
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influence of pore size distribution on the oxygen reduction reaction performance in nitrogen doped carbon microspheres†
G. A.
Ferrero
a,
K.
Preuss
b,
A. B.
Fuertes
a,
M.
Sevilla
*a and
M.-M.
Titirici
*b
aInstituto Nacional del Carbón (CSIC), P.O. Box 73, Oviedo 33080, Spain. E-mail: martasev@incar.csic.es
bSchool of Engineering and Materials Science, Queen Mary University of London Mile End Road, E1 4NS, London, UK. E-mail: m.m.titirici@qmul.ac.uk
First published on 27th January 2016
Abstract
Nitrogen-doped carbon microspheres with tunable porosity are investigated as electrocatalysts for the oxygen reduction reaction (ORR). The materials were synthesized by “nanocasting” involving the use of pyrrole as the carbon source and N-dopant, and porous silica microspheres as template. The engineered nitrogen-doped carbon particles combine several indispensable characteristics for a highly active metal-free carbon electrocatalyst: (i) a high content of nitrogen functionalities (∼8 wt%) mainly distributed in quaternary and pyridinic groups, which are highly active catalytic centers for the ORR reaction, and (ii) a high specific surface area (1200–1300 m2 g−1). Furthermore, the porosity of the N-doped microspheres can be modulated from a micro- to a mesoporous structure, i.e. from a micropore size distribution centered at ∼1 nm to a widely accessible mesoporosity with two mesopores systems (∼3 nm and ∼14 nm). The electrocatalytic activity of the N-doped carbon microspheres in the oxygen reduction reaction (ORR) was studied in both basic and acid media. Both types of materials catalyze the ORR via the efficient 4-electron process. However, the mesoporous carbon exhibits a more positive onset potential and a higher kinetic current density than the microporous microspheres and noteworthy the values are comparable to those of commercial Pt/C under basic conditions. Moreover, the mesoporous microspheres also show a better electrocatalytic activity than the microporous ones in acid medium, and a similar onset potential to that of Pt/C with a peroxide yield lower than 10%. A detailed comparison between the N-doped micro- and mesoporous microspheres reveals that the mesoporous material outperforms the microporous one not only in catalytic activity but also in durability in both electrolytes, which proves that the bimodal mesoporous structure acts as interconnected highways providing quick and full transport towards/from the catalytic sites for both reactant and products. This leads in turn to an effective metal-free carbon catalyst that can match the commercial Pt/C catalyst.
Introduction
Fuel cells have gained increasing attention as a clean and attractive alternative energy source owing to their high efficiency, high power density, and negligible pollutant emissions.1–3 Platinum-based electrocatalysts are the most widespread catalysts for electrode reactions as a result of their high activity. However, their high cost along with the sluggish kinetics in the cathodic oxygen reduction reaction (ORR) and low tolerance to fuel crossover restricts their performance and hinders the widespread commercialization of fuel cells.4–6 Recently, the development of metal-free catalysts has drawn considerable interest for cost reduction and stability enhancement without compromising the excellent catalytic activity.7,8 In this respect, N-doped porous carbon materials represent a promising alternative taking into account the easy modification of their structural and chemical properties.9–11 It has been shown that the presence of nitrogen functionalities can notably enhance the ORR activity of the carbon materials10,12 and improve the electrocatalytic durability of the electrocatalyst as a result of the strong electron donor behavior of nitrogen.10,13 In addition, the incorporation of N-groups can create catalytic charge active sites by breaking the electroneutrality of the carbon material.8 Nevertheless, the achievements reported so far for metal-free N-doped carbon catalysts are still insufficient for their commercialization, especially when the electrolyte used is acid, such as in proton exchange membrane fuel cells (PEMFCs).14–18The optimization of the electrocatalytic activity of carbon catalysts requires an appropriate design not only of the chemical properties but also of the structural properties (i.e. morphology, particle size and pore structure), which allows an improvement of the mass-transport kinetics of reactant and product towards ORR catalytic sites.14,16,19–21 Thus, a high density of catalytically active sites reduces kinetic limitations. However, the active sites need to be fully accessible to the reactants to guarantee their utilization.12 In this regard, contradictory results can be found in the literature on the role of pore size. Thus, some authors have shown that a porosity made up of large mesopores, instead of micropores, can facilitate a more efficient contact with reactants and reduce mass-transport limitations,12,22–24 whereas other authors have reported a high electrocatalytic activity by using microporous carbons with a large specific surface area.25–27 Therefore, a fundamental understanding of the influence of pore size is required in order to provide a rational design of high-performing metal-free carbon catalysts.
In this paper, we examine the role of the pore size upon the ORR catalytic activity of N-doped carbon. Thus, we report on metal-free ORR carbon catalysts based on two types of N-doped carbon microspheres (diameter ∼ 1 μm), which have different pore structures: (i) a microporous network made up of pores of ∼1 nm and (ii) a porosity formed by mesopores centered at ∼3 nm and ∼14 nm. Since both types of microspheres possess analogous chemical composition, morphology, electronic conductivity, surface area and particle size, differing only in their pore size distribution, the experiments allowed assessing precisely the influence of pore size on the electrocatalytic behavior. The electrocatalytic activity of the two types of N-doped microspheres was analyzed in both basic and acid media, since testing in acid medium is essential from a practical standpoint.28 The experiments were carried out by using a rotating disk electrode (RDE) and complemented with a rotating ring-disk electrode (RRDE) which allowed assessing the reaction mechanism and quantifying the amount of H2O2 produced.
Experimental section
Preparation of the porous N-doped carbon microspheres
The N-doped carbon particles were produced by nanocasting using porous silica microspheres as templates and pyrrole (Aldrich, 99%) as N-containing carbon precursor. The porous silica particles used as template were synthesized as reported in detail elsewhere.29 In order to modulate the pore size of the carbon microspheres, two types of silica samples were used as template. On one hand, mesoporous silica microspheres of around 1 μm and a narrow pore size distribution centered at ∼2.5 nm were synthesized as reported previously at 25 °C.30 On the other hand, silica microspheres with a porosity made up of mesopores of around 3.6 nm were obtained by means of an additional hydrothermal treatment at 80 °C. A detailed description of the synthesis procedure and structural characteristics of both types of silica particles used as template are given in the ESI† along with the textural properties and microscopy pictures (see Fig. S1†).The synthesis of the N-doped carbon microspheres involved the following steps. Firstly, the silica pores were infiltrated with a solution of FeCl3 in ethanol (2 M) in order to attain around 0.30 g FeCl3 per 1 g silica. Then, the FeCl3-impregnated silica sample was exposed, in a closed vessel, to pyrrole vapors at 25 °C for 22 h. In this way, the adsorbed pyrrole is rapidly polymerized and polypyrrole is formed within the silica pores. The polypyrrole–silica composite thus obtained was heat-treated under N2 up to the carbonization temperature (i.e. 850 °C) at a rate of 3 °C min−1 for 1 h. Finally, the carbonized sample was treated with hydrofluoric acid (48%) to dissolve the silica framework and the generated iron nanoparticles. The resulting carbon residue was collected by filtration, washed with distilled water, and dried at 120 °C for several hours. Depending on the type of silica used as template, two types of carbon microspheres were obtained: (a) microporous N-doped carbon microspheres denoted as N-CS (template: silica synthesized at 25 °C) and (b) mesoporous N-doped carbon microspheres denoted as N-CSH (template: silica particles synthesized at 80 °C).
