
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-based nanostructured materials for advanced lithium–sulfur batteries
Juan
Balach
 *a,
Julia
Linnemann
bc,
Tony
Jaumann
b and
Lars
Giebeler
*b
*a,
Julia
Linnemann
bc,
Tony
Jaumann
b and
Lars
Giebeler
*b
aDepartment of Chemistry, National University of Río Cuarto-CONICET, Route 36 Km 601, X5804ZAB, Río Cuarto, Argentina. E-mail: jbalach@exa.unrc.edu.ar
bLeibniz Institute for Solid State and Materials Research (IFW) Dresden e.V., Helmholtzstr. 20, 01069 Dresden, Germany. E-mail: l.giebeler@ifw-dresden.de
cChair of Analytical Chemistry II – Electrochemistry & Nanoscale Materials Group, Ruhr-University Bochum, ZEMOS, Universitätsstr. 150, 44801 Bochum, Germany
First published on 19th October 2018
Abstract
Since the resurgence of interest in lithium–sulfur (Li–S) batteries at the end of the 2000s, research in the field has grown rapidly. Li–S batteries hold great promise as the upcoming post-lithium-ion batteries owing to their notably high theoretical specific energy density of 2600 W h kg−1, nearly five-fold larger than that of current lithium-ion batteries. However, one of their major technical problems is found in the shuttling of soluble polysulfides between the electrodes, resulting in rapid capacity fading and poor cycling stability. This review spotlights the foremost findings and the recent progress in enhancing the electrochemical performance of Li–S batteries by using nanoscaled metal compounds and metals. Based on an overview of reported functional metal-based materials and their specific employment in certain parts of Li–S batteries, the underlying mechanisms of enhanced adsorption and improved reaction kinetics are critically discussed involving both experimental and computational research findings. Thus, material design principles and possible interdisciplinary research approaches providing the chance to jointly advance with related fields such as electrocatalysis are identified. Particularly, we elucidate additives, sulfur hosts, current collectors and functional interlayers/hybrid separators containing metal oxides, hydroxides and sulfides as well as metal–organic frameworks, bare metal and further metal nitrides, metal carbides and MXenes. Throughout this review article, we emphasize the close relationship between the intrinsic properties of metal-based nanostructured materials, the (electro)chemical interaction with lithium (poly)sulfides and the subsequent effect on the battery performance. Concluding the review, prospects for the future development of practical Li–S batteries with metal-based nanomaterials are discussed.
 Juan Balach | Juan Balach received his bachelor and Ph.D. degree from National University of Río Cuarto, Argentina, in 2007 and 2011, respectively. After a postdoctoral research stay (2012) in the Department of Colloid Chemistry at the Max Planck Institute of Colloids and Interfaces, Germany, he joined the Leibniz Institute for Solid State and Materials Research (IFW) Dresden, Germany, during 2013–2016. Since 2016, he is a Research Associate of the National Scientific and Technical Research Council (CONICET) at the National University of Río Cuarto. His interests focus on energy materials, including porous nanocarbons and inorganic nanostructured materials for supercapacitors and next-generation batteries: Li–S batteries, Li–air batteries and aqueous batteries. |
 Julia Linnemann | Julia Linnemann is a research assistant at the Chair of Analytical Chemistry II – Electrochemistry and Nanoscale Materials at the Ruhr-University Bochum, Germany. Supported by the German Academic Scholarship Foundation, she studied chemistry at the Technische Universität (TU) Dresden, Germany and the University of Wollongong, Australia. Following a short-term position at the Project House Light & Electronics of Evonik Industries in Hsinchu, Taiwan, she received a dissertation fellowship of the German Federal Environmental Foundation in 2015 to conduct a doctoral research project on the electrochemical synthesis of metal–organic materials and films for energy storage applications at the Leibniz Institute for Solid State and Materials Research (IFW) Dresden, Germany. |
 Tony Jaumann | Tony Jaumann earned his M. Sc. in Chemistry at the TU Dresden in collaboration with the University College London in 2013. He received his doctoral degree at the TU Dresden about silicon anodes in Li-ion and Li–S batteries in 2016. After a postdoctoral stay at the Leibniz Institute for Solid State and Materials Research (IFW) Dresden, Germany, until 2017, he currently works on batteries for industrial applications. |
 Lars Giebeler | Dr Lars Giebeler studied chemistry at the Universities of Gießen and Leipzig and received his doctoral degree from the Materials Sciencés Institute at TU Darmstadt under supervision of Prof. Hartmut Fueß. After his postdoctoral research at the KU Leuven (Prof. Johan Martens) and at the TU Darmstadt (Prof. Christian Hess), he joined the Leibniz Institute for Solid State and Materials Research (IFW) Dresden in 2009 and has become group leader in 2011. His research interests focus on active (nanosized) materials for technically relevant applications, operando diffraction and spectroscopy techniques. |
1. Introduction
Our techno-society has crossed the “line of no return” altering traditional lifestyle. The forthcoming technological innovations, which embrace plug-in (hybrid) electric vehicles, aerospace transportation, smart-grid and Internet of Things (IoT) applications, are in relentless pursuit of high-energy rechargeable power sources with reliable/sustainable performance and safety tolerance beyond the state-of-the-art rechargeable lithium-ion (Li-ion) batteries.1,2Undoubtedly, Li-ion battery advances have prompted an unprecedented growth in the portable-power industry. Li-ion battery technologies have been reliant on the usage of intercalation chemistry in transition metal-based lithium containing oxide/phosphate cathodes such as Li(Ni,Mn,Co)O2 (NMC), Li(Ni,Co,Al)O2 (NCA), LiMn2O4 (LMO) and LiFePO4 (LFP), where their physical constraints in specific energy densities are less than 400 W h kg−1 on the cell level even with high-energy NMC (811) cathodes and silicon anodes.3 This energy density is insufficient to meet the upcoming specific energy requirements for “green” electric vehicles and backup energy storage systems capable of coping with the fluctuations of supply from renewable sources (e.g. wind, tidal and solar energies).2 Furthermore, the aforementioned intercalation-type cathodes present some critical downsides such as high costs and safety concerns that may restrict their further implementation in large-scale power source systems. Therefore, explorations of alternative electrochemical systems which offer higher specific capacity/energy density at low cost are dearly needed for a paradigm change in energy storage due to the ever-increasing demands.
Lithium–sulfur (Li–S) batteries have been touted as one of the most plausible platforms to fulfill the energy demand of tomorrow. The pairing of a high specific capacity lithium anode (3800 mA h g−1) and sulfur cathode (1675 mA h g−1) affords a remarkably high theoretical specific energy and volumetric energy of, respectively, 2600 W h kg−1 and 2800 W h L−1 (assuming a complete reaction between sulfur and lithium to form lithium sulfide (Li2S)), outperforming by far existing Li-ion batteries as shown in Fig. 1.4–7 In addition to its high specific capacity, sulfur as an active cathode material has a low environmental impact and it is daily produced in ton quantities as a by-product of the hydrodesulfurization process in crude-oil refineries, making it abundant and cost-effective for industrial applications on a large scale.8 While emerging battery companies like Sion Power9 and Oxis Energy10 make their first steps in the field of sulfur-based energy systems, the Li–S battery technology faces numerous drawbacks leading to a poor service life that drastically hinders the step towards mass production and large-scale commercialization of the battery.
 | ||
| Fig. 1 Schematic comparison of the theoretical and practical gravimetric energy densities of various rechargeable battery systems. Expected mid-class to small electric car range based on reported Tesla Model S and Audi e-tron performances.11,12 Adapted with permission from ref. 7. Creative Commons Attribution 2.0 International License (http://creativecommons.org/licenses/by/2.0). | ||
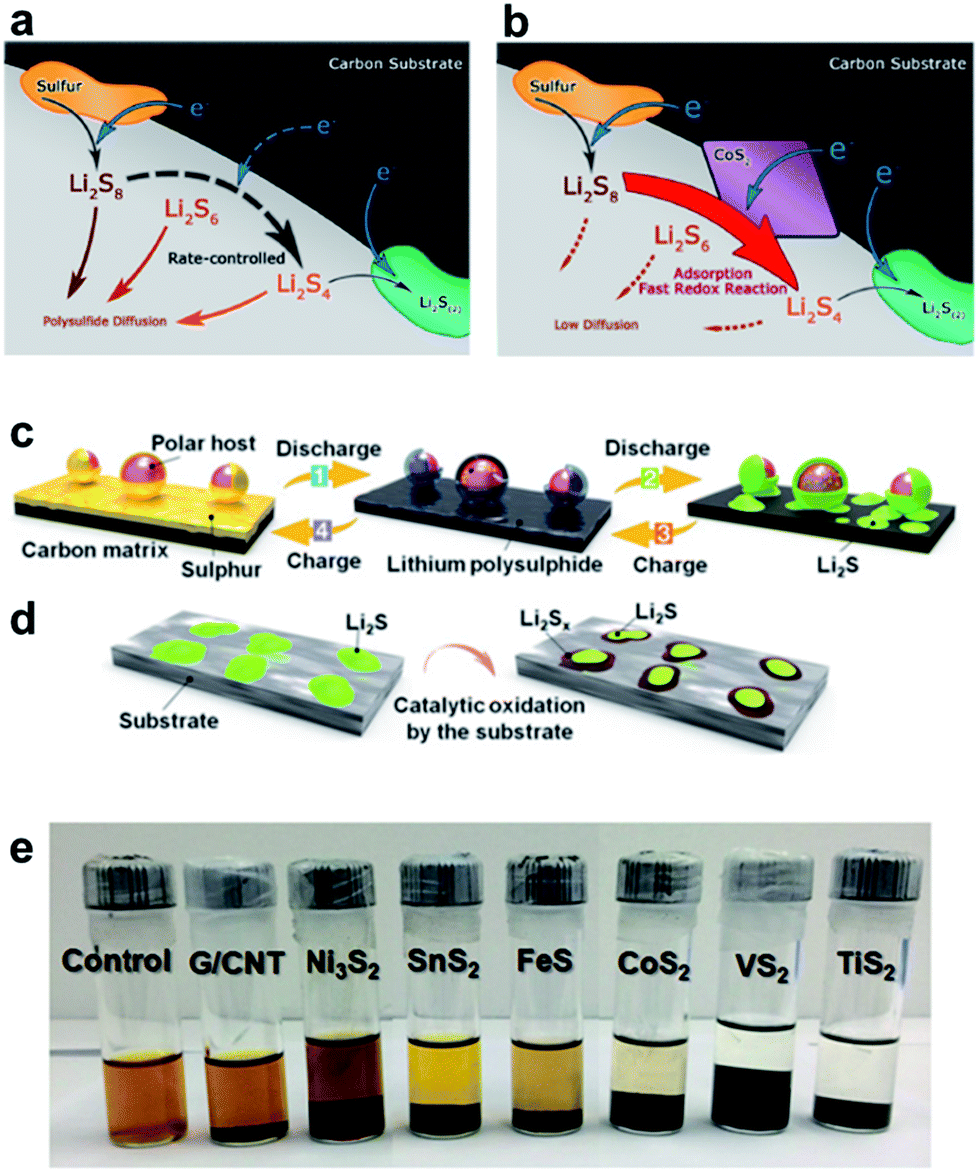
The overall redox reaction of Li/S coupling can be written as S8 + 16Li+ + 16e− ↔ 8Li2S↓, with the average voltage potential of the full cell being 2.15 V vs. Li/Li+. However, the total conversion reaction hides a multielectron process with many equilibrium reactions between sulfur and lithium polysulfide (LiPS) intermediates of various chain lengths (Fig. 2a).13 During the initial discharge of the cell, the octet sulfur (S8) in its solid phase is gradually lithiated to form long-chain LiPSs (Li2Sn; 4 ≤ n ≤ 8) which are highly soluble in commonly used ether-based electrolytes. In the subsequent discharge process, long-chain LiPSs are reduced to insoluble and poorly conductive Li2S2 and Li2S species. Essentially, the discharge process described above involves the typical two-step sulfur reduction reactions corresponding to two plateaus in the voltage profile as displayed in Fig. 2b.14 However, the formation of S3˙− radicals via disproportionation or decomposition reactions of S6− anions has also been proposed.15 The formation of soluble LiPS intermediates is one of the principal issues in the performance of sulfur-based rechargeable batteries since they are prone to escape out of the cathode scaffold driven by electric field and LiPS concentration gradient forces, leading to the loss of active sulfur material. Furthermore, the dissolved long-chain LiPSs easily diffuse through the polymeric porous separator to the negative electrode and they are reduced to Li2S2 and further irretrievably consumed to form solid Li2S at the anode by a spontaneous reaction with metallic lithium, causing lithium anode contamination/passivation, active material loss and increase of cell resistance. The unreacted soluble LiPS species then diffuse back to the cathode side during cell charging and are oxidized again to long-chain LiPSs. This phenomenon generates a constant movement of sulfur species between the two electrodes that is generally known as the “shuttle effect”.16,17 Although this LiPS shuttling is mainly responsible for the massive degradation of the battery life, there are other critical concerns inherent to the chemical features of sulfur. For instance, the insulating nature of elemental sulfur (σ = 5 × 10−30 S cm−1 at 25 °C) constrains its complete utilization. Another problem is the difference in density between sulfur (2.03 g cm−3) and its reduced discharge product Li2S (1.67 g cm−3) which entails a large volumetric expansion (≈80%) during lithiation, leading to the degradation/pulverization of the positive electrode under mechanical strain.18 Nazar and co-workers developed a breakthrough approach to physically encapsulate sulfur, enhance its redox kinetics and buffer the volumetric expansion of sulfur during lithiation which consists of infiltrating conductive mesoporous carbon with molten sulfur.19 The encouraging improvements of the cell performance obtained by the encapsulation of sulfur into the pores/cavities of conductive carbon matrices have triggered intensive research on using diverse porous carbon (nano)structures as host matrices (carbon nanoparticles, microporous carbons, mesoporous carbons, hierarchical carbons, carbon spheres, hollow carbon spheres, carbon nanotubes (CNTs), carbon nanofibers (CNFs), graphene, reduced graphene oxide (rGO) and the mix of them).20–24 However, the solid-to-liquid transformation of the active material and the weak interaction of non-polar pristine carbons with polar LiPSs often lead to the irremediable leak of LiPS species out of the cathode scaffold (specially at areal sulfur loadings higher than 4 mg cm−2),25 losing the initial intimate contact with the carbon matrix and favoring the agglomeration of Li2S/sulfur particles both at the separator/cathode interface and on the anode surface. Especially the latter reaction degrades the performance and the lifespan of the battery. Additionally, recent reviews have given a detailed overview on the functionality of almost all parts of a Li–S battery and how to improve them.26–30
 | ||
| Fig. 2 (a) Stepwise reduction pathway of octet sulfur (S8) to solid Li2S2 and Li2S products, including intermediate LiPSs (Li2Sn; 3 ≤ n ≤ 8).17 (b) Representative Li–S cell configuration and the characteristic charging/discharging voltage profile based on the stoichiometric redox chemistry between lithium and sulfur.22 (a) Reproduced with permission from ref. 17. Copyright 2014, Elsevier. (b) Reproduced with permission from ref. 22. Copyright 2015, Elsevier. | ||
The use of additives in ether-based electrolytes, LiNO3 for example, to form a passivation film on the lithium anode and suppress undesired side reactions,31,32 the utilization of heteroatom-doped carbons and polymers (e.g.: poly(3,4-ethylenedioxythiophene) (PEDOT), polyaniline (PANI) and polypyrrole (PPy)) with combined ionic and electronic conductivity to enhance both physical and chemical confinement of sulfur-based species,4,33–35 and the addition of conductive porous carbon interlayers between the separator and the cathode to intercept and re-activate migrating LiPS intermediates36 have also been proven to be viable approaches to enhance the electrochemical performance of Li–S cells. However, these methods in fact retard the diffusion of soluble LiPS species but they do not tackle the root cause. Beyond the conventional encapsulation of active sulfur into porous carbonaceous host matrices, in the last few years significant advances have been made to address the challenges discussed by using diverse metal-based nanostructured materials with specific chemical affinity to lithium (poly)sulfides.37,38 Metal-containing compounds with a tailored polar surface have been described as efficient “polar” or “chemisorptive” sulfur host materials to enhance the adsorption of LiPS intermediates, to intensify and achieve faster redox reactions.39,40 These metal-based compounds can furtherly function as redox mediators40 possessing the ability to accelerate the kinetics of redox reactions of soluble LiPSs to insoluble Li2S2/Li2S and vice versa, e.g. by reducing charge transfer resistance.
The scope of this review is to summarize the foremost findings and the recent progress towards achieving high sulfur utilization and long lifespan of Li–S batteries by using additives, sulfur hosts, and functional interlayers/hybrid separators comprising metal-based nanostructured materials, namely metal oxides, metal sulfides, metal–organic frameworks, metals, metal hydroxides, metal nitrides, metal carbides and MXenes. In particular, we emphasize the close relationships between the intrinsic properties of metal-based nanomaterials and the chemical interaction with lithium (poly)sulfides and the subsequent effect on the electrochemical performance of Li–S batteries. In an attempt to provide a guiding route towards the rational design of sulfur cathodes with high practical specific energy, the potential for the future development of practical Li–S batteries with metal-based nanomaterials is discussed.
2. Metal oxides
Metal oxides have been used for more than a decade to trap and arrest soluble LiPSs at the positive electrode and thus mitigate the inexorable diffusion of the active material between electrodes. The difference in electronegativity between oxygen and metal atoms induces a strong surface polarity in the metal oxide which serves to effectively interact, or even react via a thiosulfate mechanism, with polar LiPS species. The use of metal oxides as additives, sulfur hosts, and components in functional interlayers/hybrid separators as well as the relationship between their intrinsic properties and the electrochemical performance of Li–S cells are described in this section.2.1 Metal oxides as additives
One of the early studies on using metal oxides as additives for improving the performance of Li–S batteries was reported by Ahn and co-workers in 2004.41 The authors stated that by adding 15 wt% of nanosized Mg0.6Ni0.4O (particle size ≈ 50 nm; surface area ≈ 8 m2 g−1) as an additive, the initial specific capacity of the Li–S cells increases by up to 60% in comparison to the cells without the additive (from 741 mA h g−1 to 1185 mA h g−1 at 0.1C) due to the improvement of the LiPS adsorption. Despite the initial high capacity achieved by the cell with the Mg0.6Ni0.4O additive, the capacity steadily decreases to around 1000 mA h g−1 after 50 cycles revealing a relatively poor LiPS retention.Later, Ahn and co-workers also used a similar strategy but employing γ-Al2O3 nanoparticles as an additive.42 By adding 10 wt% of γ-Al2O3 nanoparticles (≈150 nm in diameter) to sulfur cathodes (sulfur content = 50 wt%), the cells revealed an increase in specific capacitance (402 mA h g−1 without the additive vs. 660 mA h g−1 with the additive at 0.06C). This improvement was attributed to a LiPS adsorption effect between sulfur-related species and the porous γ-Al2O3 nanoparticles.
Zhang et al. provided an interesting route to suppress the diffusion of LiPSs and enhance the performance of Li–S batteries by introducing Mg0.8Cu0.2O nanoparticles (ranging from 20 to 40 nm) into a crystalline V2O5/sulfur composite cathode.43 The composite cathode containing 10 wt% of additive and a sulfur content of ≈38 wt% showed an initial specific capacity of 545 mA h g−1 with a capacity retention of 77.5% after 30 cycles at a current density of 0.2 mA cm−2, while the cathode without the additive delivered only 227 mA h g−1 after 30 cycles. The authors claimed that the Mg0.8Cu0.2O nanoparticles not only have a positive LiPS adsorption effect but also present a catalytic effect to promote the LiPS redox reaction. However, the role of the crystalline V2O5 used as the sulfur host was not discussed in this study. Although the cyclability was relatively stable, the low sulfur utilization (≈32%) still needs to be improved for industrial applications.
Nazar and co-worker also studied the surface adsorption and pore absorption of LiPSs by using high-surface area mesoporous SiO2 and TiO2 as sorption reagents.44,45 For instance, Ji et al. fabricated a cathode electrode comprised of elemental sulfur (60 wt%), mesoporous carbon (25 wt%), mesoporous silica (SBA-15; 10 wt%) and polyvinylidene difluoride (PVDF) binder (5 wt%) with a sulfur loading of 1.2 mg cm−2.44 The Li–S cell containing SBA-15 demonstrates higher specific capacity and better capacity retention than the cell without the additive. The improved performance of the Li–S system was attributed to the resulting hydrophilic pores of mesoporous silica with Si–O groups which serve as week binding sites to reversibly adsorb/absorb hydrophilic LiPS intermediates. The retained LiPSs are released near the end of discharge to further reduce them in the pores of the conductive mesoporous carbon network. In this way, the LiPSs remain immobilized in the positive electrode during almost all the discharge process, limiting the LiPS migration to the anode side and keeping the active material available for further utilization.
Subsequently, Evers et al. carried out further research studies to optimize the cathode composition by using three different morphologies of mesoporous TiO2 (anatase, brookite and rutile phases) as additives.45 While the LiPS sorption/release mechanism of mesoporous TiO2 works in a similar manner to mesoporous SiO2, the higher electropositivity of titania is more effective in adsorbing LiPSs than silica. As a consequence, an improved capacity retention was found for the Li–S cells with α-TiO2 (rutile) as the additive (specific surface area = 275 m2 g−1; pore size = 5.2 nm) compared to the cells containing SBA-15 (specific surface area = 918 m2 g−1; pore size = 5.6 nm) at a low amount of additive (3.6 wt%).
Bearing in mind the properties of porous silica to adsorb/absorb soluble LiPS species, Lapornik et al. prepared functionalized zeolite silicalite-1 as a two-in-one additive by integrating Mn2O3 nanoparticles into a microporous silicate crystal framework (denoted as MnS-1).46 The cathodes with the functionalized MnS-1 (sulfur content = 50 wt%; sulfur loading = 2 mg cm−2; additive content = 9 wt%) exhibited higher average discharge capacity and lower polarization in comparison to a cathode containing the mesoporous silica SBA-15 additive as a control system. Despite the significant differences in the physical properties (specific surface area, pore size and pore volume) between MnS-1 and SBA-15 additives, the improvement in electrochemical properties was ascribed to the influence of Mn2O3 nanoparticles in the MnS-1. However, more studies are required to determine the main role if any of the Mn2O3 in the silicate composite.
Recently, Ponraj et al. demonstrated that hydrophilic MgO nanoparticles (≈50 nm in diameter) intrinsically functionalized with surface hydroxyl groups can serve as effective additives to capture soluble LiPSs and retain them within the cathode.47 In comparison to Mn and Ti transition metals, Mg as an alkaline earth metal possesses higher electropositivity, which would aid the chemical binding to LiPS species. As a result of the strong chemical interaction between LiPS intermediates and MgO nanoparticles, sulfur cathodes prepared by simple mixing of elemental sulfur, MgO additive, Super P carbon and PVDF binder (sulfur content = 54–60 wt%; sulfur loading = 1.8–2.0 mg cm−2; additive content = 10 wt%) showed superior cycling stability, improved discharge capacity and better rate capability compared to cathodes without the additive.
If we consider that LiPS intermediates are heteropolar, an effective LiPS-catching additive should be a compound with polar surface properties. According to innovative work carried out by Xie et al., the utilization of ferroelectric BaTiO3 nanoparticles with “spontaneous polarization” could solve the shuttle effect by trapping LiPS species owing to the induced charges on the surface of BaTiO3 nanoparticles.48 In fact, the hollow carbon nanospheres/sulfur cathode with BaTiO3 nanoparticles (sulfur content = 42 wt%; sulfur loading = 2.4 mg cm−2) showed a notable improvement in the delivered capacity compared with its counterpart cathode without BaTiO3 (835 mA h g−1vs. 407 mA h g−1 after 100 cycles, respectively). However, the cells with the BaTiO3 additive also present a clear capacity fading during the initial cycles at a low current rate, usually observed in systems with polysulfide leakage.
Although the incorporation of metal oxide additives could be presented as a simple and straightforward method to improve both the specific capacity and lifespan of Li–S batteries, the noticeable and irreversible capacity decay reported in the aforementioned systems also indicates that the LiPS dissolution into the electrolyte still occurs, giving the possibility to LiPSs to diffuse out of the sulfur cathode and migrate to the lithium anode. Therefore, an alternative and more effective methodology to fully restrict the active sulfur material in the positive electrode is needed.
2.2 Metal oxides as sulfur host cathodes
The early research on the use of metal oxides as additives gave the kick start to highlight the notable properties of these metal compounds to retain LiPSs at the cathode by chemical binding and hence improve the stability and performance of the positive electrode. Metal oxides with a certain structure and porosity can provide a dual function by serving as a sole sulfur host to accommodate the active material into their cavities/pores and also facilitating the chemisorption of formed soluble LiPS intermediates. In this regard, Cui and co-workers pioneered the utilization of TiO2 as a unique support to encapsulate sulfur for positive electrodes.49 The cathode composite in question consists of a sulfur–TiO2 yolk–shell structure with internal void space which possess the advantage of both enclosing the active material into the inner cavity and affording adequate space for alleviating the large volume changes of sulfur through cycling. To prepare the yolk–shell architecture, sulfur particles (800 nm) resulting from the reaction between Na2S2O3 and HCl were coated with a thin layer of TiO2 (15 nm) via alkaline hydrolysis of a titanium diisopropoxide bis(acetylacetonate) precursor, followed by a moderate sulfur dissolution with toluene to finally form the internal void space. Compared to a sulfur–TiO2 core–shell (with no free internal space) and uncoated sulfur composite structures, the cathode with the sulfur–TiO2 yolk–shell design (sulfur content ≈ 53 wt%; sulfur loading ≈ 0.5 mg cm−2) showed a high capacity retention with a capacity decay of 0.033% per cycle after 1000 cycles. The long lifespan was principally attributed to the intact integrity of the TiO2 shell, serving as an effective reservoir to retain sulfur compounds. TiO2-based host materials with different structures have also been explored in order to promote sulfur utilization, prevent cathode degradation and enhance the kinetics of the Li/S redox reaction.50–52 For example, Xie et al. embedded molten sulfur into/onto TiO2 nanotubes to finally produce a TiO2/sulfur composite cathode (sulfur content ≈ 45 wt%; sulfur loading ≈ 1.1 mg cm−2), enabling a stable reversible capacity of 851 mA h g−1 after 100 cycles at a C-rate of 0.2 and a resultant capacity degradation of 0.068% per cycle.53 To improve the ability of TiO2 to chemically immobilize sulfur-based species, Yang et al. prepared hydrogen reduced TiO2 microspheres as a promising host material.51 The functionalized TiO2 microspheres with an increased polar surface area due to oxygen vacancies created during a mild hydrogenation process serve as surface-bound intermediates to strongly bind LiPSs. The resulting cathode (sulfur content ≈ 40 wt%; sulfur loading = 0.8–1.3 mg cm−2) showed a capacity of 928 mA h g−1 after 50 cycles at a current density of 200 mA g−1, corresponding to a capacity degradation of 1.99% per cycle. Although the cycling performance was relatively stable, the lifespan of 50 cycles needs to be improved.TiO2 has proven to restrict the active material loss due to the adsorption effect of LiPSs. However, the semiconducting nature of TiO2 also lessens the conductivity of the cathode. To circumvent this hurdle, Nazar and co-workers54 as well as Cui and co-workers55 suggested almost at the same time to use the highly conducting Magnéli phase Ti4O7 as a sulfur host material. The structure of metallic conductive Magnéli Ti4O7 (≈2 × 103 S cm−1)56 is comprised of two-dimensional shear planes of Ti–O octahedral with polar O–Ti–O units, which can function as LiPS anchor sites (Fig. 3a). Nazar and co-workers prepared Magnéli Ti4O7 by heating a titanium ethoxide–polyethylene glycol mixture at 950 °C under an argon atmosphere.54 X-ray diffraction investigation and elemental microanalysis revealed that the obtained sample is composed of Ti4O7 as the primary crystalline phase together with 15.4 wt% of residual amorphous carbon. The Ti4O7 sample also has a relatively high conductivity of ≈3.2 S cm−1 and a high specific surface area of 290 m2 g−1, which are essential for electron/Li+-ion transport and interfacial interaction with LiPSs, respectively. After melt-infiltration of sulfur, the Ti4O7–sulfur composite cathode (sulfur content = 48 wt%; sulfur loading ≈ 0.825 mg cm−2) provided an initial specific capacity of 1070 mA h g−1 with a reasonable capacity degradation of 0.08% per cycle after 250 cycles at 0.5C. This fade rate is half of the capacity degradation obtained for a cell with a Vulcan XC72 carbon–sulfur composite cathode used as a reference. Further X-ray photoelectron spectroscopy (XPS) and in situ X-ray absorption near-edge spectroscopy (XANES) studies determined that Ti4O7 has a strong effect on decreasing the LiPS concentration in solution and also controls the gradual deposition of Li2S onto Ti4O7 particles via surface-mediated reduction at the interface (Fig. 3b and c). This phenomenon electrocatalytically enhances the redox reaction of LiPSs and, thus, improves the overall electrochemical performance of the cells. On the other hand, Cui and co-workers synthesized semiconducting Ti6O11 nanowires and metallic Ti4O7 nanoparticles as oxygen-deficient TinO2n−1 Magnéli phases by heating rutile TiO2 at, respectively, 950 and 1050 °C under a pure reducing hydrogen atmosphere.55 In order to study the electronic conductivity effect of the Ti-based scaffolds on the cell performance, TiO2–, Ti6O11–, and Ti4O7–sulfur composites were prepared by sulfur impregnation of the host samples and further heating at 155 °C in a vacuum oven. As a consequence of the highest conductivity of Ti4O7 (relative conductivity order: Ti4O7 > Ti6O11 > TiO2), the Li–S cells with Ti4O7–sulfur composite cathodes (sulfur content ≈ 51 wt%; sulfur loading = 1–3 mg cm−2) showed the best cycling performance with an initial capacity of 1044 mA h g−1 and an outstanding capacity retention of 99% over 100 cycles at 0.1C, which correspond to one of the lowest capacity degradation values (0.01% per cycle) reported so far.57–59 Further density functional theory (DFT) calculations combined with XPS studies determined that the low-coordinated Ti sites of Ti4O7 highly favor the adsorption of sulfur-based intermediates and selective Li2S deposition (Fig. 3d). Therefore, Li–S cells with superior performance can be achieved by combining the unique polar surface and the inherent electronic conductivity of Ti4O7 for, respectively, strong LiPS binding and kinetically enhanced redox electron transfer.54,55
 | ||
| Fig. 3 Magnéli titanium oxide as a sulfur host for Li–S batteries. (a) A schematic illustration of the electron density transfer between TiOx and Li2S4 (green = Li, yellow = S, blue = Ti, and red = O).54 (b) High-resolution S 2p XP spectra of Li2S4 (top), Li2S4/Ti4O7 (middle), and Li2S4/VC carbon (bottom). Black dotted line = experimental data, red line = fitted data, and solid/dotted lines in other colors = fitted individual components.54 (c) Operando XANES results showing the distribution of sulfur species upon discharge for Li–S cells with Ti4O7/S-60 (solid lines + symbols) and VC carbon/S-60 cathodes (dashed lines). Ti4O7/S-60 presents a lower concentration of LiPS compared with VC carbon/S-60. Black = Li2S; blue = LiPS showed as the sum of Li2S6 and Li2S4; red = elemental sulfur.54 (d) DFT optimized structures and adsorption energies of sulfur species on Ti4O7 (1–20) and TiO2 (110) surfaces. Gray = Ti; pink = O; yellow = S; purple = Li.55 (a–c) Reproduced with permission from ref. 54. Copyright 2014, Nature Publishing Group. (d) Reproduced with permission from ref. 55. Copyright 2014, American Chemical Society. | ||
More recently, Wei et al. proposed a cathode scaffold for Li–S batteries based on mesoporous Magnéli Ti4O7 microspheres.60 The relatively high surface area (197 m2 g−1) and the interconnected mesopores (20.4 nm) of the Magnéli Ti4O7 microspheres are able to accommodate up to 70 wt% of sulfur into their inorganic matrix. The ensuing Ti4O7 microspheres/sulfur cathodes (sulfur content = 56 wt%; sulfur loading ≈ 0.5 mg cm−2) showed a high discharge capacity of 1318 mA h g−1 at a C-rate of 0.1 and a stable cyclability comprising a capacity degradation of 0.03% per cycle over 400 cycles at a rate of 0.2C.
