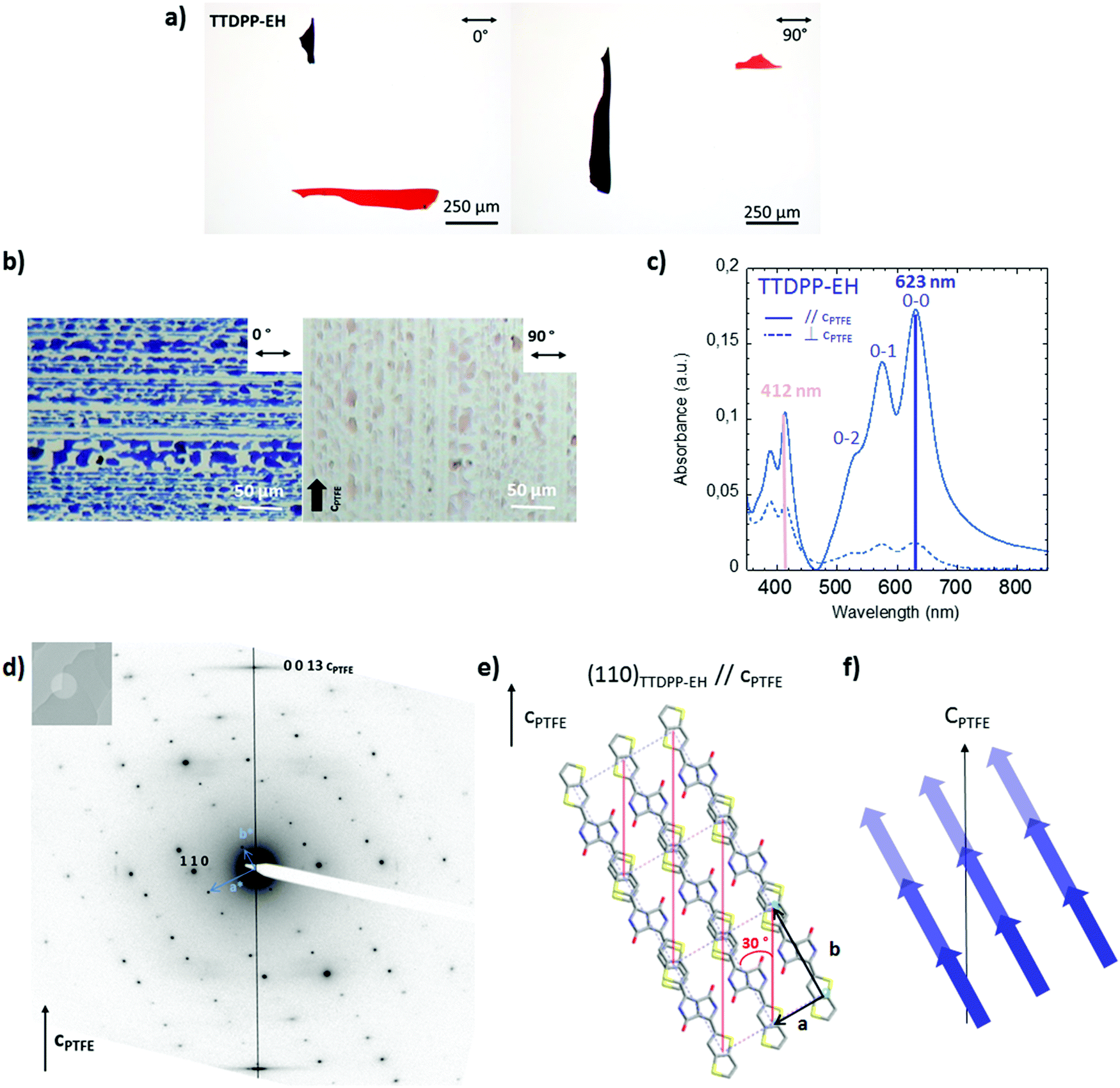
| Fig. 3 (a) Polarization colour dependent single crystal images under an optical microscope. (b–f) Optical and structural characterizations of thermally evaporated TTDPP-C6 crystals on top of oriented PTFE substrates. (b) POM images and (c) polarized UV-Visible absorption spectra with light polarization oriented parallel or perpendicular to cPTFE. (d) SAED pattern of a crystal oriented parallel to the PTFE fibers, corresponding to the [001] zone axis. (e) Top view of the TTDPP-C6 crystal orientation along the PTFE chains. The scheme highlights that the (110)TTDPP-C6 planes (represented in red) are parallel to cPTFE. The long molecular axis is tilted ±10° away from the (1 1 0) plane direction. (f) Schematic illustration of the possible intermolecular couplings between molecules’ nearest neighbors with respect to the PTFE chain direction. | ||
The crystal structure and orientation of the crystals on the PTFE substrate were studied by electron diffraction (ED). A typical SAED pattern of the crystallites is shown in Fig. 3d. The first reflections observed at 10.3, 4.9 and 4.6 Å are indexed on the basis of the XRD crystal structure as 0 1 0, 1 0 0 and 1 1 0 for the most intense one, respectively. The electron beam is thus oriented parallel to the [0 0 1] zone axis of the crystal. The structure and the (0 0 1) contact plane of the crystals grown on PTFE and on silicon oxide are similar (cf. Fig. S7, ESI†). More interestingly, the equatorial orientation of the 1 1 0 reflections indicates that the (1 1 0) planes are parallel to the PTFE c axis (vertical in Fig. 3d) and are perpendicular to the plane of the film. As seen in Fig. 3e, these planes contain the conjugated backbones of the molecule. Thus, the optical transition responsible for the color in the visible range (i.e. S0 → S1), that has its vector oriented in the plane of the molecules, along the long molecular, is also oriented along the PTFE chains. This explains why the absorption spectrum is polarized and why the maximum of absorption is obtained in the parallel direction of the PTFE polymer chains. The strong intermolecular interactions (π overlap in mixed stacked), occurring along the TTDPP-C6 chains of molecules, might also contribute to the absorption at right angle of the PTFE polymer chains.