Materials characterization
The morphology of the powders was examined by scanning (SEM, Zeiss DSM 942) and transmission (TEM, JEOL-2000 FXII) electron microscopy. The size of the particles was determined by measuring around 100 particles in the SEM images in order to obtain statistically significant results. Nitrogen sorption isotherms were recorded at −196 °C using a Micromeritics ASAP 2020 volumetric physisorption system. The Brunauer–Emmett–Teller (BET) surface area was deduced from an analysis of the isotherm in the relative pressure range of 0.04–0.20. The total pore volume was calculated from the amount of nitrogen adsorbed at a relative pressure of 0.99. The micropore volume (Vmi), the micropore surface area (Smi) and the pore size distributions corresponding to the N-CS sample were determined by applying the QSDFT (Quenched-Solid Density Functional Theory) method to the nitrogen adsorption data, assuming a slit pore model. In the case of N-CSH sample, the mesopore size distribution was calculated by means of the Kruk–Jaroniec–Sayari (KJS) method,31 and the micropore (Vmi) and mesopore volumes (Vm) were estimated using the αs-plot method. The reference adsorption data used for the αs analysis of the carbon sample correspond to a non-graphitized carbon black sample.32 Bulk elemental analysis (C, H, N and O) of the samples was carried out on a LECO CHN-932 microanalyzer. X-ray photoelectron spectroscopy (XPS) was performed on a Specs spectrometer, using Mg Kα (1253.6 eV) radiation delivered by a double anode at 150 W. Binding energies for the high resolution spectra were calibrated by setting C 1s to 284.6 eV. X-ray diffraction (XRD) patterns were obtained on a Siemens D5000 instrument operating at 40 kV and 20 mA and using Cu Kα radiation (λ = 0.15406 nm). The electrical conductivity of the carbon powders was determined on a home-made apparatus (four-probe method) by pressing the powders between two plungers into a hollow nylon cylinder (inner diameter of 8 mm), and applying a pressure of 7.1 MPa. The vertical displacement of the plunger was measured by means of a manual cathetometer.Electrochemical measurements
Electrochemical measurements were conducted using an AUTOLAB PGSTAT 101 and a Multi AUTOLAB M101 (CH Instruments). The metal-free carbon catalysts inks were prepared by ultrasonically dispersing 1.5 mg of catalyst in a solution containing 100 μL Nafion (5 wt%) solution and 900 μL deionized water. The Pt/C catalyst (20 wt% Pt on graphitized carbon, Sigma-Aldrich) ink was prepared in the same way, using the same amount of catalyst (i.e. 1.5 mg). The above prepared catalyst inks were deposited onto a polished glassy carbon electrode (using an α-Al2O3 slurry, 50 nm) and dried under room temperature. The catalyst loadings on RRDE and RDE were 0.1 mg cm−2 (20 μgPt cm−2 for the Pt/C catalyst).33,34A conventional three-electrode cell was employed, incorporating Ag/AgCl (3 M KCl) as the reference electrode, a Pt wire as the counter electrode and the catalyst film coated RRDE or RDE as the working electrode. The electrolyte was 0.5 M H2SO4 solution or 0.1 M KOH solution. All the experiments were carried out at 20 °C. Before testing, an O2/N2 flow was bubbled through the electrolyte in the cell for 30 min to saturate it with O2/N2. The measured potentials vs. Ag/AgCl (3 M KCl) were converted to the reversible hydrogen electrode (RHE) scale according to the Nernst equation:
| ERHE = EAg/AgCl + 0.059pH + EoAg/AgCl | (1) |
Cyclic voltammetry (CV) was performed from 0 to 1.2 V vs. RHE in 0.1 M KOH and 0.5 M H2SO4, with a sweep rate of 100 mV s−1.
RDE linear sweep voltammetry (LSV) measurements were conducted from 1.2 to 0 V vs. RHE in 0.1 M KOH and 0.5 M H2SO4 at a scan rate of 10 mV s−1 under disk rotation rates of 400, 800, 1200, 1600, 2000 and 2400 rpm. The working electrode was a 3.0 mm diameter GC rotating disk electrode.
The apparent number of electrons transferred in the ORR reaction on the carbon catalysts was determined by the Koutecky–Levich equation given by:
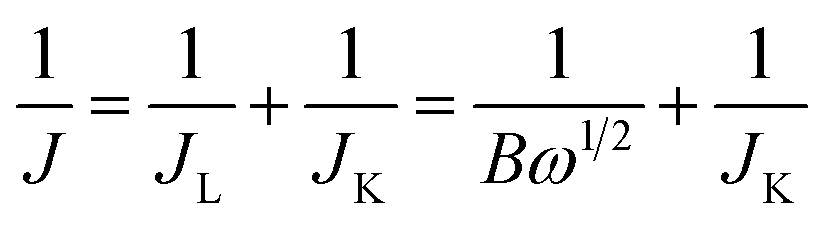 | (2) |
| B = 0.62nFC0(D0)2/3V1/6 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), C0 is the bulk concentration of O2 (1.2 × 10−3 mol L−1 for both 0.5 M H2SO4 solution and 0.1 M KOH solution), D0 is the diffusion coefficient of O2 (1.4 × 10−5 cm2 s−1 for 0.5 M H2SO4 solution and 1.9 × 10−5 cm2 s−1 for 0.1 M KOH solution) and V is the kinetic viscosity of the electrolyte (0.01 cm2 s−1 for both 0.5 M H2SO4 solution and 0.1 M KOH solution).35–37
485 C mol−1), C0 is the bulk concentration of O2 (1.2 × 10−3 mol L−1 for both 0.5 M H2SO4 solution and 0.1 M KOH solution), D0 is the diffusion coefficient of O2 (1.4 × 10−5 cm2 s−1 for 0.5 M H2SO4 solution and 1.9 × 10−5 cm2 s−1 for 0.1 M KOH solution) and V is the kinetic viscosity of the electrolyte (0.01 cm2 s−1 for both 0.5 M H2SO4 solution and 0.1 M KOH solution).35–37
