
Anion responsive and morphology tunable tripodal gelators†
Amrita Ghosh*a,
Priyadip Dasb,
Rahul Kaushika,
Krishna Kumar Damodaranc and
D. Amilan Josea
aDepartment of Chemistry, NIT Kurukshetra, Haryana-136119, India. E-mail: amritaghosh2003@gmail.com
bInstitute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel
cDepartment of Chemistry, Science Institute, University of Iceland, Dunhagi 3, 107 Reykjavík, Iceland
First published on 26th August 2016
Abstract
We report the syntheses and characterization of three isomers of trisamide tripodal derivatives. All these compounds act as low-molecular-weight gelators and the variation of the gel morphology with respect to the solvent combination has been analysed. The anion binding studies revealed that these tris-amide derivatives also act as suitable receptors for appropriate analytes. In this work, we have explored the role of geometry or extent of receptor–anion interaction on the gel architecture in the presence of anions such as F−, CN−, CH3COO− and H2PO4−. We have discussed the anion responsive gel formation of one isomer after incorporation of anions in the solution phase as well as into the gel morphology.
Introduction
Stimuli-responsive soft matter systems constitute a fascinating class of materials with promising applications in various fields. Stimuli-responsive supramolecular gels based on low molecular weight gelators (LMWGs)1–10 fall into this category. LMWGs are formed by the immobilisation of solvent molecules in the 3-D fibrous network of the gelators, which self-assemble via non-covalent interactions11–13 such as hydrogen bonding, van der Waals interactions, π–π stacking, halogen bonding, lipophilic and electrostatic interactions. Stimuli-responsive LMWGs have attracted tremendous attention over the past decade due to their potential applications in drug delivery, tissue engineering, sensing as well as catalysis.6,14–18 The properties of stimuli-responsive LMWGs can be either switched on/off by an external stimulus,8,19 where the construction and deconstruction of the gel networks can be triggered by pH, redox, light and salts/ions.20–32 The understanding and use of supramolecular interactions will enable us to control and tune the materials/gel properties such as elasticity and supramolecular structure.33–35 Specifically, such responsive systems, for example anion recognition are highly desirable for the development of sensor devices. The tuning of gel properties based on anion binding is an area of current interest because the conformational changes in the supramolecular architecture of LMWGs can be controlled by host–guest recognition, which should also be useful for the organization and modulation of associated structures.36–42 The recognition and binding of anions play critical role in life science, medicine, catalysis, and environmental chemistry.43–46 This resulted in the development of a myriad of receptors46–51 with hydrogen donor groups such as amide, urea, thiourea, pyrrole and indole indicating the importance of anion recognition.Amide groups have proved to be highly efficient in designing potential receptors for anion binding.49 The N–H group of the amide moiety is a good hydrogen bond donor, where the polarization by π-electron with the neighbouring carbonyl moiety plays a crucial role.36,52 The carbonyl functionality of the amide group is also capable of acting as a hydrogen bond acceptor functionality involving the OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O atom.46 On the other hand, amide based gelators are capable of forming organo/hydrogels,13,27,53–62 which is formed by the self-assembly of the gelator into fibril structure via cooperative and unidirectional hydrogen bonding involving amide units.
O atom.46 On the other hand, amide based gelators are capable of forming organo/hydrogels,13,27,53–62 which is formed by the self-assembly of the gelator into fibril structure via cooperative and unidirectional hydrogen bonding involving amide units.
We are interested in designing supramolecular gels based on amide derivatives of trimesic acid, which have been excellent platform in anion recognition for appropriate analytes.63–65 However, in anion recognition there are two possible types of binding of trimesic derivatives to the anionic analyte. This includes one with nonspecific binding involving three different anionic analyte and another option where the tripodal ligand may behave as a “Scorpionate ligand” with three –N(H) hydrogens to be involved simultaneously in binding to a single anionic species. Examples of both type of binding mode for related ligands are available in the literature.66 Recently, Ghosh and co-workers have shown a tripodal amide derivative of trimesic acid forms capsule upon anion or hydrated anion complexation for anions such as F−, Cl−, Br− and CH3COO−. However, most studies in this aspect are restricted either to the 1HNMR or X-ray structural studies.37,65 Efforts have been made to study the effect of anion binding to fluorescence responsive gelators37,40,67 and a fluoride-responsive gelator based on a cyano-substituted amide as a colorimetric sensor have been reported.68 Interestingly, there are few reports on supramolecular gels based on trimesic acid amide derivative69–71 with very low minimum gel concentration (MGC).69 In this work, we would like to combine the anion binding properties and gelation properties of trimesic amides, which may lead to gel capsule formation. We are particularly interested in using supramolecular interactions (anion binding) to tune gel properties and understand the changes in morphology and supramolecular structural assembly.35,72–74 In order to achieve this task, we have designed fluorescent dye labelled tris-amide derivatives and selected quinoline moiety as a fluorogenic unit (Scheme 1). These three isomers are used to analyse their role in gelation properties and selected one isomer to explore the changes in presence of anionic guests such as fluoride (F−), cyanide (CN−) acetate (CH3COO−) and phosphate (H2PO4−) both in solution and gel phase.