C.4.2. TTDPP-EH oriented films on PTFE. A similar approach has been conducted on the TTDPP-EH oriented films on PTFE. The determination of the orientation of the crystals on the substrate will help us to understand how the absorption bands are polarized. The blue crystalline film aligned on PTFE, shown in Fig. 4b, turns pale pink under polarized light. The color in thin films differs from those observed with the single crystals (Fig. 4a), as a result of the different crystallite sizes and thicknesses. TTDPP-EH single crystals turned red-violet to orange-red with the light polarization oriented parallel or perpendicular to the long axis of the crystals. However, the crystals were too absorbent and too small to be directly studied under polarized light spectroscopy. For this compound, the absorption spectra in oriented thin films are not polarized exactly as that of the TTDPP-C6. As discussed previously, the solid state absorption TTDPP-EH spectra are composed of only one broad low energy band (maximum located at 623 nm) and a high energy band (maximum located at 412 nm). The spectra are characteristics of J aggregates only. Under polarized light, both bands are affected (see Fig. 4c). The low energy band is highly polarized and has its maximum intensity when the light is oriented along the cPTFE axis (dichroic ratio = 10.1). This transition is much more polarized than for the C6 derivatives. The high energy band of EH is polarized in the same direction as the low energy band but with a lower dichroic ratio (2.6). The change in color seems to be related in that case to the intensity ratio between the low and high energy bands. As for the structural analysis, the SAED pattern shown in Fig. 4d is similar to that of the TTDPP-C6 derivatives on PTFE. The crystalline domains are oriented in the same manner. The ED patterns indicate a (0 0 1) contact plane and the (1 1 0) planes containing the thienothiophene units. The molecular axis is oriented at ±30° to the (1 1 0) planes. The main transition dipole moment can then be assumed to majorly contribute to the absorption along cPTFE.