For the RRDE test, the disc potential was scanned at 10 mV s−1, while the ring potential was held at 1.5 V vs. RHE in order to oxidize any H2O2 produced.38,39 The working electrode was a 5 mm GC disk electrode and a Pt ring electrode (375 μm gap). The H2O2 collection coefficient at the ring (N = 0.249) was provided by the manufacturer. The following equations were used to calculate n (the apparent number of electrons transferred during ORR) and % H2O2 (the percentage of H2O2 released during ORR):40
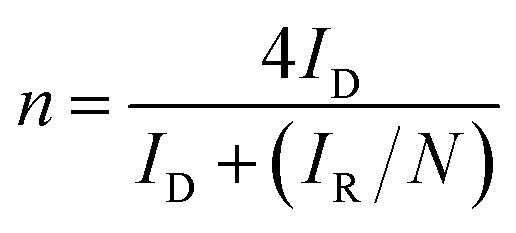 | (4) |
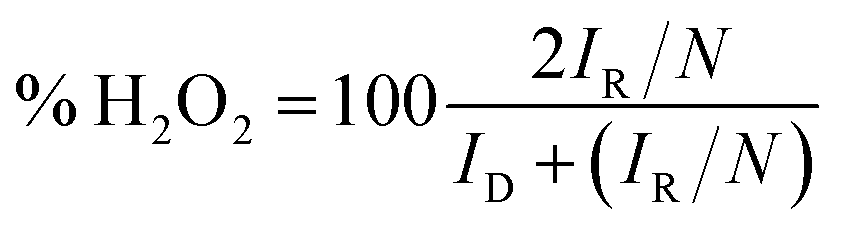 | (5) |
The stability of the catalyst was assessed by means of an US Department of Energy's accelerated durability test protocol by cycling the catalysts between 0.6 and 1.0 V (vs. RHE) at 50 mV s−1 under N2 atmosphere in 0.1 M KOH and 0.5 M H2SO4.41,42
Results and discussion
Structural and chemical properties of the N-doped carbon microspheres
The morphology and structure of the N-doped carbon microspheres were first investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The SEM and TEM images displayed respectively in Fig. 1a and b reveal that the N-CS carbon particles consist of uniform microspheres with a smooth surface and a diameter of 1.2 ± 0.1 μm. The nitrogen-doped carbon microspheres (see Fig. 1c and d) obtained from hydrothermally-treated silica particles (N-CSH) consist, as well, of uniform microspheres with a diameter slightly superior (1.7 ± 0.1 μm). Interestingly, the TEM image from N-CSH sample reveals the existence of larger pores which are radially aligned (Fig. 1d). The N2 sorption isotherms and the pore size distributions (PSDs) of the nitrogen-doped carbon microspheres are displayed in Fig. 2. They clearly illustrate the differences existing on the type of porosity. Thus, the N-CS microspheres exhibit a type I isotherm profile (see Fig. 2a), which is characteristic of microporous materials. This is confirmed by the corresponding PSD in Fig. 2b which shows that the porosity of the N-CS sample is essentially made up of micropores (∼70% of the pore volume, see Table 1) centered at around 1 nm. By contrast, the isotherm profile of the N-CSH carbon microspheres exhibits two capillary condensation steps which suggest that the porosity is made up of two pore systems. Indeed, the PSD of this sample reveals that its porosity is distributed into two mesopore systems with sizes centered at ∼3.2 nm and ∼13.7 nm. The presence of the second pore size system is in accordance with the textural characteristics deduced by TEM inspection (see Fig. 1d). In addition, the percentage of pore volume that corresponds to micropores is lower than 5%, which confirms that the porosity of the N-CSH sample us made up exclusively by mesopores. The textural properties summarized in Table 1 reveal that the carbon microspheres possess high BET surface areas of 1280 m2 g−1 and 1160 m2 g−1 for N-CS and N-CSH respectively, and large pore volumes of 0.75 cm3 g−1 (N-CS) and 1.43 cm3 g−1 (N-CSH). | ||
| Fig. 1 (a) SEM and (b) TEM images of the N-CS carbon microspheres, and (c) SEM (inset: high magnification) and (d) TEM images of the N-CSH carbon microspheres. | ||
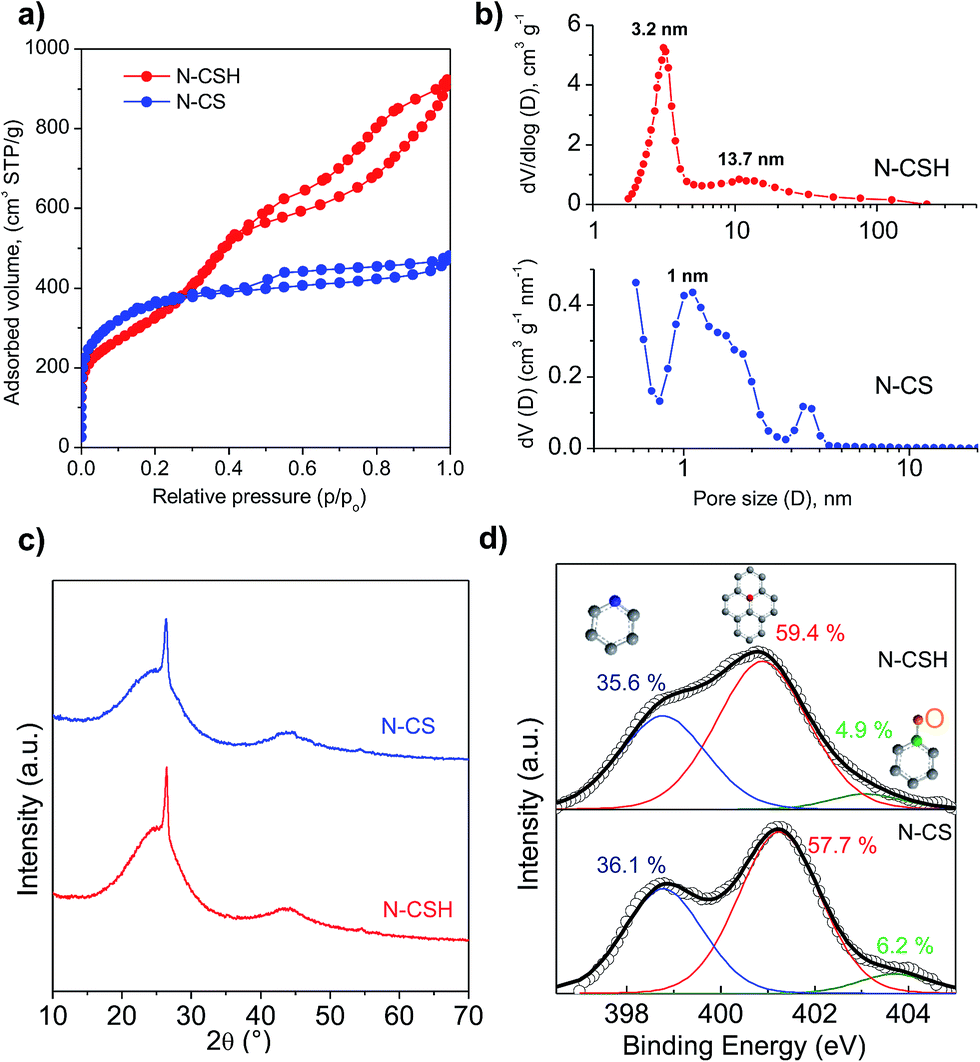 | ||
| Fig. 2 (a) Nitrogen sorption isotherms (b) pore size distributions calculated by Kruk–Jaroniec–Sayari (N-CSH) and QSDFT methods (N-CS), (c) XRD patterns of N-CS and N-CSH, and (d) XPS N 1s core level spectrum corresponding to the N-CS sample. | ||
| Sample code | S BET (m2 g−1) | V p (cm3 g−1) | V mi (cm3 g−1) | V meso (cm3 g−1) | S mi (m2 g−1) | C (wt%) | N (wt%) | O (wt%) | Conductivity (S m−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Pore volume was determined at p/p0 = 0.99. b The micropore volume and the micropore surface area of N-CS were obtained by the QSDFT method, while the micropore volume of N-CSH was determined through the αs-plot method applied to the N2 adsorption branch; the percentage of pore volume that corresponds to the micropores is given in parentheses. c The mesopore volume was obtained from the difference between the pore volume (Vp) and micropore volume (Vmi). | |||||||||
| N-CS | 1280 | 0.75 | 0.52 (70) | 0.24 | 1060 | 76.4 | 8.8 | 13.5 | 2 |
| N-CSH | 1160 | 1.43 | 0.06 (4.2) | 1.37 | — | 78.7 | 8.0 | 12.4 | 10 |
Fig. 2c shows the XRD patterns for the nitrogen-doped carbon microspheres. In both samples, a sharp peak appears at around 2θ = 26°, which is assigned to the (002) diffraction of graphitic carbon (Fig. 2c). This peak is superimposed on a broad band corresponding to amorphous carbon and it reveals the presence of a small amount of graphitized carbon embedded inside an amorphous matrix. This type of carbon is formed due to the conversion of a small amount of amorphous carbon into more ordered carbon by the catalytic action of iron particles embedded inside the carbon matrix.43–46 The percentage of graphitic carbon is similar for both types of carbon microspheres and is around 5–7.3 wt%, as determined by means of thermogravimetric analysis (see Fig. S2†).