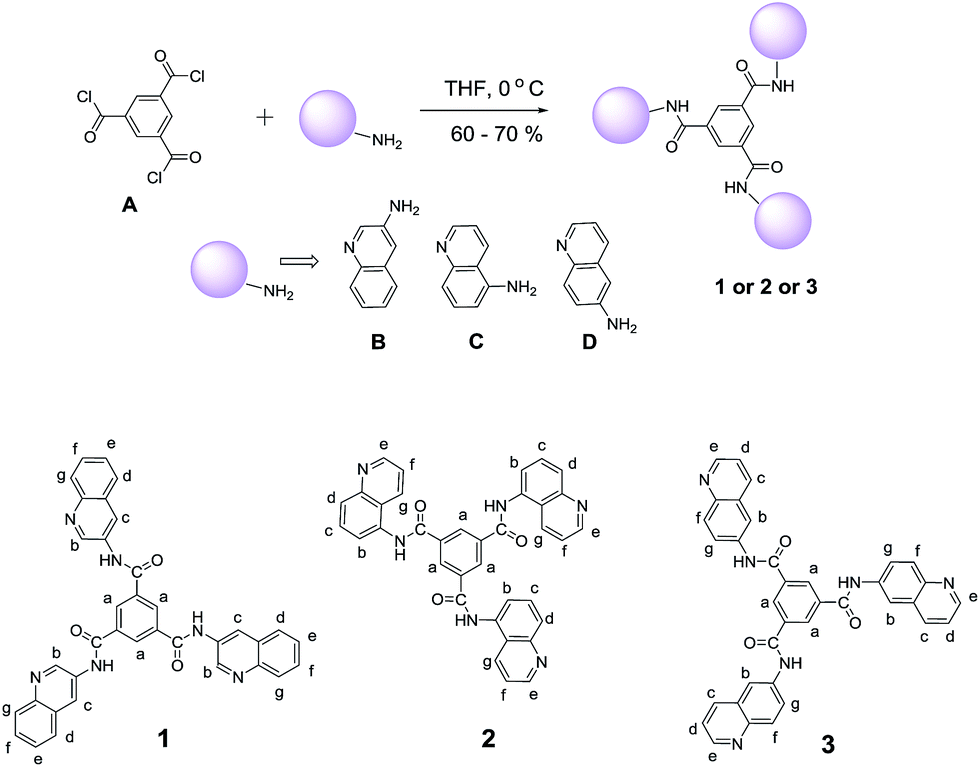 | ||
| Scheme 1 Syntheses and structures of the three compounds 1, 2 and 3. | ||
Experimental section
Materials and method
Amino quinoline derivatives and 1,3,5-tricarboxylic acid were purchased from Aldrich Chemical Co. (USA) and used as received. Solvents and triethylamine used for synthesis, were dried and distilled before use according to standard procedures. Other solvents purchased are from S.D. Fine Chemicals (India) were used without any further purification. Spec-grade solvents were used for all spectral and photophysical studies.Synthesis
All three amides were synthesized by following a general method. The amino quinolines (compounds B/C/D) (1.6 g, 11.1 mmol) was taken in a 500 mL three necked round bottom flask, fitted with a reflux condenser and dinitrogen gas inlet. It was dissolved in 300 mL of dry tetrahydrofuran (THF). To this solution ∼15 mL of dry triethylamine was added. Temperature of the reaction mixture was maintained at approximately 0 °C in an ice bath. After stirring for 15 min, a solution of A (benzene-1,3,5-tricarboxylic acid chloride, 1.0 g, 3.7 mmol) in THF (∼20–25 mL) was added slowly through a pressure equalizing funnel. Then the reaction mixture was stirred in ice cold condition for 1 h followed by room temperature stirring. After 24 h the reaction mixture was refluxed for 3 h and was cooled to room temperature. The reaction mixture was filtered (Whatman filter paper no. 1) but no significant residue could be obtained. The filtrate was then evaporated under reduced pressure to yield a colorless semi-solid material. This was further treated with 5% NaHCO3 solution for 12 h at room temperature, to yield a white precipitate, which was filtered and the residue obtained was washed thoroughly with double distilled water, dried in air and then over P2O5 for further use.Results and discussion
Synthesis
The three new chromogenic sensor molecules (1–3) could be achieved in good yield following a simple one-step reaction procedure. All these receptors were characterized by standard analytical and spectroscopic techniques. Detailed synthetic procedure and analytical data agreed well with the proposed formulation for the desired compounds.Gel formation studies
Self-assembly of gelator molecules in aqueous medium are achieved through critical balance of hydrophobic effect and hydrogen bonding. As mentioned above there are few reports on hydrogelators based on pyridine-based trimesic amides.69 This led us to check the gelation properties of compounds 1–3. Preliminary studies revealed that compounds 1 and 3 were able to form gel in DMSO/water (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v) and ethylene glycol. The compound 2 formed gel in ethylene glycol and benzyl alcohol.
:2, v/v) and ethylene glycol. The compound 2 formed gel in ethylene glycol and benzyl alcohol.