Motivated by the interesting properties of metal oxides and aiming for a more effective material to catalyze the LiPS redox reaction, Nazar and co-workers were the first group to develop ultra-thin δ-MnO2 nanosheets as a host material to confine LiPS intermediates at the cathode side by specific chemical interactions.61 Based on XPS studies the authors established that, at the beginning of the discharge process, MnO2 nanosheets have the ability to oxidize the initially reduced higher-order LiPSs to thiosulfate groups at the surface of the host material. As the reduction process continues, the newly formed and soluble long-chain LiPSs are moored to the surface thiosulfate groups (S2O32−) which serve as transfer mediators to form a slightly soluble, intermediate polythionate complex (I) and insoluble short-chain LiPSs (i.e., Li2S2 or Li2S) via an internal disproportionation reaction (eqn (1)). It is worth mentioning that a polythiosulfate complex (II) could also be generated through a similar reaction (eqn (2)).62,63
 | (1) |
 | (2) |
The authors suggested that the formation of the surface-bound polythionate complex lessens the active material loss during cycling by the early induced disproportionation conversion of higher-order LiPS intermediates to insoluble lower-order LiPS species. A visual confirmation of LiPS entrapment obtained at different depths of discharge further evidenced the strong affinity of MnO2 to sulfur-based species (Fig. 4d). At the end of discharge (after 12 h), the electrolyte solution of the optically accessible cell with a MnO2–sulfur cathode presents a pale yellow color, while the solution of the cell in the absence of MnO2 turned bright greenish yellow due to solubilized LiPSs in the electrolyte (Fig. 4c). As a result, MnO2-containing cathodes (sulfur content ≈ 56 wt%; sulfur loading = 0.7–1.0 mg cm−2) demonstrated a high electrochemical performance with a low capacity decay rate of 0.032% per cycle over 2000 cycles at 2C (Fig. 4e).
 | ||
| Fig. 4 Visual confirmation of LiPS trapping at different depths of discharge for (a) sulfur–Ketjen black and (b) sulfur–MnO2 cells.61 (c) Long-term cycling performance of the sulfur–MnO2 nanosheet composite cathode.61 (a–c) Reproduced with permission from ref. 61. Copyright 2015, Nature Publishing Group. | ||
Analogous to a sulfur–TiO2 yolk–shell structure,49 Liang et al. synthesized sulfur–MnO2 yolk–shell composite cathodes by a mild redox reaction between sulfur and KMnO4 in an aqueous solution at room temperature, followed by a partial dissolution of the sulfur core with toluene.64 The resultant high-performance cathodes with spherical-like sulfur particles (around 300–400 nm) and improved sulfur loading (sulfur content ≈ 64 wt%; sulfur loading ≈ 1.6 mg cm−2) demonstrated that it is possible to reach a high initial capacity of 1380 mA h g−1 at a low rate of 0.05C (82% of the theoretical capacity) and a reversible capacity of 315 mA h g−1 after 1700 cycles at 2C, being equivalent to a low capacity decay of 0.039% per cycle. This notable cell performance was ascribed to the distinctive features of the MnO2 shell to intrinsically adsorb LiPS species and chemically bind them by in situ formation of thiosulfate/polythionate groups as well as to the physical confinement provided by the yolk–shell nanoarchitecture.49,61 Since the KMnO4 precursor used for producing MnO2 is less expensive than typical Ti-based precursors used for TiO2/Ti4O7, the proposed MnO2–sulfur composite cathode could be viable for large-scale production and practical application in Li–S batteries.
Wang et al. investigated the interaction of MnO2 with octahedral sulfur and various Li2Sn intermediates (with n = 1, 2, 4, 6 and 8) by using theoretical calculations.65 The authors found that even the fresh cathode forms relatively weak S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O chemical bonds between terminal S atoms from the opened S8 ring and O atoms on the MnO2 surface, while linear LiPS intermediates, formed upon lithiation, present stronger chemical bonds as a consequence of additional Li–O chemical bonds. Interestingly, due to the poor stability of Li2S, the subsequent decomposition into S and Li atoms with SO and Li–O bonds was predicted after full lithiation of sulfur. However, this phenomenon has not been experimentally detected.
O chemical bonds between terminal S atoms from the opened S8 ring and O atoms on the MnO2 surface, while linear LiPS intermediates, formed upon lithiation, present stronger chemical bonds as a consequence of additional Li–O chemical bonds. Interestingly, due to the poor stability of Li2S, the subsequent decomposition into S and Li atoms with SO and Li–O bonds was predicted after full lithiation of sulfur. However, this phenomenon has not been experimentally detected.
To shed light on the fundamental surface mechanism involved between metal oxides and sulfur species and further understand its correlation with the Li–S cell stability, Liang et al. conducted a series of electrochemical studies using high surface area transition metal oxides—Fe2O3, Co3O4, V2O3, NiO, Cu2O, CuO, CoO, VO2, MnO2, V2O5 and NiOOH—to adsorb and/or activate (poly)sulfide intermediates via thiosulfate formation.66 By combining cyclic voltammetry and surface spectroscopy studies, it was possible to elucidate that metal oxides with redox potentials between 2.4 V < E < 3.2 V vs. Li/Li+ oxidize LiPSs to active thiosulfate (such as CuO, VO2 and MnO2) and those oxides with potentials higher than 3.2 V vs. Li/Li+ (e.g. V2O5 and NiOOH) additionally over-oxidize LiPSs to inactive sulfate, while metal oxides with redox potentials lower than 2.1 V vs. Li/Li+ (Fe2O3, Co3O4, V2O3, NiO, Ti4O7, Cu2O, CoO and TiO2) only bind LiPSs by polar interactions rather than by oxidation of LiPS intermediates (Fig. 5a). To provide a proof-of-concept, three metal oxide–graphene-based sulfur cathodes (sulfur content = 60 wt%; sulfur loading around 1.2–1.5 mg cm−2) containing Co3O4, VO2 and V2O5 with different redox potentials (1.11, 2.79 and 3.40 V vs. Li/Li+, respectively) were electrochemically compared under long-term cycling tests (Fig. 5b). After 280 cycles at a C-rate of 0.5, the cell with a sulfur/VO2–graphene cathode displays the best cycling performance compared to sulfur/V2O5–graphene and sulfur/Co3O4–graphene cathodes. Unlike VO2, V2O5 not only oxidizes LiPSs to thiosulfate/polythionate but also forms electrochemically inactive sulfate species which obstruct the access to the host surface and thereby lessen the reversible oxidation/reduction of active sulfur intermediates. In contrast, the sulfur/Co3O4–graphene exhibits the lowest capacity retention due to the lack of thiosulfate/polythionate formation and actually the cell failed after 250 cycles. Furthermore, the authors demonstrated that the side sulfate formation could be avoided by restricting the charge potential to 2.5 V instead of the initially used 3.0 V. Further theoretical studies performed by Zhang et al. revealed that the resulting strong chemical bonds between V2O5 and Li2S4 can induce the destruction/decomposition of the Li2S4 compound, lessening the capacity retention of the Li–S cell.67 This theoretical observation correlates well with the above experimental results described for V2O5.66
 | ||
| Fig. 5 (a) Chemical reactivity of different metal oxides with LiPSs displayed as a function of the redox potential vs. Li/Li+.66 (b) Comparison of the cycling performance at 0.5C for S/V2O5/graphene (red), S/VO2/graphene (blue), and S/Co3O4/graphene (black) cathodes.66 (a and b) Reproduced with permission from ref. 66. Copyright 2015, Wiley-VCH. | ||
Metal oxides, such as TiO2, Ti4O7, VO2 and MnO2,68–71 were proved to be an efficient intermediary to limit the dissolution of LiPSs through chemical interactions due to their polar properties. However, there are other oxides that have been considered as sulfur host materials with the aim to improve the stability of Li–S batteries, such as SiO2,72,73 Mg0.6Ni0.4O,74,75 CoO,76 Co3O4,77,78 NiCo2O4,79 and MoO2.80 As an example, Qu et al. proposed conductive, mesoporous MoO2 as a sulfur-hosting oxide to enhance the performance of Li–S cells.80 Combining the high conductivity and the physical properties of MoO2 (relative conductivity ≈ 190 S cm−1;81 surface area = 70 m2 g−1; pore size ≈ 12 nm) together with the ability of the oxide to anchor LiPSs via strong S–O binding interactions, the sulfur-infiltrated mesoporous MoO2 cathode (sulfur content = 30.4 wt%; sulfur loading ≈ 1 mg cm−2) exhibited a reversible capacity of 570 mA h g−1 after 250 cycles at a C-rate of 0.1C, which corresponds to a capacity decay rate of 0.19% per cycle. While conductive MoO2 could be a promising oxide to limit the shuttle effect and activate sulfur species, the upsurges of both sulfur content and sulfur loading are highly required for practical cells.
While metal oxide-based host cathodes are very promising to confine LiPSs species and avoid their leak to the anode side, these materials still present some concerns in terms of their inherent low electrical conductivity and high relative density.
2.3 Metal oxide/porous carbon hybrid scaffolds
Metal oxide-derived host materials capable of binding LiPSs through chemical interactions are, indeed, very promising candidates for enhancing the stability and electrochemical properties of Li–S batteries. However, their general insulating nature and high relative density drastically decrease the capacity retention and energy density of the cells, respectively, when they are used as sole sulfur host materials. It is worth mentioning that in spite of using metal oxide materials as unique supports to store sulfur, most of the studies described in Subsection 2.2 also utilized some conductive additives (e.g. carbon black). A more attractive approach to effectively encapsulate sulfur without compromising the conductivity of the cathode matrix could be the integration of metal oxides into conductive and (porous) carbonaceous materials. In the last few years, several studies have shown improvements of the cathode performance by modifying all types of conductive carbon substrates (i.e. carbon black, CNTs, CNFs, graphene, rGO, porous carbons, heteroatom-doped carbons, etc.)24,82 with diverse metal oxides, such as La2O3,83,84 SiO2,85–87 indium tin oxide (ITO),88 TiO2,89–98 TiO,99,100 Ti4O7,101 MnO2,102–112 Mn3O4,59,113 MgO,84,114 VO2,115 V2O3,116 Co3O4,117 CeO2,84,118,119 ZrO2,120–122 Nb2O5,123 SnO2,124 ZnO,125,126 α-Fe2O3,127 Fe3O4,128 NiO–NiCo2O4,129 NiFe2O4,58 MoO2,130 MoO3,131 Mo4O11,132 Al2O3,84 CaO,84 Y2O3,133 and Nd2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 134 and complex perovskites like Ba0.5Sr0.5Co0.8Fe0.2O3−δ.135Table 1 summarizes the most significant studies on metal oxide–conductive carbon composites used as sulfur host materials for Li–S batteries in terms of sulfur cathode parameters (sulfur content and areal sulfur loading) and electrochemical performance.
134 and complex perovskites like Ba0.5Sr0.5Co0.8Fe0.2O3−δ.135Table 1 summarizes the most significant studies on metal oxide–conductive carbon composites used as sulfur host materials for Li–S batteries in terms of sulfur cathode parameters (sulfur content and areal sulfur loading) and electrochemical performance.
| Metal oxide–conductive carbon | Initial capacity [mA h g−1] | Reversible capacity [mA h g−1] | Current ratea | Cycle number | Degradation rate per cycle [%] | Sulfur contentb [wt%] | Sulfur loading [mg cm−2] | Ref. |
|---|---|---|---|---|---|---|---|---|
| a 1C = 1674 mA g−1. b Mass percentage of sulfur on the whole cathode excluding the Al or Ni substrate. c Capacity degradation rate is estimated from the figure since authors did not provide the specific value in the reference. d GO = graphene oxide. e CNTs = carbon nanotubes. f LiNO3-free electrolyte was used for the tested battery. g RFC = resorcinol-formaldehyde carbon. | ||||||||
| La2O3/N-doped meso-carbon | 1241 | ≈880c | 0.2C | 100 | ≈0.291c | 48 | N/A | 83 |
| La2O3-Kapok tree fibers | 1013c | 870c | 0.5C | 300 | 0.047 | 63–70 | 0.7–1.2 | 84 |
| SiO2-mildly reduced GOd | ≈1425c | 763 | 0.1C | 50 | ≈0.929c | N/A | N/A | 72 |
| ITO-carbon nanofiber mat | 1136 | 1000 | 0.2C | 300 | 0.040 | 40 | 2.0 | 88 |
| ITO-carbon nanofiber mat | 866 | 710 | 0.2C | 500 | 0.036 | 57 | 4.0 | 88 |
| TiO2 nanowire-graphene | N/A | 1053 | 0.2C | 200 | N/A | 62 | 3.2 | 89 |
| Hollow carbon nanofiber@TiO2 | 1040 | 650 | 0.5C | 200 | 0.187 | 54 | 1.6 | 90 |
| Hollow carbon nanofiber@TiO2 | 970 | 380 | 1C | 500 | 0.122 | 54 | 1.6 | 90 |
| TiO2/graphene | 871 | 732 | 1C | 400 | 0.040 | 44 | 1.0 | 91 |
| TiO2/N-doped graphene | 1069 | 918 | 1C | 500 | 0.028 | 59 | 1.3–1.8 | 92 |
| TiO@hollow carbon spheres | 1066 | 630 | 0.5C | 500 | 0.082 | 56 | 1.5 | 99 |
| MnO2@hollow carbon fibers | 1147 | ≈840c | 0.2C | 100 | ≈0.268c | 50 | 3.5–3.9 | 102 |
| MnO2–GO–CNTse | 1150 | 964 | 0.2C | 100 | 0.162 | 64 | 2.8 | 104 |
| Mn3O4–carbon cloth | 593 | 355 | 2C | 3000 | 0.013 | ≈62 | 2.8 | 59 |
| MgO-Kapok tree fibers | ≈1035c | ≈930c | 0.5C | 300 | 0.034 | 63–70 | 0.7–1.2 | 84 |
| V2O3–carbon microspheres | 1177 | 921 | 0.5C | 100 | 0.217 | ≈45 | 1.5–1.6 | 116 |
| CeO2/Ketjen black carbon | 905 | 710 | 1C | 300 | 0.072 | 60 | N/A | 118 |
| Nb2O5-meso-carbon | 1289 | 913 | 0.5C | 200 | 0.146 | 48 | 1.5 | 123 |
| Mo4O11–graphenef | ≈1190c | ≈880c | 0.1C | 80 | ≈0.323c | 49 | 0.5 | 132 |
| α-Fe2O3/graphene | ≈670c | ≈370c | 2C | 500 | 0.090 | 48 | 0.6 | 127 |
| Yolk–shell carbon@Fe3O4 | 1104 | 855 | 0.1C | 200 | 0.113 | 64 | 5.5 | 128 |
| ZrO2-holey CNTs | 1138 | 878 | 0.5C | 200 | 0.114 | 36 | N/A | 121 |
| NiFe2O4–CNTs | 890 | 850 | 1C | 500 | 0.009 | 54.7 | 1.0–1.2 | 58 |
| Nd2O3–RFCg | 1168 | 907 | 0.5C | 300 | 0.074 | 44.6 | 2.2–3.0 | 134 |
| Ba0.5Sr0.5Co0.8Fe0.2O3−δ/CNT | 793 | 632 | 0.5C | 400 | 0.062 | 70 | 2.6–5.3 | 135 |
In general, the use of an insulating material (i.e., metal oxides) should increase the resistance of the electrode due to a deficiency in electron transport. Actually, if the metal oxide has the ability to strongly trap insulating LiPS species, it is expected to encounter an accumulation in electronically inactive zones which should reduce the utilization of the active material and also the capacity retention. However, despite the insulating nature of most metal oxides, several studies have reported significant improvements in the electrochemical performance of ternary metal oxide/carbon/sulfur electrodes compared with conventional sulfur/carbon composite electrodes. Therefore, the initially adsorbed LiPSs should be later transferred from the oxide surface to the conductive substrate to finally undergo the electrochemical reaction. Intrigued by this observation, Cui and co-workers studied the competitive processes of adsorption of LiPS species on oxides and diffusion of LiPSs from the oxide surface to the conductive carbon matrix.84 To fabricate the oxide/porous carbon flake nanostructures, Kapok tree fibers (KFs) were used as both the bio-template and carbon source (Fig. 6a). While various nonconductive oxides were used in this study, the MgO- and La2O3-containing carbon material/sulfur composite electrodes showed the best electrochemical performance with high capacities and good capacity retention over 300 cycles (Fig. 6b). As an oxide selection criterion for the design of LiPSs/oxide interfaces for advanced Li–S batteries, the authors proposed polar sulfur hosts with strong binding to LiPS intermediates, a high surface area and, preferably, good surface diffusion properties. An interesting approach in terms of high performance and long cycling stability at high sulfur loading (>3 mg cm−2) was reported by Yao et al. They used conductive tin-doped indium oxide—also well known as ITO—nanoparticles to decorate a carbon nanofiber (CNF) host material (Fig. 6c).88 For the cathode preparation, a dissolved Li2S8 polysulfide solution commonly termed as the catholyte was used as the starting material instead of conventional solid sulfur or Li2S components.136–138 Preliminary surface analysis using energy dispersive X-ray (EDX) spectroscopy and scanning electron microscopy (SEM) showed that Li2S and intermediate LiPSs deposited preferentially on ITO instead of carbon substrates during, respectively, discharge and charge processes, indicating stronger affinity of LiPSs to polar oxygen-rich ITO than to nonpolar carbon. As a consequence of the controlled nucleation and deposition of solid sulfur/Li2S species, ITO-CNF/Li2S8 catholyte hybrid electrodes (sulfur content = 40 wt%; sulfur loading = 2.0 mg cm−2) revealed an enhanced electrochemical performance with a low capacity decay rate of 0.040% per cycle over 300 cycles at 0.2C. It was also shown that when combining solid sulfur and the Li2S8 catholyte, the hybrid cathode with a high sulfur loading (4.0 mg cm−2) can deliver a reversible capacity of 710 mA h g−1 after 500 cycles (Fig. 6d), representing a low capacity decay rate of 0.036% per cycle. Another representative example was reported by Li et al., who proposed the preparation of a rationally designed hybrid host composite by filling highly conductive hollow CNFs with polar MnO2 nanosheets (MnO2@HCNFs).102 For such a purpose, SiO2-coated MnO2 nanowires and resorcinol-formaldehyde resins were used as the hard template and carbon source, respectively. After pyrolysis of the composite and subsequent NaOH-etching of the SiO2 coating, sulfur was infiltrated into the hollow MnO2@HCNF host via the melt-diffusion route, while the outer conductive and porous carbon layer aids in driving electron and Li+ ion transport during charge/discharge cycling. The polar cavity filled with MnO2 nanosheets serves as a specific polysulfide container capable of mitigating the polysulfide dissolution and also promoting the sulfur-based redox activity. The electrochemical evaluation of the sulfur-infiltrated MnO2@HCNF cathode (sulfur content ≈ 50 wt%; sulfur loading = 3.5–3.9 mg cm−2) revealed an initial discharge capacity of 1147 mA h g−1 and stable cycling performance for over 100 cycles at 0.2C. Furthermore, the extended cycling performance of sulfur–MnO2@HCF at 0.5C proved a good areal capacity retention of 2.3 mA h cm−2 after 300 cycles. The integrated structure of MnO2-filled HCNFs certainly improves the lifespan of the cells by chemical binding of sulfur-intermediates to the MnO2 nanosheets.
 | ||
| Fig. 6 (a) A schematic illustration of biotemplated fabrication of oxides/carbon nanostructures using the Kapok tree fibers as both template and carbon sources. (b) Cycling performance of various metal oxides/KF/S composite electrodes at 0.5C.84 (c) A schematic illustration of the preparation of a LiPS-ITO micropattern glassy carbon cathode showing the LiPS deposition. (d) Cycling performance at 0.5C of an ITO-CNF/Li2S8 catholyte hybrid electrode.88 (e) Long-term cycling performance of MnO2- and Mn3O4@carbon cloth/sulfur cathodes. (f–i) A scheme showing the structural changes of both MnO2 and Mn3O4 crystals upon interaction with LiPS.59 (a and b) Reproduced with permission from ref. 84. Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/). (c and d) Reproduced with permission from ref. 88. Copyright 2014, Nature Publishing Group. (e–i) Reproduced with permission from ref. 59. Copyright 2017, The Royal Society of Chemistry. | ||
Nanocrystalline NiFe2O4 is a soft magnetic material with an inverse spinel structure.139 This kind of ferrite material has been explored as an anode material for LIBs owing to its electrochemical ability to react with 8 moles of Li, delivering a high theoretical capacity of 915 mA h g−1.140 In 2015, Fan et al. used a hybrid CNT/NiFe2O4/sulfur cathode material for the first time.58 The one-dimensional CNTs and two-dimensional NiFe2O4 nanosheet components confer, respectively, electron conductivity and LiPS anchor sites to the designed three-dimensional (3D) host material. The latter sulfur nanoparticles (5–20 nm) attached onto the CNT/NiFe2O4 surface serve as the active energy storage component. The resulting 3D hybrid CNT/NiFe2O4/sulfur composite cathode (sulfur content ≈ 54 wt%; sulfur loading ≈ 1.1 mg cm−1) delivered a high initial capacity of 1350 mA h g−1 at 0.1C and a capacity of ≈850 mA h g−1 over 500 cycles at 1C with only 0.009% capacity loss per cycle, one of the best values reported so far.55,57,59 Although the capacity retention was outstanding, the low sulfur loading in the hybrid cathode needs to be increased to meet the standard for practical applications. It is noted here, that, despite the promising benefits showed by the NiFe2O4 nanosheets, no further studies on NiFe2O4-containing sulfur cathodes have been reported up to now.
Recently, Li et al. suggested an interesting ternary-type MnO2/graphene oxide/carbon nanotube (MnO2/GO/CNT) scaffold with a three-dimensional architecture and synergistic functions.104 The proposed sulfur cathode complex consists of (i) innermost one-dimensional CNTs serving as the conductive backbone for the composite, (ii) two-dimensional petal-like MnO2/GO nanosheets attached on the sidewalls of the CNT-based backbone having dual-efficient polysulfide-adsorption capability,61,141 and (iii) outmost nanosized sulfur-active components fixed onto the MnO2/GO surface. The hybrid sulfur cathode (sulfur content = 64 wt%; sulfur loading ≈ 2.8 mg cm−2) demonstrated discharge specific capacities of 1500, 1300, 1150 and 1048 mA h g−1 at, respectively, 0.05, 0.1, 0.2 and 0.5C, a reasonable capacity decay of 0.162% per cycle after 100 cycles, and high coulombic efficiency (≈99%). The authors attributed the enhanced performance of the Li–S cells to the features and synergistic effects of the components in the ternary composite, such as the relatively high specific surface area (≈156 m2 g−1) able to tolerate the volume changes caused by discharged products, the conductive CNT-frame for long-range electron transport and the strong chemisorption of the MnO2 to LiPSs. More recently, Guo et al. proposed a Mn3O4 composite with nano-wall arrays as a sulfur-hosting material.59 The binder-free Mn3O4@carbon cloth/S cathode (sulfur content ≈ 62 wt%; sulfur loading = 2.8 mg cm−2) was prepared by the direct growth of Mn3O4 nanoparticles on a carbon cloth via an impregnation-hydrothermal decomposition route using KMnO4 as both Mn and O source and subsequent sulfur melt diffusion at 155 °C. High reversible specific capacities of ≈1000 and 950 mA h g−1 are achieved at rates of 0.1 and 0.5C, respectively. Notably, the battery showed a high coulombic efficiency (higher than 98%) and outstanding capacity retention (60%) over 3000 cycles at 2C with a decay as low as ≈0.013% per cycle, one of the longest cycle lives reported so far.61,142 In contrast, the control cell with a MnO2@carbon cloth/S cathode exhibited a capacity retention of 24% after 1500 cycles, under similar cell conditions (Fig. 6e). Such stable cell operation at relatively high sulfur loading was attributed to the good stability of the Mn3O4 structure upon cycling. As illustrated in Fig. 8 and Fig. 6f, Mn4+ cations in a MnO2 crystal are reduced to Mn2+ upon interaction with LiPS species. The resulting oxide with Mn2+ cations might be dissolved into the electrolyte during cell cycling (Fig. 6g), weakening the structure of MnO2 and thus losing the capability to retain the active material. On the other hand, the Mn3O4 structure (Fig. 6h) consists of edge sharing MnO6 octahedra (Mn2+) that are corner linked to MnO4 tetrahedra (Mn4+). Based on SEM and XPS analyses and considering minimal reorganization theory, the authors proposed a simultaneous MnO4 tetrahedral expansion and a MnO6 octahedral contraction by the respective reduction of Mn4+ and oxidation of Mn2+ to Mn3+ upon LiPS interaction rather than the formation of Mn2+ ions (Fig. 6i). Thus, the Mn3O4 structure is less prone to suffer from damage/disintegration.
2.4 Metal oxides in functional interlayers and separator coatings
If we consider the number of publications on the topic of Li–S batteries, most of the studies are dedicated to the engineering design of sulfur cathodes using diverse host matrices and the synthesis of novel electrolytes that prevent the diffusion of LiPSs—around 65% and 13% of the Li–S battery-based publications, respectively.143–145 Although previous studies have made great advances in understanding the chemistry involved in the Li/S couple and thus maximized Li–S cell's performance, the inexorable dissolution of high-order LiPSs in conventional ether-based electrolytes and their further diffusion/migration towards the lithium anode seem to be barely avoidable.In an effort to tackle the LiPS leakage, Manthiram's group proposed in 2012 the modification of the cell configuration by the insertion of a free-standing carbon interlayer between the separator and the sulfur cathode as a LiPS-trapping conductive membrane.146,147 The novelty of this “interlayer” concept resides in the multiple functionalities that are present at the conductive and porous membrane. Firstly, the porous interlayer works as a reservoir to intercept and retain the dissolved LiPS in the cathode side. Secondly, due to its high electrical conductivity, it serves as an upper-current collector to reduce the resistance of the cathode by boosting the electron/ion transport. Thirdly, its accessible porous structure offers a physical space to shock-absorb the huge volume changes of the trapped sulfur-based species during cell cycling, preventing interlayer and cathode degradation.36,148 In other words, the interlayer acts as a secondary sulfur (unfilled) cathode or as an extension of the primary sulfur cathode whose functions are triggered during cell operation by the early capture and storage of the migrating sulfur species and further reutilization of the sequestered active material. Inspired by this pioneering work, two years later the same group used a similar in situ LiPS-trapping concept by integrating a carbon interlayer in a commercial polypropylene separator.149–151 The designed functional carbon-coated separators not only incorporate the features shown by free-standing carbon interlayers but also the manufacturing coating process allows to decrease the thickness and, thus, the weight of the carbon layer, resulting in a cell with higher specific energy density. In comparison with the conventional Li–S cell configuration, the innovative Li–S cells containing an interlayer or a separator coating produced from conductive carbon nanostructures (i.e., CNTs, graphene oxide, rGO, etc.),152–156 porous (doped) carbons157–162 or conducting polymers163–167 have considerably improved sulfur utilization, capacity retention and cycle life. However, since bare carbon materials only provide for weak interaction with polar LiPS species, in the past few years, there has been increased interest to incorporate diverse metal oxide nanomaterials as one of the components of the functional separator coatings/interlayers in order to improve the LiPS affinity/utilization via chemisorption and/or electrocatalytic effects. The explored metal oxides include TiO2,168–174 SnO2,175,176 MnO2,142,177 MnO,178 BaTiO3,179 RuO2,180 CeO2,181 Mg0.6Ni0.4O,182 Li4Ti5O12,183 LiAlO2,184 V2O5,185,186 SiO2,187 La2O3,188 Y2O3189 and NiO.190 A summary of representative metal oxide-containing functional interlayers/hybrid separators developed recently is shown in Table 2. The values of this table should be taken with care as the capacity and reversibility strongly depend on the applied cell parameters such as the amount and type of electrolyte, electrode thickness, sulfur mass loading, sulfur composition, binder and separator. In order to provide a comparable picture we added some important parameters such as the mass loading of the interlayer/coating, sulfur ratio within the whole cathode (excluding the Al or Ni substrate), sulfur mass loading and C-rates.