The bulk nitrogen content of the carbon microspheres was determined by elemental analysis and the results are listed in Table 1. It can be observed that the carbon samples have high and analogous amounts of nitrogen, i.e. 8.0–8.8 wt%. As the catalytic activity towards the ORR reaction is not only determined by the amount of N, but also by the type of N-structures,47,48 X-ray photoelectron spectroscopy (XPS) was used to investigate the nature of the nitrogen functional groups existing in the carbon microspheres. The XPS N 1s core level spectra of both nitrogen-doped microspheres are displayed in Fig. 2d. The samples can be deconvoluted into three peaks that are assigned to pyridinic nitrogen, quaternary nitrogen and pyridine-N-oxide.49,50 The main contributions correspond to quaternary and pyridinic nitrogen groups, being respectively 57.7–59.4% and 36.1–35.6%, while pyridine-N-oxide represents only 6.2–4.9% of the total amount of nitrogen for N-CS and N-CSH. This result is important because the quaternary and pyridinic groups have been suggested as the main ORR active sites.11,19,51–53 As can be seen, both types of carbon microspheres display a similar distribution of the nitrogen functionalities. In addition, the XPS general spectra of both microspheres show that there is no presence of residual iron species (see Fig. S3†). This result is also confirmed by the thermogravimetric analyses in Fig. S2,† which show no residue in the carbon samples. The absence of residual iron was further corroborated by elemental energy-dispersive X-ray (EDX) (see Fig. S4†). It is to be expected as the samples were washed with hydrofluoric acid. This is a relevant result since it has been shown that a small amount of iron, even traces, might enhance the electrocatalytic activity of N-doped carbon materials.54
Electrocatalytic activity of the N-doped carbon microspheres in the ORR reaction
Cyclic voltammetry (CV) at a scan rate of 100 mV s−1 and RDE linear sweep voltammetry (LSV) at a scan rate of 10 mV s−1 were conducted in 0.1 M KOH and 0.5 M H2SO4 electrolytes for the N-doped porous carbon microspheres. The experiments carried out under nitrogen protection show good charge propagation throughout the porous carbon framework, as evidenced by the quasi-rectangular corners of the cyclic voltammograms (see Fig. 3). This is the result of the good electronic conductivity (see Table 1) and accessible porous structure of the materials. On the other hand, the experiments performed under oxygen atmosphere in KOH show that the N-doped microspheres have a significant electrocatalytic activity towards ORR, as inferred by the obvious cathodic peak (Fig. 3a and b). This well-defined peak appears at 0.837 V for N-CSH (see Fig. 3a), while for N-CS the value is slightly shifted to lower values (0.797 V, see Fig. 3b), which suggests the effectiveness of the mesoporous microspheres over the micropores ones. This result also shows a more efficient electrochemical reduction reaction of oxygen in comparison with that of previously reported N-doped carbon microspheres (from ∼0.82 V to ∼0.77 V).24,55–58 However, in 0.5 M H2SO4, a pronounced cathodic peak is no visible, demonstrating the superior ORR catalytic activity of the materials in an alkaline medium, as already observed in the literature (see Fig. 3c and d).16,59 The slower kinetics of the ORR reaction in acidic medium compared to alkaline medium is corroborated by the linear sweep voltammograms (LSV) in the O2-saturated electrolytes in Fig. S5.† Thus, an obvious plateau can be discerned in the polarization curves in the diffusion-limited region in KOH electrolyte, while it is absent in H2SO4 electrolyte, which is indicative of kinetic control in acidic medium even at high overpotentials.15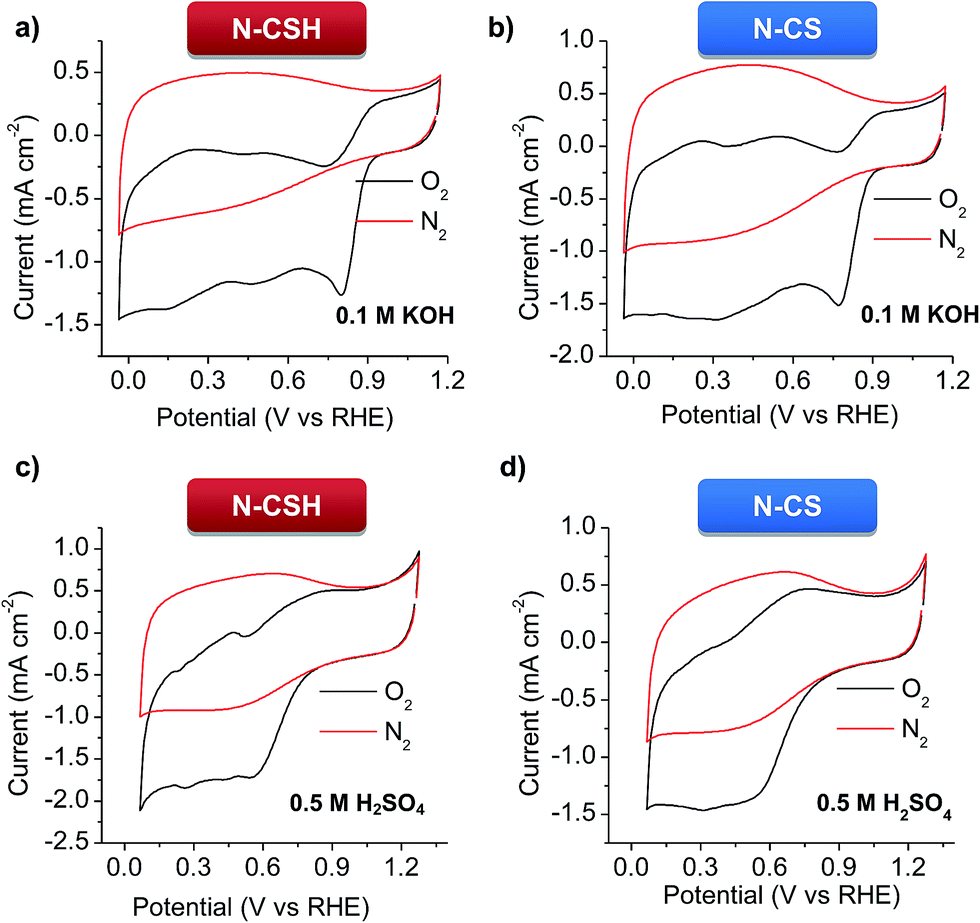 | ||
| Fig. 3 Cyclic voltammograms of the N-doped carbon microspheres in N2- and O2-saturated electrolytes at a scan rate of 50 mV s−1. (a and b) 0.1 M KOH, and (c and d) 0.5 M H2SO4. | ||