Gelation properties of these compounds were systematically scanned with 16 different solvents and out of all these possibilities, gels were obtained in two organic solvents (ethylene glycol and benzyl alcohol) as well as in a solvent combination (DMSO/water) (Table 1). The poor solubility of compounds 1 and 3 in water prompted us to use DMSO as a solubilising agent, resulting in a mixture of DMSO/water (8:2 v/v) and we fixed this composition for gelation studies by making compounds soluble at elevated temperature. It may be noted that the hydrogelators are poorly soluble in water and sometimes suitable co-solvent is required to solubilize the gelator in aqueous medium.69,75,76 In a typical experiment, the mixture was heated at ∼90 °C and on subsequent cooling to room temperature for ∼1 h, results gel formation, which was confirmed by simple inversion test. The thermo reversible nature of these gels was established by repeating the heating and cooling cycle. DMSO/water gels are prepared from an 8:2 (v/v) mixture of the corresponding solvents. The minimum gel concentrations (MGC) of these gelators were less than 1.0 wt%. All the three compounds showed excellent gelation properties in organic solvents, ethylene glycol and benzyl alcohol (for compound 2). In ethylene glycol, 1 can be classified as the supergelator, as the MGC is well below 1.0 wt%, (which is 0.25 wt%) (Table 2).
| S. No. | Solvents | 1 | 2 | 3 |
|---|---|---|---|---|
| a G = gelator, WG = weak gelator and NG = non-gelator.b As in other percentages no gel formed. | ||||
| 1 | Nitrobenzene | NG | NG | NG |
| 2 | Methyl salicylate | NG | NG | NG |
| 3 | Ethylene glycol | G | G | G |
| 4 | 1,2-Dichloro benzene | NG | NG | NG |
| 5a | Bromo benzene | NG | NG | NG |
| 6a | Chloro benzene | NG | NG | NG |
| 7 | DMF | NG | NG | NG |
| 8 | Benzyl alcohol | NG | G | WG |
| 9a | Benzene | NG | NG | NG |
| 10a | Toluene | NG | NG | NG |
| 11a | p-Xylene | NG | NG | NG |
| 12a | 1,4-Dioxane | NG | NG | NG |
| 13a | 1,2-Dichloroethane | NG | NG | NG |
| 14 | DMSO/water (8:2)b |
G | NG | G |
| 15 | DMSO | NG | NG | NG |
| 16 | Water | NG | NG | NG |
| Gelator/solvent | MGC/wt% (w/v) | Tgel/°C (at MGC) |
|---|---|---|
| 1/DMSO/water (8:2, v/v) |
0.9 | 78 |
| 1/ethylene glycol | 0.25 | 150 |
| 2/benzyl alcohol | 0.5 | 80 |
| 2/ethylene glycol | 1.5 | 152 |
| 3/DMSO/water (8:2, v/v) |
0.95 | 52 |
| 3/ethylene glycol | 1.5 | 148 |
In benzyl alcohol 2 forms translucent super gel (MGC is 0.5 wt%) whereas in ethylene glycol it is capable of forming quite a good transparent gel (MGC is 1.5 wt%) (Fig. 1). Compound 3 forms very unstable weak gelatinous mass in benzyl alcohols but in ethylene glycol it can form good stable gel where MGC is 1.5 wt%. All the gels are opaque, stable over a period of several months, and display thermo reversible properties.
 | ||
| Fig. 1 Gelation of 1, 2 and 3 in various solvents and solvent combination. | ||
Tgel measurement
We have investigated the relative thermal stability of the gels, formed under different solvent conditions by analysing the temperature at which the gel was converted into a liquid phase (sol form) (Tgel). For this, the gel from the respective gelator was (1.0 mL) prepared in a 15 mm diameter test tube. A locally made glass ball weighing 0.10 g was placed on the gel surface. The test tube was then heated in a temperature regulated oil bath and the temperature at which the ball touched the bottom of the test tube was recorded (Tgel). The Tgel values obtained for different gelators are summarized in Table 2. Tgel values revealed that thermal stability of the gel obtained from DMSO/water (8:2 v/v) of 1 is higher than 3, which may be due to the position of the quinoline nitrogen that could form a stable hydrogen bonded network. The relative thermal stability of the gels obtained from 1, 2 and 3 in ethylene glycol are comparable.
Structural morphology analysis of gels by SEM and TEM
The structural morphology of the gels of 1, 2 and 3 were studied by SEM analysis. The gels prepared from DMSO/water (8:2, v/v) were filtered and dried under high vacuum. A small portion of the dried gel was placed on the pin mount with graphite planchets on top and coated with gold. The SEM images of the gels formed by these gelators in different conditions are shown in Fig. 2 and 3.
 | ||
| Fig. 2 Representative SEM micrographs of (A) 1 in DMSO/water 8:2 (0.9 wt%) displaying entangled fibers having both handednesses; inset depicts the right-handedly twisted fibers and (B) 3 DMSO/water 8:2 (1.0 wt%). | ||
 | ||
| Fig. 3 Representative SEM micrographs of (A) 1 in ethylene glycol (0.5 wt%) displaying tube/rod type fibers; inset: rod with higher magnification; (B) 2 in benzyl alcohol (0.5 wt%); (C) 2 in ethylene glycol (1.5 wt%); (D) 3 in ethylene glycol (1.5 wt%). | ||
Analysis of the SEM images of gelator 1 obtained from DMSO/water (8:2, v/v) revealed the presence of several micrometer long entangled fibrous networks comprising twisted fibers of both handedness and these type of fibres have been reported (Fig. 2A).77,78
However, a clear rod/tubular structures with relatively thick wall were observed in the SEM images (Fig. 3A) for gels obtained from ethylene glycol. The gel formed by gelator 3 in DMSO/water (8:2, v/v) displayed tape type morphology with width ranging from 1 to 8 μm (Fig. 2B).59
The SEM images of the gel formed by 2 (obtained from benzyl alcohol) showed a complicated network (Fig. 3B); appearance of scattered colonies, displaying microthin plate-type morphology.79 However, gels of 2 from ethylene glycol were slender fibers of tape morphology (Fig. 3C). SEM images of the gel of 3 in ethylene glycol (Fig. 3D) displayed a tape like typical fibrous network with varying thickness. These results clearly indicate the role of solvents in determining the gel morphology. The gel obtained from DMSO/water (8:2) was distinctly different from that of the organogel. The morphology of 1 seems to be quite interesting and we have done further investigation to develop a better image of the twisted architecture and tubular structure of both the DMSO/water (8:2, v/v) and organogel by performing transmission electron microscopy (TEM) analysis (Fig. 4).