 | ||
| Fig. 4 (a) Polarization colour dependent single crystals image under an optical microscope. (b–f) Optical and structural characterizations of thermally evaporated TTDPP-EH crystals on top of oriented PTFE substrates. (a) POM images and (b) polarized UV-Visible absorption spectra with light polarization oriented parallel or perpendicular to cPTFE. (c) SAED pattern of a crystal oriented parallel to the PTFE fibers, corresponding to the [001] zone axis. (d) Top view of the TTDPP-EH crystal orientation along the PTFE chains. The scheme highlights that the (110) TTDPP-EH planes (represented in red) are parallel to cPTFE. The long molecular axis of the molecule is tilted ±30° away from the (1 1 0) plane direction. (e) Schematic illustration of the S0 → S1 transition dipole orientations with respect to the PTFE chain direction. | ||
The above results demonstrate that crystalline thin films of DPP-co-thieno[3,2-b]thiophene present outstanding polarized optical properties in a broad visible range. Also the crystals can be aligned on a PTFE substrate that helps to elucidate the orientation of the optical transitions. It is worth mentioning that there is no matching of the cell parameters between the PTFE crystals ((1 0 0) surface) and the TTDPP-C6 crystals. The orientation mechanism via an epitaxy process seems improbable. The observed molecular orientation suggests a preferential nucleation at numerous steps or surface ridges running parallel to the PTFE friction direction, as also observed for a minor population of sexithiophene crystals on PTFE,23a or tetracene and pentacene crystals.23b One could also think of anchoring of the side chains of the DPP molecules into the grooves of the PTFE (see Scheme S9, ESI†). The situation differs for TTDPP-EH crystals since d110 = 4.93 Å (very close to d−110 = 4.9 Å of PTFE). An orientation mechanism via epitaxy, in this case, could explain why the crystals are better aligned and the polarization degree of absorption is higher than that for TTDPP-C6.
Discussion
Analysis of the above results helps rationalize the correlation of the polarized absorption properties and structure. The introduction of the thieno[3,2-b]thiophene units onto the DPP core leads to highly stable and colorful compounds, showing absorption in a broad UV-Vis range. While the side chain nature has no impact on the optical properties in solution, these properties differ in the solid state depending on the side chain nature.The molecular backbone of TTDPP-EH stays quite planar, but the bulky side groups prevent proper molecular packing. There is no intermolecular overlap between the electron rich TT group and the electron poor DPP groups. XRD analysis evidences a cofacial slipping of the molecules (along b axis) which leads to limited excitonic coupling (π overlap only at the tips of the molecules between two TT rings). Although the energy shift towards low energy was very low and the band broadened, we attribute this exciton band to a J-like character.
In sharp contrast, the TTDPP-C6 derivative shows stronger intermolecular couplings than TTDPP-EH (a larger bathochromic shift of the low energy band and the appearance of an H band) due to the more cofacial stacking of the molecules and a denser crystal packing. In the present case, the origin of the coexistence of J and H bands in aggregates containing only one molecule per unit cell can be analyzed in terms of structure and specific interactions in the crystal structure. It is important to note that polymorphism has been excluded by the combination of DSC and structural analysis. A comparison of the TTDPP-EH case with no joint presence of J–H aggregates in its absorption spectrum is also instructive.
The S0 → S1 transition band is strongly polarized in the long molecular axis direction for TTDPP-EH, whereas it can also be seen at right angle for the TTDPP-C6. Clearly other coupling processes must contribute to a significant red shift observed for the C6 derivative. Referring to the C6 crystal structure, a partial overlap of the electron-rich TT and electron-deficient DPP units (mixed stack) is observed and the stacking distance is small (3.36 Å), which is not the case for TTDPP-EH. Thus intermolecular charge transfer may take place within the C6 derivative in the solid state. This type of phenomenon has been reported and validated by complementary techniques (time-resolved fluorescence spectroscopy and transient absorption spectroscopy) for other DPP derivatives that show a good π overlap of the electron-rich (thiophene or phenyl) and electron-poor (DPP) parts in their structure.15,24 For instance, Mauck et al. showed that such strong intermolecular couplings lead to charge transfer (CT) that can impact the singlet exciton fission rate.25 Many authors, including Spano and co-workers,26 and Hoffman and co-workers,8a have shown that CT interactions can have a strong effect on the solid state absorption spectra and in particular in J- and H-aggregate behavior. Therefore, a CT-mediated short-range coupling could be at the origin of both an important bathochromic shift linked with the appearance of the blue-shifted band at 523 nm of TTDPP-C6. Thus, the differences in colour and optical properties of both compounds could find their origin in their different coupling sources (coulombic for TTDPP-EH, and CT mediated or a combination thereof for TTDPP-C6).26 Further photophysical and theoretical studies, out of the scope of this work, would be needed to better identify the excitonic origins and their coupling interactions of our so-called J- and H-like aggregate bands.