The ORR activity of N-CSH, N-CS and commercial Pt/C (20 wt%) catalyst was compared by evaluating the LSV in O2-saturated electrolytes (see Fig. 4a and b). It should be noted that, to provide a realistic picture, the same amount of N-doped carbon catalyst and Pt/C catalyst was used (0.1 mg cm−2). In basic media, the N-doped mesoporous carbon microspheres exhibit the same onset potential and more positive half-wave potential (E1/2) than commercial Pt/C, which points out their high activity towards ORR. Specifically, the N-CSH catalyst shows an ORR onset potential at 0.927 V compared with 0.907 V for the microporous microspheres (N-CS) and 0.927 V in the case of commercial Pt/C catalyst (comparable to that found by other authors25,60,61) (see Fig. 4a). Noteworthy, the value of E1/2 is only ∼50–60 mV more negative than the best-performing metal-free carbon catalysts reported so far.62,63 In addition, the N-CSH microspheres exhibit a higher diffusion-limited current density than that of N-CS (i.e., 3.4 vs. 3.2 mA cm−2), although both are lower than that of commercial Pt/C (4.2 mA cm−2, in agreement with other Pt/C reported values for the same mass loading of catalyst64–67). These results show improved accessibility of the active sites in the case of the mesoporous microspheres compared to the microporous ones despite their slightly larger diameter (i.e. larger diffusion path). Compared to N-doped mesoporous nanospheres reported very recently with similar nitrogen content but different distribution of the nitrogen functionalities (lower proportion of N-quaternary),56 the onset potential of the microspheres here developed is more positive (0.857 V for the mesoporous nanospheres, despite their higher mass loading), which suggests that N-quaternary species have a higher catalytic activity towards ORR than N-pyridinic species, in accordance with their results and those of other authors.7,68 The sharp increase and rapid saturation of the current registered on both microspheres samples suggests that the diffusion-controlled process can be related to an efficient 4 electron pathway. This is confirmed by the Koutecky–Levich analyses displayed in Fig. S6a,† where the slopes are parallel to that of an ideal four-electron process. The RDE experiments performed in acid medium (Fig. 4b) confirm that the N-doped carbon microspheres exhibit a lower activity than in KOH medium (Fig. 4a). Nevertheless, the carbon catalysts still possess a relatively good ORR activity. Thus, both types of carbon microspheres exhibit the same onset potential as Pt/C (0.809 V). Note that this value was obtained for a mass loading of 0.1 mg cm−2 (20 μgPt cm−2) and differences on the onset potential can be observed depending on the catalyst loading used.69,70 In the case of the N-CSH sample, with a mesoporous structure, the half-wave potential is located at around 0.529 V and the maximum current density is 3.29 mA cm−2 at 0.069 V, both values being superior to those of the microporous N-CS sample (i.e. 0.5 V and 2.84 mA cm−2), but inferior to those of Pt/C (see Fig. 4b). The good performance of the N-CSH sample can be attributed to its easily accessible porosity, which is formed by two interconnected systems of mesopores (see Fig. 2b) that facilitate the mass-transfer processes during the electrochemical reaction, minimizing thereby mass-transfer limitations. The Koutecky–Levich analyses show that the slopes are more similar to a 4 electron process (see Fig. S6b†), which suggests a favorable direct reduction of O2 to H2O.
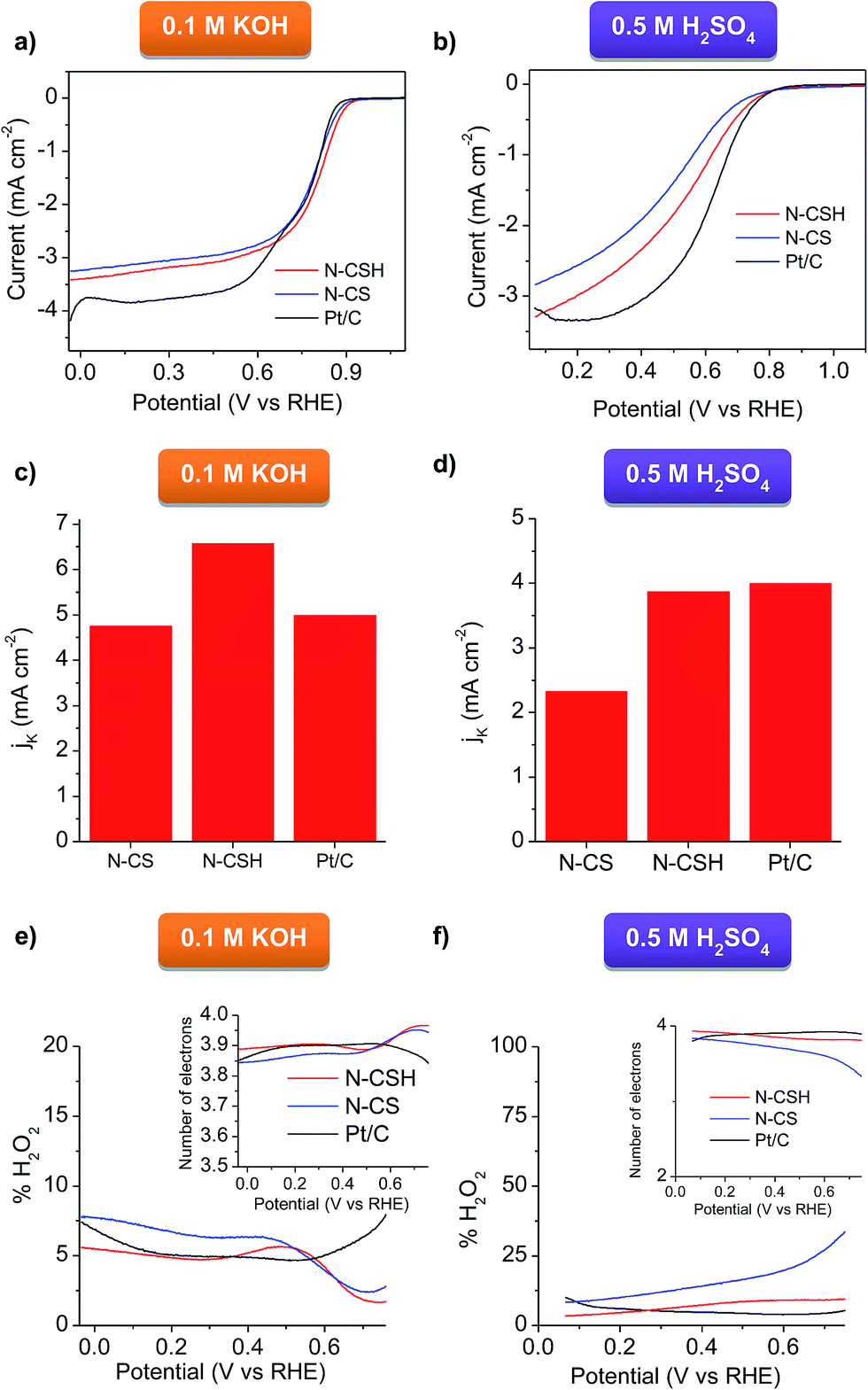 | ||
| Fig. 4 RDE polarization curves at 1600 rpm for the N-CSH and N-CS samples compared to 20 wt% Pt/C in (a) 0.1 M KOH and (b) 0.5 M H2SO4, (c) electrochemical activity given as the kinetic-limiting current density (JK) at 0.67 V in 0.1 M KOH and (d) at 0.48 V in 0.5 M H2SO4, and peroxide yields of the carbon microspheres in (e) 0.1 M KOH and (f) 0.5 M H2SO4 (inset: variation of the number of electrons with potential). | ||
The kinetic current densities (JK) deduced from the Koutecky–Levich equation are represented in Fig. 4c for the KOH electrolyte. The JK has a value of 6.6 mA cm−2 for N-CSH at 0.677 V, which is notably superior to the values deduced for N-CS (4.8 mA cm−2) and Pt/C (5 mA cm−2), and comparable – or even higher – to the values reported in the literature for nitrogen-doped carbon materials.68,71 Similar results are obtained in acidic media, where N-CSH exhibits a higher value of limiting kinetic current density than N-CS. Thus, the N-CSH carbon has a calculated electrochemical kinetic current density (JK) value of 3.9 mA cm−2 at 0.48 V, almost twice the value obtained at the same potential for N-CS (2.3 mA cm−2), which confirms the better electrocatalytic behavior of N-CSH in comparison to N-CS. Moreover, this calculated value is almost the same as that obtained for commercial Pt/C (4 mA cm−2) (see Fig. 4d). These results reveal the superior electrocatalytic activity of the N-doped mesoporous carbon microspheres regardless of the electrolyte used, even compared to commercial Pt/C.