 | ||
| Fig. 4 Representative HR-TEM micrographs (A) of DMSO/water (8:2) gel formed by 1 showing the occasional twisted fiber and (B) organogel formed by 1 showing rod/tubular morphology with thick walls. | ||
The twisted architecture was clearly seen for the gel obtained from 1 in DMSO/water (8:2, v/v) mixture in the individual fibres in HR-TEM analysis (Fig. 4A), while the rod/tube like structure with thick wall like morphology (Fig. 4B) was confirmed for the gel formed by 1 in ethylene glycol (Fig. 4B).
Anion binding studies in solution phase
As mentioned in the introduction, the amide moieties are important receptors for anion binding. Thus, we have studied the anion binding behaviour of 1 in solution and gel state.The dilute solutions (∼10−5 M) of compounds 1–3 in DMSO were almost colourless. However, this changed to a very pale yellow colour upon addition of excess of either of the following salts solutions (in DMSO): n-Bu4NF− (TBAF), n-Bu4NCH3COO− (TBAA), n-Bu4NH2PO4− (TBAP) and n-Bu4NCN− (TBACN). Here we are reporting only the absorption and emission titration of receptor 1 with the anions. For the other two isomers (2 and 3) the scanning with different anions was done in solution phase (SI).
Electronic spectrum of the compound 1 in DMSO was found to change in the presence of these four anions added as the DMSO solution. Systematic changes in spectral pattern on addition of increasing amount of any of these four anions are shown in Fig. 5A–D. Significant changes in the UV-Vis absorption spectra of 1 were obtained for anions such as F−, CH3COO−, H2PO4− and CN−. Systematic titration of 1 with F− displayed a steady decrease in the absorption bands at 274, 323 and 337 nm along with the formation of new peak at 380 nm, a clear isosbestic point was observed at 342 nm (Fig. 5A). Similar type of titration profile was found in the presence of varying [CN−]; however, the intensity of the absorption band at 380 nm is less in case of CN− (Fig. 5D). Fig. 5A and D indicated the possible deprotonation of the –NH proton.80 On the other hand, no such new absorption peak at 380 nm was observed for CH3COO− and H2PO4− ions, though decrease in the absorption band intensities at 274, 320 and 337 nm (Fig. 5B and C) was only observed.
 | ||
| Fig. 5 UV-Vis titration of receptor 1 (4.5 × 10−5 M) in DMSO solution with (A) F− (0–8.0 × 10−4 M) (B) CH3COO− (0–8.5 × 10−5 M) (C) H2PO4− (0–8.3 × 10−5 M) (D) CN− (0–8.3 × 10−5 M). | ||
Steady state luminescence spectra of 1 with various anions were recorded and are shown in Fig. 6. For all the compounds, emission band maxima appeared at ∼416 nm, the characteristic band maxima for the naphthalene unit. A steady increase in emission intensity was observed on increasing anions [X−] and is shown in Fig. 6; while extent of changes was most prominent for F− and CN− as compared to the situation for CH3COO− and H2PO4−. One would expect that the extent of changes in emission intensity is synonymous to the extent of binding between receptor 1 and the respective anionic analyte. This trend also matched well with the spectrophotometric titrations. The growth in the emission intensities were observed for all the cases after addition of anions.81–83 From a mechanistic point of view, anion has the potential to form hydrogen bonds with the amide group of the molecules. Presumably this has added to the overall rigidity of the fluorophore which restrict the rotational freedom in the adduct form and reduced the possibility of the non-radiative decay process. It is being reflected in the emission growth.