Conclusions
A detailed structural analysis of two new compounds based on diketopyrrolopyrrole and thieno[3,2-b]thiophene with potential for opto-electronic application helps illustrate the impact of the crystal structure on electronic coupling. A limited change in the structure of the side chains grafted on the DPP core has an impact on their packing and properties. In particular both the slip angle and the molecular shift distances in a stack of molecules, defined by the excitonic theory, are affected. The bulky branched ethylhexyl groups hamper a good packing of the conjugated backbone and lead to an important shift of the molecules along the molecular axis and also along the short axis. In sharp contrast linear chains can favor strong intermolecular interactions resulting in a probable intermolecular charge transfer between the electron-poor and electron-rich units. The TTDPP-C6 molecules investigated here, with their strong intermolecular couplings, could be of interest in this context. More generally, the strategy of using branched alkyl side chains to provide better solubility of the conjugated core can be detrimental for optical properties. The architecture of the side chains is thus to be considered carefully in the chemical design of new compounds. Finally, polarized, air stable, and color tunable films have been obtained. Whereas a color change induced by external stimuli has been observed in DPP derivatives,27 the sensitivity to light polarization behavior in the DPP crystals is reported for the first time. Such structure–property relationships should open up a way for further uses of TTDPP units.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Laure Biniek thanks the Université de Strasbourg and Investissement d’Avenir (IdEx 2015 Attractivité) as well as Fédération Régionale Matériaux et Nanosciences Alsace (FRMNA-AAP jeunes chercheurs 2015) for financial support. Marc Schmutz and Christian Blanck from the microscopy platform of the Charles Sadron Institute are acknowledged for technical support on TEM analysis. Christophe Melart from ICPEES is acknowledged for technical support on TGA characterization. Martin Brinkmann, Nicolas Leclerc and Bernard Lotz are gratefully acknowledged for fruitful discussions and their careful reading of the manuscript.Notes and references
- (a) B. Tieke, A. R. Rabindranath, K. Zhang and Y. Zhu, Beilstein J. Org. Chem., 2010, 6, 830 CrossRef PubMed; (b) S. Qu and H. Tian, Chem. Commun., 2012, 48, 3039 RSC; (c) C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Funct. Mater., 2013, 25, 1859 CrossRef PubMed; (d) D. Chandran and K.-S. Lee, Macromol. Res., 2013, 21, 272 CrossRef; (e) Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684 RSC; (f) M. Kaur and D. H. Choi, Chem. Soc. Rev., 2015, 44, 58 RSC; (g) M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280 CrossRef; (h) W. Li, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Acc. Chem. Res., 2016, 49, 78 CrossRef PubMed.
- (a) J. H. Park, E. H. Jung, J. W. Jung and W. H. Jo, Adv. Mater., 2013, 25, 2583 CrossRef PubMed; (b) T. Bura, S. Beaupré, O. A. Ibraikulov, M.-A. Légaré, J. Quinn, P. Lévêque, T. Heiser, Y. Li, N. Leclerc and M. Leclerc, Macromolecules, 2017, 50, 7080 CrossRef; (c) B. Sun, W. Hong, Z. Yan, H. Aziz and Y. Li, Adv. Mater., 2014, 26, 2636 CrossRef PubMed; (d) I. Bulut, P. Chávez, A. Mirloup, Q. Huaulmé, B. Heinrich, A. Hébraud, S. Méry, R. Ziessel, T. Heiser, P. Lévêque and N. Leclerc, J. Mater. Chem. C, 2016, 4, 4296 RSC; (e) R. Sevincȩk, S. Çelik, M. Aygün, S. Alp and S. Isik, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, 1546 CrossRef PubMed; (f) T. Mukhopadhyay, B. Puttaraju, S. P. Senanayak, A. Sadhanala, R. Friend, H. A. Faber, T. D. Anthopoulos, U. Salzner, A. Meyer and S. Patil, ACS Appl. Mater. Interfaces, 2016, 38, 25415 CrossRef PubMed; (g) S. Militzer, T. M. P. Tran, P. J. Mésini and A. Ruiz-Carretero, ChemNanoMat, 2018 DOI:10.1002/cnma201800192.