| Metal oxides in interlayers | Initial capacity [mA h g−1] | Reversible capacity [mA h g−1] | Current ratea | Cycle number | Degradation rate per cycle [%] | Interlayer mass [mg cm−2] | Sulfur contentb [wt%] | Sulfur loading [mg cm−2] | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a 1C = 1674 mA g−1. b Mass percentage of sulfur on the whole cathode including the interlayer weight. Those values with asterisk represent the sulfur content without the interlayer weight since the authors did not provide such information in the paper. c Capacity/degradation rate was estimated from the figure since the authors did not provide the specific number in the paper. d The battery was tested in a pouch cell configuration. | |||||||||
| CNT@TiO2 | 1258 | 541 | 0.5C | 1000 | 0.055 | 0.7 | 37 | 1.7 | 173 |
| CNT@TiO2 | 969 | 783 | 0.2C | 100 | 0.192 | 0.7 | 39 | 3.0 | 173 |
| TiO2–CNFs | 1238 | 770 | 0.2C | 300 | 0.126 | 1.6 | 45 | 3.0 | 174 |
| SnO2@hollow carbon spheres | 996 | 832 | 0.5C | 100 | 0.165 | 0.2 | ≈48 | ≈2.0 | 175 |
| V2O5–CNFs | 816 | 576 | 3C | 1000 | 0.029 | 1.0 | ≈52 | ≈ 2.0 | 186 |
| NiO/rGO | 1500 | 700 | 0.1 A g−1 | 150 | 0.355 | 1.06 | ≈51 | 4.0 | 190 |
| NiO/rGO–Sn | 1690 | 868 | 0.1 A g−1 | 150 | 0.324 | 1.4 | ≈47 | 4.0 | 190 |
| Metal oxides in separator coatings | Initial capacity [mA h g−1] | Reversible capacity [mA h g−1] | Current ratea | Cycle number | Degradation rate per cycle [%] | Interlayer mass [mg cm−2] | Sulfur contentb [wt%] | Sulfur loading [mg cm−2] | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| TiO2/carbonized bacterial cellulose | 1000c | 752 | 0.5C | 100 | 0.248c | N/A | 70* | 2.5 | 171 |
| MnO2/GO/CNTs | 1025 | 293 | 1C | 2500 | 0.029 | 0.1 | 60–80 | 1.1–2.4 | 142 |
| Hollow CNF@MnO2 | 800c | 593 | 1C | 400 | 0.065 | N/A | 70* | 2.1 | 177 |
| MnO–Ketjen black carbon | 1059 | 901 | 1C | 200 | 0.075 | 0.125 | ≈57 | 1.5–2.0 | 178 |
| BaTiO3 | 1122 | 929 | 0.5C | 50 | 0.344 | 2.4 | 41 | 3.0 | 179 |
| RuO2–mesoporous carbon | 695 | 665 | 0.5C | 200 | 0.022 | 0.3 | ≈63 | ≈2.0 | 180 |
| CeO2–Ketjen black carbon | 1004 | 625 | 1C | 500 | 0.075 | 0.3 | ≈49 | ≈1.9 | 181 |
| V2O5d | 890c | 800c | C/15 | 250 | 0.040 | N/A | 60* | 3.0 | 185 |
| SiO2 nanoparticles | 937 | 603 | 0.2C | 200 | 0.178 | 0.124 | 48 | ≈1.3 | 187 |
| La2O3–Ketjen black carbon | 966 | 720 | 1C | 200 | 0.127 | 0.4 | 42 | 1.5 | 188 |
| Y2O3–Ketjen black carbon | 1054 | 816 | 1C | 200 | 0.113 | N/A | 60* | 1.3 | 189 |
V2O5 was one of the first polar metal oxides to be introduced into an interlayer for Li–S cells. Li et al. deposited electronically conductive V2O5 onto one side of commercial polypropylene battery separators (Celgard 3401 and 3501).185 The V2O5 interlayer acts as both a solid-state Li+ ion conductor and a polysulfide anion barrier. By blocking the LiPS diffusion to the Li anode, the cell composed of a nanoporous carbon foam–sulfur composite cathode (sulfur content ≈ 60 wt%; sulfur loading = 3.0 mg cm−2) attested a stable cyclability for over ≈1 year with an average capacity of 800 mA h g−1 representing an estimated degradation rate of 0.040% per cycle. Instead of a free-standing interlayer or a separator coating, Xiao et al. directly coated the surface of a porous CNTs/sulfur cathode with a porous graphene/TiO2 layer.168 The added interlayer corresponds to 7.8 wt% of the whole cathode. While the porous graphene provides an electrically conductive network able to physically trap soluble and escaping sulfur species, the TiO2 in the interlayer further promotes the chemical anchorage of LIPSs via S–Ti–O interactions.50,63 Using this advanced cathode with a coupled graphene/TiO2 interlayer (sulfur content = 51.2 wt%; sulfur loading = 0.51 mg cm−2), cells cycled over 1000 times exhibited ultralow capacity decay rates of 0.010 and 0.018% per cycle, at C-rates of 2 and 3C, respectively.55,57–59 Similar to Li et al.,104 Wang and co-workers also employed a ternary MnO2/GO/CNT nanostructured architecture. In this case the designed ternary system was layered onto a polypropylene separator (Celgard 2400), acting as a LiPS-trapping shield (Fig. 7a).142 The ultrathin functional interlayer denoted as G/M@CNT (thickness of 2 μm and areal density of 0.104 mg cm−2) facilitates electron transport through the high conductivity CNTs and enables the chemisorption of migrating LiPS intermediates by strong interactions between LiPSs with polar oxygen groups in the GO sheets and MnO2 nanoparticles. The improved Li–S cell with a functional interlayer@pristine separator (Table 2) demonstrated a notable cycling performance over 2500 cycles with a low capacity degradation of 0.029% per cycle at 1C, while the cell with a pristine separator only reached ≈700 cycles before cell failure (Fig. 7b).
 | ||
| Fig. 7 (a) Schematic configuration of the Li–S cells with a pristine separator (left) and a G/M@CNT-coated separator (middle). Photographs of the G/M@CNT-coated separator (right). (b) Long-term cycling performance of cells with pristine and G/M@CNT-coated separators.142 (c) A schematic illustration of the RuO2-MPC-HS structure (left) and the combined cycling performance of the Li–S cell with RuO2-MPC-HS (right).180 (d) A schematic diagram of the poled BaTiO3 (BTO) effect toward LiPS rejection. (e) LiPS diffusion test. PE-poled BTO separator showed a better rejection of Li2S8 solution (left bottle) compared with the PE separator.179 (a and b) Reproduced with permission from ref. 142. Copyright 2017, Wiley-VCH. (c) Reproduced with permission from ref. 180. Copyright 2016, The Royal Society of Chemistry. (d and e) Reproduced with permission from ref. 189. Copyright 2016, Wiley-VCH. | ||
Among typically used metal oxides (e.g. V2O5, TiO2, and MnO2), in the last few years, new metal oxides have been proposed to confine and re-use the sulfur active material. For instance, electrically conductive and catalytically active RuO2 nanoparticles (≈2 nm) were used to improve the LiPS redox reaction kinetics and hence the sulfur (re)utilization.180 As a proof of concept, a multifunctional RuO2 nanoparticle-decorated mesoporous carbon-coated hybrid separator (denoted as RuO2-MPC-HS) was used to boost the electrochemical performance of Li–S batteries (Fig. 7c). The hybrid separator not only provides an electron transport network but also serves as an effective LiPS-net to early trap and retain the active material in the positive electrode. As a consequence of the electrocatalytic effect resulting from the RuO2 nanoparticles, a simple-mixed sulfur/carbon black cathode (sulfur content ≈ 63 wt%; sulfur loading ≈ 2.0 mg cm−2) delivered a high initial capacity of 1276 mA h g−1 at 0.1C and remarkable cycling stability with a low degradation rate of 0.022% per cycle over 200 cycles at 0.5C (Fig. 7c). Dipole-aligned BaTiO3 particles, already used as an additive in Li–S cells,48 were utilized by Yim et al. to coat one side of a commercial poly(ethylene) separator with the aim to reject polar LiPS species during migration to the lithium anode (Fig. 7d).179 Li–S cells with a LiNO3-free electrolyte comprising a poled BaTiO3-coated separator, previously activated in an electric field, demonstrated a notable reduction of the over-charging behavior typically observed during charge processes, providing an initial coulombic efficiency of 79.6%, while cells with an non-poled BaTiO3-coated separator and a pristine separator exhibited coulombic efficiencies of 42.3 and 26.3%, respectively. Such behavior was also visualized by a LiPS rejection test (Fig. 7e). The enhanced coulombic efficiency in the absence of the LiNO3 additive is explained by the poling effect of the BaTiO3-coating which effectively repels negatively charged LiPSs by electrostatic repulsion. A cycling performance investigation carried out at 0.5C exposed an initial capacity of 1122 mA h g−1 for the cell with a poled BaTiO3-coated separator (cathode sulfur content = 41 wt%; sulfur loading = 3.9 mg cm−2). It is noted that the test was limited to only 50 cycles revealing an ending capacity of 929 mA h g−1. Additionally, the BaTiO3-coating also avoids thermal shrinkage of the polymeric separator at high temperatures, improving cell's safety. A Li–S cell with a flexible, freestanding ternary hollow NiO/rGO–Sn interlayer sandwiched between the separator and sulfur cathode was recently proposed by Li et al.190 In this multifunctional interlayer each component synergistically serves a specific purpose: (i) the rGO constructs a 3D highly conductive network, (ii) the hollow NiO tightly wrapped by rGO nanosheets provides a physical place to store soluble LiPSs and buffers volume changes and (iii) the Sn, in tandem with NiO, chemically interacts with LiPS intermediates to immobilize them in the interlayer, as concluded according to XPS analyses. The cell with the ternary interlayer showed a slight improvement in capacity compared with the control cell containing a NiO/rGO interlayer (Table 2). Note, however, that the added Sn increases the mass of the ternary interlayer by roughly 32%, which is detrimental to the whole sulfur content and hence cell's energy density.158,191
Undoubtedly, the reconfiguration of the Li–S battery by either integrating a functional interlayer or using a hybrid functional separator is a promising approach to hinder the migration of soluble LiPS intermediates, to indirectly protect the lithium anode from side reactions, to reactivate dead sulfur-based species, to decrease internal cell resistance and thus to enhance the overall electrochemical performance of the Li–S batteries. Nonetheless, special attention should be paid to the added weight of the interlayer/separator coating since this parameter could be counter-productive in terms of energy density.
In summary, the use of metal oxides improves the Li–S cell performance by constraining the LiPS shuttle phenomenon. Further screening of novel nanostructured metal oxides for advanced sulfur composite cathodes and, most importantly, the fundamental understanding of how LiPS species chemically interact with these oxide materials are critical to make a significant leap forward to high-performance Li–S batteries.
3. Metal sulfides
(Transition) metal sulfides (TMSs) are the most reported metal chalcogenides as co-components in Li–S batteries. Many of them are widely available and exhibit unique properties such as semi-metallic to metallic characteristics, magnetic moments and polar bonds within the molecular structure. Their adsorption capabilities for many gases are well known in heterogeneous catalysis, in particular for hydrodesulfurization.192 TMSs are also used in many other applications such as magnetism, fuel cells, electrochemical water splitting and battery electrode materials. Metal (di)sulfides (Fe, Co, Ni, Mo, Cr, W, Cu, and Mn)193,194 have been studied as both intercalation and conversion electrodes for positive and negative electrodes in secondary lithium batteries.195 There are some excellent reviews focusing on metal chalcogenides as electrode materials themselves.196,197 Herein, we will review the beneficial interaction of LiPSs with metal sulfides as co-components to improve the electrochemical performance of Li–S batteries.At the beginning of this decade, metal sulfides found their way as additive, coating or host materials for sulfur composite cathodes and for functional separators to improve the active material utilization and cycle life of Li–S batteries. They are supposed to enhance electronic and ionic conductivity within the electrode, improve charge transfer processes and most importantly exhibit the capability to capture sulfur species and thus prevent shuttling between the cathode and anode. It is believed that the adsorption of LiPSs and their redox-reaction on the conductive electrode surface can govern the overall reaction kinetics, in particular when the LiPS concentration is very high like in high-energy batteries and thus diffusion processes are very fast.198
If the electrode surface is non-polar as it is for conventional carbon, the adsorption of polar LiPS intermediates is energetically unfavorable and slow. In this regard, the addition of polar/ionic compounds by doping carbon (i.e. with nitrogen) was proven to enhance the electrochemical performance. The adsorption on a metal sulfide is thereby best described by Lewis base–acid interactions where the LiPSs provide a free electron pair binding to the metal cation (Lewis acid). After successful LiPS adsorption, charge transfer reactions and the reversal oxidation of Li2S to Li2Sn may be the rate determining step. The decomposition of Li2S during charging was proven to be successfully catalyzed by several metal sulfides199 and the potential for catalysis may be somehow related to the electronic conductivity of the metal sulfide. A demonstrative scheme for the catalytic sulfur reduction with CoS2 as the catalyst is shown in Fig. 8a and b in which the rate controlling step is highlighted as the charge transfer to the adsorbed LiPSs.198 Zhou et al. proposed a similar scheme in which the catalytic decomposition and oxidation of Li2S was found to be an important step to reach high efficiency and reversibility (Fig. 8c and d).199
 | ||
| Fig. 8 Scheme for the redox-reaction of sulfur to Li2Sn on the electrode surface (a) without and (b) with the CoS2 catalyst.198 A general scheme for the decomposition and oxidation of Li2S to form soluble Li2Sn (c) without and (d) with the catalyst, and (e) a visual adsorption capability of different metal sulfides to capture LiPSs.199 (a and b) Reproduced with permission from ref. 198. Copyright 2016, American Chemical Society. (c and d) Reproduced with permission from ref. 199. Copyright 2016, National Academy of Sciences. | ||
It was shown for TiOx that the electronic conductivity of the co-component can improve the cycle life and the efficiency of the sulfur cathode.55 It is thus important to consider the physical properties of chalcogenides and to study both effects: (i) capability to adsorb LiPSs and (ii) electronic conductivity to accelerate charge transport processes. Further improvement of ionic conductivity may play a critical role. Very often the capability to capture LiPSs is evaluated visibly or with spectroscopy based on the adsorption of a brownish LiPS solution with and without the sulfide (Fig. 8e). However, a standardized procedure to measure the adsorption capability of LiPSs (i.e. in mg Li2Sn per mg host material), as conducted by Pang et al., is still not well established but could simplify the identification of promising metal sulfide additives for sulfur cathodes.200Table 3 provides an overview of the electronic conductivity and the affinity of some chalcogenides to Li2S4/Li2S determined through first principles DFT calculations from various reports. Considering all reports, the highest binding energy to Li2S is found for TiS2 and VS2. As a comparison, graphitic carbon which is frequently used to encapsulate or make an electrical contact with sulfur exhibits only low capability to capture short and long chained LiPSs. These findings conform to a recent study from Chen et al. who found the strongest anchoring effect for VS2 followed by TiS2 based on theoretical calculations.201 In a comparative study, Zhou et al. experimentally confirmed the best performance with VS2 followed by TiS2 and CoS2.199 Although VS2 seems to offer superior properties as an additive in sulfur cathodes, most reports deal with TiS2 and CoS2. Interestingly, MoS2 also shows strong affinity to Li2S at the edge of the crystal facet and exceeds the values of all other metal sulfides, whereas the terrace side of MoS2 exhibits only low capability. This dependency of the exposed side of the crystal facet to the LiPS adsorption was experimentally and theoretically studied by Wang et al. using differently shaped MoS2 crystals to boost LiPS redox-reactions.202 In addition, Zhou et al. proposed that the Li atom within the Li2S4 molecule binds to the negatively polarized sulfide within the CoS2 structure while the LiPSs are nucleophilic and bind to the Co atom.203 These findings prove that the exposed interfacial facet of the nanocrystal is highly important and may open pathways to tailor the adsorption capabilities of LiPSs not only by the type of metal sulfide but also by engineering the crystal shape.
| Material | Electrical conductivity, σ [S cm−1] | Binding energy to Li2S4/Li2S determined by DFT calculations [eV] | Mechanism of lithium storage in the range, 1.5–2.6 V vs. Li/Li+ | Ref. |
|---|---|---|---|---|
| Graphite | 1–1000 | 0.34 (Li2S4) | No reaction | 198, 209 and 210 |
| 0.29 (Li2S) | ||||
| WS2 | 6.7 | 0.8 (Li2S4) | No reaction | 211, 193 and 207 |
| 1.45 (Li2S) | ||||
| NiS2 (111) | 2–55 | 2.06 (Li2S4) | Intercalation/conversion < 1.8 V | 212 |
| TiS2 | 30–50 | 2.99 (Li2S) | Intercalation < 2.5 V | 213 and 214 |
| ZrS2 | 1.32 | 2.7 (Li2S) | Intercalation/conversion | 214 and 215 |
| VS2 | 0.1 | 2.94 (Li2S) | Intercalation | 214 and 216 |
| FeS2 | 0.6 | N/A | N/A | 217 |
| SnS2 (001) | 1.8 × 10−4 (semiconductor) | 1.26 (Li2S4) | Intercalation | 218 and 219 |
| Bi2S3 (001) | 1.8 × 10−7 | 2.52 (Li2S4) | Conversion < 1.73 V | 220 and 221 |
| MoS2 | 1000 | 0.87 terrace site | No reaction | 220 |
| 4.48 Mo-edge (Li2S) | 202 | |||
| CoS2 (111) | 6–5000 | 1.97 (Li2S4) | No reaction | 217, 222, 198 and 208 |
| Co9S8 | 290 | 2.74 (Li2S) (002) | No reaction | 222 |
| 1.71 (Li2S4) (002) | 200 | |||
| CuS | 870 | N/A | N/A | 223 |
| Cu2S | 6700 | N/A | N/A | 217 |
| ZnS2 | 1 × 10−6 | N/A | N/A | 217 |
In order to understand the interaction of sulfur species with TMSs, it is also of high importance to consider the structural changes of the metal sulfide during lithium insertion within the potential range of sulfur (1.7–2.6 V vs. Li/Li+) as they significantly affect the physical properties. Some metal sulfides (i.e. VS2 and TiS2) intercalate lithium ions up to a certain potential, some undergo a conversion reaction (i.e. FeS2, NiS2, and MoS2)204–206 often to Li2S and metal cations and some merely show a reaction as in the case of WS2 and CoS2.207,208 The different types of lithium insertion are presented in Table 3.
Metal sulfides were found to be efficient compounds to enhance the adsorption of LiPSs and enhance and afford faster redox reactions. A brief overview of the achievements in improving Li–S batteries with different TMSs is shown in Table 4. Again, the values reported in this table should be taken with care as the capacity and reversibility strongly depend on the applied cell parameters such as the amount and type of electrolyte, electrode thickness, sulfur mass loading, sulfur composition, binder and separator. In order to provide a comparable picture we added some important parameters such as the sulfur ratio within the electrode, mass loading and C-rates. As concluded from Table 4, most reports deal with cobalt sulfides and titanium sulfides likely because of their wide availability, high electronic conductivity and high affinity to LiPS species. The properties of these metal sulfides were proven to lower the overpotential for Li2S oxidation and to enhance energy efficiency compared to other metal sulfides such as FeS, SnS2 and Ni2S3.199 Both, cobalt and titanium sulfides as co-components will be reviewed first.
| Material | Initial capacity [mA h g−1] | Degradation rate per cycle [%] | Sulfur contentb [wt%] | Sulfur loading [mg cm−2] | Ref. |
|---|---|---|---|---|---|
| a 1C = 1674 mA g−1. For clarification: interlayer is placed between the cathode and separator, coating was placed onto the separator, host corresponds to the carrier material for sulfur and additive means simple addition to the sulfur cathode composite. b Mass percentage of sulfur on the cathode excluding current collector substrate. | |||||
| CoS2 interlayer | 1240 at 0.2C | 0.17 at 0.2C | 64 | 1.55 | 227 |
| Co9S8 host | 1130 at 0.05C | 0.045 at 0.5C | 75 | 1.5 | 200 |
| Co3S4 host | 1012 at 0.2C | 0.079 at 1C | 53 | 4.7 | 228 |
| CoS2 additive | 1368 at 0.5C | 0.034 at 2C | 75 | 0.5 | 198 |
| CoS2 additive | 1326 at 0.1C | 0.047 at 1C | 56 | 2.3 | 203 |
| Co9S8-Celgard | 1385 at 0.1C | 0.039 at 1C | 70 | 2.0 | 233 |
| TiS2 additive | 1000 per g (S + TiS2) at 0.1C | 0.1 at 0.1C | 48 | N/A | 235 |
| TiS2 additive | 1000 per g (S + TiS2) at 0.1C | 1.3 at 0.1C | 45 | N/A | 238 |
| TiS2 encapsulation | 1156 at 0.2C | 0.058 at 0.5C | 35 | 2 | 214 |
| MoS2 additive | 1270 at 0.2C | 0.07 at 0.2C | 38 | 3.9 | 246 |
| MoS2 additive | 1339 at 0.2C | 0.08 at 0.5C | N/A | 2 | 202 |
| MoS2 coating | 950 at 0.2C | 0.083 at 0.5C | 65 | N/A | 247 |
| SnS2 additive | 1237 at 0.2C | 0.127 at 0.2C | 51 | N/A | 249 |
| SnS2 additive | 1400 at 0.1C | 0.058 at 0.5C | 62 | 2.4 | 219 |
| Bi2S3 additive | 1480 at 0.1C | 0.028 at 0.5C | 46 | 2.2–3.3 | 221 |
| α-NiS2 host | 1540 at 0.067C | 0.019 at 0.33C | 50 | 2 | 251 |
| NiS2 | 1203 at 0.1C | 0.04 at 2C | 39 | 2.0–3.3 | 212 |
| WS2 host | 1581 at 0.1C | 0.0072 at 2C | 55 | 2 | 57 |
| WS2 interlayer | 1454 at 0.02C | 0.055 at 0.5C | 70 | 4 | 254 |
3.1 Cobalt sulfide
There are several known Co–S phases with different crystal structures (i.e. CoS, CoS2, Co3S4 and Co9S8). Their unique physical properties such as high electrical conductivity (up to 5000 S cm−1 at room temperature)222 and magnetic moment lead to applications in (electro)-catalysis,224–226 as an anode material for lithium ion batteries, in magnetic applications and secondary alkaline batteries.208The application of various cobalt sulfides as co-components in sulfur cathodes recently gained increasing attention. Until now, CoS2, Co3S4 and Co9S8 supported sulfur cathodes have been reported.198,200,203,227–233 Yuan et al. prepared CoS2 (cattierite type) particles (20–200 nm) through a hydrothermal method deposited in graphene layers as a sulfophilic host material for sulfur cathodes with CoS2 compositions ranging from 0–30 wt%.198 It was shown that an increasing amount of CoS2 accelerates the electrochemical reaction, decreases liquid–solid polarization and positively affects the LiPS redox kinetics. Furthermore, the adsorption capability of LiPSs was visually proven. The best performance was achieved with 15 wt% CoS2 at an initial discharge capacity of 1368 mA h g−1 and 1005 mA h g−1 after 150 cycles (75 wt% sulfur loading at 0.5C) while the discharge capacity without CoS2 was only 843 mA h g−1 and 513 mA h g−1 after 150 cycles. DFT calculations with Li2S4 molecules confirmed the strong interfacial interaction of CoS2 and LiPSs rather than chemical adsorption (Table 4). The calculations also indicated enhanced charge transfer processes on the molecular level when Li2S4 was adsorbed to the (111) CoS2 plane. Pang et al. report about a graphene-like metallic Co9S8 nanosheet structure as a host material for a high sulfur content.200 Co9S8 with a surface area of 108 m2 g−1 was prepared through microwave solvothermal methods. Capacities up to 1130 mA h g−1 at C/20 were achieved and the rate capability was very high with almost no capacity drop from 0.5 to 2C even though the sulfur content was as high as 75 wt% in the cathode highlighting the positive effect of Co9S8. After 400 cycles at 2C, about 75% of the capacity is retained (Fig. 9a). The intrinsic adsorptivity of Co9S8 (normalized to the surface area) for LiPSs is almost five times higher than that observed for materials such as Ti4O7 or meso-TiO2 frequently reported as LiPS capturing materials (Fig. 8d). In fact, DFT calculations showed that at the (008) facets, only positively charged Co atoms are exposed. The binding energy of Li2S2 can reach to 6.06 eV, one of the highest values reported so far. Furthermore, a high mass loading of 4.5 mg cm−2 at a C-rate of 0.5 with a reversible areal capacity of 2.5 mA h cm−2 was demonstrated. These results highlight the large potential of Co9S8 and cobalt sulfides in general as additives for sulfur-cathodes. Zhou et al. synthesized N-doped carbon hosts with and without embedded Co or CoS2 nanoparticles by carbonization of a metal–organic framework (ZIF-67).203 A capacity of 1326 mA h g−1 at 0.1C (56 wt% sulfur) and the best reversibility was achieved with CoS2 nanoparticles (Fig. 9b). After 250 cycles the electrode with CoS2 shows a capacity of 702 mA h g−1 while the electrode with Co/N-doped carbon exhibits 589 mA h g−1 and the bare carbon host only 446 mA h g−1. The enhanced performance of the sulfur cathode is attributed to the synergistic effect of CoS2 and N-doping within a porous carbon material to accelerate sulfur redox coupling which was clearly evidenced by the visual adsorption of LiPSs. Even after a short exposure of 1 h of the CoS2–carbon composite to a LiPS solution, the entire LiPS solution turned colorless whereas the Co–carbon or carbon host needed about 72 h to anchor the LiPS species (Fig. 9c). Instead of the preparation of CoS2 composite cathodes, Ma et al. inserted interlayers between the cathode and separator, made of hydrophilic porous carbons and CoS2, in order to prevent the diffusion of LiPS intermediates to the anode and, thus, to reduce the shuttle effect.227 The cycle stability could be significantly improved due to lower charge transfer resistance and the adsorption capability of the modified CoS2 interlayer.
 | ||
| Fig. 9 Some chosen studies dealing with cobalt sulfides in sulfur cathodes: (a) Galvanostatic cycling of a Co9S8/S (75 wt% S) composite.200 (b) Galvanostatic cycling at 0.5C (within the 56 wt% S cathode) and (c) the capability to adsorb LiPSs of different carbon host materials with and w/o CoS2.203 (d) SEM and TEM pictures of carbon/Co3S4 polyhedra as a host material for sulfur and their electrochemical performance at (e) high mass loading and (f) long-term cycling (53 wt% S within the cathode).228 (a) Reproduced with permission from ref. 200. Copyright 2016, The Royal Society of Chemistry. (b and c) Reproduced with permission from ref. 203. Copyright 2016, Elsevier. (d–f) Reproduced with permission from ref. 228. Copyright 2017, Elsevier. | ||
Xu et al. prepared hollow Co3S4 polyhedra with a porous shell as a host material for sulfur within free-standing activated carbon nanofibers (ACNFs) (Fig. 9d).228 By comparing ACNFs with and without Co3S4 polyhedra as a sulfur host, enhanced rate capability, reversibility and smaller polarization were confirmed. A high areal capacity of 13 mA h cm−2 at 13.5 mg cm−2 sulfur mass and a current rate of 0.3C was achieved (Fig. 9e and f).
According to Song et al., an areal capacity higher than 4 mA h cm−2 is required for Li–S batteries to outperform commercial Li-ion batteries.234 Furthermore, a capacity of 953 mA h g−1 at 1C and 610 mA h g−1 after 450 cycles with a relatively high loading of 2.5 mg cm−2 were demonstrated. Here again, the outperforming electrochemical performance was mainly attributed to the physical properties of Co3S4.
3.2 Titanium sulfide
Among the several titanium compounds to capture LiPSs, the most frequently reported compounds are titanium oxides which undergo strong Ti–S interactions. As discussed above, this material was already successfully employed as a performance enhancing additive in many Li–S batteries.50,51,53 In recent years, TiS2 also turned out to be a promising additive for Li–S batteries.235 TiS2 is already known since the 1970s as a layered intercalation cathode material for rechargeable lithium batteries236 and was already commercialized in the first generation of Li-ion batteries.237 TiS2 exhibits semi-metallic to metallic behavior depending on the state of lithiation and therewith fulfills the requirements as an additive for sulfur composites: (i) polarity and (ii) high electrical conductivity.As one of the first groups, Garsuch et al. tested ball-milled sulfur/carbon/TiS2 composite electrodes and observed improvements in the cycle life with the addition of TiS2.235 The optimum composition was found to be 20 wt% of TiS2. However, the specific capacity normalized to the active mass was reduced when TiS2 was added. It was proposed that the addition of TiS2 can enhance ionic and electrical conductivity in the cathode composite, but the surface needs to be tailored to electrically contact all sulfur. Su et al. also partially replaced the carbon additive by TiS2 within sulfur composite electrodes.238 Similar to the observations made by Garsuch et al.,235 the capacity based on the active mass was reduced by TiS2 addition, but the cycling stability increased.
Seh et al.214 used a different design to incorporate TiS2 in Li2S cathodes. They encapsulated Li2S (particle size < 1 μm) in TiS2 with different thicknesses ranging from 10–50 nm through an in situ reaction of TiCl4 with Li2S particles followed by a heat treatment to crystallize TiS2. It was found that the charge transfer resistance and the potential barrier in the first charging process significantly decreased with TiS2 encapsulation. The initial capacity increased from 708 mA h g−1 to 806 mA h g−1 compared to bare Li2S particles with an average capacity loss of 0.058% per cycle. The reason was found to be the high conductivity of TiS2 and the high affinity of Li2S/Li2Sn to TiS2 by several experimental techniques and DFT theoretical calculations. They also encapsulated Li2S with ZrS2 and VS2. The performance of the materials was comparable to the one with TiS2 indicating that these materials also show high affinity for LiPS species. The resulting electronic conductivities were 4.0 × 10−9 and 3.8 × 10−9 S cm−1, whereas the Li2S@TiS2 structure showed the highest conductivity of 5.1 × 10−3 S cm−1.
Ma et al.239 prepared a TiS2 foam infiltrated with sulfur by an in situ reaction of a commercially available Ti metal foam with sulfur at 700 °C in a sealed quartz tube. The structure of the final electrode material can be sub-divided into three parts: a Ti metal core surrounded by a TiS2 film and sulfur. The 3D hybrid structure can store up to 40 mg cm−2 of sulfur and exhibits a capacity of up to 30 mA h cm−2 at a total electrode weight (including the current collector) and about 260 mA h g−1 as a cathode composite. The capacity retention under these conditions was still impressively high and accounts to less than 0.3% per cycle.
Matsuyama et al. prepared amorphous TiS3/S/C composite electrodes and found poor performance in Li–S batteries with liquid electrolytes when adding TiS3 to the electrode.240 It was attributed to LiPS dissolution which is in contrast to other reports214,235–239 as it evidences that the capability of TiS3 to capture LiPSs seems to be very low. However, a remarkable improvement could be achieved with solid electrolytes.
3.3 Molybdenum sulfide
Another promising metal sulfide additive is MoS2 offering high electrical conductivity and the binding energies of Li2S to the Mo-edge in the MoS2 structure of up to 4.48 eV (Table 3).202,220,241–245 Note that MoS2 is not active in the potential range of sulfur (1.8–2.6 V vs. Li/Li+). Dirlam et al. fabricated sulfur/MoS2 and sulfur co-polymer/MoS2 composite electrodes by facile dispersion of 2D MoS2 sheets in molten sulfur and ball milling.246 A considerably enhanced cycle life and sulfur utilization were found with the composite prepared through dispersion in molten sulfur. Ghazi et al. coated MoS2 onto the separator instead of a direct addition to the sulfur cathode.247 The modified side of the separator faced a conventional sulfur cathode, a mixture of carbon and sulfur, during cell tests vs. Li/Li+. Greatly improved reversibility with a decay of only 0.083% per cycle at 0.5C was achieved exceeding the performance of a reference separator made of graphene oxide.MoS2 itself is also considered as a promising intercalation cathode as well as a conversion anode material. Recently, some groups used molybdenum sulfides as the initial precursor material to form a sulfur-based composite material after the first initial discharging process.194,206 Balach et al. studied the irreversible electrochemical decomposition of MoS2 to Li2S and Mo nanoparticles as a sulfur-based cathode showing typical sulfur electrochemical characteristics and performed ex situ measurements.206 In contrast to commonly used ether-based electrolytes for Li–S batteries, the group successfully conducted reversible cycling in carbonate-based electrolytes which are actually well known to be incompatible with LiPSs. Despite using a cathode with an ultrahigh Li2S loading of 10.7 mg cm−2, the cell delivered an average areal capacity of 7.5 mA h cm−2 at a C-rate of 0.1. Furthermore, the MoS2-derived Li2S cathode was coupled with a lithiated silicon anode to assemble a Li–S full-cell providing an initial capacity of 780 mA h g−1. It was found that the polymeric gel-like solid-electrolyte interface (SEI) formed during the initial discharging process keeps LiPSs tightly embedded in the Mo/carbon matrix and thereby prevents the formed LiPSs from a dissolution into the electrolyte and finally a diffusion to the metal anode. This strategy may allow the usage of carbonate-based electrolytes which may allow the application of (safe) alternative anode materials (i.e. Si and Sn) instead of lithium metal.