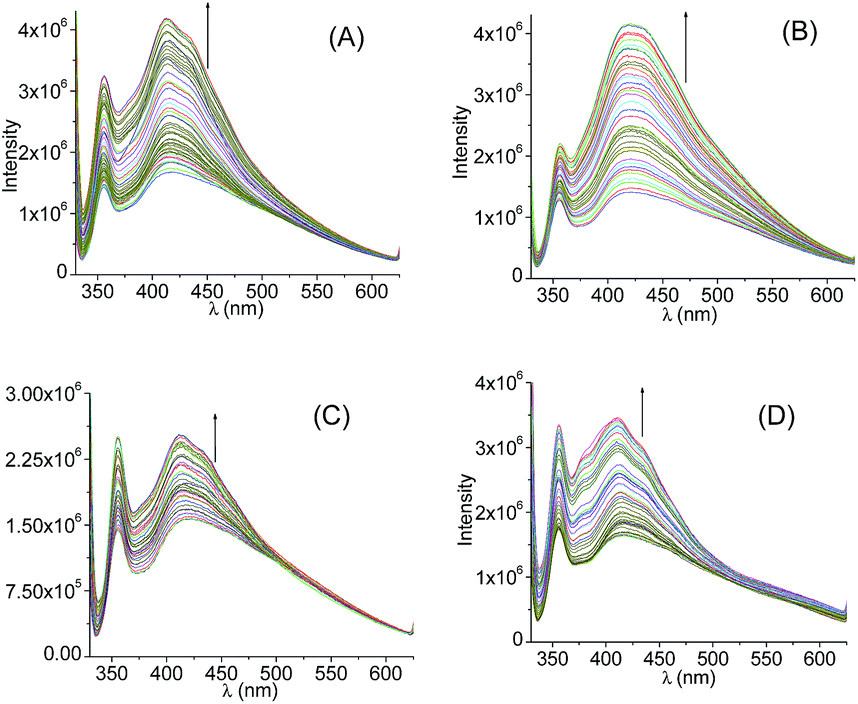 | ||
| Fig. 6 Emission titration of receptor 1 (4.5 × 10−5 M) in DMSO solution with (A) F− (0–10.0 × 10−4 M) (B) CH3COO− (0–11.5 × 10−5 M) (C) H2PO4− (0–10.3 × 10−5 M) (D) CN− (0–11.3 × 10−5 M); excitation wavelength: 274 nm. | ||
Attempt to evaluate affinity constant for binding of 1 with these three anions were not possible using various standard expressions—as calculations using different methodologies and procedures (SI) yielded different values, which added to the uncertainties and the reliability of the binding constants thus evaluated. However, the understanding about binding stoichiometry has emerged out of these exercises. Our attempt using non-linear expression to check the binding stoichiometry revealed that binding stoichiometry was anything else other than 1:1.
To rationalize the binding phenomenon further we have carried out 1HNMR experiment in absence and presence of varying [F−] with all the three respective receptors and three sets of 1HNMR spectra are shown in Fig. 7A–C. Responses, probed by differences in the chemical shift values of the 1HNMR signals for three receptors, were different in the presence of added F−.
 | ||
| Fig. 7 Partial 1HNMR (500 MHz) spectra of 1 (A) in the presence of TBAF in DMSO (d6) at room temperature. 2 (B) and 3 (C). | ||
This could only be explained based on the difference in orientation of the different isomers of the quinoline moiety in the respective receptors. Let us first discuss the chemicals shifts observed for receptor 1 in the presence of added F−. Apart for the Ha, –N(H)Amide and Hc hydrogen atoms, an upfield shifts were observed for all other hydrogen atoms belongs to the quinoline moiety. As expected, due to the H-bonding with the F−, a downfield shift was observed –N(H)Amide hydrogen atoms—however it was broadened and could not located beyond a certain [F−]. Weaker H-bond formation between the Hc and F−, could account for the slight downfield shift observed for Hc hydrogen atoms. For all other remaining hydrogen atoms of the quinoline moiety an upfield shift was observed and the exact reason for such shift is difficult to predict.
Based on the scorpionate mode of binding (1:1) the predicted structure for the H-bonded adduct should have F− situated at the top of the phenyl ring of the trimesic amide derivative and as all the protons of the three phenyl remained equivalent even after binding to the fluoride ion, one can also predict a symmetric orientation of the fluoride ion with respect to the three phenyl hydrogens.
However, possibility of the 1:1 binding could be nullified on the basis of the fluorescence titrations results discussed above. Possibility of the 1:2 stoichiometry for the binding mode could also be ignored based on the results of the 1H NMR studies; as this would cause some asymmetric chemical shift for the phenyl and –N(H)Urea hydrogen atoms. Thus, results of our studies are likely to support the 1:3 binding stoichiometry for the host–guest complex formation. Presumably, presence of the fluoride ion provides a deshielding effect on three phenyl H-atoms and thus responsible for the little downfield shift. For compound 2, similar explanation could be offered for Ha, –N(H)Amide and Hg hydrogen atoms; while the extent of shift is lesser for Hg (Δδ (ppm) = 0.055) due to a weaker Hg⋯F− hydrogen bond compared to that for Ha (Δδ (ppm) = 0.11) for the respective Ha⋯F− in receptor 1. For receptor 3, the extent of downfield shift for Hb hydrogen atom was more (Δδ (ppm) = 0.13) than that was observed for Hg hydrogen atom in case of receptor 2 and this perhaps indicative of a weaker H-bond formation with the F−. Further, this may also be attributed to the comparable binding affinity observed for receptor 1 and receptor 3. Therefore, based on the results of the fluorescence and 1HNMR studies a 1:3 host–guest assembly between receptors and F− seem to be most plausible.
The effect of anion in gel formation
In this study, the gel formation is due to the molecular self-assembly process which is controlled by the molecular parameters of the monomeric unit of the gelator molecule. This depends on several non-covalent interaction such as H-bonding, π–π interaction, electrostatic interaction (dipole dipole interaction) and hydrophobic interaction. These interaction energies govern the stabilities and the average size of the self assembled structures. The morphology and the topologies of the self-assembled structure arise from the self-assembly properties of the gelator and their specific interactions. We envisage that the π–π interaction between the aromatic moieties and the intra-molecular H-bonding involving the amide moiety are accountable for the self-assembly process leading to the gel formation.The evaluation of the anion binding properties in solution prompted us to study the effect of anion in gel formation. In case of these gels, the most important interaction that dominates in achieving the specific supramolecular architecture is of dipole–dipole type interaction (–N(H)Amide–OCAmide or –N(H)Amide–Nquinolin); apart from other non-bonded interactions like π–π stack, van der Waals, hydrogen bonding interactions.