- (a) I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328 CrossRef PubMed; (b) P. Brocorens, A. Van Vooren, M. L. Chabinyc, M. F. Toney, M. Shkunov, M. Heeney, I. McCulloch, J. Cornil and R. Lazzaroni, Adv. Mater., 2009, 21, 1193 CrossRef; (c) L. Biniek, S. Fall, C. L. Chochos, O. Boyron, N. Leclerc, P. Lévêque and T. Heiser, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1861 CrossRef; (d) S. Fall, L. Biniek, Y. Odarchenko, D. V. Anokhin, G. de Tournadre, P. Lévêque, N. Leclerc, D. A. Ivanov, O. Simonetti, L. Giraudet and T. Heiser, J. Mater. Chem. C, 2016, 4, 286 RSC; (e) L. San Miguel, W. W. Porter III and A. J. Matzger, Org. Lett., 2007, 9, 1005 CrossRef PubMed.
- H. Liu, E. A. B. Kantchev, H. S. Tan and T. B. Norsten, J. Mol. Eng. Mater., 2013, 1, 1250003 CrossRef.
- (a) J. Mei, K. R. Graham, R. Stalder, S. P. Tiwari, H. Cheun, J. Shim, M. Yoshio, C. Nuckolls, B. Kippelen, R. K. Castellano and J. R. Reynolds, Chem. Mater., 2011, 23, 2285 CrossRef; (b) E. R. Draper, B. Dietrich and D. J. Adams, Chem. Commun., 2017, 53, 1864 RSC; (c) G. Sheel Thool, K. Narayanaswamy, A. Venkateswararao, S. Naqvi, V. Gupta, S. Chand, V. Vivekananthan, R. R. Koner, V. Krishnan and S. Prakash Singh, Langmuir, 2016, 32, 4346 CrossRef PubMed; (d) E. D. Głowacki, H. Coskun, M. A. Blood-Forsythe, U. Monkowius, L. Leonat, M. Grzybowski, D. Gryko, M. Schuette White, A. Aspuru-Guzik and N. S. Sariciftci, Org. Electron., 2014, 15, 3521 CrossRef PubMed; (e) C. Fu, P. J. Beldon and Dmitrii F. Perepichka, Chem. Mater., 2017, 29, 2979 CrossRef.
- (a) J. Mizuguchi, T. Imoda, H. Takahashi and H. Yamakami, Dyes Pigm., 2006, 68, 47 CrossRef; (b) J. Dhar, D. P. Karothu and S. Patil, Chem. Commun., 2015, 51, 97 RSC; (c) C. Fu, P. J. Beldon and D. F. Perepichka, Chem. Mater., 2017, 29, 2979 CrossRef.
- (a) H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. Shakya Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. J. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2011, 133, 3272 CrossRef PubMed; (b) I. Meager, R. S. Ashraf, S. Rossbauer, H. Bronstein, J. E. Donaghey, J. Marshall, B. C. Schroeder, M. Heeney, T. D. Anthopoulos and I. McCulloch, Macromolecules, 2013, 46, 5961 CrossRef; (c) X. Liu, J. Huang, J. Xu, D. Gao, W. Zhang, K. Shi and G. Yu, RSC Adv., 2016, 6, 35394 RSC; (d) P. Josse, A. Labrunie, C. Dalinot, S. M. McAfee, S. Dabos-Seignon, J. Roncali, G. C. Welch, P. Blanchard and C. Cabanetos, Org. Electron., 2016, 37, 479 CrossRef.
- (a) P. M. Kazmaier and R. Hoffmann, J. Am. Chem. Soc., 1994, 116, 9684 CrossRef; (b) C. Kitamura, Y. Abe, T. Ohara, A. Yoneda, T. Kawase, T. Kobayashi, H. Naito and T. Komatsu, Chem. – Eur. J., 2010, 16, 890 CrossRef PubMed; (c) A. Liess, A. Lv, A. Arjona-Esteban, D. Bialas, A.-M. Krause, V. Stepanenko, M. Stolte and F. Würthner, Nano Lett., 2017, 17, 1719 CrossRef PubMed; (d) M. Surin, E. Hennebicq, C. Ego, D. Marsitzky, A. C. Grimsdale, K. Müllen, J.-L. Brédas, R. Lazzaroni and P. Leclère, Chem. Mater., 2004, 16, 994 CrossRef.
- J. C. Wittmann and P. Smith, Nature, 1991, 352, 414 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- M86-E01078 APEX2 User Manual, Bruker AXS Inc., Madison, USA, 2006 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef PubMed.