Wang et al.202 showed that the atomic sites on the crystal surface of a metal sulfide additive are highly important to capture LiPSs. They prepared differently shaped crystal surfaces with varying amounts of terrace or edge sites with MoS2 nanostructures (nanoparticles and vertically aligned 2D sheets) on CNFs and studied their electrochemical behavior as a positive current collector for LiPSs. It was experimentally found that the exposed crystal facet (Mo-rich or S-rich edge) of the MoS2 particle is highly important for an improved operation mode of Li–S batteries and as a catalyst. The best performance was achieved with vertically aligned MoS2 sheets which contain high amounts of Mo-rich edges. This observation was confirmed by DFT calculations. The high affinity of Li2S to the Mo-edge of the MoS2 structure with a binding energy of 4.48 eV was reported while the sulfur-edge only offered 0.87 eV.
3.4 Other metal sulfides
There are several other metal sulfides which were investigated to boost the electrochemical performance of Li–S batteries, such as SnS2,219,248,249 FeS2,250 Bi2O3,221 NiS2,212,251,252 NbS2,253 WS2,57,254 MnS,255 CuS,223 VS2256,257 and ZnS.258 Very interesting results have been obtained recently with WS2 independently by two different groups (Fig. 10). Using WS2 as a host or an additive in Li–S batteries, remarkable reversibility and sulfur utilization (about 95%) were reported by Lei et al.57 (Fig. 10a–c) and Park et al.254 (Fig. 10d and e), respectively. Lei et al. used C@WS2 as a host material for sulfur and obtained a discharge capacity of 1581 mA h g−1 at 0.1C with only 0.0072% capacity loss per cycle over 1500 cycles, the best degradation rate value reported so far.57 By conducting DFT calculations, they found that particular short chain sulfides (Li2S2 and Li2S) chemically interact with WS2. For example, the binding energy of Li2S4 is just 0.8 eV whereas for Li2S 1.45 eV was determined. Since these binding energies are lower compared to other metal sulfides (Table 4), sulfides with moderate binding energies in the range of 0.8 eV < Eb < 2.0 eV were proposed to be the best choice for high performance Li–S batteries. This description is in agreement with the work of Park et al.,254 who proposed a disproportion of long-chain PSs to short-chain PSs after trapping at the edge sites of WS2. Both groups experimentally confirmed the high adsorption capability for LiPSs by visualization in a glass vial (Fig. 10c).
 | ||
| Fig. 10 (a) Schematic illustration of the preparation of the CNFs/WS2 host material for sulfur, (b) its electrochemical performance (55 wt% sulfur; 2 mg cm−2; 1.7–2.7 V vs. Li/Li+) and (c) a visual demonstration of the adsorption capability for LiPSs with (bottom) and without WS2 (top) over galvanostatic discharge.57 (d) Schematic illustration of the faster reaction kinetics with the WS2 support and (e) the electrochemical performance with and without the WS2 support in various cell configurations.254 (a–c) Reproduced with permission from ref. 57. Copyright 2016, Wiley-VCH. (d and e) Reproduced with permission from ref. 254. Copyright 2017, Wiley-VCH. | ||
Li and co-workers prepared hollow carbon spheres filled with sulfur and different compositions of SnS2 nanoparticles ranging from 5 to 7 nm in size.249 It was found that SnS2 nanoparticles enhance the life time of the cell, decrease charge transfer resistance, increase the diffusion of Li+ ions in the Li2S composite and anchor LiPSs within the cathode. The optimum SnS2 concentration was found to be 10 wt%. Li et al. prepared both SnS2/S/C and SnO2/S/C composite electrodes.219 The SnS2-based composite showed considerably higher capacity and slightly enhanced reversibility than the SnO2-based composite, although the DFT calculated binding energy of Li2S4 is higher to SnO2 than to SnS2 which is actually an indication of better reversibility. As a reason it was stated that the binding energy between Li2S4 and SnO2 of 3.25 eV may have been too high causing the disruption of the Li2S4 molecule which has been suggested by other groups as well.57 The charge transfer resistance was lower in the case of SnS2 highlighting that strong interaction/adsorption may not be the most important parameter to enhance the electrochemical performance. More importantly, a balance between electrical conductivity, the charge transfer process and moderate LiPS adsorption may be crucial for improving the cell performance.
Another interesting metal sulfide used in Li–S batteries is FeS2. It is widely available, very cheap and can retain the low cost advantage of Li–S batteries. For example, Zhang et al. showed that FeS2 used as an additive can chemically adsorb LiPSs and prevent diffusion to the anode.250 It was evidenced that the binding of LiPSs involves the formation of a Li2FeS2+n complex through a radical reaction. By increasing the amount of FeS2 from 0 to 15 wt% within the electrode composite, the cycle life of the Li–S battery could be increased from 50 cycles to 200 cycles, which is attributed to the efficient adsorption of LiPSs within the FeS2-containing cathode.
Bi2S3 was tested by Li et al. in sulfur composite electrodes for Li–S batteries prepared through a melting technique at 280 °C.221 It was found that this compound also seems to have very good capability to capture LiPSs, thereby anchoring LiPSs within the composite. The excellent affinity of LiPSs to Bi2S3 was confirmed by first principles DFT calculations. They studied different amounts in the range of 10–20 wt% of Bi2S3 and found the optimal performance in terms of capacity retention at 14 and 19 wt% of Bi2S3. The first discharge capacity was up to 1500 mA h g−1 at 0.1C. However, it should be noted that Bi2S3 contributes to the capacity in the chosen voltage window.
There are further studies dealing with NiS2,212,251 MnS,255 CuS223 and ZnS258 as additive or host materials for sulfur composite electrodes. Except for CuS, all of these studies reported enhanced electrochemical performance in the presence of these metal sulfides. Among these reports, NiS2 seems to be a promising co-component for sulfur cathodes. In the same manner, it is mostly attributed to capturing LiPSs and anchoring them within the cathode. The major physical properties and their effect in sulfur composite electrodes are summarized in Tables 3 and 4, respectively.
It should be noted that chalcogenides also comprise selenides and tellurides. However, to the best of our knowledge, no reports about metal selenides and tellurides appeared yet utilizing these kinds of chalcogenides as hosts, additives or interlayers in sulfur cathodes or Li–S cells to enhance sulfur redox-reactions or anchoring LiPSs. This area may be worth exploring in the future.
4. Transition metal carbides (TMCs) and nitrides (TMNs) including 2-dimensional materials (MXenes)
4.1 Transition metal carbides (TMC)
In most transition metal carbides (TMCs) based on metals of the groups 6–8 of the periodic table of elements, carbon atoms are placed in interstitial sites in the metallic lattice. Thus, TMCs like e.g. TiC,259–265 WC261,266 and NbC267 exhibit metallic properties such as high electronic conductivity in the order of 104 S cm−1 and were recently investigated to enhance the performance of Li–S batteries. In this context, the effect of TMCs on the homogenous deposition of insoluble Li2S at the sulfur electrode scaffold during discharging is regarded as a crucial issue.259,262,265In 2016, titanium carbide nanoparticles started to be applied in sulfur electrodes.259–261 In that regard, Salem et al. proposed that TMCs offer superior properties for electron transfer reactions involving LiPSs compared to transition metal oxides due to the greater density of states near the Fermi level as a result of the favorable interaction of d-electrons of the metal with the sp-electrons of the carbon.261 Accordingly, WC and TiC nanoparticles with a diameter of 100 nm were investigated as an electrocatalyst for the LiPS reaction by experimental and theoretical methods. The improvement of the corresponding batteries was found to be based on the enhanced electron transfer reaction and the capability to adsorb LiPS intermediates, with better results for TiC than for WC. Experimental studies comparing TiO2/carbon composites to analogous TiC/carbon composites confirmed the advantage of TiC over TiO2 components.259,263 Using graphene or nanoparticle/graphene composites as a sulfur host, Peng et al. observed an increased number of nucleation sites of Li2S with TiO2 nanoparticles but inhibited lateral growth.259 However, TiC nanoparticles enabled a high number of nucleation sites and full surface coverage with Li2S films of increased thickness due to enhanced radial growth of Li2S. Furthermore, reduced charge transfer resistance and a shift in the peak potential in cyclic voltammetry were measured suggesting that conductive TiC facilitates both the liquid–liquid transformation of LiPSs and the liquid–solid nucleation/growth of Li2S. After 100 cycles at 0.2C, a reversible capacity of 670 mA h g−1 was obtained for a considerable sulfur loading of 3.5 mg cm−2. Besides TiC nanoparticle/graphene host259 and interlayer264 materials, TiC nanoparticles were combined with CNFs262,265 and mesoporous CMK-3263 for application as sulfur host materials.
Cai et al. synthesized nanocrystalline NbC by a magnesiothermic reaction at 600 °C and coated the material on a membrane to employ it as an interlayer in Li–S batteries.267 Using a cathode with a sulfur loading of 1.5 mg cm−1, a reversible capacity of 988 mA h g−1 was achieved after 100 cycles at 0.2C and a capacity of ≈500 mA h g−1 after 1500 cycles at 2C, corresponding to a degradation rate of 0.04% per cycle. WC was used as an additive for the positive electrode and compared to WO3 showing that batteries with WC exhibit a much higher discharge capacity in the region of the second voltage plateau and an improved cycling stability.266 The difference in the capacity becomes even more distinct for higher current rates. The authors concluded that WC promotes the disproportionation of LiPSs and thus enables the repeated utilization of “recycled” long-chain LiPSs in the reduction process. This catalytic property is attributed to strong sulfophilic surface moieties capturing soluble LiPS species by representing tungsten disulfide-like surfaces because nanoscale layers of specifically adsorbed S atoms on WC were evidenced by XPS measurements. A comparative study on TMC nanoparticle/CNF electrodes revealed the superior performance of tungsten semicarbide (W2C), reaching a capacity of 1128 mA h g−1 after 200 cycles at 0.2C and a degradation rate of 0.07% per cycle, over Mo2C and TiC.265 In line with DFT calculations of stable configurations of Li2S6 on the three metal carbides, W2C nanoparticles are assumed to function as an oxidation and reduction catalyst, where Li2Sn diffusion from the active sites to the carbon matrix is facilitated by a moderate adsorption energy of W2S to sulfidic species, resulting in the homogenous deposition of sulfur species on the entire carbon matrix.
4.2 2-Dimensional carbides of the MXene class
The materials class of MXenes comprises 2-dimensional (2D) transition metal carbides, carbonitrides and nitrides, which often exhibit hydrophilic surfaces containing exposed redox-active transition metal atoms and electrical conductivity in the range of 104 S cm−1.268 The name MXene refers to the similarities to graphene and the precursor phases of layered ternary carbides and nitrides (MAX phases).269 MXenes are described by the general formula Mn+1XnTx (n = 1–3), where M represents group 4 to 6 transition metals, X carbon and/or nitrogen and T terminal surface groups, mostly hydroxyl (–OH), oxo (–O) or fluoro (–F) groups, with n+1 layers of M covering n layers of X. The mixture of –OH, –O and –F terminations on the surface results from the synthesis methods of selective etching of certain metal atoms forming layers which interleave the layers of TMC and TMN, in which hydrofluoric acid is applied.268Liang et al. proposed the use of 2D Ti2C as a host material for sulfur for the first time and demonstrated promising results.270 Exfoliated and delaminated Ti2C nanosheets were prepared with surface areas of 20.2 m2 g−1 and 67.9 m2 g−1, respectively. Compared to conventional porous carbon hosts, this surface area is very low and intuitively a poor electrochemical performance would be expected as a good electrical contact cannot be established with sulfur. However, the delaminated Ti2C with infiltrated sulfur shows an excellent electrochemical performance. The discharge capacity was measured to up to 1400 mA h g−1 at 0.05C (sulfur content = 56 wt%; sulfur loading = 1 mg cm−2) and the decay rate over 650 cycles at 0.5C was only 0.05% per cycle. The authors attributed the superior performance to the chemisorption of LiPS intermediates on the Ti2C surface which creates S–Ti–C bonds facilitating electron transfer and redox reaction kinetics. This assumption was confirmed by XPS measurements showing evidence for such redox behavior. It is important to note that host materials with low surface areas are able to provide high rate performance for sulfur cathodes. These materials are attractive for high energy batteries as they help to increase the tap density and therewith the volumetric as well as specific energy density. Following up on the work of Liang et al., several other studies investigated MXenes and corresponding composites as a sulfur host material,271–274 separator coating275,276 or applied MXenes in both functions.277
Bao et al. reported a TiC@mesoporous carbon composite infiltrated with sulfur for positive electrodes of Li–S batteries.271 High discharge capacities of up to 1225 mA h g−1 (at 0.5C and 58 wt% sulfur within the entire cathode) were achieved with a capacity loss of 0.19% per cycle at 0.5C. The enhanced performance compared to the control electrode was explained by the hydrophilic surface characteristics of Ti3C2Tx. However, Liang et al. proposed in a continued work to their first paper about MXenes that the strong interaction of LiPSs and the surface groups is more complex and originates from a dual mode mechanism.274 Initially, a cleavage of Ti–OH occurs and results in the formation of thiosulfates. The created vacancies on Ti3C2 are filled by a Lewis-base reaction of LiPSs to form Ti–S bonds. A demonstrative representation is shown in Fig. 11a.
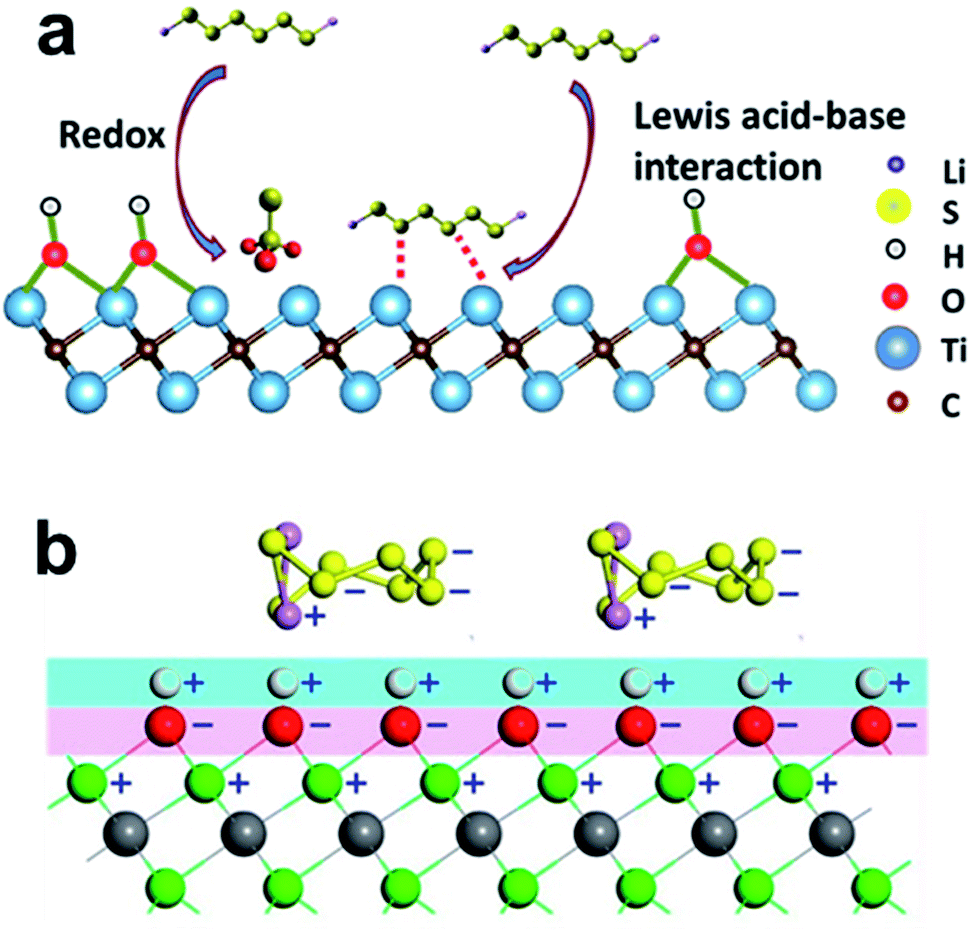 | ||
| Fig. 11 (a) A schematic representation of the dual mode mechanism of the strong interaction of LiPSs on Ti3C2OH MXenes274 and (b) scheme of charged atoms in LiPS and MXenes, where “+” represents the electropositive atoms and “−” represents the electronegative atoms with Ti: green, S: yellow, Li: Purple, O or F: red and H: white.278 (a) Reproduced with permission from ref. 274. Copyright 2016, Wiley-VCH. (b) Reproduced with permission from ref. 278. Copyright 2017, American Chemical Society. | ||
For the application as a sulfur host material, the utilization of the functional surfaces of 2D exfoliated MXene materials may be interfered by the usually observed stacking of the metal carbide sheets through van der Waals forces and hydrogen bonding. Accordingly, rGO nanosheets were employed as spacers yielding a 3D morphology with accessible 2D surfaces of multilayer Ti3C2Tx nanosheets sandwiched between rGO layers. After solution infiltration of sulfur, the composite achieved an initial capacity of 1144 mA h g−1 at 0.5C which decreased to 878 mA h g−1 after 300 cycles corresponding to a degradation rate of 0.08% per cycle.273 Furthermore, the same group reported a crumpled N-doped MXene nanosheet host material which was synthesized by thermal annealing of a coagulated precipitate of Ti3C2Tx flakes and positively charged melamine as an N-source and spacer. With a high sulfur loading of 5.1 mg cm−2, the reversible capacity and the degradation rate after 500 cycles at 0.2C were 588 mA h g−1 and 0.05% per cycle. The interaction of Li- and N-atoms was proven by XPS measurements conducted after the discharge.272 The suggestion to use MXenes for separator coatings in Li–S batteries is based on the ability to obtain very thin and homogenous closed layers of electrically conducting 2D nanosheets with highly polar surface sites.275,276 Comparing a Ti3C2Tx covered glass fiber separator to a graphene coated one, Lin et al. observed a higher initial discharge capacity for the graphene layer but lower cycling stability.275 Corresponding ab initio calculations showed that Ti3C2 exhibits much stronger interactions with LiPSs than graphene, whereas the Ti–S interactions are, however, weakened due to strongly polar F- or OH-functions. Therefore, the authors expect an additional performance improvement, if the number of such functional groups would be reduced.
Further computational studies applying DFT calculations enabled a more differentiated view on the role of surface functionalities on MXenes for Li–S batteries.278–282 For bare Ti2C surfaces, Rao et al. calculated distances of S atoms of LiPS and Ti atoms of MXenes in the range of Ti–S bond lengths in TiS2 crystals, corresponding to strong interactions.278 Moreover, it was found for defect sites (representing the surface partially uncovered with functional groups) that the interaction of Ti and S atoms is strong enough to break the covalent S–S bond that constitutes the S chain of LiPS.280,281 In contrast to Lin et al.,275 this was interpreted as a drawback because active sulfur material is irreversibly lost.280,281 However, continued trapping of sulfur is not assumed as the adsorption energy of a second S atom adsorbed on the previously trapped S atom is smaller than the formation energy of octet sulfur.280 The strong attraction of Ti and S is reduced for O- and F-termination groups as the repulsive force from O and F, which have more electrons around their surfaces, increases.278 As seen in Fig. 11b, the repulsive forces will be slightly shielded by H atoms, if the surface is functionalized with OH groups.278 However, H atoms can be relatively easily replaced in line with the known behavior of an increasing number of O groups and a decreasing number of OH groups observed, if long-chain LiPS are introduced. While the interaction of LiPS with F terminations is relatively weak suggesting an anchoring mechanism, the interaction with Ti2CO2 is certainly stronger due to attractions between Li and O atoms leading to elongation of Li–S bonds.281 The electronic conductivity of MXenes is not affected by LiPS adsorption as the band gaps do not obviously change or are even narrowed as for F-doped surfaces.278
In summary, 2D MXene materials are very promising for application in Li–S batteries as they combine the properties of high electrical conductivity and surfaces suitable for anchoring or decomposing LiPS species. In this regard, it would be interesting to investigate the potential electrocatalytic function of MXenes towards the conversion of LiPS intermediates. The nanosheet morphology of MXene materials enables their use for thin separator coatings and to achieve a high exposure of their functional surface to the sulfur species, if restacking is omitted. Until now, nearly all reports employing MXenes for Li–S batteries have dealt with titanium carbide-based materials. So, for future research, 2D derivatives of further TMCs, transition metal carbonitrides and nitrides might be highly interesting. Accordingly, the following section discusses “conventionally” nanostructured representatives of the latter material's class.
4.3 Transition metal nitrides (TMNs)
Transition metal nitrides (TMN) of the groups 4 to 6 of the periodic table of elements, are, similar to the discussed TMCs, interstitial compounds with high electronic conductivity, good chemical stability and polar metal–nitride (M–N) bonds. In particular, TiN283–294 and VN295–301 have recently gained considerable attention and were investigated with promising results. In 2016, Mosavati et al. suggested to apply TiN nanoparticle powder as a material for the positive electrode to promote LiPS conversion reactions achieving a capacity of 1040 mA h g−1 after 100 cycles at 0.1C.283 Goodenough and co-workers prepared a mesoporous TiN host material with a specific surface area of 70 m2 g−1 through reduction of ZnTiO3 with hot ammonia gas.284 The TiN host material was infiltrated with sulfur and tested in Li–S batteries. For comparison, a TiO2 host material was prepared in a similar way and infiltrated with sulfur. A high capacity of 1121 mA h g−1 at 0.1C (50 wt% sulfur content in the cathode) and a decay rate of 0.07% per cycle over 500 cycles were achieved with TiN exceeding the performance of the TiO2 reference cathode. The results were mainly attributed to the excellent electronic conductivity, robust host framework and good adsorption capabilities for LiPSs.284 Deng et al. prepared hollow, porous TiN tubes through a sol–gel process and tested the final cathodes with a sulfur loading of 1 mg cm−2 at 52 wt% sulfur content.285 An initial capacity of 1481 mA h g−1 at 0.1C was observed and a reversible capacity of 1020 mA h g−1 at a C-rate of 0.2 was demonstrated. The capacity loss per cycle was reported to be 0.015%, which is one of the best reported values so far.55,57–59 Hao et al. prepared TiN/S composite electrodes by simple mixing of 30 nm TiN nanoparticles with sulfur.286 The demonstrated electrochemical performance was not as good as reported in other studies, but the simplicity of the approach makes it relatively attractive. A heterogeneous catalytic effect of TiN to promote the redox kinetics of LiPSs was proposed by Jeong et al. in a combined computational and experimental study.288 The very strong interaction of a cyclooctasulfur molecule on the TiN surface accounting to 6.6 eV was calculated, which is far higher than that reported for various TiOx modifications. TiN was furthermore applied for separator coatings287,290,291 in combination with TiO2290 or rGO, yielding a reversible capacity of 550 mA h g−1 at 2C after 1000 cycles in the latter case.287
Mosavati et al. tested various TMNs including WN nanoplates, Mo2N nanorods and VN nanoparticle as additives within the sulfur cathode to boost the performance of Li–S batteries.296 The differently shaped TMNs were synthesized through a wet chemical process and an annealing step. Interestingly, results at an ultrahigh sulfur loading of up to 12 mg cm−2 were demonstrated which makes the study attractive for practical application. Best performance was obtained using WN which was attributed to strong S–W–N interactions. In contrast, VN showed a quite poor performance. However, Ma et al. reported a VN host material with a highly porous hollow structure delivering a capacity of 837 mA h g−1 after 1000 cycles at 1C for a sulfur loading of 1.2 mg cm−2.298 Further studies on VN/carbon host materials researched carbon encapsulated VN nanowires,297 porous carbon/VN fibers,300 and composites of VN nanoentities and N-doped carbon.299,301 Sun et al. measured a reversible capacity of 1252 mA h g−1 after 100 cycles at 0.2C for a porous VN nanoribbon/graphene composite due to fast redox reaction kinetics.295 Ren et al. also intended to utilize the properties of a functional catalyst in Li–S batteries and synthesized cobalt-doped VN yolk–shell nanospheres encapsulated in a thin layer of N-doped carbon.299 Investigating a third TMN species, mesoporous Co4N spheres achieved by nitridation of Co3O4 were applied as a host material giving a capacity of 1100 mA h g−1 at 0.5C after 100 cycles for a sulfur content of 72 wt%.302
Undoubtedly, TMNs, TMCs and, specially, MXenes for sulfur cathodes are a very young topic with raising interest.263 We believe that this material class is an attractive candidate to improve the capacity retention and lifespan of Li–S batteries.
5. Metal–organic frameworks (MOFs) and other metal-complex based compounds
Metal complexes consist of a metallic center and surrounding ligands typically having a lone pair of electrons to form coordinative bonds to metal ions or atoms. In MOF structures, a network of repeating coordination entities features (potential) spatial voids, called pores, in a thermally stable crystalline framework structure comprising metal ions or metal complexes as centers and organic ligands, called linkers, with two or more functional groups to form coordinative bonds to several centers.303 The shape and possible chemical functionalities of the highly regular MOF pores depend on the particular metallic center(s), the organic linker(s) and the resulting framework structure and thus, these properties are designable. Due to its large pores with small apertures and the polar character of the metal center–ligand bonds, Demir-Cakan et al. used a mesoporous chromium trimesate denoted as MIL-100(Cr) (Table 5) as the first example of a MOF-based sulfur host in Li–S batteries.304 In this study, the cycling stability increases remarkably compared to mesoporous carbon or polar silica materials as the confinement in the MOF pores strongly suppresses the diffusion of LiPS from the host matrix. Further research also focuses on utilizing the Lewis acid function of coordinately unsaturated metal sites of certain MOFs to interact with LiPS anions305–309 as well as the Lewis base function of certain linker molecules to interact with Li+ cations.308,310,311 In this section, we will summarize recent findings concerning the use of MOFs and other metal complexes in Li–S batteries highlighting remarkable achievements regarding the cycle life and the underlying mechanistic principles of general relevance in understanding the role of metal-containing compounds in Li–S batteries. In this regard, the highly ordered and tunable framework structure allows for well-designed systematic studies. Table 5 aims to give an overview on widely researched compounds of this material class and their structural properties related to performance parameters achieved in Li–S batteries. Moreover, we discuss application-relevant aspects regarding the thermal, chemical and electrochemical stability of MOFs as well as their electrically insulating nature with respect to the influence of particle size and conjunction to conductive additives or matrices in composite materials on capacity and rate capability.| Metal–organic framework (MOF) | Framework structure | Surface area and pore volume | Initial capacity [mA h g−1] | Reversible capacity [mA h g−1] | Current ratea | Cycle number | Degradation rate per cycle [%] | Sulfur contentb [wt%] | Sulfur loading [mg cm−2] | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a 1C = 1674 mA g−1. b Current collector substrate excluded. | ||||||||||
| MIL-100 (Cr): [Cr3F(H2O)3O(BTC)2]n |

|
1485 m2 g−1 | 1580 | 450 | 0.1C | 60 | 1.2 | N/A | N/A | 304 |
| 0.95 cm3 g−1 | ||||||||||
| HKUST-1(Cu): [Cu3(BTC)2]n |
|
1500 m2 g−1 | 1498 | 500 | 0.1C | 170 | 0.39 | 20 | 0.5 | 305 |
| 0.67 cm3 g−1 | 431 | 286 | 0.5C | 300 | 0.11 | 30 | N/A | 312 | ||
| N/A | 1263 | 681 | 0.2C | 500 | 0.09 | 40 | 1.0 | 313 | ||
| 143 m2 g−1 | ≈1050 | ≈780 | 0.2C | 1000 | ≈0.03 | N/A | N/A | 314 | ||
| 0.16 cm3 g−1 | ||||||||||
| ZIF-8 (Zn): [Zn(MeIm)2]n |
|
N/A | ≈1200 | 510 | 0.1C | 100 | ≈0.6 | 14 | N/A | 315 |
| 0.70 cm3 g−1 | 738 | 553 | 0.5C | 300 | 0.083 | 30 | N/A | 312 | ||
| 1309 m2 g−1 | 1600 | 380 | 0.05C/0.1C | 25 | 2.5 | N/A | N/A | 316 | ||
| 0.64 cm3 g−1 | ||||||||||
| N/A | ≈1200 | 598 | 0.2C | 50 | ≈1.0 | 40 | 10 | 313 | ||
| 919 m2 g−1 | ≈1250 | 750 | 0.2C | 1000 | ≈0.04 | N/A | N/A | 314 | ||
| 0.70 cm3 g−1 | ||||||||||
| MOF-5 (Zn): [Zn4O (BDC)3]n |

|
684 m2 g−1 | 1476 | 609 | 0.2C | 200 | 0.29 | 35 | 0.6 | 317 |
| 0.42 cm3 g−1 | ||||||||||
| N/A | ≈1200 | 746 | 0.2C | 50 | ≈0.76 | 40 | 1.0 | 313 | ||
| Cu–TDPAT: [Cu3(TDPAT)(H2O)3]n |

|
1473 m2 g−1 | 820 | 745 | 1C | 500 | 0.02 | 40 | 1.2 | 308 |
| 0.55 cm3 g−1 | ||||||||||
In Li–S batteries, MOFs are mostly employed as a sulfur host material. In this regard, it is important to introduce sulfur properly into the pores of a MOF which is commonly realized by melt diffusion into an activated MOF material. In some cases, vapor phase infusion,318 infiltration of sulfur dissolved in CS2313 or encapsulation of sulfur nanoparticles by MOF synthesis in solution314 have been used. Wang et al. observed a much lower cycling stability, if they use HKUST-1 (copper benzene tricarboxylate)305 or ZIF-8 (zeolitic imidazolate framework)315 as MOF-based additives by just mechanically mixing them with sulfur instead of applying the melt diffusion process, suggesting sulfur confined inside the framework pores as a key aspect for increasing the cycle life. In line with these conclusions, the ability of the positive electrode to confine LiPSs was found to be more relevant than the electrode conductivity by comparing a Ni–MOF to an isostructural Co–MOF.306 While these MOFs only differ in the metal ions, the interaction of the Ni ions and LiPSs is stronger than that for Co ions as investigated by DFT calculations leading to improved cycling performance for the Ni–MOF host (under similar initial capacities) even though the electronic conductivity of the Co–MOF is higher. This finding suggests that performance enhancement due to electrocatalytic processes related to enhanced charge transfer, as reported for other metal-containing materials discussed in this review, does not apply to MOF hosts.