To examine how ion–dipole interaction affect the gel architectures, gel studies were carried out for gelator 1 in the presence of tetrabutylammonium salts of four different anionic analytes (F−, CN−, CH3COO− and H2PO4−), which were scanned to bind with 1 in solution phase through ion–dipole interactions (hydrogen bonding interaction). The effect of anion binding on the gelation process was monitored by repeating the gelation experiment of 1 (9 to 9.6 mg) in DMSO/water (1 mL, 8:2, v/v) in the presence of 10 molar equivalents of each of the four different above mentioned anions (Fig. 8).84
 | ||
| Fig. 8 (A) Representative SEM micrographs of the gel formed by 1 in DMSO/water 8:2 (0.9 wt%) in the presence of different anions. (B) Photographs of test tubes showing gelation of compound 1 in DMSO/water 8:2 (0.9 wt%) in the presence of different anions. | ||
Gelator 1 and 10 equivalents of TBAF were added to a 10 mm diameter glass test tube containing DMSO/water (1 mL, 8:2, v/v) and the mixture was gently heated until bath temperature attained 90 °C and a clear solution was found. After cooling to the room temperature gel was obtained, confirmed by inversion test (Fig. 8). Similar experiments were repeated for CN−, CH3COO− and H2PO4− and stable gels were obtained in all cases (Fig. 8). Both water and DMSO are known to interact strongly with higher dipole moment through dipole–dipole or dipole–induced dipole interactions. Such interactions are expected to be even more significant for interaction with anions such as F−, CN−, CH3COO− and H2PO4−, having higher charge density.
Thus, the more effective solvation of these anions in DMSO/water medium makes it a less efficient competitor for the gelator –NH groups. Accordingly, the intermolecular hydrogen bonding is only partly influenced and the gel is still preserved. However, these interactions are strong enough to influence the gel strength and morphology for the respective gelators as estimated by the lower Tgel values; as well as the difference in the nature of the fibril structure evident in the SEM (Fig. 8). Tgel values for gel obtained from 1 in the presence of 10 equivalent of different anions were found to be lower by at least 5/6 °C (Tgel for F−, CN−, CH3COO− and H2PO4− was evaluated as 70, 72, 73, 72 °C; while accuracy for the temperature measured is ±0.5 °C), respectively from the values obtained without anions. Fig. 8 revealed that no distinct helical twist was observed for the gel obtained from 1 in DMSO/water (8:2, v/v) in the presence of any of these four anionic analytes.
However, for further increase in F− concentration (e.g. 12 equivalent), a new solution was made, which then did not form a gel on cooling. Only gelatinous precipitate was observed after cooling the hot clear solution (Fig. 9).
 | ||
| Fig. 9 Photographs of gel to solution transition in the presence of excess anions. | ||
The reason is with higher equivalents of fluoride ion, the ion dipole interactions involving the F− and the receptor molecule (1) was strong enough to disrupt the gel formation (Fig. 9). It has been reported that after a certain concentration of anion the gel formation may disrupt.85,86
FT-IR spectra were recorded to gain an insight to the hydrogen bonding interaction of the amide CO group for compound 1 in its bulk solid form (monomeric form), gel form (in DMSO/water (8:2, v/v)) and with excess fluoride anion. In the bulk solid the amide band appears at 1672 cm−1 whereas in the gel state this band was shifted to 1654 cm−1. This moderate shift of about 18 cm−1 indicates that the CO groups are involved in the intramolecular hydrogen bonding in the self-assembled gel state. When excess (12 equivalents) of TBAF was used the amide CO band was observed at 1670 cm−1 (ESI Fig. 12†). This result suggests that the hydrogen bonding in the self assembled gelator molecule diminishes in presence of excess TBAF, because fluoride is highly electronegative and takes part in hydrogen bond formation.