To quantify the amount of H2O2 generated during the ORR process and confirm the ORR mechanism, a RRDE electrode was used. Fig. 4e compares the peroxide yield for N-CS and N-CSH in 0.1 M KOH. These results reveal that the N-CSH sample has a better selectivity. Nevertheless, for both samples, the H2O2 yield is lower than 10% (∼5% for N-CSH and ∼7.5% for N-CS). The superior performance offered by N-CSH over N-CS is further confirmed in acid media. Thus, the H2O2 yield for N-CSH is lower than 10% over the whole potential range, whereas that of N-CS is ∼10% for high overpotentials and increases over 15% for low overpotentials (see Fig. 4f). As shown by the number of electrons transferred in the reaction (inset of Fig. 4e and f), both carbon samples catalyze the efficient four-electron pathway. Needless to say the good electrocatalytic activity offered by Pt/C, with a number of electrons transferred of ∼4 in both media. Anyway, the N-CSH mesoporous carbon exhibits not only the same number of electrons transferred as Pt/C, but also a similar % of H2O2 produced, which corroborates the excellent electrocatalytic activity of N-CSH towards ORR in both media. The high ORR activity of the catalysts developed in this work may be attributed to the combine effect of a large number of quaternary and pyridinic nitrogen groups,19,52,53 and the well-designed porosity which facilitates fast access to the active sites, especially for larger pore size.72–74 Furthermore, the comparison between both types of carbon microspheres reveals that the N-doped carbon microspheres with mesopores possess a higher electrocatalytic activity towards ORR compared with those with a micropore size distribution owing to enhanced accessibility to the active sites.
The N-doped carbon microspheres were also exposed to fuel molecules (methanol) for testing possible poisoning effects, problem encountered by direct methanol fuel cells (DMFC) working with platinum catalyst.75 To compare the stability of the different electrocatalysts, the chronoamperometric response was measured by injecting pure methanol into the O2-saturated 0.1 M KOH electrolyte (see Fig. 5a) and the RDE polarization curve was measured in the presence of 1 M methanol in acidic media (see Fig. 5b). As can be seen in Fig. 5a corresponding to KOH electrolyte, no current decay is observed for N-CSH, while a small current decay of 15% is recorded for N-CS. However, the Pt/C catalyst suffers a 40% current decay. In acidic medium, as evidenced by Fig. 5b, there is a small shift of the polarization curve towards higher overpotentials (downshift of 10 mV and 20 mV for N-CSH and N-CS, respectively) when the ORR is carried out in the presence of methanol. These results suggest that the presence of methanol hardly affects the ORR catalytic performance of the N-doped carbon microspheres, especially in the case of the N-CSH sample, contrarily to the currently commercialized Pt/C catalyst.
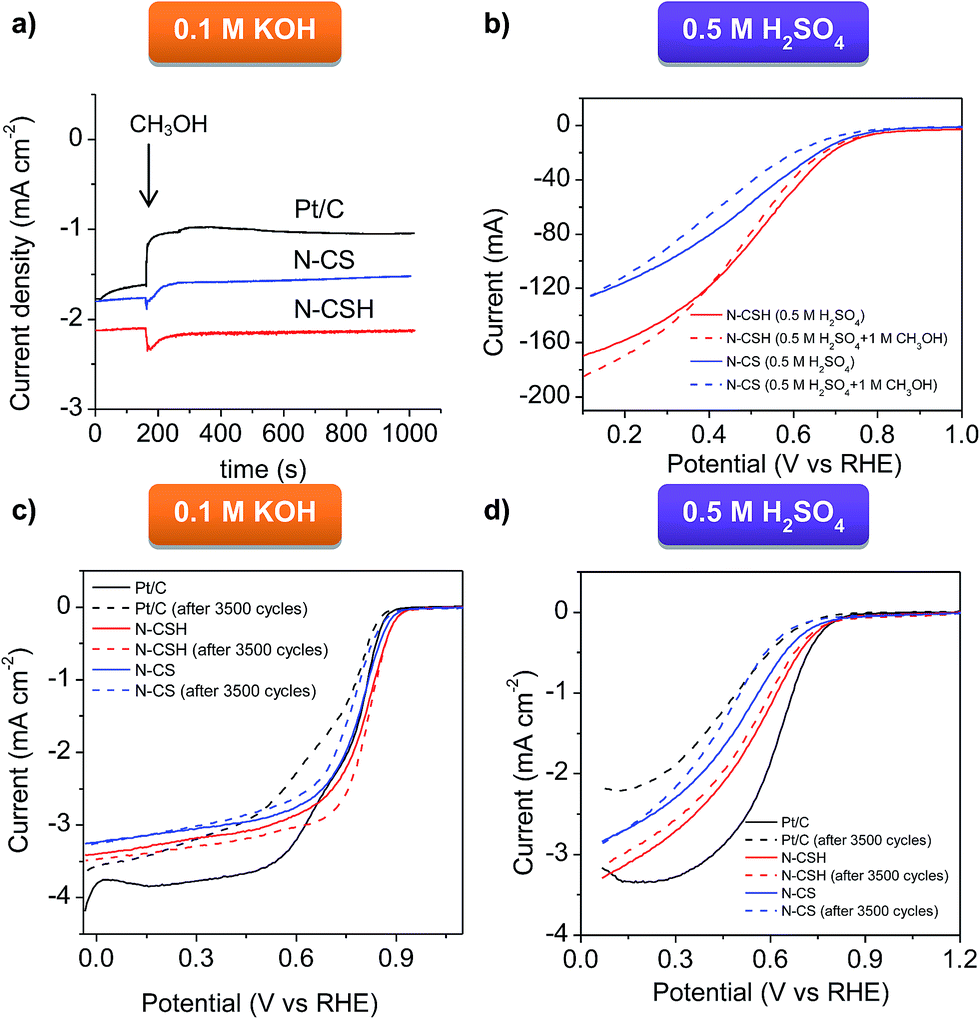 | ||
| Fig. 5 (a) Chronoamperometric responses over 1000 s at a constant rotation speed of 1600 rpm in O2-saturated KOH solution for the N-doped carbon microspheres and Pt/C catalyst (the arrow symbolizes the addition of methanol), (b) polarization curves of the N-doped carbon microspheres in O2-saturated 0.5 M H2SO4 with and without 1 M methanol, and LSVs for the N-doped carbon microspheres and Pt/C catalyst at 1600 rpm in O2-saturated (c) 0.1 M KOH and (d) 0.5 M H2SO4 before and after 3500 potential cycles. | ||
The stability of the catalysts is another critical feature of the ORR process.76 Accordingly, it was assessed by an accelerated durability test protocol (see Experimental section for specific details). The ORR stability of N-CSH, N-CS and Pt/C was analyzed by recording linear sweep voltammograms in KOH and H2SO4 saturated with O2 before and after the cycling protocol. Fig. 5c displays the polarization curves before and after 3500 cycles in KOH electrolyte. No significant variations are observed in the onset and half-wave potentials in the case of the N-CSH sample, whereas for N-CS both parameters downshift by 10 mV and 40 mV respectively. Since the decay of the half-wave potential in the polarization curves before and after a long cycling test is a measurement of the stability of the catalyst,77,78 the excellent durability of the N-doped materials in basic medium is undeniable. Besides, both materials exhibit better electrocatalytic stability than commercial Pt/C, which exhibits a decrease on the onset and half-wave potentials of 10 and 90 mV respectively. The results obtained in 0.5 M H2SO4, shown in Fig. 5d, reveal that the onset potential of N-CSH does not experience any modification, while the half-wave potential slightly decreases (by ∼10 mV). However, for the N-CS sample, the onset potential decreases by 70 mV, with a similar diminution on the half-wave potential. On the other hand, Fig. 5d makes abundantly clear that commercial Pt/C is the least stable, with a diminution of 70 mV on the onset potential and 140 mV on the half-wave potential. These results prove the superior electrocatalytic stability of the N-doped microspheres in both electrolytes compared to commercially available Pt/C. On the other hand, the better stability of N-CSH in comparison to N-CS may be explained by their different pore structure. Thus, the N-CSH carbon possesses a bimodal mesoporous structure that enhances the mass-transport kinetics of reactant and products towards/away from the catalytic sites in such a way that the active sites are fully accessible during the whole accelerated durability test protocol.79 On the contrary, the results obtained for the microporous sample N-CS suggest a slower transport of reactants and products, giving rise to the accumulation of the products on the active sites. Furthermore, accumulation of products might also be favored by the enhanced adsorption potential existing in micropores.