As mentioned earlier, the electrical conductivity of MOFs is generally very low. Therefore, it is assumed that MOF host cathodes are based on electron tunneling through an insulating layer with a thickness of several nanometers to a conductive carbon matrix.304 Thus, a threshold amount of conductive additive or the use of MOF/conductive matrix composite materials is necessary. Electrochemical processes involving charge transfer only occur near the interface of MOF particles and conductive material, where electrons, sulfur and Li+ ions from the electrolyte are available.312 A rotating-ring disk electrode (RRDE) study on the mechanism of the conversion reaction in Li–S batteries conducted by Lu et al. reveals how the MOF host electrodes may possibly work.319 They show that the electrochemical steps of the sulfur reduction exhibit fast reactions kinetics with 4 to 5 transferred electrons accounting for about one quarter of the total capacity. The complete conversion can be only achieved via chemical reactions, such as disproportionation and chain growth, which reform the electrochemically reducible LiPS species and exhibit slow reaction kinetics. In this respect, low-dielectric solvents as 1,3-dioxolane/dimethoxyethane mixtures and the related poor stabilization of certain ionic species play a significant role. Likely, the electrochemical processes occur near the MOF/conductive material interface while the chemical processes can also occur further away utilizing the electronically uncontacted sulfur located in the host matrix. However, the strong confinement of LiPSs in the MOF host ensures that the chemical conversion steps occur at the cathode. Thus, re-generated reducible LiPSs diffuse to interfaces at the conductive material where such species are consumed by electrochemical reduction during discharge. In other words, LiPSs diffuse following the concentration gradient within the MOF host to the electrochemical reaction interface while the competing diffusion process to the bulk electrolyte outside the host matrix is suppressed due to the stabilizing interactions between LiPSs and the MOF matrix. Accordingly, in various articles, an initial fade in capacity over the first cycles is ascribed to sulfur on the outer surface of MOF crystals which was not introduced into the pores and therefore causes un-confined LiPS.306,313,315 Nevertheless, an activation process with increasing capacity in the initial period until reaching a maximum also often occurs and is attributed to proceeding wetting of the MOF interior by dissolved LiPSs.306,312,320,321
The mechanistic understanding also explains further characteristics observed in investigations on MOFs as sulfur hosts. For instance, cathode composites made from MOFs gown on CNTs showed higher capacities, especially at high current rates, compared with conventional mixed sulfur-infused MOFs/CNT positive electrodes.313,316 The MOF/conductive additive conjunction and thus the contact area determine the capacity at certainly high enough current rates at which the kinetical limitation caused by slow chemical reactions restricts sulfur utilization. According to the described mechanism, the rate capability is enhanced, if a high interfacial area of the sulfur-hosting MOF phase and electron conducting phase is provided and short diffusion lengths are realized, ensuring fast transport of LiPSs to further sulfur species inside the MOFs for chemical reactions as well as fast transport of re-formed reducible LiPSs to the electron transferring interface. Thus, improved capacity and rate capability are obtained for smaller MOF crystal sizes or an increased amount of conductive additive.304,312,313,320 Furthermore, Zhou et al. reported that the considerably varying charge transfer resistance for different MOF hosts does not affect the performance of Li–S batteries.312 This observation emphasizes the rate-determining role of chemical reactions and transport in the inner MOF pores further off the interface. In conclusion, the proper functioning of a sulfur electrode based on a MOF host material especially relies on the superior trapping ability of MOF pores enabling high capacity by confining soluble chemically reactive LiPSs and re-formed reducible LiPSs close to both the MOF-based host matrix and the electrochemical reaction interface. The physical and chemical LiPS-trapping abilities of the MOF structure are able to prevent LiPS leakage even in the presence of large quantities of such species due to increased and fast formation of LiPSs, which have to undergo slow chemical reactions to provide for further discharge. Therefore, an excellent recovery after applying high current rates can be achieved.
Besides the physical confinement of LiPSs in MOF pores, Wang et al. intended to make use of the Lewis acidic function of coordinatively unsaturated (open) Cu2+ sites of a well-known copper benzene tricarboxylate (Cu-BTC) framework (HKUST-1, Table 5) to bind LiPS anions.305 The initial capacity of ≈1500 mA h g−1 decreased to 500 mA h g−1 after 50 cycles at 0.1C and remained at around 500 mA h g−1 for another 120 cycles, corresponding to an overall degradation rate of 0.4% per cycle. Later, it was also shown that a high density of Cu-rich surface defects improves the capacity and the long term stability.322 A comprehensive study on Ni–BTB–BP (BTB = benzene-1,3,5-tribenzoate; BP = 4,4′-bipyridyl), a MOF with a high pore volume of 2.15 cm3 g−1 and well-connected meso- (diameter: 2.8 nm) and micropores (diameter: 1.4 nm), was reported by Xiao and co-workers.306 Ni–BTB–BP with Ni2+ centers coordinates LiPS anions as axial ligands achieving a degradation rate of 0.11% per cycle for 100 cycles at 0.1C. XPS measurements revealed a lowered binding energy of Ni2+ due to interaction with LiPS anions, while DFT investigations showed that a sulfur atom on one end of the LiPS chain coordinates to Ni2+ centers of the MOF with binding energies increasing with the chain length.306 By computational screening of 16 metal-substituted analogues of MOF-74 (with a 2,5-dioxido-1,4-benzenedicarboxylate base), which are known for the highest density of open metal sites, a Ni–organic framework (MOF-74 (Ni)) was identified as a promising sulfur host regarding the ability to anchor Li2S4 and Li2S species.323 As seen in Fig. 12, the sulfur atoms of the LiPSs interact with the metal ion centers of MOFs while terminal Li atoms are localized adjacent to oxygen atoms which are the nearest neighbors of the unsaturated metal sites. As the interactions of LiPSs and the MOFs are much stronger than that of elemental sulfur and the MOFs, Lewis acid–base interactions are assumed for LiPSs and van der Waals interactions for S8.323 Accordingly, the shifting of the S2p signal to lower energies in XPS measurements, a higher sublimation temperature of sulfur, and color changes of the infused MOF powders in experimental investigations have been reported for sulfur–MOF composite cathodes.304,305,308,317,318 Wang et al. investigated the effect of the number of available Lewis acidic sites.307 They used mixed metal–organic frameworks (MMOFs) consisting of Zr6(OH)4O4 clusters linked by porphyrin ligands which then can contain additional metal ions chelated by planar N atoms of the porphyrin molecules. Thus, three MOF compounds only differing in the porphyrin center were tested as sulfur hosts providing no, one (FeCl) or two (Cu2+) Lewis acidic sites. For the Fe- and Cu-containing MMOFs, high cycling stability, rate capability and recovery after applying higher C-rates were obtained. Yet, Cu2+ and its two Lewis acidic sites per ion were shown to be superior to FeCl and achieved a capacity degradation of 0.07% per cycle from the 10th to the 200th cycle at 0.5C with a reversible capacity of 704 mA h g−1. In addition to MOFs, the Lewis acidic sites of other coordination compounds, such as Na2Fe[Fe(CN)6],324 a Prussian blue analogue, make these compounds interesting as sulfur host materials.324,325
 | ||
| Fig. 12 Lowest energy structures for adsorbed (a) S8, (b) intact and (c) dissociated Li2S4, and (d) Li2S in MOF-74 (Ni) investigated by DFT calculations. Purple, red and black spheres represent Ni, O, and C atoms in the MOF, and blue and yellow represent Li and S.323 (a–d) Reproduced with permission from ref. 323. Copyright 2017, American Chemical Society. | ||
Besides the Lewis acidic functionality, the LiPS trapping capability of MOFs can be tuned by introducing Lewis base properties due to the organic linker molecules. Park et al. compared isostructural zirconium–organic frameworks MOF-867 (Table 5),310 achieving 790 mA h g−1 reversible capacity, and UiO-67, achieving only 600 mA h g−1, which are based on a similar linker comprising two sp2 nitrogen atoms (MOF-867) or no nitrogen atoms (UiO-67). The same trend was observed for IRMOF-10 compounds with and without N-containing linkers. In an in situ spectroelectrochemical investigation, the adsorption intensities for the N-containing MOF-867 host cathode increased during discharge and returned to their initial intensities during charging while the adsorption intensities of the UiO-67 cathode remains unchanged during the whole time. XPS and FTIR measurements provided further proof for the Lewis acid–Lewis base interactions of Li ions of Li2S4 and sp2-hybridized nitrogen atoms of the organic ligand. The concept of Lewis base ligands for chemical adsorption of LiPSs was also applied to functional separator coatings311,326,327 in Li–S batteries, e.g. using a 2D coordination framework comprising phosphate groups.311 Regarding MOFs as a sulfur host material, the combination of both open metal sites and N-containing linkers in the cage-like Cu–TDPAT (TDPAT = 2,4,6-tris(3,5-dicarboxylphenylamino)-1,3,5-triazine) framework (Table 5) achieved an outstanding cycling stability with a reversible capacity of 745 mA h g−1 at 1C after 500 cycles, corresponding to a degradation rate of ≈0.02% per cycle.308 The MOF host material was filled with ≈50 wt% of sulfur which results in a sulfur content of 40 wt% in the cathode (excluding the current collector) and a sulfur loading of 1.2 mg cm−2.
Zhou et al.312 reported that the capacity fading in Li–S batteries employing MOF hosts had seem to be directly related to the aperture of the pores with enhanced stability for smaller “pore entrances” (ZIF-8 (Zn): 3.4 Å,328 MOF-5 (Zn): 8.0 Å,329 MIL-53 (Al): 8.5 Å,330 and HKUST-1 (Cu): 9.0 Å331). However, the examined MOF materials also differ in other properties, e.g. structure type, metallic center, linker molecules and crystal size. Moreover, in contrast, Mao et al. observed a reduced cycle life for ZIF-8 (Zn) and MOF-5 (Zn) compared to HKUST-1 (Cu) based electrodes associated with more sulfur dispersed on the external MOF surface of ZIF-8 (Zn) and MOF-5 (Zn) crystals ascribed to obstructed sulfur infiltration during material processing due to small pore apertures.313 Thus, the comprehensive understanding of the influence of the size of MOF pore windows remains unclear at this time. Concerning the particle size of MOF host materials, an optimum size of 200 nm was found for ZIF-8 balancing capacity and cycling stability (Fig. 13).320 Opposing size dependencies are observed for these properties, as a high capacity depends on high sulfur utilization during the conversion reactions while high cycle life requires moderate crystal sizes to diminish the significance of leaching of sulfur species at the external crystal surface. Morphological and also structural properties may also play a significant role regarding the potential sulfur loading.
 | ||
| Fig. 13 ZIF-8 (Zn) samples with different crystal sizes displaying (a–e) SEM images, (f) XRD patterns and (g) statistical results of the performance as a sulfur host material in Li–S batteries at 0.5C.320 Reproduced with permission from ref. 320. Copyright 2015, The Royal Society of Chemistry. | ||
As elucidated when describing the mechanism of conversion in MOF-based sulfur cathodes, interfacial processes at the conductive component profoundly affect the rate capability and the battery capacity. For instances, Mao et al. fabricated self-standing, binder-free cathodes by introducing sulfur into MOF crystals synthesized by chemical conversion of metal hydroxide entities at a 3D conductive network of CNTs.313 Employing HKUST-1 (Cu), with a sulfur loading of 1 mg cm−2, an initial capacity of 1263 mA h g−1 at 0.2C is achieved with a fading rate of 0.08% per cycle over 500 cycles and excellent recovery to 1102 mA h g−1 after applying current rates up to 10C. For increased electrode thickness leading to a sulfur amount of 11.33 mg cm−2 (68 wt%), the areal capacity equals 7.45 mA h cm−2 corresponding to a gravimetric capacity of 658 mA h g−1. As also observed for a ZIF-8(Zn)/MWCNT electrode,316 the conjunction provided between the MOF crystals and interpenetrating CNTs is a key feature of this kind of composite allowing for high battery performance due to proper adhesion and a large number of connection points.
Another successful strategy to increase the area for interfacial charge transfer is to wrap MOF particles with conductive materials.305,315,318,324,332–334 At the same time, this approach may further hinder the leaching of LiPSs and thus improve the cycling stability.324,332 Zhao et al. wrapped MIL-101 (Cr) crystals with graphene sheets achieving higher discharge capacity with smaller polarization.332 For MIL-100 (V)/rGO nanosheets, the main advantage of the composite compared to MIL-100 (V) is the rate performance.318 By wrapping Na2Fe[Fe(CN)6] crystals with the conducting polymer PEDOT, the initial capacity at 0.1C increased from 1020 mA h g−1 to 1291 mA h g−1 with a degradation rate of 0.15% per cycle over 100 cycles for high sulfur loadings of 64–66 wt% in the electrode.324 As investigated by EIS, the PEDOT coating reduces the charge transfer resistance, thus enabling high sulfur utilization even at high sulfur loading.
In conclusion, many MOFs and comparable coordination compounds exhibit exceptional ability to demobilize LiPSs because of high porosity in combination with small pore/window sizes as well as the Lewis acid function of open metal sites and the Lewis base function of organic linker molecules. Thus, MOF materials applied in Li–S batteries can greatly enhance their cycle life and enable the further reduction of formed LiPS species during the discharge. Due to the low conductivity or rather the non-conductive nature of MOFs, the development of MOF/conductive network composite structures and the improvement of the interfacial processes between these two components are crucial to boost the cell capacity, especially at high sulfur loading, and rate capability. The combination of discussed approaches may represent a promising starting point for further progress in the field, respectively applying tailored MOFs with suitable linker and unsaturated metal center chemistry, wrapping MOF particles with conductive sheet materials and growing MOFs on conductive matrix materials—perhaps even beyond carbon. The suitability of certain MOFs to be used for sulfur electrodes is further related to the chemical and electrochemical stability of the respective MOF compound. Recently, a comprehensive study on MOF-5 as a sulfur host material revealed that a large decrease of the capacity during the first cycles is caused by an initial electrochemical process which irreversibly oxidizes part of the active sulfur via its reaction with carbonate groups to form passive sulfate species.317 Similar large initial capacity decays and XPS signals corresponding to a sulfate-like environment were also reported for other MOFs based on carboxylate linkers.304,305,307 In addition to performance enhancement, a decisive role for the potential commercial application of MOFs in Li–S batteries is expected for the development of cost-effective and scalable methods to produce MOF materials tightly adhering to conductive components, e.g. electrodeposition335 and in situ synthesis on metal oxide support surfaces under hydrothermal conditions.336
6. Metals
This section examines metallic components influencing the electrochemical performance of Li–S batteries. Metals as chemical elements, alloys or metallic allotropes of non-metals or metalloids are characterized by typical physical properties, such as high electrical and thermal conductivity, corresponding to a certain type of chemical bonding. Accordingly, metallic bonding is based on electrostatic forces between metal cations forming a metal lattice and delocalized electrons forming an electron cloud or, in a more detailed way, by the band model considering orbitals. Regarding their chemical properties, metals are able to form e.g. sulfidic metal compounds based on covalent bonds or coordination compounds like MOFs, which are already discussed in Section 5. Thus, the issue of the electrochemical stability of metals is important not only in regard of electrode stability, but also in regard of the possible formation of metal compounds due to side reactions during battery operation. For instance, metal sulfides on the surface of metal components may influence cycling stability, electrocatalytic activity and capacity contributions from corresponding side reactions.The advantageous effects of metals on the performance of Li–S batteries are discussed to rely mainly on the electrocatalysis of the LiPS conversion as well as on the adsorption and confinement of LiPS.337–352 Furthermore, the influence on the morphology of insoluble Li2S deposits may play an important role in cathode stability.347,348,353 Several studies on metal/carbon composite sulfur electrodes also address the significance of interactions of the carbon matrix with metallic parts e.g. in metal nanoparticle/graphene host materials.341,354,355 Remarkably, Li–S batteries employing nickel with a high sulfur loading corresponding to 40 mg cm−2 achieved a reversible capacity of about 670 mA h g−1 after 100 cycles at 0.2C.356 The positive electrode of the battery consists of a catalytic carbon-coated Ni foam current collector and a Li2S6-catholyte. Hence, the active material is initially present in the form of diluted LiPS subsequently taking part in the conversion reaction. Further concepts to apply metal components in sulfur electrodes comprise metal/carbon composite host materials and separator coatings, and metal additives along with metal in the form of electrode decoration or dopants. As summarized by Table 6, research activities include mainly nickel and cobalt-based materials as well as noble metals like platinum and metallic main group elements. Accordingly, in this section, the role of metal current collectors is described followed by metal components based on nickel, cobalt and further metals or metal alloys.
| Way of metal employment | Initial capacity [mA h g−1] | Reversible capacity [mA h g−1] | Current ratea | Cycle number | Degradation rate per cycle [%] | Sulfur contentb [wt%] | Sulfur loading [mg cm−2] | Ref. |
|---|---|---|---|---|---|---|---|---|
| a 1C = 1674 mA g−1. b Mass percentage of sulfur on the whole cathode excluding the Al or Ni substrate. c The capacity/degradation rate is estimated from the figure since authors did not provide the specific number in the paper. d Mass percentage of sulfur on the cathode including the nickel foam current collector with a carbon shell. e NP = nanoparticle. f CNT = carbon nanotube. g rGO = reduced graphene oxide. h To activate the electrode, a lower C-rate was applied for a few initial cycles. | ||||||||
| Ni foam current collector, Li2S8 catholyte | ≈1080c | ≈870c | 0.1C | 50 | ≈0.39c | N/A | 0.152 | 337 |
| C-coated Ni foam current collector, Li2S6 catholyte | 1024 | 669 | 0.2C | 100 | 0.35 | 60d | 40 | 356 |
| S-NPe on Ni foam current collector | ≈990c | 775 | 0.5C | 200 | ≈0.1c | N/A | 0.84 | 357 |
| S/Ni composite as active material | 1469 | 758 | 0.5C | 200 | 0.24 | 29 | 0.8–1.1 | 358 |
| NiSx-alloy-coated S/Ni on Ni foam current collector | 1029 | 800 | 0.167C | 100 | 0.22 | N/A | 3.68 | 359 |
| Ni-NP/graphene/N-doped CNTf Li2S6 catholyte | 1150 | 908 | 0.5C | 100 | 0.21 | 50 | 0.81 | 340 |
| Ni-NP/graphene host material | 1092 | 832 | 0.2C | 500 | 0.05 | 49 | 1.0–1.5 | 354 |
| Co–N-doped graphitic C host material | 1137 | 930 | 0.2C | 300 | 0.06 | 29 | 1.4 | 341 |
| Cellular Co-NP/N-doped C host material | 685 | 514 | 2C | 850 | 0.03 | 94 | 3.6 | 345 |
| Co/N-doped C nanofiber/rGOg separator coating | 865 | 616 | 0.5C | 500 | 0.05 | 78 | 1.0–1.2 | 350 |
| Pt-NP/C host material | 1158 | 575 | 0.5C | 200 | 0.25 | 39 | 1.0 | 348 |
| Pd3Co-NP cathode additive | 648 | 544 | 1C | 200 | 0.08 | 60 | 1.13 | 360 |
| Ir/C separator coating | 1508 | 689 | 0.2C | 100 | 0.54 | 60 | 0.8 | 349 |
| Fe-NP/graphitic C host material | 980 | 500 | 0.8Ch | 450 | 0.11 | 56 | 1.2 | 361 |
| Cu-NP/C host material | 1050 | 630 | 0.06C | 500 | 0.08 | 40 | 1.0 | 362 |
| Au-NP/C host material | 1107 | 771 | 0.1C | 100 | 0.30 | 54 | 1.3 | 353 |
| Ti-particle film on the cathode | 1255 | 722 | 0.5C | 100 | 0.42 | 56 | N/A | 363 |
| Al-particle film on the cathode | 1257 | 977 | 0.5C | 100 | 0.22 | 56 | N/A | 363 |
| Te-doped S | ≈780c | 673 | 3Ch | 400 | 0.03 | 58 | 1.0–1.2 | 355 |
6.1 The role of metal current collectors in the positive electrode
In 2014, a detailed analysis of the literature on Li–S batteries revealed that only 6% of the publications deal with the issues of binder, separator or current collector materials while the vast majority of articles (64%) discuss thin film sulfur electrodes.364 However, as summarized in this section, there is a huge capability to increase the performance and the sulfur loading of Li–S batteries by using advanced metal current collectors for the positive electrodes.Comparing different current collector materials (nickel foam, carbon foam, non-woven carbon, and vertically aligned carbon nanotubes), Barchasz et al. observed a significantly increased discharge capacity and cycle life for nickel foam and attributed the effect to the high specific surface area and the stable morphology of the current collector.365 Following studies on nickel foam current collectors366 and interlayers367 also discussed the accommodation of active material and the corresponding internal electron transport network as well as the trapping of LiPS as reasons for the improvement. Similarly, the high relevance of the current collector morphology was demonstrated by Cheng et al. who realized increased sulfur loadings and improved sulfur utilization by using 3D aluminum foam/carbon nanotube scaffolds.368 The sulfur composite achieved an initial discharge capacity of 860 mA h g−1 with a sulfur loading of 7.0 mg cm−2 at 0.1C, while the commonly sulfur-flat aluminum foil cathode yields only 534 mA h g−1 for a mass loading of 4.61 mg cm−2. As a further current collector providing for an electron transport micro-network, interwoven stainless steel was investigated.369 Introducing only sulfur with no additional carbon additive or host material, the corresponding Li–S batteries showed a reversible capacity of 420 mA h g−1 after 250 cycles at 0.1C.
Regarding the metal used as a current collector material, Raguzin et al. found aluminum and platinum foils to be inert towards the electrochemical reactions in Li–S batteries with sulfur/carbon black cathode materials obtained by melt diffusion.370 However, in the voltage range of 1.0–3.0 V, nickel foil is electrochemically active (Ni(0) → Ni(I/II)3S2 → Ni(II)S) and contributes to the measured capacity resulting in etching and therefore a lower cycling stability and a voltage drop for nickel current collectors. Consequently, after 30 cycles, the assembled cell predominantly behaves as a Ni3S2/Li battery supplying a voltage of 1.4 V. Earlier studies on nickel foam current collectors also indicated the involvement of Ni in the electrochemical conversion reaction observing NiS on the foam surface after several cycles when discharged below 1.5 V.371,372 The effect of side reactions with nickel can be minimized by adding Si or SiO2 as dopants or narrowing the cut off voltage.370 Zhao et al. potentiostatically electrodeposited sulfur nanodots from a 0.1 M Na2S aqueous solution on a nickel foam and then applied the obtained composite as a positive electrode in Li–S batteries.357 Such devices achieved a reversible capacity of 775 mA h g−1 after 200 cycles at 0.5C in the smaller voltage range of 1.7–2.6 V for a comparably low sulfur loading of 0.84 mg cm−2.
A further important aspect of the role of metallic nickel in Li–S batteries was indicated by Hassoun et al. in 2012, when they reported enhanced electrode kinetics for thin nickel coatings on sulfur/carbon electrodes.373 Later on, Babu et al. provided a comprehensive study on the electrocatalytic activity of metals to enhance the reaction kinetics of LiPS conversion.337 For this purpose, 50 to 200 nm thick metal films of aluminum, gold, Ni or platinum were coated on stainless steel or aluminum foils by electron beam evaporation and employed as positive electrodes in Li2S8-catholyte-based Li–S batteries. Such a battery design consists of a Li- or Li+-containing negative electrode and an electronically conductive positive metal electrode and comprises sulfur in the form of dissolved LiPS in the electrolyte. Thus, the positive electrode is made up of a bare current collector and the catholyte with an active S-containing redox species. While Al-coated foils were found to be inactive for LiPS conversion, Pt- and Ni-coated electrodes showed electrocatalytic properties with increasing peak currents and stable peak positions in cyclic voltammograms for increasing scan rates, as well as reduced peak separation. In the voltage range from 1.5 V to 3.0 V, the best performance was obtained for a macroporous 3D nickel foam current collector achieving a reversible capacity of ≈900 mA h g−1 after 50 cycles at 0.1C with a Li2S8 concentration of 0.06 mol L−1 corresponding to a sulfur loading of 0.152 mg cm−2.337 Following studies on nickel foam current collector/LiPS catholyte electrodes demonstrated that the sulfur loading can be tremendously increased for such electrodes.356,374 By incorporating nickel foam into a carbon shell of interwoven CNTs entangled with a carbon nanofiber network, a 6 M Li2S6 catholyte corresponding to a sulfur mass loading of ≈40 mg cm−2 could be used to achieve an initial capacity of 1024 mA h g−1 (41 mA h cm−2) and a reversible capacity of 669 mA h g−1 (27 mA h cm−2) after 100 cycles at 0.2C in the voltage range of 1.7–2.8 V.356
In summary, metallic current collector materials for positive electrodes in Li–S batteries can enhance the performance and enable high sulfur loading mainly due to improved electron transport resulting from a suitable 3D morphology of highly conductive metals and electrocatalytic activity of certain metals, e.g. nickel, in LiPS conversion reactions. Possible side reactions with the metal may require to reduce the voltage range to achieve stable electrochemical characteristics over long cycle times. The possibly limited voltage range and larger mass of some metals have to be considered for the design of commercial Li–S batteries as they may cause a significant decrease in the energy density of the devices.
6.2 Metals as additives and metal/porous carbon hybrid scaffolds
Former studies on S/Ni composites, in which the electrodes were prepared from binder- and conductive carbon-containing slurries, also showed that interactions of nickel and sulfur lead to the formation of sulfide species like Ni2S3.338,358 Moreover, the presence of Ni fibers (3 wt%) changed the morphology of sulfur from smooth to rough agglomerated particles.338 Zhu et al. obtained a reversible capacity of 758 mA h g−1 at 0.5C after 200 cycles with a sulfur/RANEY® nickel alloy (incl. NixAlyOz) composite in the voltage range of 1.7–2.8 V, observing also better rate capability and subsequent capacity recovery as well as higher sulfur loading compared to a sulfur/carbon composite electrode.358
The electrocatalytic properties of metallic nickel have also been exploited by using Ni-decorated carbon materials as sulfur hosts in composites obtained by melt-diffusion.354,375 The capacity achieved employing MWCNTs with 27 wt% of Ni is higher than that achieved for MWCNTs without nickel decoration, especially if the current rate is increased.375 After 200 cycles at 0.5C a capacity of 545 mA h g−1 remained for a sulfur loading of 1.0 mg cm−2. It should be noted that metal residues and impurities caused by the synthesis of certain carbon materials, e.g. CNTs and MOF-derived carbons,376 may account for a considerable part of the performance enhancement of the carbon materials by affecting electrode kinetics.
Moreover, for metal/carbon composites, the carbon matrix may have a significant influence on the interactions between the metallic component and LiPSs. According to DFT calculations published by Yao et al.,354 the adsorption of sulfur clusters on nickel/graphene is stronger compared to a nickel slab surface while the adsorption on copper/graphene is weaker than that on copper, and the adsorption on tin/graphene is comparable to that on tin.354
As seen in Fig. 14, the graphene substrate can change the metal's valence band center by influencing the electron density distribution, thus tuning the metal–S interaction. While significantly affecting a transition metal with localized d states, the effect may be rather insignificant for a main group metal with extended p states. If defects are present in the graphene substrate, the metal–S interaction is more similar to that in a free-standing metal slab. Also, for such metal surfaces, the adsorption strength on Ni is higher than that on Cu or Sn. Smaller sulfur clusters show lower adsorption energies on all considered surfaces meaning that sulfur tends to form dispersive smaller clusters on metal surfaces rather than gathering into larger clusters. Experimentally, nickel nanoparticle (10 at%)/graphene employed as a sulfur host material achieved an initial capacity of 1092 mA h g−1 degrading at a rate of only 0.05% per cycle to a reversible capacity of 832 mA h g−1 after 500 cycles at 0.2C for a sulfur loading of 1.0–1.5 mg cm−2. As predicted by the DFT calculations, the capacities and rate capabilities are advantageous for Ni/graphene compared to Sn and Cu, with all three metal/graphene composites showing better capacity retention than bare graphene. Nickel nanoparticle/graphene composites were also used for positive Li–S battery electrodes containing sulfur in the form of a LiPS-catholyte.339,340,377 Mosavati et al. found higher discharging capacities for smaller nickel particles of 20 nm compared to 40 nm or 100 nm.377 After the 40th cycle, a passivation layer was observed on the particle surface, which appears to be thinner for nickel particle sizes of 40 nm and 100 nm and may consist of Li2S and NiSx.
 | ||
| Fig. 14 Calculated adsorption energies of sulfur clusters Sx (x = 1, 2, 4, and 8) on the surfaces of (a) metals and (b) metal/graphene systems, (c) energy level interactions between metal surfaces and sulfur clusters (blue, red and green bars denote the d-band or p-band centers of the Ni d band, Cu d band, and Sn p band; yellow bars denote the p orbitals of sulfur clusters) and (d) the stable configurations of sulfur clusters adsorbed on a nickel surface.354 (a–d) Reproduced with permission from ref. 354. Copyright 2018, American Chemical Society. | ||
In summary, the performance enhancement obtained with metallic nickel components may be related to the electrocatalytic properties of Ni towards the liquid–solid conversion of LiPSs, improved electrical conductivity, and trapping of LiPSs due to chemical adsorption and porous morphologies. Accordingly, several research studies observed decreased charge transfer resistances for Ni-containing materials in EIS studies,338,339,358,359,375 reduced polarization of Li–S batteries,339 and the decoloring of an LiPS solution due to an added Ni/carbon composite.340 Furthermore, we discussed several findings indicating the formation of nickel sulfides or alloys as an important aspect of the underlying mechanism of the improved nickel-based sulfur electrodes.