Conclusions
New tripodal compounds 1–3 were prepared with amide functionality equipped with different derivatives of quinoline moiety. These compounds (1–3) act as excellent low-molecular-weight gelators (both organo and hydrogels). The compound 1 was investigated in the presence of anions, in both solution and gel phase. We have shown that 1 binds with anions (F−/CN−/CH3COO−/H2PO4−) strongly in solutions. The anion binding studies in gel state revealed that we could tune the gel morphology with respect to the solvent combination. Increasing anion concentration resulted in the conversion of gels to solution due to the ion dipole interactions between anions and the polarisable –N(H)Amide hydrogen atoms, which dominate over the dipole–dipole interactions between amide derivatives. This work offers an approach to design novel gelators as stimuli responsive soft materials, which could lead to possible applications in sensor devices for anion recognition and pharmaceutical industries.Acknowledgements
The authors acknowledge Dr Amitava Das for his continuous encouragement and support in this work. The authors thank CSIR-CSMCRI for the instrument support and research facilities. The Director and the Department of Chemistry, NIT Kurukshetra, Haryana are also acknowledged for the research facilities. AG wish to thank SERB-DST, India for financial support under project grant SB/FT/CS-193/2013.References
- S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC
.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 2005, 3615–3631 CrossRef
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed
- M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS PubMed
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080–2088 RSC
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS PubMed
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed
- Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, 2006 Search PubMed
- F. Fages, F. Voegtle and M. Zinic, Top. Curr. Chem., 2005, 256, 77–131 CrossRef CAS PubMed
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC
- A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS PubMed
- C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernández, Angew. Chem., Int. Ed., 2014, 53, 700–705 CrossRef CAS PubMed
- A. Maity, F. Ali, H. Agarwalla, B. Anothumakkool and A. Das, Chem. Commun., 2015, 51, 2130–2133 RSC
- K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664–6676 CrossRef CAS
- J. Eastoe, M. Sanchez-Dominguez, P. Wyatt and R. K. Heenan, Chem. Commun., 2004, 2608–2609 RSC
- S.-I. Kawano, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2004, 126, 8592–8593 CrossRef CAS PubMed
- H.-J. Kim, J.-H. Lee and M. Lee, Angew. Chem., Int. Ed., 2005, 44, 5810–5814 CrossRef CAS PubMed
- K. J. C. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. van Esch, Angew. Chem., Int. Ed., 2004, 43, 1663–1667 CrossRef CAS PubMed
- I. Hwang, W. S. Jeon, H.-J. Kim, D. Kim, H. Kim, N. Selvapalam, N. Fujita, S. Shinkai and K. Kim, Angew. Chem., Int. Ed., 2007, 46, 210–213 CrossRef CAS PubMed
- Y. Kuang, Y. Gao and B. Xu, Chem. Commun., 2011, 47, 12625–12627 RSC
- O. Kotova, R. Daly, C. M. G. dos Santos, M. Boese, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2012, 51, 7208–7212 CrossRef CAS PubMed
- A. Westcott, C. J. Sumby, R. D. Walshaw and M. J. Hardie, New J. Chem., 2009, 33, 902–912 RSC
- M.-O. M. Piepenbrock, N. Clarke and J. W. Steed, Soft Matter, 2010, 6, 3541–3547 RSC
- R. Liu, A. H. Milani, J. M. Saunders, T. J. Freemont and B. R. Saunders, Soft Matter, 2011, 7, 9297–9306 RSC
- R. Liu, A. H. Milani, T. J. Freemont and B. R. Saunders, Soft Matter, 2011, 7, 4696–4704 RSC
- P. Das, S. Yuran, J. Yan, P. S. Lee and M. Reches, Chem. Commun., 2015, 51, 5432–5435 RSC
- C. E. Stanley, N. Clarke, K. M. Anderson, J. A. Elder, J. T. Lenthall and J. W. Steed, Chem. Commun., 2006, 3199–3201 RSC
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Commun., 2008, 2644–2646 RSC
- L. Applegarth, N. Clark, A. C. Richardson, A. D. M. Parker, I. Radosavljevic-Evans, A. E. Goeta, J. A. K. Howard and J. W. Steed, Chem. Commun., 2005, 5423–5425 RSC
- Z. Dzolic, M. Cametti, A. Dalla Cort, L. Mandolini and M. Zinic, Chem. Commun., 2007, 3535–3537 RSC
- H. Maeda, Y. Haketa and T. Nakanishi, J. Am. Chem. Soc., 2007, 129, 13661–13674 CrossRef CAS PubMed
- S. Wang, W. Shen, Y. Feng and H. Tian, Chem. Commun., 2006, 1497–1499 RSC
- Q. Liu, Y. Wang, W. Li and L. Wu, Langmuir, 2007, 23, 8217–8223 CrossRef CAS PubMed
- H. Maeda, Chem.–Eur. J., 2008, 14, 11274–11282 CrossRef CAS PubMed
- M. Yamanaka, T. Nakamura, T. Nakagawa and H. Itagaki, Tetrahedron Lett., 2007, 48, 8990–8993 CrossRef CAS