Conclusions
In summary, we provide a synthetic strategy towards N-doped carbon microspheres with an engineered pore structure and structural features that lead to a high-performing ORR catalyst. Depending on the template used, the porosity of the carbon microspheres was tuned from a micro- to a mesoporous network. Such synthesized N-carbons were tested as metal-free electrocatalyst for ORR in basic (0.1 M KOH) and acid (0.5 M H2SO4) electrolytes. As both kinds of microspheres have a similar chemical composition, electronic conductivity, specific surface area and particle size, the only major difference being their pore size distribution, we could inferred the effect of pore size on the electrocatalytic activity. The N-doped mesoporous carbon microspheres clearly exhibited a better electrocatalytic performance than the microporous carbon microspheres in terms of ORR catalytic activity, selectivity and durability in both media. The outstanding performance of the mesoporous microspheres is the result of the combination of several features: (i) a high nitrogen content (∼8 wt%), quaternary- and pyridinic-N being the major contributions, (ii) an accessible mesoporosity (two mesopore systems centered at ∼3 nm and ∼14 nm) that enhances the mass-transport of the species towards and away from the catalytic active sites and (iii) a large specific surface area (1160 m2 g−1) and pore volume (1.43 cm3 g−1). Moreover, these N-doped mesoporous carbon microspheres outperformed commercial Pt/C catalyst in basic media, showing similar onset potential but higher half-wave potential and kinetic current density. Additionally, their electrocatalytic performance in acidic medium is comparable to that of Pt/C, showing analogous onset potential and kinetic current density. Furthermore, contrarily to platinum, both types of carbon microspheres are unaffected by methanol crossover and are robust against cycling, which makes them excellent metal-free carbon catalyst for ORR. Finally, the percentage of hydrogen peroxide produced in the case of the mesoporous microspheres is lower than 10% in H2SO4 electrolyte and 6% in KOH electrolyte (both values being lower than those of the microporous carbon), proving their electrocatalytic selectivity towards an efficient four electron mechanism.Acknowledgements
This research work was supported by the Spanish Ministerio de Economía y Competitividad, MINECO (MAT2012-31651), Fondo Europeo de Desarrollo Regional (FEDER) and FICYT Regional Project (GRUPIN14-102). G. A. F. thanks the MINECO for his predoctoral contract and M. S. thanks the Spanish Ministerio de Ciencia e Innovación for her Ramón y Cajal contract. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- Y.-J. Wang, D. P. Wilkinson and J. Zhang, Chem. Rev., 2011, 111, 7625–7651 CrossRef CAS PubMed.
- J. Zhang, PEM fuel cell electrocatalysts and catalyst layers: fundamentals and applications, Springer Science & Business Media, 2008 Search PubMed.
- D. Yu, E. Nagelli, F. Du and L. Dai, J. Phys. Chem. Lett., 2010, 1, 2165–2173 CrossRef CAS.
- N. Alonso-Vante, ChemPhysChem, 2010, 11, 2732–2744 CrossRef CAS PubMed.
- X. Yu and S. Ye, J. Power Sources, 2007, 172, 145–154 CrossRef CAS.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- D.-W. Wang and D. Su, Energy Environ. Sci., 2014, 7, 576–591 CAS.
- P. Trogadas, T. F. Fuller and P. Strasser, Carbon, 2014, 75, 5–42 CrossRef CAS.
- Y. Shao, J. Sui, G. Yin and Y. Gao, Appl. Catal., B, 2008, 79, 89–99 CrossRef CAS.
- K.-H. Wu, D.-W. Wang, D.-S. Su and I. R. Gentle, ChemSusChem, 2015, 8, 2772–2788 CrossRef CAS PubMed.
- N. Daems, X. Sheng, I. F. J. Vankelecom and P. P. Pescarmona, J. Mater. Chem. A, 2014, 2, 4085–4110 CAS.
- V. V. Strelko, V. S. Kuts and P. A. Thrower, Carbon, 2000, 38, 1499–1503 CrossRef CAS.
- P. Chen, L.-K. Wang, G. Wang, M.-R. Gao, J. Ge, W.-J. Yuan, Y.-H. Shen, A.-J. Xie and S.-H. Yu, Energy Environ. Sci., 2014, 7, 4095–4103 CAS.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 CAS.
- Z. Mo, S. Liao, Y. Zheng and Z. Fu, Carbon, 2012, 50, 2620–2627 CrossRef CAS.
- V. Vishnyakov, Vacuum, 2006, 80, 1053–1065 CrossRef CAS.
- J. Yin, Y. Qiu, J. Yu, X. Zhou and W. Wu, RSC Adv., 2013, 3, 15655–15663 RSC.
- J. Yin, Y. Qiu and J. Yu, J. Electroanal. Chem., 2013, 702, 56–59 CrossRef CAS.
- A. Anastasopoulos, J. Davies, L. Hannah, B. Hayden, C. Lee, C. Milhano, C. Mormiche and L. Offin, ChemSusChem, 2013, 6, 1973–1982 CrossRef CAS PubMed.
- N. Brun, S. A. Wohlgemuth, P. Osiceanu and M. M. Titirici, Green Chem., 2013, 15, 2514–2524 RSC.
- I.-Y. Jeon, D. Yu, S.-Y. Bae, H.-J. Choi, D. W. Chang, L. Dai and J.-B. Baek, Chem. Mater., 2011, 23, 3987–3992 CrossRef CAS.
- N. Gavrilov, I. A. Pašti, M. Mitrić, J. Travas-Sejdić, G. Ćirić-Marjanović and S. V. Mentus, J. Power Sources, 2012, 220, 306–316 CrossRef CAS.