The majority of the materials is derived from the zeolitic imidazolate framework [Co(MeIm)2]n (MeIm = 2-methylimidazole),341–343,345,346,351,383 known as ZIF-67 (Co), a MOF with an open tetrahedral structure and a pore diameter of 11.6 Å.384 The carbonization of chemically precipitated ZIF-67 (Co) is performed with temperatures of 500–900 °C either under an inert atmosphere (N2 or Ar)341,342,345,351,383 or under reductive conditions (H2/Ar mixture)343,346 yielding composites of metallic cobalt nanoparticles and N-doped carbon matrices. After this pyrolysis step, the obtained composites show BET surface areas of around 200–300 m2 g−1,342,343,346,383 pore volumes of around 0.3 cm2 g−1,343,346 and a cobalt content of around 40 wt%,342,343,346 which was further reduced by chemical etching in some of the studies.341,343,345,351,383 He et al. reported etched uniform particles in a rhombic dodecahedral shape with sizes of around 350 nm which are made up of graphitic carbon co-doped with cobalt and nitrogen.341 After liquid infiltration of Li2S nanoparticles, a reversible capacity of 930 mA h g−1 and a degradation rate of 0.06% per cycle were determined after 300 cycles at 0.2C for a sulfur loading of 1.4 mg cm−2 and a sulfur content of 29 wt%. Li et al. achieved a capacity of 850 mA h g−1 and a capacity loss of 0.21% per cycle after 200 cycles at also 0.2C for a sulfur loading of 1.0 mg cm−2 and a sulfur content of 49 wt%.342 In this study, the Co nanoparticles embedded in the N-doped carbon polyhedrons were not removed by etching and sulfur was introduced by melt diffusion. Wrapping etched ZIF-67(Co)-derived polyhedrons with rGO nanosheets yielded a host material delivering a capacity of 949 mA h g−1 after 300 cycles at 0.18C corresponding to a degradation rate of 0.07% per cycle with 1.0 mg cm−2 sulfur.343 As extension to the combination with functional carbon materials, Liu et al. pyrolyzed and etched a bimetallic Co/Zn-ZIF assembled with GO sheets and poly(vinylpyrrolidone) (PVP) to synthesize N-doped porous carbon nanosheets with embedded cobalt nanoparticles (11 wt%) as a host material for sulfur and lithium,341 thus serving as both electrodes. With a specific surface area of 500 m2 g−1 and a pore volume of 1.0 cm3 g−1, these materials in a Li–S battery showed reversible capacities of 633 mA h g−1 and 619 mA h g−1 after 200 cycles at 1C and 2C for a sulfur loading of 0.8–1.0 mg cm−2. The issue of quite low sulfur loadings was addressed by the direct formation of ZIF-67(Co) onto the surface of CoAl layered double hydroxide (LDH) via in situ nucleation and directed epitaxial growth followed by thermolysis and etching of this sacrificial template. The obtained Co-nanoparticle/N-doped carbon composite showed a cellular morphology with a hierarchical micro-mesoporous honeycomb-like architecture (see Fig. 15a) and a specific surface area of 460 m2 g−1. For a sulfur loading of 3.6 mg cm−2 and a high sulfur content of 94 wt%, this host material achieved 514 mA h g−1 after 850 cycles at 2C corresponding to a degradation rate of only 0.03% per cycle. For a cathode with 7.5 mg cm−2 of sulfur, still, a capacity of about 400 mA h g−1 was achieved after 300 cycles at 1C, demonstrating high rate performance and cycling stability.345 Wang et al. realized a capacity of 679 mA h g−1 after 50 cycles at 0.5C for a sulfur loading of 5.2 mg cm−2 by using a battery design with a 10 μm ZIF-67(Co)-derived interlayer coated on the surface of the sulfur/carbon electrode.383
 | ||
| Fig. 15 (a) Schematic illustration and corresponding SEM images of the synthesis of cellular Co nanoparticle/N-doped carbon composites from CoAl-LDH templates and the ZIF-67 (Co) precursor,345 and (b) schematic illustration and TEM image of a ZIF-67 (Co)-derived cobalt nanoparticle/N-doped carbon composite and its interaction with LiPSs during charging and discharging.342 (a) Reproduced with permission from ref. 345. Copyright 2017, American Chemical Society. (b) Reproduced with permission from ref. 342. Copyright 2016, The Royal Society of Chemistry. | ||
Besides ZIF-67(Co), other cobalt-containing precursors have been used to synthesize both sulfur host materials344,352,378 and separator coatings.350 Zhang et al. used a pyrolysis process to obtain a porous 3D-matrix consisting of graphene nanosheets and MWCNTs with Co-nanoparticles (36.5 wt%) wrapped on the top of the nanotubes or distributed randomly on the graphene sheets using GO, urea and Co(NO3)2 salt as starting materials.378 Li–S batteries employing this sulfur host material achieved an initial capacity of 1374 mA h g−1, which decreased to 837 mA h g−1 over 200 cycles at 0.1C for a sulfur loading of 1.3–1.6 mg cm−2, and a reversible capacity of 336 mA h g−1 for a sulfur loading of 4.7 mg cm−2.
The positive effect of the metallic cobalt components on the performance of Li–S batteries is mainly ascribed to the electrocatalytic properties of cobalt and enhanced LiPS adsorption. Most reported materials contain nitrogen as a doping heteroatom besides cobalt. Based on XPS measurements, several groups identified pyridinic, pyrrolic and graphitic nitrogen in carbon matrices and furthermore observed both metallic and divalent cobalt.342,350,378,383 The thereby indicated interactions of Co, N and C atoms may strengthen the adsorption ability of the composites towards LiPSs and promote the conversion reaction as shown in Fig. 15b. According to DFT calculations, the adsorption energy for LiPSs follows the order C–Co–N > C–Co > C–N > C implying that C–Co–N serves as a conductive Lewis base matrix.341 The incorporation of N atoms and Co nanoparticles modulates the electron density of a carbon surface through a displacement of charge from Co atoms to other atoms nearby.344 The increased electron density of graphitic N atoms leads to the formation of bonds with Li atoms.351 In line with these findings, the color of Li2S4 or Li2S6 solutions faded when adding the Co/N-doped carbon composite materials.346,350,352,378 For a Co/N-doped carbon host material, Zhong et al. reported a degenerating rate performance of the corresponding Li–S batteries, if the Co/N ratio was higher or lower than 1:7.2.352 Furthermore, possible O-functional groups343,346,350,352,378 and cobalt sulfide layers343,346 may also affect the adsorption of LiPSs. Regarding the electrocatalytic properties of cobalt components, lower overpotentials343,352,383 and charge transfer resistances343,346,350,378 for the conversion reactions as well as smaller peak separation in cyclic voltammograms352,383 were observed for carbon materials with Co compared to similar materials without Co. It should be noted that the presence of cobalt during the thermolysis synthesis also affects the structure of the carbon component as it catalyzes graphitization. Therefore, the absence of cobalt components may not be the only difference in the resulting battery materials.
The performance achieved with cobalt composite materials can be further enhanced by using conductive scaffold materials like graphene and CNTs, which also additionally lowers the charge transfer resistance determined for the conversion reactions.343,344,351,379 The synergistic effects of N-doped carbon matrices and metallic cobalt nanoparticles or doping atoms as well as enhanced charge transport enable exceptionally low capacity fading over many cycles due to LiPS-adsorption and electrocatalytic properties. By thermolysis of suitable precursors like ZIF-67 (Co) and sometimes chemical etching, Co/N-doped carbon composites in the form of polyhedrons, nanorods, nanofibers and nanosheets were obtained and used as a sulfur host material or separator coating. For a possible commercial application of such materials, increasing the sulfur loading seems to be a crucial next step.
Tao et al. used a wet chemical method to decorate sulfur with porous Pt structures which prevent sulfur microparticles from agglomeration and grain growth during long-term aging.385 Due to the high morphological integrity and enhanced electrochemical reaction kinetics attributed to good electrical conductivity, a reversible capacity of 680 mA h g−1 was obtained after 80 cycles at 0.1C. Furthermore, the Pt-decorated sulfur has a higher tap density than pristine sulfur enabling a higher volumetric capacity. The role of Pt in the mechanism of the sulfur electrode in Li–S batteries was investigated more in detail by Thangavel et al. using a Pt/conductive carbon positive electrode containing 80 wt% of platinum powder and a 2 mM Li2S8-catholyte.347 The decreased overpotential and peak separation for the sulfur/sulfide conversion in cyclic voltammetry and reduced charge transfer resistance were attributed to the electrocatalytic function of platinum. Assumingly, the oxidation of the platinum surface encourages stronger interactions with LiPSs and involves Pt–S sulfidic bond formation. Potentiostatic chronocoulometric measurements accompanied by UV-Vis characterization show that the surface coverage on platinum is higher than that on carbon. Moreover, the Pt-catalyst leads to instantaneous nucleation and 3D growth, while progressive nucleation on carbon restricts to 2D growth of solid Li2S2/Li2S species as concluded from cyclic voltammograms and Avrami theory. Lin et al. obtained Li–S batteries with a reversible capacity of 503 mA h g−1 after 200 cycles at 0.5C using a commercial platinum nanoparticle/carbon composite as a sulfur host with a rather low platinum content of 1 wt%.348 XPS measurements proved chemisorptive interactions of platinum and LiPSs, while EIS investigations suggest that platinum promotes a more favorable deposition of Li2S2 and Li2S as the charge transfer resistance in the mid-frequency region commonly assigned to the properties of the polymeric-like SEI is decreased.
Performance enhancement accompanied by electrochemical data indicating the improvement of the electrode kinetics was also achieved with further group 8–10 transition metals, such as the Pd3Co alloy (15 wt% nanoparticle additive),360 iridium (10 wt% or 25 wt% nanoparticles on Ketjen black as a host or separator coating),349 and iron (nanoparticles embedded in N-doped CNFs mixed with graphene as a separator coating387 and Fe/Fe3C nanoparticles with a graphene shell on a cotton textile as a host).389 Zhang et al. sputtered aluminum or titanium on the surfaces of sulfur/carbon electrodes to realize an improvement due to enhanced electrical conductivity, filled interspaces and related confinement as well as improved electrode kinetics.363 As seen in Table 6, aluminium provided for better results than titanium, which might also be related to the higher amount of deposited aluminium. Magnetron sputtered aluminium was also used to decorate functional carbon interlayers between the sulfur electrode and the separator.386 3 wt% of gold nanoparticles was decorated onto a sulfur/carbon electrode by a wet chemical method resulting in a capacity of 771 mA h g−1 after 100 cycles at 0.1C.353 Considering DFT calculations and XPS measurements, the improved electrochemical performance and kinetics are attributed to the controlled deposition of LiPSs by mediation of gold nanoparticles which suppress the formation of thick aggregates of thus less active materials.353 For a carbon host decorated with 10 wt% of copper nanoparticles, the structural and binding energy data suggest the formation of copper sulfidic compounds as the underlying mechanism for the improvement of the corresponding Li–S batteries.362
In addition to metal additives and metal composite materials, another approach is to alter the electrochemical properties of sulfur by doping. Metallic tellurium powder was heated with sulfur in a sealed tube to establish 1–5 wt% Te-content changing the binding energy of sulfur and tellurium, shifting the TGA curve to higher temperatures but without affecting the XRD pattern and binding energy related to Te–Te bonds. The highly uniform doping is assumed to improve the electrical conductivity and redistribute the electron density of the sulfur sites to facilitate the lithiation/delithiation process, which was also demonstrated by first principles calculations. Te-doped sulfur/carbon electrodes provided a capacity of 673 mA h g−1 after 400 cycles at 3C corresponding to a degradation rate of 0.026% per cycle for a sulfur loading of 1.0–1.2 mg cm−2.355 Selenium also seems suitable as a doping element as a sulfur-rich S1−xSex/carbon composite (x ≤ 10) delivered a capacity of 1090 mA h g−1 after 200 cycles at 0.12C.388
In conclusion, several metals and alloys are suitable to catalyze the electrochemical conversion of sulfur or LiPSs to Li2S. The proper adsorption of LiPS intermediates on the surface of many metals constitutes an initial step of the electrocatalytic process and lessens the shuttle effect. For certain metals, the formation of metal compounds such as sulfides during charge and discharge may also significantly influence the surface electrochemistry of the metal-containing battery materials. There are several Li–S battery design concepts to involve metals as well as many synthetic approaches to obtain nanostructured metallic components and metal/carbon composites, making these materials very promising for high-performance electrodes in Li–S batteries.
7. Metal hydroxides
Nanostructured metal hydroxides with hydrophilic groups and a functional polar surface have been recently investigated as promising cathode host materials for Li–S batteries, such as Co(OH)2 nanosheets,390 Ni(OH)2 nanoparticles,125,391,392 Ni(OH)2 hollow spheres,393 Ni3(NO3)2(OH)4 shells,394 layered double hydroxides,395,396 and so on. In 2015, Nie et al. reported Co(OH)2 nanosheets as a conceptually new metal-containing nanostructured material to obstruct the LiPS shuttling and prolong the service life of Li–S cells.390 The positive electrode consists of a sulfur/conductive carbon black (S/CB) composite uniformly coated with Co(OH)2 nanosheets (S/CB@Co(OH)2). This novel S/CB@Co(OH)2 cathode with a protective Co(OH)2 layer provided higher capacities and better capacity retention, especially at high current rates, compared with a S/CB cathode without a Co(OH)2 coating used as a control cell (capacity retentions of, respectively, 71.2% and 20.2% after 200 cycles at 1C). The improved cell performance was attributed to the metal hydroxide coating which inhibits the shuttle diffusion of LiPS species by the effective entrapment/reutilization of the active material. Nie et al. also described a similar cathode concept but in this case the S/CCB composite surface was covered with Ni(OH)2 nanoparticles (1–2 nm) instead of Co(OH)2 nanosheets.391 After 200 cycles at 1C, the prepared S/CB@Ni(OH)2 cathode showed a capacity retention of around 70%, with the initial and ending capacities of, respectively, 810 and 590 mA h g−1. Interestingly, these values of specific capacities and capacity retention are very similar to those obtained previously with a S/CB@Co(OH)2 cathode.390 A particular effect of different metal atoms (i.e. Co or Ni) in the hydroxide nanomaterial may play a rather unspecific role in the interaction with sulfur-based species. Jiang et al. proposed a Ni3(NO3)2(OH)4 shell to effectively encapsulate sulfur in the form of a S/CB composite and expand the lifespan of the cathode.394 This thin-layered Ni-based hydroxide is able to irreversibly react with Li+ ions during the initial discharge/charge cycles to further form a stable and shelly (Li, Ni)-mixed hydroxide protective film onto the S/CB composite (Fig. 16a). The formed thin film with functional polar/hydrophilic groups offers a good permeability to Li+ and at the same time serves as a chemical anchor layer for LiPSs. As a result, an advanced hybrid cathode (sulfur content = 62.4%; sulfur loading = 1.8–2.5 mg cm−2) with a high reversible capacity of ≈1250 mA h g−1 and a high coulombic efficiency of ≈98% after 500 cycles at 0.2C is achieved. In contrast, the S/CB composite in the absence of the Ni-based hydroxide exhibits both a low capacity and a low coulombic efficiency of, respectively, ≈200 mA h g−1 and ≈52% after only 300 cycles (Fig. 16b). Lou's group suggested an interesting sulfur host based on double-shelled nanocages with Co(OH)2 and Ni, Co-based layered double hydroxides as, respectively, inner and outer shells (denoted as CH@Ni,Co-LDH).395 The outer LDH shell is a class of synthetic anionic clay with a 2D lamellar structure whose chemical formula, based on the used molar Ni/Co ratio of 1:2, is expressed as [Ni2+1/3Co3+2/3(OH)2][NO32−1/3]·mH2O. The as-obtained CH@Ni,Co-LDH composite with a hollow polyhedral structure and a specific surface area of 117 m2 g−1 is able to accommodate a sulfur content of 75 wt%. The resulting cathode with a high sulfur loading of 3 mg cm−2 (sulfur content of the whole cathode = 52.5 wt%) demonstrated stable cyclability at both 0.1 and 0.5C, with ending capacities of, respectively, 653 and 491 mA h g−1 at the 100th cycle and corresponding capacity degradation rates of 0.356 and 0.343% per cycle. The good performance of the cathode was ascribed to the structure of the novel hydroxide-based host capable of accommodating a large amount of active sulfur material and its singular hydroxy-functionalized polar surfaces with strong binding affinity to LiPSs; however the latter was not experimentally demonstrated in this work. Recently, Zhang and co-workers also proposed the use of layered double hydroxides, but in this case the authors engineered Ni,Fe-layered double hydroxides (size < 5 nm) embedded in an N-doped mesoporous graphene framework (Ni,Fe-LDH@NG) serving as “sulfophilic” and “lithiophilic” components, respectively.396 Interestingly, the subtle Ni,Fe-LDH@NG composite was coated on one side of a commercial polypropylene (Celgard 2400) separator instead of adding it to the sulfur cathode. Despite the use of a simple ball-milled sulfur/carbon cathode with a sulfur content of 63 wt% and a high areal sulfur loading of 4.3 mg cm−2, the cell with the Ni,Fe-LDH@NG-coated separator (added coating mass of 0.3 mg cm−2) revealed a high initial capacity of 1078 mA h g−1 (areal capacity = 4.6 mA h cm−2) at a current density of 1 mA cm−2; while the reversible capacity, after 100 cycles, was sustained at 800 mA h g−1 (3.4 mA h cm−2). In contrast, the reference cell with a pristine separator showed a capacity of 400 mA h g−1 (1.5 mA h cm−2) after only 60 cycles, under similar cell conditions (Fig. 16c). The stable cell operation at such a high sulfur loading was attributed to the cooperative “sulfophilic” and “lithiophilic” domains of the Ni,Fe-LDH@NG complex which cooperatively chemisorbs LiPS intermediates by either “lithiophilic” (via Li–N bonds) or “sulfophilic” (via S–Fe bonds) interactions and catalyzes efficiently interfacial redox reactions, as it was supported by XRD and XPS studies. The cooperative interface of Ni,Fe-LDH@NG is schematically illustrated in Fig. 16d. As a somewhat exotic system, a MgBO2(OH)/CNT composite was used to functionalize a usual Celgard 2400 separator and enabled high sulfur retention, rapid redox kinetics and Li+ ion transport along with a high mechanical stability, especially at elevated temperatures of up to 140 °C.397
 | ||
| Fig. 16 (a) A schematic illustration showing the working mechanisms of NNH in the sulfur cathodes. (b) Cycling performance of cathodes with and without the (Li, Ni)-mixed hydroxide denoted as S8@CB@NNH and S8@CB, respectively. The inset shows their utilization ratio of active sulfur.394 (c) Cycling performance of cells with pristine and LDH@NG-coated separators. (d) A scheme showing the cooperative interface of LDH@NG, where the adsorption and redox of LiPSs are facilitated by the binding of Li and S surface species.396 (a and b) Reproduced with permission from ref. 394. Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/). (c and d) Reproduced with permission from ref. 396. Copyright 2016, Wiley-VCH. | ||
Nanostructured metal hydroxides with abundant functional polar/hydrophilic groups have proven to improve the cycling performance of Li–S batteries. However, the working mechanism of the metal hydroxide in the sulfur cathodes was not clearly explained. The interactions between LiPS species and metal hydroxides with different morphologies and chemical properties should be further examined by combining direct experimental investigations and theoretical studies. Only then we can gain new insights and identify the actual effect of the novel materials and whether the discussion on how the materials work is reasonable.
8. Future prospects and conclusions
In this condensed review, we handled numerous metal-based materials which have already proved their high impact on each part of the secondary Li–S battery cells. Their use as additives to improve cathodes, anodes or separators as well as the possibility to form an active material in situ inside a battery gives a large scope to optimize Li–S batteries to a certain extent and improve the possibility to be successfully introduced into the market.Metal-based materials are typically polar, and they may effectively adsorb or even bind LiPS intermediates. However, the literature reports a huge number of metal-based compounds with different (nano)structures and surface chemistry to electrochemically convert LiPSs. It is a great challenge to figure out the most promising metal-based compounds to rationally design electrode materials for Li–S batteries since there are numerous key factors that influence the relationship between material properties and Li–S cell performance: (i) the surface polarity to adsorb/bind LiPS intermediates, (ii) the electrocatalytic effect of the material which may act as a redox mediator in the multielectron conversion chemistry of sulfur, (iii) the electrical conductivity of the material or the composite electrode, influencing the electron transport, the cell resistance and seemingly electron transfer, and (iv) the physical and morphological features (particle size, surface area, pore size, pore volume, etc.) that have a strong impact on the contact between the active phase and both the active material and the electrolyte as well as on LiPS confinement.
In each section we discussed concepts which show highly interesting results that, despite still being far away from the practical needs of a market-ready Li–S battery, are encouraging to go beyond classical concepts and try novel, innovative experiments that help to understand reaction and deactivation mechanisms. Especially, the latter will allow us to overcome certain problems of Li–S batteries to expand the lifetime and to improve energy densities or to overcome safety and reliability concerns.
Of course, a large variety of individual materials does not facilitate an early application and market introduction of a new battery chemistry, as Li–S still is. With a powerful world's scientific community, this mega challenge will be solved with the desired outcome, affordable commercial Li–S batteries.
Many reports with outstanding and promising high-performance Li–S cells are far from practical applications. One of the issues is related to the complex multistep method used to prepare the metal-based material and/or sulfur composite cathode, making it unattractive in terms of cost-benefit for large-scale industrialization. The second concern is the low areal sulfur loading of 0.5–2.0 mg cm−2 typically used in most of the publications. For practical Li–S cells, a cathode sulfur content > 80 wt%, sulfur loadings > 6 mg cm−2 and an electrolyte/sulfur ratio < 2 mL g−1 are required to provide competitive specific energies (≈500 W h kg−1) compared to high-voltage Li-ion cells. It is highly important that Li–S batteries operate at low electrolyte amounts, which is one of the most crucial parameters to achieve high energy density. To make the big jump from lab-scale to industrial-scale fabrication of Li–S batteries, several critical parameters should be considered: (i) sulfur content, (ii) sulfur areal loading, (iii) electrolyte/sulfur ratio, (iv) used electrolyte, (v) utilization of additive(s) and its concentration in the electrolyte (e.g.: LiNO3), (vi) applied current density, (vii) voltage window, and (viii) cell configuration. Furthermore, other significant parameters which are often not addressed in detail in the academic literature but are crucial for practical aspects should be considered: e.g. electrode thickness, type and mass of the substrate, porosity and surface area of the substrate and mass of the interlayer/coating layer if any.
Beyond all positive arguments for Li–S on behalf of the possible high performance and low cost in production and sales, attention should also be paid to the end of the use of this battery type. At the moment we are starting to recognize which unexpected impacts on the environmental system are accompanying hazardous waste. We should pay attention to applying environmentally friendly and harmless substances. They should be somehow biocompatible or not bioavailable where the latter might be quite challenging if nanoscale materials are used in the battery. This consideration is additionally of great importance, when considering accidents and release of highly active nanoscale compounds, by fire, crashes or other incidents. As these materials can also largely affect the environment, before an application, we should be aware of what really happens to biology in case of accidents and whether we can avoid this by carefully choosing the right components.
Additionally, we should be able to recycle Li–S batteries on a large scale. With probably expectable low price of a Li–S battery, the recycling of sulfur-based batteries may be easier and consequently cheaper than for classical Li-ion batteries. Since a first incineration step will directly evolve carbon and sulfur compounds, sulfur in particular can be recovered by typical gas scrubbing from the exhaust. The other residues are preserved in the incineration ash and can be, e.g., (electro)chemically and fractionally reprocessed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Agencia Nacional de Promoción Científica y Tecnológica (FONCYT-PICT no. 2017-0824) and Secretaría de Ciencia y Técnica de la Universidad Nacional de Río Cuarto (SECYT-UNRC) and the German Federal Ministry of Education and Research (BMBF) through the Excellent Battery – WING center ‘‘Batteries – Mobility in Saxony’’ (grant no. 03X4637C) for partially funding this work. Additional funding is gratefully provided by the European Union/European Regional Development Fund and the Free State of Saxony via the NaSBattSy project (SAB grant no. 100234960).Notes and references
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 RSC.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- M. Hagen, D. Hanselmann, K. Ahlbrecht, R. Maça, D. Gerber and J. Tübke, Adv. Energy Mater., 2015, 5, 1401986 CrossRef.
- P. Adelhelm, P. Hartmann, C. L. Bender, M. Busche, C. Eufinger and J. Janek, Beilstein J. Nanotechnol., 2015, 6, 1016–1055 CrossRef CAS PubMed.
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS PubMed.
- I. Kovalev, Y. Mikhaylik and T. Weiß, High energy rechargeable Li–S battery development at Sion Power and BASF, Li–S Workshop, Dresden, 2013 Search PubMed.
- R. V. Noorden, Nature, 2014, 507, 26–28 CrossRef PubMed.
- Tesla Model S, https://www.tesla.com/models, accessed September 2018.
- Audi e-tron, https://www.audi.com/en/models/e-tron.html, accessed September 2018.
- D. Zheng, G. Wang, D. Liu, J. Si, T. Ding, D. Qu, X. Yang and D. Qu, Adv. Mater. Technol., 2018, 3, 1700233 CrossRef.
- R. Xu, J. Lu and K. Amine, Adv. Energy Mater., 2015, 5, 1500408 CrossRef.
- Q. Wang, J. Zheng, E. Walter, H. Pan, D. Lv, P. Zuo, H. Chen, Z. D. Deng, B. Y. Liaw, X. Yu, X. Yang, J.-G. Zhang, J. Liu and J. Xiao, J. Electrochem. Soc., 2015, 162, A474–A478 CrossRef CAS.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969–A1976 CrossRef CAS.
- M. R. Busche, P. Adelhelm, H. Sommer, H. Schneider, K. Leitner and J. Janek, J. Power Sources, 2014, 259, 289–299 CrossRef CAS.
- Y. Yang, G. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018–3032 RSC.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- H. Kim, H.-D. Lim, J. Kim and K. Kang, J. Mater. Chem. A, 2014, 2, 33–47 RSC.
- Z. Li, Y. Huang, L. Yuan, Z. Hao and Y. Huang, Carbon, 2015, 92, 41–63 CrossRef CAS.
- J.-G. Wang, K. Xie and B. Wei, Nano Energy, 2015, 15, 413–444 CrossRef CAS.
- S. Wu, R. Ge, M. Lu, R. Xu and Z. Zhang, Nano Energy, 2015, 15, 379–405 CrossRef CAS.
- S. Rehman, K. Khan, Y. Zhao and Y. Hou, J. Mater. Chem. A, 2017, 5, 3014–3038 RSC.
- H.-J. Peng, J.-Q. Huang, X.-B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 7, 1700260 CrossRef.
- R. Fang, S. Zhao, Z. Sun, D.-W. Wang, H.-M. Cheng and F. Li, Adv. Mater., 2017, 29, 1606823 CrossRef PubMed.
- X. Chen, T. Hou, K. A. Persson and Q. Zhang, Mater. Today, 2018 DOI:10.1016/j.mattod.2018.04.007.
- L. Kong, H.-J. Peng, J.-Q. Huang and Q. Zhang, Nano Res., 2017, 10, 4027–4054 CrossRef CAS.
- J.-Q. Huang, Q. Zhang and F. Wei, Energy Storage Mater., 2015, 1, 127–145 CrossRef.
- L. Borchardt, M. Oschatz and S. Kaskel, Chem.–Eur. J., 2016, 22, 7324–7351 CrossRef CAS PubMed.
- S. S. Zhang, J. Electrochem. Soc., 2012, 159, A920–A923 CrossRef CAS.
- S. S. Zhang, Electrochim. Acta, 2012, 70, 344–348 CrossRef CAS.
- Y. Yang, G. Yu, J. J. Cha, H. Wu, M. Vosgueritchian, Y. Yao, Z. Bao and Y. Cui, ACS Nano, 2011, 5, 9187–9193 CrossRef CAS PubMed.
- Y. Fu and A. Manthiram, Chem. Mater., 2012, 24, 3081–3087 CrossRef CAS.
- W. Zhou, Y. Yu, H. Chen, F. J. DiSalvo and H. D. Abruña, J. Am. Chem. Soc., 2013, 135, 16736–16743 CrossRef CAS PubMed.
- J.-Q. Huang, Q. Zhang and F. Wei, Energy Storage Mater., 2015, 1, 127–145 CrossRef.
- X. Liu, J.-Q. Huang, Q. Zhang and L. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef PubMed.
- H. J. Peng, J. Q. Huang and Q. Zhang, Chem. Soc. Rev., 2017, 46, 5237–5288 RSC.
- D. Liu, C. Zhang, G. Zhou, W. Lv, G. Ling, L. Zhi and Q.-H. Yang, Adv. Sci., 2017, 5, 1700270 CrossRef PubMed.
- Z.-W. Zhang, H.-J. Peng, M. Zhao and J.-Q. Huang, Adv. Funct. Mater., 2018, 28, 1707536 CrossRef.
- M.-S. Song, S.-C. Han, H.-S. Kim, J.-H. Kim, K.-T. Kim, Y.-M. Kang, H.-J. Ahn, S. Dou and J.-Y. Lee, J. Electrochem. Soc., 2004, 151, A791–A795 CrossRef CAS.
- Y. J. Choi, B. S. Jung, D. J. Lee, J. H. Jeong, K. W. Kim, H. J. Ahn, K. K. Cho and H. B. Gu, Phys. Scr., 2007, 2007, 62 CrossRef.
- Y. Zhang, X. Wu, H. Feng, L. Wang, A. Zhang, T. Xia and H. Dong, Int. J. Hydrogen Energy, 2009, 34, 1556–1559 CrossRef CAS.
- X. Ji, S. Evers, R. Black and L. F. Nazar, Nat. Commun., 2011, 2, 325 CrossRef PubMed.
- S. Evers, T. Yim and L. F. Nazar, J. Phys. Chem. C, 2012, 116, 19653–19658 CrossRef CAS.
- V. Lapornik, N. N. Tusar, A. Ristic, R. K. Chellappan, D. Foix, R. Dedryvère, M. Gaberscek and R. Dominko, J. Power Sources, 2015, 274, 1239–1248 CrossRef CAS.
- R. Ponraj, A. G. Kannan, J. H. Ahn and D.-W. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 4000–4006 CrossRef CAS PubMed.
- K. Xie, Y. You, K. Yuan, W. Lu, K. Zhang, F. Xu, M. Ye, S. Ke, C. Shen and X. Zeng, Adv. Mater., 2017, 29, 1604724 CrossRef PubMed.
- Z. W. Seh, W. Li, J. J. Cha, G. Zheng, Y. Yang, M. T. McDowell, P.-C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331 CrossRef PubMed.
- Z. Liang, G. Zheng, W. Li, Z. W. Seh, H. Yao, K. Yan, D. Kong and Y. Cui, ACS Nano, 2014, 8, 5249–5256 CrossRef CAS PubMed.
- Z.-Z. Yang, H.-Y. Wang, L. Lu, C. Wang, X.-B. Zhong, J.-G. Wang and Q.-C. Jiang, Sci. Rep., 2016, 6, 22990 CrossRef CAS PubMed.
- Q. Sun, K. Chen, Y. Liu, Y. Li and M. Wei, Chem.–Eur. J., 2017, 23, 16312–16318 CrossRef CAS PubMed.
- K. Xie, Y. Han, W. Wei, H. Yu, C. Zhang, J.-G. Wang, W. Lu and B. Wei, RSC Adv., 2015, 5, 77348–77353 RSC.
- Q. Pang, D. Kundu, M. Cuisinier and L. Nazar, Nat. Commun., 2014, 5, 4759 CrossRef CAS PubMed.
- X. Tao, J. Wang, Z. Ying, Q. Cai, G. Zheng, Y. Gan, H. Huang, Y. Xia, C. Liang, W. Zhang and Y. Cui, Nano Lett., 2014, 14, 5288–5294 CrossRef CAS PubMed.
- J. Smith, F. Walsh and R. Clarke, J. Appl. Electrochem., 1998, 28, 1021–1033 CrossRef CAS.
- T. Lei, W. Chen, J. Huang, C. Yan, H. Sun, C. Wang, W. Zhang, Y. Li and J. Xiong, Adv. Energy Mater., 2017, 7, 1601843 CrossRef.
- Q. Fan, W. Liu, Z. Weng, Y. Sun and H. Wang, J. Am. Chem. Soc., 2015, 137, 12946–12953 CrossRef CAS PubMed.
- J. Guo, X. Zhang, X. Du and F. Zhang, J. Mater. Chem. A, 2017, 5, 6447–6454 RSC.
- H. Wei, E. F. Rodriguez, A. S. Best, A. F. Hollenkamp, D. Chen and R. A. Caruso, Adv. Energy Mater., 2017, 7, 1601616 CrossRef.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- N. Wiberg, A. Holleman and E. Wiberg, Inorganic Chemistry, Academic Press, San Diego, 101st edn, 2001 Search PubMed.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, J. Electrochem. Soc., 2015, 162, A2567–A2576 CrossRef CAS.
- X. Liang and L. F. Nazar, ACS Nano, 2016, 10, 4192–4198 CrossRef CAS PubMed.
- X. Wang, G. Li, J. Li, Y. Zhang, A. Wook, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 2533–2538 RSC.