- C. Yang, B. Wu, Y. Chen and K. Zhang, Macromol. Rapid Commun., 2015, 36, 750–754 CrossRef CAS PubMed
- M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed
- N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS
- P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205–241 RSC
- L. R. Eller, M. Stȩpień, C. J. Fowler, J. T. Lee, J. L. Sessler and B. A. Moyer, J. Am. Chem. Soc., 2007, 129, 11020–11021 CrossRef CAS PubMed
- P. A. Gale, J. R. Hiscock, C. Z. Jie, M. B. Hursthouse and M. E. Light, Chem. Sci., 2010, 1, 215–220 RSC
- S. O. Kang, R. A. Begum and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 45, 7882–7894 CrossRef CAS PubMed
- D. A. Jose, D. K. Kumar, B. Ganguly and A. Das, Inorg. Chem., 2007, 46, 5817–5819 CrossRef CAS PubMed
- R. Li, Y. Zhao, S. Li, P. Yang, X. Huang, X.-J. Yang and B. Wu, Inorg. Chem., 2013, 52, 5851–5860 CrossRef CAS PubMed
- J. E. A. Webb, M. J. Crossley, P. Turner and P. Thordarson, J. Am. Chem. Soc., 2007, 129, 7155–7162 CrossRef CAS PubMed
- M. de Loos, A. G. J. Ligtenbarg, J. van Esch, H. Kooijman, A. L. Spek, R. Hage, R. M. Kellogg and B. L. Feringa, Eur. J. Org. Chem., 2000, 2000, 3675–3678 CrossRef
- L. Feng and K. A. Cavicchi, Soft Matter, 2012, 8, 6483–6492 RSC
- V. T. Freitas, P. P. Lima, R. A. S. Ferreira, E. Pecoraro, M. Fernandes, V. de Zea Bermudez and L. D. Carlos, J. Sol-Gel Sci. Technol., 2013, 65, 83–92 CrossRef CAS
- O. Kotova, R. Daly, C. M. G. dos Santos, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Inorg. Chem., 2015, 54, 7735–7741 CrossRef CAS PubMed
- S. Mukhopadhyay, G. Krishnamoorthy and U. Maitra, J. Phys. Chem. B, 2003, 107, 2189–2192 CrossRef CAS
- A. Ghosh, D. A. Jose and R. Kaushik, Sens. Actuators, B, 2016, 229, 545–560 CrossRef CAS
- D. K. Kumar, D. A. Jose, P. Dastidar and A. Das, Langmuir, 2004, 20, 10413–10418 CrossRef CAS PubMed
- H. Chen, Y. Feng, G.-J. Deng, Z.-X. Liu, Y.-M. He and Q.-H. Fan, Chem.–Eur. J., 2015, 21, 11018–11028 CrossRef CAS PubMed
- J. Liu, Y. Feng, Z.-X. Liu, Z.-C. Yan, Y.-M. He, C.-Y. Liu and Q.-H. Fan, Chem.–Asian J., 2013, 8, 572–581 CrossRef CAS PubMed
- Z.-X. Liu, Y. Feng, Z.-C. Yan, Y.-M. He, C.-Y. Liu and Q.-H. Fan, Chem. Mater., 2012, 24, 3751–3757 CrossRef CAS
- Y. Liu, X. Wu, C. He, R. Zhang and C. Duan, Dalton Trans., 2008, 5866–5868 RSC
- T. H. Noh, W. Hong, H. Lee and O.-S. Jung, Dalton Trans., 2015, 44, 787–794 RSC
- M. Arunachalam and P. Ghosh, Org. Lett., 2010, 12, 328–331 CrossRef CAS PubMed
- M. Arunachalam and P. Ghosh, Inorg. Chem., 2010, 49, 943–951 CrossRef CAS PubMed
- K. Ghosh and S. Panja, RSC Adv., 2015, 5, 12094–12099 RSC
- Y. Zhang and S. Jiang, Org. Biomol. Chem., 2012, 10, 6973–6979 CAS
- D. K. Kumar, D. A. Jose, P. Dastidar and A. Das, Chem. Mater., 2004, 16, 2332–2335 CrossRef CAS
- S. Banerjee, N. N. Adarsh and P. Dastidar, Soft Matter, 2012, 8, 7623–7629 RSC
- N. Shi, G. Yin, H. Li, M. Han and Z. Xu, New J. Chem., 2008, 32, 2011–2015 RSC
- J. W. Steed, Chem. Commun., 2006, 2637–2649 RSC
- D. J. Abdallah and R. G. Weiss, Chem. Mater., 2000, 12, 406–413 CrossRef CAS
- T. Becker, C. Yong Goh, F. Jones, M. J. McIldowie, M. Mocerino and M. I. Ogden, Chem. Commun., 2008, 3900–3902, 10.1039/b807248e
- U. Maitra, S. Mukhopadhyay, A. Sarkar, P. Rao and S. S. Indi, Angew. Chem., Int. Ed., 2001, 40, 2281–2283 CrossRef CAS
- F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef CAS
- P. Sahoo, D. Krishna Kumar, D. R. Trivedi and P. Dastidar, Tetrahedron Lett., 2008, 49, 3052–3055 CrossRef CAS
- P. Sahoo, R. Sankolli, H.-Y. Lee, S. R. Raghavan and P. Dastidar, Chem.–Eur. J., 2012, 18, 8057–8063 CrossRef CAS PubMed
- P. Sahoo, N. N. Adarsh, G. E. Chacko, S. R. Raghavan, V. G. Puranik and P. Dastidar, Langmuir, 2009, 25, 8742–8750 CrossRef CAS PubMed
- P. Xue, R. Lu, J. Jia, M. Takafuji and H. Ihara, Chem.–Eur. J., 2012, 18, 3549–3558 CrossRef CAS PubMed
- P. Jiang and Z. Guo, Coord. Chem. Rev., 2004, 248, 205–229 CrossRef CAS
- A. Tamayo, C. Lodeiro, L. Escriche, J. Casabó, B. Covelo and P. González, Inorg. Chem., 2005, 44, 8105–8115 CrossRef CAS PubMed
- Z.-Q. Wu, X.-B. Shao, C. Li, J.-L. Hou, K. Wang, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2005, 127, 17460–17468 CrossRef CAS PubMed
- Z. Dzolic, M. Cametti, A. Dalla Cort, L. Mandolini and M. Zinic, Chem. Commun., 2007, 3535–3537 RSC
- T. H. Kim, M. S. Choi, B.-H. Sohn, S.-Y. Park, W. S. Lyoo and T. S. Lee, Chem. Commun., 2008, 2364–2366 RSC
- H. Yang, T. Yi, Z. Zhou, Y. Zhou, J. Wu, M. Xu, F. Li and C. Huang, Langmuir, 2007, 23, 8224–8230 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16345a |
| This journal is © The Royal Society of Chemistry 2016 |