- (a) A. B. Tamayo, M. Tantiwiwat, B. Walker and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 15543 CrossRef; (b) M. A. Naik, N. Venkatramaiah, C. Kanimozhi and S. Patil, J. Phys. Chem. C, 2012, 116, 26128 CrossRef; (c) M. Stolte, S.-L. Suraru, P. Diemer, T. He, C. Burschka, U. Zschieschang, H. Klauk and F. Würthner, Adv. Funct. Mater., 2016, 26, 7415 CrossRef.
- B. Barszcz, K. Kędzierski, H. Y. Jeong and T.-D. Kim, J. Lumin., 2017, 185, 219 CrossRef.
- H. Liu, H. Jia, L. Wang, Y. Wu, C. Zhan, H. Fu and J. Yao, Phys. Chem. Chem. Phys., 2012, 14, 14262 RSC.
- S. Wood, J. Wade, M. Shahid, E. Collado-Fregoso, D. D. C. Bradley, J. R. Durrant, M. Heeney and J.-S. Kim, Energy Environ. Sci., 2015, 8, 3222 RSC.
- (a) M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371 Search PubMed; (b) A. S. Davidov, Theory of Molecular Excitons, Plenum Press, New York, 1971 CrossRef; (c) M. R. Philpott, Some Modern Aspects of Exciton Theory, in Advances in Chemistry and Physics, ed. I. Prigogine and S. A. Rice, J. Wiley, Hoboken, NJ, 1973, vol. 23 Search PubMed; (d) T. E. Kaiser, I. G. Scheblykin, D. Thomsson and F. Würthner, J. Phys. Chem. B, 2009, 113, 15836 CrossRef PubMed; (e) M. Mas-Montoya and R. A. J. Janssen, Adv. Funct. Mater., 2017, 1605779 CrossRef.
- TTDPP-C6 s and TTDPP-EH structures have been deposited at CCDC 1839874 and 1839875,† respectively. Checkcif files are also available as ESI†.
- C. B. Nielsen, A. J. P. White and I. McCulloch, J. Org. Chem., 2015, 80, 5045 CrossRef PubMed.
- M. Kirkus, L. Wang, S. Mothy, D. Beljonne, J. Cornil, R. A. J. Janssen and S. C. J. Meskers, J. Phys. Chem. A, 2012, 116, 7927 CrossRef PubMed.
- S. Ghosh, S. Cherumukkil, C. H. Suresh and A. Ajayaghosh, Adv. Mater., 2017, 1703783 CrossRef PubMed.
- (a) J. C. Wittmann, C. Straupé, S. Meyer, B. Lotz, P. Lang, G. Horowitz and F. Garnier, Thin Solid Films, 1997, 303, 207 CrossRef; (b) M. Brinkmann, S. Graff, C. Straupé, J.-C. Wittmann, C. Chaumont, F. Nuesch, A. Aziz, M. Schaer and L. Zuppiroli, J. Phys. Chem. B, 2003, 107, 10531 CrossRef; (c) J.-F. Moulin, M. Brinkmann, A. Thierry and J.-C. Wittmann, Adv. Mater., 2002, 14, 436 CrossRef.
- J. Mizuguchi and S. Homma, J. Appl. Phys., 1989, 66, 3104 CrossRef.
- (a) P. E. Hartnett, E. A. Margulies, C. M. Mauck, S. A. Miller, Y. Wu, Y.-L. Wu, T. J. Marks and M. R. Wasielewski, J. Phys. Chem. B, 2016, 120, 1357 CrossRef PubMed; (b) C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749 CrossRef PubMed.
- (a) H. Yamagata, D. S. Maxwell, J. Fan, K. R. Kittilstved, A. L. Briseno, M. D. Barnes and F. C. Spano, J. Phys. Chem. C, 2014, 118, 28842 CrossRef; (b) N. J. Hestand and F. C. Spano, Chem. Rev., 2018 DOI:10.1021/acs.chemrev.7b00581.
- S. Ying, M. Chen, Z. Liu, M. Zheng, H. Zhang, S. Xue and W. Yang, J. Mater. Chem. C, 2017, 5, 5994 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis details, X-ray structural analysis. CCDC 1839874 and 1839875. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8tc02022a |
| This journal is © The Royal Society of Chemistry 2018 |