- X. Liang, C. Y. Kwok, F. Lodi-Marzano, Q. Pang, M. Cuisinier, H. Huang, C. J. Hart, D. Houtarde, K. Kaup, H. Sommer, T. Brezesinski, J. Janek and L. F. Nazar, Adv. Energy Mater., 2016, 6, 1501636 CrossRef.
- Q. Zhang, Y. Wang, Z. W. Seh, Z. Fu, R. Zhang and Y. Cui, Nano Lett., 2015, 15, 3780–3786 CrossRef CAS PubMed.
- K. Luan, S. Yao, Y. Zhang, R. Zhuang, J. Xiang, X. Shen, T. Li, K. Xiao and S. Qin, Electrochim. Acta, 2017, 252, 461–469 CrossRef CAS.
- L. Ni, Z. Wu, G. Zhao, C. Sun, C. Zhou, X. Gong and G. Diao, Small, 2017, 13, 1603466 CrossRef PubMed.
- K. Cao, H. Liu, Y. Li, Y. Wang and L. Jiao, Energy Storage Mater., 2017, 9, 78–84 CrossRef.
- G. Radhika, R. Subadevi, K. Krishnaveni, W.-R. Liu and M. Sivakumar, J. Nanosci. Nanotechnol., 2018, 18, 127–131 CrossRef CAS PubMed.
- B. Campbell, J. Bell, H. Hosseini Bay, Z. Favors, R. Ionescu, C. S. Ozkan and M. Ozkan, Nanoscale, 2015, 7, 7051–7055 RSC.
- M. Xue, Y. Zhou, J. Geng, P. Zeng, Y. Xu, Y. Wang, W. Tang, P. Wu, S. Wei and Y. Zhou, RSC Adv., 2016, 6, 91179–91184 RSC.
- H. Tang, S. Yao, M. Jing, X. Wu, J. Hou, X. Qian, D. Rao, X. Shen, X. Xi and K. Xiao, J. Alloys Compd., 2015, 650, 351–356 CrossRef CAS.
- Y. Zhang, Y. Zhao, A. Yermukhambetova, Z. Bakenov and P. Chen, J. Mater. Chem. A, 2013, 1, 295–301 RSC.
- S. Wu, Y. Wang, S. Na, C. Chen, T. Yu, H. Wang and H. Zang, J. Mater. Chem. A, 2017, 5, 17352–17359 RSC.
- H. Cheng, S. Wang, D. Tao and M. Wang, Funct. Mater. Lett., 2014, 07, 1450020 CrossRef.
- H. Wang, T. Zhou, D. Li, H. Gao, G. Gao, A. Du, H. Liu and Z. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 4320–4325 CrossRef CAS PubMed.
- A. Iqbal, Z. Ali Ghazi, A. Muqsit Khattak and A. Ahmad, J. Solid State Chem., 2017, 256, 189–195 CrossRef CAS.
- Q. Qu, T. Gao, H. Zheng, Y. Wang, X. Li, X. Li, J. Chen, Y. Han, J. Shao and H. Zheng, Adv. Mater. Interfaces, 2015, 2, 1500048 CrossRef.
- B. Hu, L. Mai, W. Chen and F. Yang, ACS Nano, 2009, 3, 478–482 CrossRef CAS PubMed.
- L. Zhang, Y. Wang, Z. Niu and J. Chen, Carbon, 2019, 141, 400–416 CrossRef CAS.
- F. Sun, J. Wang, D. Long, W. Qiao, L. Ling, C. Lv and R. Cai, J. Mater. Chem. A, 2013, 1, 13283–13289 RSC.
- X. Tao, J. Wang, C. Liu, H. Wang, H. Yao, G. Zheng, Z. W. Seh, Q. Cai, W. Li, G. Zhou, C. Zu and Y. Cui, Nat. Commun., 2016, 7, 11203 CrossRef CAS PubMed.
- R. Wang, K. Wang, S. Gao, M. Jiang, M. Zhou, S. Cheng and K. Jiang, Nanoscale, 2017, 9, 14881–14887 RSC.
- H. Wu, Q. Tang, H. Fan, Z. Liu, A. Hu, S. Zhang, W. Deng and X. Chen, Electrochim. Acta, 2017, 255, 179–186 CrossRef CAS.
- Z. Li, N. Zhang, Y. Sun, H. Ke and H. Cheng, J. Energy Chem., 2017, 26, 1267–1275 CrossRef.
- H. Yao, G. Zheng, P.-C. Hsu, D. Kong, J. J. Cha, W. Li, Z. W. Seh, M. T. McDowell, K. Yan, Z. Liang, V. K. Narasimhan and Y. Cui, Nat. Commun., 2014, 5, 3943 CrossRef CAS PubMed.
- G. Zhou, Y. Zhao, C. Zu and A. Manthiram, Nano Energy, 2015, 12, 240–249 CrossRef CAS.
- Z. Zhang, Q. Li, S. Jiang, K. Zhang, Y. Lai and J. Li, Chem.–Eur. J., 2015, 21, 1343–1349 CrossRef CAS PubMed.
- L. Gao, M. Cao, Y. Q. Fu, Z. Zhong, Y. Shen and M. Wang, J. Mater. Chem. A, 2016, 4, 16454–16461 RSC.
- M. Yu, J. Ma, H. Song, A. Wang, F. Tian, Y. Wang, H. Qiu and R. Wang, Energy Environ. Sci., 2016, 9, 1495–1503 RSC.
- S. R. Azari, M. S. Rahmanifar, M. F. El-Kady, A. Noori, M. F. Mousavi and R. B. Kaner, J. Iran. Chem. Soc., 2017, 14, 2579–2590 CrossRef CAS.
- J.-Y. Hwang, H. M. Kim, S.-K. Lee, J.-H. Lee, A. Abouimrane, M. A. Khaleel, I. Belharouak, A. Manthiram and Y.-K. Sun, Adv. Energy Mater., 2016, 6, 1501480 CrossRef.
- G. D. Park, J. Lee, Y. Piao and Y. C. Kang, Chem. Eng. J., 2018, 335, 600–611 CrossRef CAS.
- J. Song, J. Zheng, S. Feng, C. Zhu, S. Fu, W. Zhao, D. Du and Y. Lin, Carbon, 2018, 128, 63–69 CrossRef CAS.
- M. Fang, Z. Chen, Y. Liu, J. Quan, C. Yang, L. Zhu, Q. Xu and Q. Xu, J. Mater. Chem. A, 2018, 6, 1630–1638 RSC.
- X. Shang, P. Guo, T. Qin, M. Liu, M. Lv, D. Liu and D. He, Adv. Mater. Interfaces, 2018, 5, 1701602 CrossRef.
- X. Zhao, Q. Li, T. Yu, M. Yang, K. Fink and X. Shen, Sci. Rep., 2016, 6, 19448 CrossRef CAS PubMed.
- Y. Chen, S. Choi, D. Su, X. Gao and G. Wang, Nano Energy, 2018, 47, 331–339 CrossRef CAS.
- H. Tang, S. Yao, S. Xue, M. Liu, L. Chen, M. Jing, X. Shen, T. Li, K. Xiao and S. Qin, Electrochim. Acta, 2018, 268, 158–167 CrossRef.
- Z. Li, J. Zhang and X. W. D. Lou, Angew. Chem., Int. Ed., 2015, 54, 12886–12890 CrossRef CAS PubMed.
- J. Zhang, Y. Shi, Y. Ding, W. Zhang and G. Yu, Nano Lett., 2016, 16, 7276–7281 CrossRef CAS PubMed.
- Y. Li, D. Ye, W. Liu, B. Shi, R. Guo, H. Zhao, H. Pei, J. Xu and J. Xie, ACS Appl. Mater. Interfaces, 2016, 8, 28566–28573 CrossRef CAS PubMed.
- W. Sun, X. Ou, X. Yue, Y. Yang, Z. Wang, D. Rooney and K. Sun, Electrochim. Acta, 2016, 207, 198–206 CrossRef CAS.
- Y. Guo, N. T. Wu and Y. Zhang, ECS Trans., 2017, 75, 141–150 CrossRef CAS.
- L. Ni, G. Zhao, G. Yang, G. Niu, M. Chen and G. Diao, ACS Appl. Mater. Interfaces, 2017, 9, 34793–34803 CrossRef CAS PubMed.
- G. Yuan, H. Jin, Y. Jin and L. Wu, J. Solid State Electrochem., 2017, 22, 693–703 CrossRef.
- Y. Li, B. Shi, W. Liu, R. Guo, H. Pei, D. Ye, J. Xie and J. Kong, Electrochim. Acta, 2018, 260, 912–920 CrossRef CAS.
- M. He, P. Zuo, H. Zhang, J. Hua, Y. Ma, C. Du, X. Cheng, Y. Gao and G. Yin, Electrochim. Acta, 2018, 259, 440–448 CrossRef CAS.
- M. Chen, Q. Lu, S. Jiang, C. Huang, X. Wang, B. Wu, K. Xiang and Y. Wu, Chem. Eng. J., 2018, 335, 831–842 CrossRef CAS.
- Z. Li, R. Soc. Open Sci., 2018, 5, 171824 CrossRef PubMed.
- X. Chen, L. Yuan, Z. Hao, X. Liu, J. Xiang, Z. Zhang, Y. Huang and J. Xie, ACS Appl. Mater. Interfaces, 2018, 10, 13406–13412 CrossRef CAS PubMed.
- M. Xiang, H. Wu, H. Liu, J. Huang, Y. Zheng, L. Yang, P. Jing, Y. Zhang, S. Dou and H. Liu, Adv. Funct. Mater., 2017, 27, 1702573 CrossRef.
- Y. Song, W. Zhao, X. Zhu, L. Zhang, Q. Li, F. Ding, Z. Liu and J. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 15733–15741 CrossRef CAS PubMed.
- R. Tang, X. Li, Z. Ding and L. Zhang, RSC Adv., 2016, 6, 65162–65170 RSC.
- L. Liu, Q. Yang, M. Jiang, S. Wang, B. Liu, D. Fang, J. Huang, Q. Wang, L. Dong and C. Xiong, J. Mater. Res., 2017, 33, 1226–1235 CrossRef.
- X. Qian, L. Jin, L. Zhu, S. Yao, D. Rao, X. Shen, X. Xi, K. Xiao and S. Qin, RSC Adv., 2016, 6, 111190–111196 RSC.
- L. Ma, R. Chen, G. Zhu, Y. Hu, Y. Wang, T. Chen, J. Liu and Z. Jin, ACS Nano, 2017, 11, 7274–7283 CrossRef CAS PubMed.
- C. Wan, W. Wu, C. Wu, J. Xu and L. Guan, RSC Adv., 2015, 5, 5102–5106 RSC.
- Y. Zhou, C. Zhou, Q. Li, C. Yan, B. Han, K. Xia, Q. Gao and J. Wu, Adv. Mater., 2015, 27, 3774–3781 CrossRef CAS PubMed.
- Y. Li, J. Zhu, R. Shi, M. Dirican, P. Zhu, C. Yan, H. Jia, J. Zang, J. He and X. Zhang, Chem. Eng. J., 2018, 349, 376–387 CrossRef CAS.
- Y. Tao, Y. Wei, Y. Liu, J. Wang, W. Qiao, L. Ling and D. Long, Energy Environ. Sci., 2016, 9, 3230–3239 RSC.
- M. Wang, L. Fan, X. Wu, D. Tian, J. Cheng, Y. Qiu, H. Wu, B. Guan, N. Zhang, K. Sun and Y. Wang, J. Mater. Chem. A, 2017, 5, 19613–19618 RSC.
- X. Gu, C.-J. Tong, B. Wen, L.-M. Liu, C. Lai and S. Zhang, Electrochim. Acta, 2016, 196, 369–376 CrossRef CAS.
- Y. Kong, J. Luo, C. Jin, H. Yuan, O. Sheng, L. Zhang, C. Fang, W. Zhang, H. Huang, Y. Xia, C. Liang, J. Zhang, Y. Gan and X. Tao, Nano Res., 2017, 11, 477–489 CrossRef.
- C. Zheng, S. Niu, W. Lv, G. Zhou, J. Li, S. Fan, Y. Deng, Z. Pan, B. Li, F. Kang and Q.-H. Yang, Nano Energy, 2017, 33, 306–312 CrossRef CAS.
- J. He, L. Luo, Y. Chen and A. Manthiram, Adv. Mater., 2017, 29, 1702707 CrossRef PubMed.
- L. Hu, C. Dai, H. Liu, Y. Li, B. Shen, Y. Chen, S. J. Bao and M. Xu, Adv. Energy Mater., 2018, 8, 1800709 CrossRef.
- X. Wu, Y. Du, P. Wang, L. Fan, J. Cheng, M. Wang, Y. Qiu, B. Guan, H. Wu and N. Zhang, J. Mater. Chem. A, 2017, 5, 25187–25192 RSC.
- L. Luo, X. Qin, J. Wu, G. Liang, Q. Li, M. Liu, F. Kang, G. Chen and B. Li, J. Mater. Chem. A, 2018, 6, 8612–8619 RSC.
- L. Zhou, N. Ding, J. Yang, L. Yang, Y. Zong, Z. Liu and A. Yu, ACS Sustainable Chem. Eng., 2016, 4, 3679–3687 CrossRef CAS.
- Z. Ding, X. Li, P. Zhang, J. Yu and Y. Hua, New J. Chem., 2017, 41, 12726–12735 RSC.
- X. Li, L. Zhang, Z. Ding and Y. He, J. Electroanal. Chem., 2017, 799, 617–624 CrossRef CAS.
- L. Kong, X. Chen, B.-Q. Li, H.-J. Peng, J.-Q. Huang, J. Xie and Q. Zhang, Adv. Mater., 2017, 30, 1705219 CrossRef PubMed.
- Y. Fu, Y.-S. Su and A. Manthiram, Angew. Chem., 2013, 125, 7068–7073 CrossRef.
- C. Zu, Y. Fu and A. Manthiram, J. Mater. Chem. A, 2013, 1, 10362–10367 RSC.
- K. Han, J. Shen, S. Hao, H. Ye, C. Wolverton, M. C. Kung and H. H. Kung, ChemSusChem, 2014, 7, 2545–2553 CrossRef CAS PubMed.
- S. D. Shenoy, P. A. Joy and M. R. Anantharaman, J. Magn. Magn. Mater., 2004, 269, 217–226 CrossRef CAS.
- C. Vidal-Abarca, P. Lavela and J. L. Tirado, J. Phys. Chem. C, 2010, 114, 12828–12832 CrossRef CAS.
- S. Yuan, Z. Guo, L. Wang, S. Hu, Y. Wang and Y. Xia, Adv. Sci., 2015, 2, 1500071 CrossRef PubMed.
- W. Kong, L. Yan, Y. Luo, D. Wang, K. Jiang, Q. Li, S. Fan and J. Wang, Adv. Funct. Mater., 2017, 27, 1606663 CrossRef.
- X. Fang and H. Peng, Small, 2015, 11, 1488–1511 CrossRef CAS PubMed.
- R. Chen, T. Zhao and F. Wu, Chem. Commun., 2015, 51, 18–33 RSC.
- R. Demir-Cakan, Li-S Batteries, World Scientific Publishing Co., New Jersey, 2017 Search PubMed.
- Y.-S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed.
- Y.-S. Su and A. Manthiram, Chem. Commun., 2012, 48, 8817–8819 RSC.
- J. Balach, T. Jaumann, M. Klose, S. Oswald, J. Eckert and L. Giebeler, J. Phys. Chem. C, 2015, 119, 4580–4587 CrossRef CAS.
- S.-H. Chung and A. Manthiram, J. Phys. Chem. Lett., 2014, 5, 1978–1983 CrossRef CAS PubMed.
- S.-H. Chung and A. Manthiram, Adv. Funct. Mater., 2014, 24, 5299–5306 CrossRef CAS.
- S.-H. Chung and A. Manthiram, Adv. Mater., 2014, 26, 7352–7357 CrossRef CAS PubMed.
- J.-Q. Huang, Z.-L. Xu, S. Abouali, M. Akbari Garakani and J.-K. Kim, Carbon, 2016, 99, 624–632 CrossRef CAS.
- H.-S. Kang and Y.-K. Sun, Adv. Funct. Mater., 2016, 26, 1225–1232 CrossRef CAS.
- H. J. Peng, D. W. Wang, J. Q. Huang, X. B. Cheng, Z. Yuan, F. Wei and Q. Zhang, Adv. Sci., 2016, 3, 1500268 CrossRef PubMed.
- J. Sun, Y. Sun, M. Pasta, G. Zhou, Y. Li, W. Liu, F. Xiong and Y. Cui, Adv. Mater., 2016, 28, 9797–9803 CrossRef CAS PubMed.
- H. Yao, K. Yan, W. Li, G. Zheng, D. Kong, Z. W. Seh, V. K. Narasimhan, Z. Liang and Y. Cui, Energy Environ. Sci., 2014, 7, 3381–3390 RSC.
- Z. Zhang, G. Wang, Y. Lai, J. Li, Z. Zhang and W. Chen, J. Power Sources, 2015, 300, 157–163 CrossRef CAS.
- J. Balach, T. Jaumann, M. Klose, S. Oswald, J. Eckert and L. Giebeler, Adv. Funct. Mater., 2015, 25, 5285–5291 CrossRef CAS.
- J. Balach, T. Jaumann, M. Klose, S. Oswald, J. Eckert and L. Giebeler, J. Power Sources, 2016, 303, 317–324 CrossRef CAS.
- J. Song, Z. Yu, M. L. Gordin and D. Wang, Nano Lett., 2016, 16, 864–870 CrossRef CAS PubMed.
- J. Balach, H. K. Singh, S. Gomoll, T. Jaumann, M. Klose, S. Oswald, M. Richter, J. Eckert and L. Giebeler, ACS Appl. Mater. Interfaces, 2016, 8, 14586–14595 CrossRef CAS PubMed.
- C. Jin, W. Zhang, Z. Zhuang, J. Wang, H. Huang, Y. Gan, Y. Xia, C. Liang, J. Zhang and X. Tao, J. Mater. Chem. A, 2017, 5, 632–640 RSC.
- S. H. Chung and A. Manthiram, Adv. Mater., 2014, 26, 7352–7357 CrossRef CAS PubMed.
- I. Bauer, S. Thieme, J. Brückner, H. Althues and S. Kaskel, J. Power Sources, 2014, 251, 417–422 CrossRef CAS.
- J.-Q. Huang, Q. Zhang, H.-J. Peng, X.-Y. Liu, W.-Z. Qian and F. Wei, Energy Environ. Sci., 2014, 7, 347–353 RSC.
- Y. Chen, N. Liu, H. Shao, W. Wang, M. Gao, C. Li, H. Zhang, A. Wang and Y. Huang, J. Mater. Chem. A, 2015, 3, 15235–15240 RSC.
- Z. Zhang, Z. Zhang, J. Li and Y. Lai, J. Solid State Electrochem., 2015, 19, 1709–1715 CrossRef CAS.
- Z. Xiao, Z. Yang, L. Wang, H. Nie, M. e. Zhong, Q. Lai, X. Xu, L. Zhang and S. Huang, Adv. Mater., 2015, 27, 2891–2898 CrossRef CAS PubMed.
- G. Liang, J. Wu, X. Qin, M. Liu, Q. Li, Y.-B. He, J.-K. Kim, B. Li and F. Kang, ACS Appl. Mater. Interfaces, 2016, 8, 23105–23113 CrossRef CAS PubMed.
- G. Xu, J. Yuan, X. Tao, B. Ding, H. Dou, X. Yan, Y. Xiao and X. Zhang, Nano Res., 2015, 8, 3066–3074 CrossRef CAS.
- F. Li, G. Wang, P. Wang, J. Yang, K. Zhang, Y. Liu and Y. Lai, J. Electroanal. Chem., 2017, 788, 150–155 CrossRef CAS.
- Y. An, Z. Zhang, H. Fei, S. Xiong, B. Ji and J. Feng, ACS Appl. Mater. Interfaces, 2017, 9, 12400–12407 CrossRef CAS PubMed.
- L. Yang, G. Li, X. Jiang, T. Zhang, H. Lin and J. Y. Lee, J. Mater. Chem. A, 2017, 5, 12506–12512 RSC.
- T. Zhao, Y. Ye, C.-Y. Lao, G. Divitini, P. R. Coxon, X. Peng, X. He, H.-K. Kim, K. Xi, C. Ducati, R. Chen, Y. Liu, S. Ramakrishna and R. V. Kumar, Small, 2017, 13, 1700357 CrossRef PubMed.
- J. Liu, L. Yuan, K. Yuan, Z. Li, Z. Hao, J. Xiang and Y. Huang, Nanoscale, 2016, 8, 13638–13645 RSC.
- N. Hu, X. Lv, Y. Dai, L. Fan, D. Xiong and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 18665–18674 CrossRef CAS PubMed.
- Y. Lai, P. Wang, F. Qin, M. Xu, J. Li, K. Zhang and Z. Zhang, Energy Storage Mater., 2017, 9, 179–187 CrossRef.
- X. Qian, L. Jin, D. Zhao, X. Yang, S. Wang, X. Shen, D. Rao, S. Yao, Y. Zhou and X. Xi, Electrochim. Acta, 2016, 192, 346–356 CrossRef CAS.
- T. Yim, S. H. Han, N. H. Park, M.-S. Park, J. H. Lee, J. Shin, J. W. Choi, Y. Jung, Y. N. Jo, J.-S. Yu and K. J. Kim, Adv. Funct. Mater., 2016, 26, 7817–7823 CrossRef CAS.
- J. Balach, T. Jaumann, S. Muhlenhoff, J. Eckert and L. Giebeler, Chem. Commun., 2016, 52, 8134–8137 RSC.
- X. Qian, D. Zhao, L. Jin, S. Yao, D. Rao, X. Shen, Y. Zhou and X. Xi, RSC Adv., 2016, 6, 114989–114996 RSC.
- H. Tang, S. Yao, J. Mi, X. Wu, J. Hou and X. Shen, Mater. Lett., 2017, 186, 127–130 CrossRef CAS.
- D. An, L. Shen, D. Lei, L. Wang, H. Ye, B. Li, F. Kang and Y.-B. He, J. Energy Chem., 2018 DOI:10.1016/j.jechem.2018.05.002.
- F. Li, F. Qin, G. Wang, K. Zhang, P. Wang, Z. Zhang and Y. Lai, RSC Adv., 2018, 8, 1632–1637 RSC.
- W. Li, J. Hicks-Garner, J. Wang, J. Liu, A. F. Gross, E. Sherman, J. Graetz, J. J. Vajo and P. Liu, Chem. Mater., 2014, 26, 3403–3410 CrossRef CAS.
- M. Liu, Q. Li, X. Qin, G. Liang, W. Han, D. Zhou, Y.-B. He, B. Li and F. Kang, Small, 2017, 13, 1602539 CrossRef PubMed.
- J. Li, Y. Huang, S. Zhang, W. Jia, X. Wang, Y. Guo, D. Jia and L. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 7499–7504 CrossRef CAS PubMed.
- X. Qian, D. Zhao, L. Jin, X. Shen, S. Yao, D. Rao, Y. Zhou and X. m. Xi, Mater. Res. Bull., 2017, 94, 104–112 CrossRef CAS.
- S. Wang, X. Qian, L. Jin, D. Rao, S. Yao, X. Shen, K. Xiao and S. Qin, J. Solid State Electrochem., 2017, 21, 3229–3236 CrossRef CAS.
- C. Li, S. Dong, D. Guo, Z. Zhang, M. Wang and L. Yin, Electrochim. Acta, 2017, 251, 43–50 CrossRef CAS.
- E. J. Berg and S. Trabesinger, J. Electrochem. Soc., 2018, 165, A5001–A5005 CrossRef CAS.
- E. M. Fernández, P. G. Moses, A. Toftelund, H. A. Hansen, J. I. Martínez, F. Abild-Pedersen, J. Kleis, B. Hinnemann, J. Rossmeisl, T. Bligaard and J. K. Nørskov, Angew. Chem., Int. Ed., 2008, 47, 4683–4686 CrossRef PubMed.
- Y. Du, X. Zhu, L. Si, Y. Li, X. Zhou and J. Bao, J. Phys. Chem. C, 2015, 119, 15874–15881 CrossRef CAS.
- V. V. T. Doan-Nguyen, K. S. Subrahmanyam, M. M. Butala, J. A. Gerbec, S. M. Islam, K. N. Kanipe, C. E. Wilson, M. Balasubramanian, K. M. Wiaderek, O. J. Borkiewicz, K. W. Chapman, P. J. Chupas, M. Moskovits, B. S. Dunn, M. G. Kanatzidis and R. Seshadri, Chem. Mater., 2016, 28, 8357–8365 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- Z. Yuan, H.-J. Peng, T.-Z. Hou, J.-Q. Huang, C.-M. Chen, D.-W. Wang, X.-B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519–527 CrossRef CAS PubMed.
- G. Zhou, H. Tian, Y. Jin, X. Tao, B. Liu, R. Zhang, Z. W. Seh, D. Zhuo, Y. Liu, J. Sun, J. Zhao, C. Zu, D. S. Wu, Q. Zhang and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 840–845 CrossRef CAS PubMed.
- Q. Pang, D. Kundu and L. F. Nazar, Mater. Horiz., 2016, 3, 130–136 RSC.
- X. Chen, H.-J. Peng, R. Zhang, T.-Z. Hou, J.-Q. Huang, B. Li and Q. Zhang, ACS Energy Lett., 2017, 2, 795–801 CrossRef CAS.
- H. Wang, Q. Zhang, H. Yao, Z. Liang, H.-W. Lee, P.-C. Hsu, G. Zheng and Y. Cui, Nano Lett., 2014, 14, 7138–7144 CrossRef CAS PubMed.
- J. Zhou, N. Lin, W. l. Cai, C. Guo, K. Zhang, J. Zhou, Y. Zhu and Y. Qian, Electrochim. Acta, 2016, 218, 243–251 CrossRef CAS.
- Y. Yamaguchi, T. Takeuchi, H. Sakaebe, H. Kageyama, H. Senoh, T. Sakai and K. Tatsumi, J. Electrochem. Soc., 2010, 157, A630–A635 CrossRef CAS.
- U. K. Sen, P. Johari, S. Basu, C. Nayak and S. Mitra, Nanoscale, 2014, 6, 10243–10254 RSC.
- J. Balach, T. Jaumann and L. Giebeler, Energy Storage Mater., 2017, 8, 209–216 CrossRef.
- J. W. Seo, Y. W. Jun, S. W. Park, H. Nah, T. Moon, B. Park, J. G. Kim, Y. J. Kim and J. Cheon, Angew. Chem., Int. Ed., 2007, 46, 8828–8831 CrossRef CAS PubMed.
- Y. Wang, J. Wu, Y. Tang, X. Lü, C. Yang, M. Qin, F. Huang, X. Li and X. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 4246–4250 CrossRef CAS PubMed.
- N. Deprez and D. S. McLachlan, J. Phys. D: Appl. Phys., 1988, 21, 101–107 CrossRef CAS.
- G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Li, Z. W. Seh and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef CAS PubMed.
- W. Ki, X. Huang, J. Li, D. L. Young and Y. Zhang, J. Mater. Res., 2007, 22, 1390–1395 CrossRef CAS.
- Y. Lu, X. Li, J. Liang, L. Hu, Y. Zhu and Y. Qian, Nanoscale, 2016, 8, 17616–17622 RSC.
- B. Liu, J. Yang, Y. Han, T. Hu, W. Ren, C. Liu, Y. Ma and C. Gao, J. Appl. Phys., 2011, 109, 053717 CrossRef.
- Z. W. Seh, J. H. Yu, W. Li, P.-C. Hsu, H. Wang, Y. Sun, H. Yao, Q. Zhang and Y. Cui, Nat. Commun., 2014, 5, 5017 CrossRef CAS PubMed.
- L. E. Conroy and K. C. Park, Inorg. Chem., 1968, 7, 459–463 CrossRef CAS.
- A. V. Murugan, M. Quintin, M.-H. Delville, G. Campet and K. Vijayamohanan, J. Mater. Chem., 2005, 15, 902–909 RSC.
- T. A. Bither, R. Bouchard, W. Cloud, P. Donohue and W. Siemons, Inorg. Chem., 1968, 7, 2208–2220 CrossRef CAS.
- A. Voznyi, V. Kosyak, A. Opanasyuk, N. Tirkusova, L. Grase, A. Medvids and G. Mezinskis, Mater. Chem. Phys., 2016, 173, 52–61 CrossRef CAS.
- X. Li, Y. Lu, Z. Hou, W. Zhang, Y. Zhu, Y. Qian, J. Liang and Y. Qian, ACS Appl. Mater. Interfaces, 2016, 8, 19550–19557 CrossRef CAS PubMed.
- C. I. Pearce, R. A. D. Pattrick and D. J. Vaughan, Rev. Mineral. Geochem., 2006, 61, 127–180 CrossRef CAS.
- X. Li, J. Liang, Y. Lu, Z. Hou, W. Zhang, Y. Zhu and Y. Qian, J. Power Sources, 2016, 329, 379–386 CrossRef CAS.
- N. Kumar, N. Raman and A. Sundaresan, Z. Anorg. Allg. Chem., 2014, 640, 1069–1074 CrossRef CAS.
- K. Sun, D. Su, Q. Zhang, D. C. Bock, A. C. Marschilok, K. J. Takeuchi, E. S. Takeuchi and H. Gan, J. Electrochem. Soc., 2015, 162, A2834–A2839 CrossRef CAS.
- R. C. Hoodless, R. B. Moyes and P. B. Wells, Catal. Today, 2006, 114, 377–382 CrossRef CAS.
- L. Zhu, D. Susac, M. Teo, K. C. Wong, P. C. Wong, R. R. Parsons, D. Bizzotto, K. A. R. Mitchell and S. A. Campbell, J. Catal., 2008, 258, 235–242 CrossRef CAS.
- Z. Luo, C. Tan, X. Zhang, J. Chen, X. Cao, B. Li, Y. Zong, L. Huang, X. Huang, L. Wang, W. Huang and H. Zhang, Small, 2016, 12, 5920–5926 CrossRef CAS PubMed.
- Z. Ma, Z. Li, K. Hu, D. Liu, J. Huo and S. Wang, J. Power Sources, 2016, 325, 71–78 CrossRef CAS.
- H. Xu and A. Manthiram, Nano Energy, 2017, 33, 124–129 CrossRef CAS.
- J. Pu, Z. Shen, J. Zheng, W. Wu, C. Zhu, Q. Zhou, H. Zhang and F. Pan, Nano Energy, 2017, 37, 7–14 CrossRef CAS.
- T. Chen, Z. Zhang, B. Cheng, R. Chen, Y. Hu, L. Ma, G. Zhu, J. Liu and Z. Jin, J. Am. Chem. Soc., 2017, 139, 12710–12715 CrossRef CAS PubMed.
- T. Chen, L. Ma, B. Cheng, R. Chen, Y. Hu, G. Zhu, Y. Wang, J. Liang, Z. Tie, J. Liu and Z. Jin, Nano Energy, 2017, 38, 239–248 CrossRef CAS.