- Y.-L. Liu, C.-X. Shi, X.-Y. Xu, P.-C. Sun and T.-H. Chen, J. Power Sources, 2015, 283, 389–396 CrossRef CAS.
- X. Zhao, H. Zhao, T. Zhang, X. Yan, Y. Yuan, H. Zhang, H. Zhao, D. Zhang, G. Zhu and X. Yao, J. Mater. Chem. A, 2014, 2, 11666–11671 CAS.
- J.-Y. Choi, R. S. Hsu and Z. Chen, J. Phys. Chem. C, 2010, 114, 8048–8053 CAS.
- Y. He, X. Han, Y. Du, B. Song, P. Xu and B. Zhang, ACS Appl. Mater. Interfaces, 2015 DOI:10.1021/acsami.5b07865.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- A. B. Fuertes and P. Tartaj, Small, 2007, 3, 275–279 CrossRef CAS PubMed.
- R. I. Nooney, D. Thirunavukkarasu, Y. Chen, R. Josephs and A. E. Ostafin, Chem. Mater., 2002, 14, 4721–4728 CrossRef CAS.
- M. Kruk, M. Jaroniec and A. Sayari, Langmuir, 1997, 13, 6267–6273 CrossRef CAS.
- M. Kruk, M. Jaroniec and K. P. Gadkaree, J. Colloid Interface Sci., 1997, 192, 250–256 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- R. Silva, D. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 135, 7823–7826 CrossRef CAS PubMed.
- Z.-Y. Wu, X.-X. Xu, B.-C. Hu, H.-W. Liang, Y. Lin, L.-F. Chen and S.-H. Yu, Angew. Chem., Int. Ed., 2015, 54, 8179–8183 CrossRef CAS.
- D. R. Lide, CRC handbook of chemistry and physics, CRC Press, 2004 Search PubMed.
- H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1996, 100, 6715–6721 CrossRef.
- J. Wu, C. Jin, Z. Yang, J. Tian and R. Yang, Carbon, 2015, 82, 562–571 CrossRef CAS.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- W. Xing, G. Yin and J. Zhang, Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Elsevier, 2014 Search PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Carbon, 2006, 44, 468–474 CrossRef CAS.
- M. Sevilla, C. Sanchis, T. Valdes-Solis, E. Morallon and A. B. Fuertes, J. Phys. Chem. C, 2007, 111, 9749–9756 CAS.
- A. Ōya and H. Marsh, J. Mater. Sci., 1982, 17, 309–322 CrossRef.
- H. Marsh, D. Crawford and D. W. Taylor, Carbon, 1983, 21, 81–87 CrossRef CAS.
- P. H. Matter, L. Zhang and U. S. Ozkan, J. Catal., 2006, 239, 83–96 CrossRef CAS.
- P. Matter and U. Ozkan, Catal. Lett., 2006, 109, 115–123 CrossRef CAS.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641–1653 CrossRef CAS.
- H. Schmiers, J. Friebel, P. Streubel, R. Hesse and R. Köpsel, Carbon, 1999, 37, 1965–1978 CrossRef CAS.
- D. Geng, Y. Chen, Y. Chen, Y. Li, R. Li, X. Sun, S. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760–764 CAS.
- X. Sheng, N. Daems, B. Geboes, M. Kurttepeli, S. Bals, T. Breugelmans, A. Hubin, I. F. J. Vankelecom and P. P. Pescarmona, Appl. Catal., B, 2015, 176–177, 212–224 CrossRef CAS.
- H. Wang, X. Bo, C. Luhana and L. Guo, Electrochem. Commun., 2012, 21, 5–8 CrossRef CAS.
- J. Masa, A. Zhao, W. Xia, Z. Sun, B. Mei, M. Muhler and W. Schuhmann, Electrochem. Commun., 2013, 34, 113–116 CrossRef CAS.
- J. Tang, J. Liu, C. Li, Y. Li, M. O. Tade, S. Dai and Y. Yamauchi, Angew. Chem., Int. Ed., 2015, 54, 588–593 CAS.
- B. Bayatsarmadi, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem.–Asian J., 2015, 10, 1546–1553 CrossRef CAS PubMed.
- T. Yang, J. Liu, R. Zhou, Z. Chen, H. Xu, S. Z. Qiao and M. J. Monteiro, J. Mater. Chem. A, 2014, 2, 18139–18146 CAS.
- J. Li, Z. Li, J. Tong, C. Xia and F. Li, RSC Adv., 2015, 5, 70010–70016 RSC.
- M. Sevilla, L. Yu, T. P. Fellinger, A. B. Fuertes and M.-M. Titirici, RSC Adv., 2013, 3, 9904–9910 RSC.
- C.-T. Hung, N. Yu, C.-T. Chen, P.-H. Wu, X. Han, Y.-S. Kao, T.-C. Liu, Y. Chu, F. Deng, A. Zheng and S.-B. Liu, J. Mater. Chem. A, 2014, 2, 20030–20037 CAS.
- Z.-Y. Wu, X.-X. Xu, B.-C. Hu, H.-W. Liang, Y. Lin, L.-F. Chen and S.-H. Yu, Angew. Chem. Int. Ed., 2015, 54, 8179–8183 CrossRef CAS PubMed.
- H.-W. Liang, X. Zhuang, S. Brüller, X. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed.
- H. Jin, H. Huang, Y. He, X. Feng, S. Wang, L. Dai and J. Wang, J. Am. Chem. Soc., 2015, 137, 7588–7591 CrossRef CAS PubMed.
- J. Liang, Y. Zheng, J. Chen, J. Liu, D. Hulicova-Jurcakova, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 3892–3896 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- H.-P. Cong, P. Wang, M. Gong and S.-H. Yu, Nano Energy, 2014, 3, 55–63 CrossRef CAS.
- C. Hu, L. Wang, Y. Zhao, M. Ye, Q. Chen, Z. Feng and L. Qu, Nanoscale, 2014, 6, 8002–8009 RSC.
- K. Parvez, S. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. Feng and K. Müllen, ACS Nano, 2012, 6, 9541–9550 CrossRef CAS PubMed.
- Y.-H. Shih, G. V. Sagar and S. D. Lin, J. Phys. Chem. C, 2008, 112, 123–130 CAS.
- L. Wang, L. Zhang and J. Zhang, Electrochem. Commun., 2011, 13, 447–449 CrossRef CAS.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- L. Zhang, Z. Su, F. Jiang, L. Yang, J. Qian, Y. Zhou, W. Li and M. Hong, Nanoscale, 2014, 6, 6590–6602 RSC.
- K. Qiu and Z. X. Guo, J. Mater. Chem. A, 2014, 2, 3209–3215 CAS.
- G. Tao, L. Zhang, L. Chen, X. Cui, Z. Hua, M. Wang, J. Wang, Y. Chen and J. Shi, Carbon, 2015, 86, 108–117 CrossRef CAS.
- V. Neburchilov, J. Martin, H. Wang and J. Zhang, J. Power Sources, 2007, 169, 221–238 CrossRef CAS.
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 CAS.
- H. Wang, K. Wang, H. Song, H. Li, S. Ji, Z. Wang, S. Li and R. Wang, RSC Adv., 2015, 5, 48965–48970 RSC.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- G. Tao, L. Zhang, L. Chen, X. Cui, Z. Hua, M. Wang, J. Wang, Y. Chen and J. Shi, Carbon, 2015, 86, 108–117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta10063a |
| This journal is © The Royal Society of Chemistry 2016 |