- C. Dai, J. M. Lim, M. Wang, L. Hu, Y. Chen, Z. Chen, H. Chen, S. J. Bao, B. Shen and Y. Li, Adv. Funct. Mater., 2018, 28, 1704443 CrossRef.
- J. He, Y. Chen and A. Manthiram, Energy Environ. Sci., 2018, 11, 2560–2568 RSC.
- J. Song, T. Xu, M. L. Gordin, P. Zhu, D. Lv, Y. B. Jiang, Y. Chen, Y. Duan and D. Wang, Adv. Funct. Mater., 2014, 24, 1243–1250 CrossRef CAS.
- A. Garsuch, S. Herzog, L. Montag, A. Krebs and K. Leitner, ECS Electrochem. Lett., 2012, 1, A24–A26 CrossRef CAS.
- M. S. Whittingham and J. A. Panella, Mater. Res. Bull., 1981, 16, 37–45 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- Y.-S. Su and A. Manthiram, J. Power Sources, 2014, 270, 101–105 CrossRef CAS.
- L. Ma, S. Wei, H. L. Zhuang, K. E. Hendrickson, R. G. Hennig and L. A. Archer, J. Mater. Chem. A, 2015, 3, 19857–19866 RSC.
- T. Matsuyama, A. Hayashi, C. J. Hart, L. F. Nazar and M. Tatsumisago, J. Electrochem. Soc., 2016, 163, A1730–A1735 CrossRef CAS.
- H. Lin, L. Yang, X. Jiang, G. Li, T. Zhang, Q. Yao, G. W. Zheng and J. Y. Lee, Energy Environ. Sci., 2017, 10, 1476–1486 RSC.
- Z. Li, S. Deng, R. Xu, L. Wei, X. Su and M. Wu, Electrochim. Acta, 2017, 252, 200–207 CrossRef CAS.
- P. Guo, D. Liu, Z. Liu, X. Shang, Q. Liu and D. He, Electrochim. Acta, 2017, 256, 28–36 CrossRef CAS.
- F. Sun, H. Tang, B. Zhang, X. Li, C. Yin, Z. Yue, L. Zhou, Y. Li and J. Shi, ACS Sustainable Chem. Eng., 2018, 6, 974–982 CrossRef CAS.
- L. Yan, N. Luo, W. Kong, S. Luo, H. Wu, K. Jiang, Q. Li, S. Fan, W. Duan and J. Wang, J. Power Sources, 2018, 389, 169–177 CrossRef CAS.
- P. T. Dirlam, J. Park, A. G. Simmonds, K. Domanik, C. B. Arrington, J. L. Schaefer, V. P. Oleshko, T. S. Kleine, K. Char, R. S. Glass, C. L. Soles, C. Kim, N. Pinna, Y.-E. Sung and J. Pyun, ACS Appl. Mater. Interfaces, 2016, 8, 13437–13448 CrossRef CAS PubMed.
- Z. A. Ghazi, X. He, A. M. Khattak, N. A. Khan, B. Liang, A. Iqbal, J. Wang, H. Sin, L. Li and Z. Tang, Adv. Mater., 2017, 29, 1606817 CrossRef PubMed.
- M. Li, J. Zhou, J. Zhou, C. Guo, Y. Han, Y. Zhu, G. Wang and Y. Qian, Mater. Res. Bull., 2017, 96, 509–515 CrossRef CAS.
- X. Li, L. Chu, Y. Wang and L. Pan, Mater. Sci. Eng., B, 2016, 205, 46–54 CrossRef CAS.
- S. S. Zhang and D. T. Tran, J. Mater. Chem. A, 2016, 4, 4371–4374 RSC.
- Z. Liu, X. Zheng, S.-l. Luo, S.-q. Xu, N.-y. Yuan and J.-n. Ding, J. Mater. Chem. A, 2016, 4, 13395–13399 RSC.
- M. Lao, G. Zhao, X. Li, Y. Chen, S. X. Dou and W. Sun, ACS Appl. Energy Mater., 2018, 1, 167–172 CrossRef CAS.
- Z. Xiao, Z. Yang, L. Zhang, H. Pan and R. Wang, ACS Nano, 2017, 11, 8488–8498 CrossRef CAS PubMed.
- J. Park, B. C. Yu, J. S. Park, J. W. Choi, C. Kim, Y. E. Sung and J. B. Goodenough, Adv. Energy Mater., 2017, 7, 1602567 CrossRef.
- J. D. Liu, X. S. Zheng, Z. F. Shi and S. Q. Zhang, Ionics, 2014, 20, 659–664 CrossRef CAS.
- Z. Cheng, Z. Xiao, H. Pan, S. Wang and R. Wang, Adv. Energy Mater., 2017, 8, 1702337 CrossRef.
- X. Zhu, W. Zhao, Y. Song, Q. Li, F. Ding, J. Sun, L. Zhang and Z. Liu, Adv. Energy Mater., 2018, 8, 1800201 CrossRef.
- L. Chen, J.-D. Liu and S.-Q. Zhang, J. Inorg. Mater., 2013, 28, 1127–1131 CrossRef CAS.
- H. J. Peng, G. Zhang, X. Chen, Z. W. Zhang, W. T. Xu, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12990–12995 CrossRef CAS PubMed.
- B. Cao, Y. Chen, D. Li, L. Yin and Y. Mo, ChemSusChem, 2016, 9, 3338–3344 CrossRef CAS PubMed.
- H. Al Salem, V. R. Chitturi, G. Babu, J. A. Santana, D. Gopalakrishnan and L. M. R. Arava, RSC Adv., 2016, 6, 110301–110306 RSC.
- F. Zhou, L. T. Song, L. L. Lu, H. B. Yao and S. H. Yu, ChemNanoMat, 2016, 2, 937–941 CrossRef CAS.
- H. Huang, J. Liu, Y. Xia, C. Cheng, C. Liang, Y. Gan, J. Zhang, X. Tao and W. Zhang, J. Alloys Compd., 2017, 706, 227–233 CrossRef CAS.
- T. Zhou, Y. Zhao, G. Zhou, W. Lv, P. Sun, F. Kang, B. Li and Q.-H. Yang, Nano Energy, 2017, 39, 291–296 CrossRef CAS.
- F. Zhou, Z. Li, X. Luo, T. Wu, B. Jiang, L.-L. Lu, H.-B. Yao, M. Antonietti and S.-H. Yu, Nano Lett., 2018, 18, 1035–1043 CrossRef CAS PubMed.
- J. Choi, T.-G. Jeong, B. W. Cho, Y. Jung, S. H. Oh and Y.-T. Kim, J. Phys. Chem. C, 2018, 122, 7664–7669 CrossRef CAS.
- W. Cai, G. Li, K. Zhang, G. Xiao, C. Wang, K. Ye, Z. Chen, Y. Zhu and Y. Qian, Adv. Funct. Mater., 2018, 28, 1704865 CrossRef.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, Chem. Mater., 2017, 29, 7633–7644 CrossRef CAS.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3907–3911 CrossRef CAS PubMed.
- W. Bao, D. Su, W. Zhang, X. Guo and G. Wang, Adv. Funct. Mater., 2016, 26, 8746–8756 CrossRef CAS.
- W. Bao, L. Liu, C. Wang, S. Choi, D. Wang and G. Wang, Adv. Energy Mater., 2018, 8, 1702485 CrossRef.
- W. Bao, X. Xie, J. Xu, X. Guo, J. Song, W. Wu, D. Su and G. Wang, Chem.–Eur. J., 2017, 23, 12613–12619 CrossRef CAS PubMed.
- X. Liang, Y. Rangom, C. Y. Kwok, Q. Pang and L. F. Nazar, Adv. Mater., 2017, 29, 1603040 CrossRef PubMed.
- C. Lin, W. Zhang, L. Wang, Z. Wang, W. Zhao, W. Duan, Z. Zhao, B. Liu and J. Jin, J. Mater. Chem. A, 2016, 4, 5993–5998 RSC.
- J. Song, D. Su, X. Xie, X. Guo, W. Bao, G. Shao and G. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 29427–29433 CrossRef CAS PubMed.
- Y. Dong, S. Zheng, J. Qin, X. Zhao, H. Shi, X. Wang, J. Chen and Z.-S. Wu, ACS Nano, 2018, 12, 2381–2388 CrossRef CAS PubMed.
- D. Rao, L. Zhang, Y. Wang, Z. Meng, X. Qian, J. Liu, X. Shen, G. Qiao and R. Lu, J. Phys. Chem. C, 2017, 121, 11047–11054 CrossRef CAS.
- E. S. Sim, G. S. Yi, M. Je, Y. Lee and Y.-C. Chung, J. Power Sources, 2017, 342, 64–69 CrossRef CAS.
- E. S. Sim and Y.-C. Chung, Appl. Surf. Sci., 2018, 435, 210–215 CrossRef CAS.
- Y. Zhao and J. Zhao, Appl. Surf. Sci., 2017, 412, 591–598 CrossRef CAS.
- X. Liu, X. Shao, F. Li and M. Zhao, Appl. Surf. Sci., 2018, 455, 522–526 CrossRef CAS.
- N. Mosavati, V. R. Chitturi, S. O. Salley and K. S. Ng, J. Power Sources, 2016, 321, 87–93 CrossRef CAS.
- Z. Cui, C. Zu, W. Zhou, A. Manthiram and J. B. Goodenough, Adv. Mater., 2016, 28, 6926–6931 CrossRef CAS PubMed.
- D.-R. Deng, T.-H. An, Y.-J. Li, Q.-H. Wu, M.-S. Zheng and Q.-F. Dong, J. Mater. Chem. A, 2016, 4, 16184–16190 RSC.
- Z. Hao, L. Yuan, C. Chen, J. Xiang, Y. Li, Z. Huang, P. Hu and Y. Huang, J. Mater. Chem. A, 2016, 4, 17711–17717 RSC.
- D.-R. Deng, J. Lei, F. Xue, C.-D. Bai, X.-D. Lin, J.-C. Ye, M.-S. Zheng and Q.-F. Dong, J. Mater. Chem. A, 2017, 5, 23497–23505 RSC.
- T.-G. Jeong, D. S. Choi, H. Song, J. Choi, S.-A. Park, S. H. Oh, H. Kim, Y. Jung and Y.-T. Kim, ACS Energy Lett., 2017, 2, 327–333 CrossRef CAS.
- J. Zhang, C. You, W. Zhang, J. Wang, S. Guo, R. Yang and Y. Xu, Electrochim. Acta, 2017, 250, 159–166 CrossRef CAS.
- T. Zhou, W. Lv, J. Li, G. Zhou, Y. Zhao, S. Fan, B. Liu, B. Li, F. Kang and Q.-H. Yang, Energy Environ. Sci., 2017, 10, 1694–1703 RSC.
- X. He, Y. Shuai, L. Na, K. Chen, Y. Zhang, Z. Zhang and F. Gan, Mater. Lett., 2018, 215, 91–94 CrossRef CAS.
- W. Zeng, Z. Wang, M. M.-C. Cheng and K. S. Ng, J. Electrochem. Soc., 2018, 165, A1011–A1018 CrossRef CAS.
- L. Chen, W. Yang, H. Zhang, J. Liu and Y. Zhou, J. Mater. Sci., 2018, 53, 10363–10371 CrossRef CAS.
- Y. Wang, R. Zhang, Y.-C. Pang, X. Chen, J. Lang, J. Xu, C. Xiao, H. Li, K. Xi and S. Ding, Energy Storage Mater., 2018, 16, 228–235 CrossRef.
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H.-M. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef PubMed.
- N. Mosavati, S. O. Salley and K. S. Ng, J. Power Sources, 2017, 340, 210–216 CrossRef CAS.
- X. Li, K. Ding, B. Gao, Q. Li, Y. Li, J. Fu, X. Zhang, P. K. Chu and K. Huo, Nano Energy, 2017, 40, 655–662 CrossRef CAS.
- L. Ma, H. Yuan, W. Zhang, G. Zhu, Y. Wang, Y. Hu, P. Zhao, R. Chen, T. Chen and J. Liu, Nano Lett., 2017, 17, 7839–7846 CrossRef CAS PubMed.
- W. Ren, L. Xu, L. Zhu, X. Wang, X. Ma and D. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 11642–11651 CrossRef CAS PubMed.
- Y. Zhong, D. Chao, S. Deng, J. Zhan, R. Fang, Y. Xia, Y. Wang, X. Wang, X. Xia and J. Tu, Adv. Funct. Mater., 2018, 28, 1706391 CrossRef.
- L. Zhu, C. Li, W. Ren, M. Qin and L. Xu, New J. Chem., 2018, 42, 5109–5116 RSC.
- D.-R. Deng, F. Xue, Y.-J. Jia, J.-C. Ye, C.-D. Bai, M.-S. Zheng and Q.-F. Dong, ACS Nano, 2017, 11, 6031–6039 CrossRef CAS PubMed.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CAS.
- R. Demir-Cakan, M. Morcrette, F. Nouar, C. Davoisne, T. Devic, D. Gonbeau, R. Dominko, C. Serre, G. Férey and J.-M. Tarascon, J. Am. Chem. Soc., 2011, 133, 16154–16160 CrossRef CAS PubMed.
- Z. Wang, X. Li, Y. Cui, Y. Yang, H. Pan, Z. Wang, C. Wu, B. Chen and G. Qian, Cryst. Growth Des., 2013, 13, 5116–5120 CrossRef CAS.
- J. Zheng, J. Tian, D. Wu, M. Gu, W. Xu, C. Wang, F. Gao, M. H. Engelhard, J.-G. Zhang, J. Liu and J. Xiao, Nano Lett., 2014, 14, 2345–2352 CrossRef CAS PubMed.
- Z. Wang, B. Wang, Y. Yang, Y. Cui, Z. Wang, B. Chen and G. Qian, ACS Appl. Mater. Interfaces, 2015, 7, 20999–21004 CrossRef CAS PubMed.
- X.-J. Hong, T.-X. Tan, Y.-K. Guo, X.-Y. Tang, J.-Y. Wang, W. Qin and Y.-P. Cai, Nanoscale, 2018, 10, 2774–2780 RSC.
- J. Xu, T. Lawson, H. Fan, D. Su and G. Wang, Adv. Energy Mater., 2018, 8, 1702607 CrossRef.
- J. H. Park, K. M. Choi, D. K. Lee, B. C. Moon, S. R. Shin, M.-K. Song and J. K. Kang, Sci. Rep., 2016, 6, 25555 CrossRef CAS PubMed.
- P. Chiochan, S. Kaewruang, N. Phattharasupakun, J. Wutthiprom, T. Maihom, J. Limtrakul, S. S. Nagarkar, S. Horike and M. Sawangphruk, Sci. Rep., 2017, 7, 17703 CrossRef PubMed.
- J. Zhou, R. Li, X. Fan, Y. Chen, R. Han, W. Li, J. Zheng, B. Wang and X. Li, Energy Environ. Sci., 2014, 7, 2715–2724 RSC.
- Y. Mao, G. Li, Y. Guo, Z. Li, C. Liang, X. Peng and Z. Lin, Nat. Commun., 2017, 8, 14628 CrossRef PubMed.
- L. Bai, D. Chao, P. Xing, L. J. Tou, Z. Chen, A. Jana, Z. X. Shen and Y. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 14328–14333 CrossRef CAS PubMed.
- Z. Wang, Z. Dou, Y. Cui, Y. Yang, Z. Wang and G. Qian, Microporous Mesoporous Mater., 2014, 185, 92–96 CrossRef CAS.
- Y. Yue, B. Guo, Z.-A. Qiao, P. F. Fulvio, J. Chen, A. J. Binder, C. Tian and S. Dai, Microporous Mesoporous Mater., 2014, 198, 139–143 CrossRef CAS.
- P. M. Shanthi, P. J. Hanumantha, B. Gattu, M. Sweeney, M. K. Datta and P. N. Kumta, Electrochim. Acta, 2017, 229, 208–218 CrossRef CAS.
- Y. Hou, H. Mao and L. Xu, Nano Res., 2017, 10, 344–353 CrossRef CAS.
- Y.-C. Lu, Q. He and H. A. Gasteiger, J. Phys. Chem. C, 2014, 118, 5733–5741 CrossRef CAS.
- J. Zhou, X. Yu, X. Fan, X. Wang, H. Li, Y. Zhang, W. Li, J. Zheng, B. Wang and X. Li, J. Mater. Chem. A, 2015, 3, 8272–8275 RSC.
- M.-T. Li, Y. Sun, K.-S. Zhao, Z. Wang, X.-L. Wang, Z.-M. Su and H.-M. Xie, ACS Appl. Mater. Interfaces, 2016, 8, 33183–33188 CrossRef CAS PubMed.
- A. E. Baumann, G. E. Aversa, A. Roy, M. L. Falk, N. M. Bedford and V. S. Thoi, J. Mater. Chem. A, 2018, 6, 4811–4821 RSC.
- H. Park and D. J. Siegel, Chem. Mater., 2017, 29, 4932–4939 CrossRef CAS.
- D. Su, M. Cortie, H. Fan and G. Wang, Adv. Mater., 2017, 29, 1700587 CrossRef PubMed.
- Z. Zhang, Y. Wang, J. Liu, D. Sun, X. Ma, Y. Jin and Y. Cui, Electrochim. Acta, 2018, 271, 58–66 CrossRef CAS.
- S. Bai, X. Liu, K. Zhu, S. Wu and H. Zhou, Nat. Energy, 2016, 1, 16094 CrossRef CAS.
- H. Zhang, C. Lin, X. Hu, B. Zhu and D. Yu, ACS Appl. Mater. Interfaces, 2018, 10, 12708–12715 CrossRef CAS PubMed.
- Q. Shi, Z. Chen, Z. Song, J. Li and J. Dong, Angew. Chem., Int. Ed., 2011, 50, 672–675 CrossRef CAS PubMed.
- Z. Zhao, X. Ma, A. Kasik, Z. Li and Y. S. Lin, Ind. Eng. Chem. Res., 2013, 52, 1102–1108 CrossRef CAS.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem.–Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- T. R. C. Van Assche, T. Duerinck, S. Van der Perre, G. V. Baron and J. F. M. Denayer, Langmuir, 2014, 30, 7878–7883 CrossRef CAS PubMed.
- Z. Zhao, S. Wang, R. Liang, Z. Li, Z. Shi and G. Chen, J. Mater. Chem. A, 2014, 2, 13509–13512 RSC.
- W.-W. Jin, H.-J. Li, J.-Z. Zou, S.-Z. Zeng, Q.-D. Li, G.-Z. Xu, H.-C. Sheng, B.-B. Wang, Y.-H. Si, L. Yu and X.-R. Zeng, RSC Adv., 2018, 8, 4786–4793 RSC.
- X.-C. Liu, Y. Yang, J. Wu, M. Liu, S. P. Zhou, B. D. A. Levin, X.-D. Zhou, H. Cong, D. A. Muller, P. M. Ajayan, H. D. Abruña and F.-S. Ke, ACS Energy Lett., 2018, 3, 1325–1330 CrossRef CAS.
- J. Linnemann, L. Taudien, M. Klose and L. Giebeler, J. Mater. Chem. A, 2017, 5, 18420–18428 RSC.
- S. Aguado, J. Canivet and D. Farrusseng, J. Mater. Chem., 2011, 21, 7582–7588 RSC.
- G. Babu, K. Ababtain, K. Y. S. Ng and L. M. R. Arava, Sci. Rep., 2015, 5, 8763 CrossRef CAS PubMed.
- H. Tang, S. Yao, M. Jing, X. Wu, J. Hou, X. Qian, D. Rao, X. Shen, X. Xi and K. Xiao, Electrochim. Acta, 2015, 176, 442–447 CrossRef CAS.
- H. Al Salem, G. Babu, C. V. Rao and L. M. R. Arava, J. Am. Chem. Soc., 2015, 137, 11542–11545 CrossRef CAS PubMed.
- R. Fang, S. Zhao, S. Pei, Y. Cheng, P. Hou, M. Liu, H.-M. Cheng, C. Liu and F. Li, Carbon, 2016, 109, 719–726 CrossRef CAS.
- J. He, Y. Chen, W. Lv, K. Wen, C. Xu, W. Zhang, Y. Li, W. Qin and W. He, ACS Nano, 2016, 10, 10981–10987 CrossRef CAS PubMed.
- Y.-J. Li, J.-M. Fan, M.-S. Zheng and Q.-F. Dong, Energy Environ. Sci., 2016, 9, 1998–2004 RSC.
- Z. Li, C. Li, X. Ge, J. Ma, Z. Zhang, Q. Li, C. Wang and L. Yin, Nano Energy, 2016, 23, 15–26 CrossRef CAS.
- M. Zhang, C. Yu, C. Zhao, X. Song, X. Han, S. Liu, C. Hao and J. Qiu, Energy Storage Mater., 2016, 5, 223–229 CrossRef.
- Y. Li, J. Fan, J. Zhang, J. Yang, R. Yuan, J. Chang, M. Zheng and Q. Dong, ACS Nano, 2017, 11, 11417–11424 CrossRef CAS PubMed.
- D. Xiao, Q. Li, H. Zhang, Y. Ma, C. Lu, C. Chen, Y. Liu and S. Yuan, J. Mater. Chem. A, 2017, 5, 24901–24908 RSC.
- N. K. Thangavel, D. Gopalakrishnan and L. M. R. Arava, J. Phys. Chem. C, 2017, 121, 12718–12725 CrossRef CAS.
- Z. Lin, X. Li, W. Huang, X. Zhu, Y. Wang and Z. Shan, ChemElectroChem, 2017, 4, 2577–2582 CrossRef CAS.
- P. Zuo, J. Hua, M. He, H. Zhang, Z. Qian, Y. Ma, C. Du, X. Cheng, Y. Gao and G. Yin, J. Mater. Chem. A, 2017, 5, 10936–10945 RSC.
- G. Chen, X. Song, S. Wang, Y. Wang, T. Gao, L.-X. Ding and H. Wang, J. Membr. Sci., 2018, 548, 247–253 CrossRef CAS.
- S. Liu, J. Li, X. Yan, Q. Su, Y. Lu, J. Qiu, Z. Wang, X. Lin, J. Huang, R. Liu, B. Zheng, L. Chen, R. Fu and D. Wu, Adv. Mater., 2018, 30, 1706895 CrossRef PubMed.
- M.-e. Zhong, J. Guan, Q. Feng, X. Wu, Z. Xiao, W. Zhang, S. Tong, N. Zhou and D. Gong, Carbon, 2018, 128, 86–96 CrossRef CAS.
- C.-Y. Fan, P. Xiao, H.-H. Li, H.-F. Wang, L.-L. Zhang, H.-Z. Sun, X.-L. Wu, H.-M. Xie and J.-P. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 27959–27967 CrossRef CAS PubMed.
- X. Yao, J. Xu, Z. Hong, G. Li, X. Wang, F. Lu, W. Wang, H. Liu, C. Liang, Z. Lin and W. Wang, J. Phys. Chem. C, 2018, 122, 3263–3272 CrossRef CAS.
- K. Xu, X. Liu, J. Liang, J. Cai, K. Zhang, Y. Lu, X. Wu, M. Zhu, Y. Liu, Y. Zhu, G. Wang and Y. Qian, ACS Energy Lett., 2018, 3, 420–427 CrossRef CAS.
- L. Luo, S.-H. Chung, C.-H. Chang and A. Manthiram, J. Mater. Chem. A, 2017, 5, 15002–15007 RSC.
- Q. Zhao, X. Hu, K. Zhang, N. Zhang, Y. Hu and J. Chen, Nano Lett., 2015, 15, 721–726 CrossRef CAS PubMed.
- X. Zhu, J. Tian, X. Liu, W. Huang, D. Luo, Z. Wang and Z. Shan, RSC Adv., 2017, 7, 35482–35489 RSC.
- Ş. Sörgel, O. Kesten, A. Wengel and T. Sörgel, Energy Storage Mater., 2018, 10, 223–232 CrossRef.
- J. H. Yom, S. M. Cho, S. W. Hwang and W. Y. Yoon, J. Electrochem. Soc., 2016, 163, A2179–A2184 CrossRef CAS.
- Y. J. Hong, K. C. Roh and Y. C. Kang, Carbon, 2018, 126, 394–403 CrossRef CAS.
- S. Zheng, F. Yi, Z. Li, Y. Zhu, Y. Xu, C. Luo, J. Yang and C. Wang, Adv. Funct. Mater., 2014, 24, 4156–4163 CrossRef CAS.
- J. Zhang, H. Li, Q. Tang, P. Bai, Y. Pan and Z. Lin, RSC Adv., 2016, 6, 114447–114452 RSC.
- J. Xiao, Adv. Energy Mater., 2015, 5, 1501102 CrossRef.
- C. Barchasz, F. Mesguich, J. Dijon, J.-C. Leprêtre, S. Patoux and F. Alloin, J. Power Sources, 2012, 211, 19–26 CrossRef CAS.
- S.-H. Chung and A. Manthiram, Electrochim. Acta, 2013, 107, 569–576 CrossRef CAS.
- K. Zhang, F. Qin, J. Fang, Q. Li, M. Jia, Y. Lai, Z. Zhang and J. Li, J. Solid State Electrochem., 2014, 18, 1025–1029 CrossRef CAS.
- X.-B. Cheng, H.-J. Peng, J.-Q. Huang, L. Zhu, S.-H. Yang, Y. Liu, H.-W. Zhang, W. Zhu, F. Wei and Q. Zhang, J. Power Sources, 2014, 261, 264–270 CrossRef CAS.
- J. X. Zhang, Z. S. Ma, J. J. Cheng, Y. Wang, C. Wu, Y. Pan and C. Lu, J. Electroanal. Chem., 2015, 738, 184–187 CrossRef CAS.
- I. Raguzin, S. Choudhury, F. Simon, M. Stamm and L. Ionov, Adv. Mater. Interfaces, 2017, 4, 1600811 CrossRef.
- J. J. Cheng, J. T. Zhu, Y. Pan, Z. S. Ma, H. J. Song, J. A. Pan, Z. Z. Li and C. Lu, ECS Electrochem. Lett., 2015, 4, A19–A21 CrossRef CAS.
- L.-J. Liu, Y. Chen, Z.-F. Zhang, X.-L. You, M. D. Walle, Y.-J. Li and Y.-N. Liu, J. Power Sources, 2016, 325, 301–305 CrossRef CAS.
- J. Hassoun, M. Agostini, A. Latini, S. Panero, Y.-K. Sun and B. Scrosati, J. Electrochem. Soc., 2012, 159, A390–A395 CrossRef CAS.
- Z. Liu, X. Zheng, N.-Y. Yuan and J.-N. Ding, J. Mater. Chem. A, 2017, 5, 942–946 RSC.
- X. Wu, S. Yao, J. Hou, M. Jing, X. Qian, X. Shen, J. Xiang and X. Xi, J. Nanosci. Nanotechnol., 2017, 17, 2482–2487 CrossRef CAS PubMed.
- P. Zuo, H. Zhang, M. He, Q. Li, Y. Ma, C. Du, X. Cheng, H. Huo, Y. Gao and G. Yin, Carbon, 2017, 122, 635–642 CrossRef CAS.
- N. Mosavati, V. R. Chitturi, L. M. R. Arava, S. O. Salley and K. Y. S. Ng, Electrochim. Acta, 2015, 185, 297–303 CrossRef CAS.
- Z. Zhang, L. L. Kong, S. Liu, G. R. Li and X. P. Gao, Adv. Energy Mater., 2017, 7, 1602543 CrossRef.
- S.-K. Park, J.-K. Lee and Y. C. Kang, Adv. Funct. Mater., 2017, 28, 1705264 CrossRef.
- Y.-Q. Lu, Y.-J. Wu, T. Sheng, X.-X. Peng, Z.-G. Gao, S.-J. Zhang, L. Deng, R. Nie, J. Światowska, J.-T. Li, Y. Zhou, L. Huang, X.-D. Zhou and S.-G. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 13499–13508 CrossRef CAS PubMed.
- H. Yu, B. Zhang, F. Sun, G. Jiang, N. Zheng, C. Xu and Y. Li, Appl. Surf. Sci., 2018, 450, 364–371 CrossRef CAS.
- W. Hu, Y. Hirota, Y. Zhu, N. Yoshida, M. Miyamoto, T. Zheng and N. Nishiyama, ChemSusChem, 2017, 10, 3557–3564 CrossRef CAS PubMed.
- J. Wang, T. Wu, S. Zhang, S. Gu, J. Jin and Z. Wen, Chem. Eng. J., 2018, 334, 2356–2362 CrossRef CAS.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. ÓKeeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS PubMed.
- X. Tao, F. Chen, Y. Xia, H. Huang, Y. Gan, X. Chen and W. Zhang, Chem. Commun., 2013, 49, 4513–4515 RSC.
- J. Zhang, H. Li, Z. Lin, Q. Tang, W. Qi, L. Wang, H. Zheng and K. Zhou, RSC Adv., 2017, 7, 39172–39177 RSC.
- X. Song, S. Wang, G. Chen, T. Gao, Y. Bao, L.-X. Ding and H. Wang, Chem. Eng. J., 2018, 333, 564–571 CrossRef CAS.
- X. Li, J. Liang, K. Zhang, Z. Hou, W. Zhang, Y. Zhu and Y. Qian, Energy Environ. Sci., 2015, 8, 3181–3186 RSC.
- Z. Gao, Y. Schwab, Y. Zhang, N. Song and X. Li, Adv. Funct. Mater., 2018, 28, 1800563 CrossRef.
- X.-Q. Niu, X.-L. Wang, D.-H. Wang, Y. Li, Y.-J. Zhang, Y.-D. Zhang, T. Yang, T. Yu and J.-P. Tu, J. Mater. Chem. A, 2015, 3, 17106–17112 RSC.
- X.-Q. Niu, X.-L. Wang, D. Xie, D.-H. Wang, Y.-D. Zhang, Y. Li, T. Yu and J.-P. Tu, ACS Appl. Mater. Interfaces, 2015, 7, 16715–16722 CrossRef CAS PubMed.
- S. Gope and A. J. Bhattacharyya, ACS Appl. Energy Mater., 2018, 1, 2942–2954 CrossRef CAS.
- C. Dai, L. Hu, M.-Q. Wang, Y. Chen, J. Han, J. Jiang, Y. Zhang, B. Shen, Y. Niu, S.-J. Bao and M. Xu, Energy Storage Mater., 2017, 8, 202–208 CrossRef.
- J. Jiang, J. Zhu, W. Ai, X. Wang, Y. Wang, C. Zou, W. Huang and T. Yu, Nat. Commun., 2015, 6, 8622 CrossRef CAS PubMed.
- J. Zhang, H. Hu, Z. Li and X. W. Lou, Angew. Chem., Int. Ed., 2016, 55, 3982–3986 CrossRef CAS PubMed.
- H.-J. Peng, Z.-W. Zhang, J.-Q. Huang, G. Zhang, J. Xie, W.-T. Xu, J.-L. Shi, X. Chen, X.-B. Cheng and Q. Zhang, Adv. Mater., 2016, 28, 9551–9558 CrossRef CAS PubMed.
- L. Kong, H.-J. Peng, J.-Q. Huang, W. Zhu, G. Zhang, Z.-W. Zhang, P.-Y. Zhai, P. Sun, J. Xie and Q. Zhang, Energy Storage Mater., 2017, 8, 153–160 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |