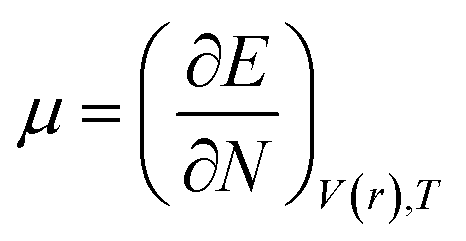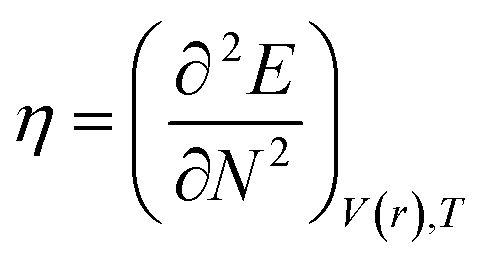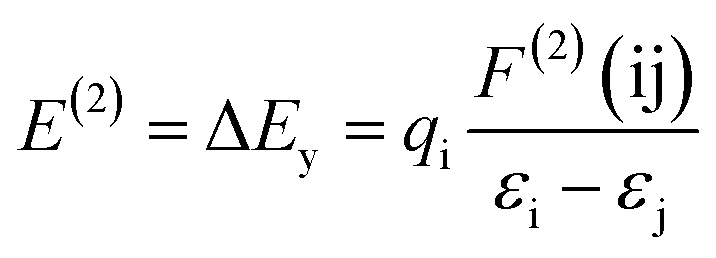DOI:
10.1039/C6RA14006H
(Paper)
RSC Adv., 2016,
6, 64818-64825
The role of solvent polarity in the electronic properties, stability and reactivity trend of a tryptophane/Pd doped SWCNT novel nanobiosensor from polar protic to non-polar solvents
Received
30th May 2016
, Accepted 2nd July 2016
First published on 4th July 2016
Abstract
Carbon nanotubes and amino acids have a high potential to offer specific advantages as the transducer and the recognition elements of biosensors. Their compatible size with biological structures makes them suitable as implantable sensors. In this work solvent effects on the electronic structure properties of a tryptophan hybrid with Pd doped single walled carbon nanotubes as a new novel biosensor were investigated. As the chemical reaction of a nanobiosensor is affected by the nature of the solvents, 5 different solvents, water, DMSO, ethanol, acetone and carbon tetrachloride are employed to study the role of the solvent polarity on the molecular stability, the optimized geometry and charge distribution of Try/Pd-SWCNT nanobiosensor. To derive the optimized geometries, the density functional theory computations were performed at the B3LYP level with the 6-31G(d) basis set. In addition, the molecular orbital calculations such as natural bond orbitals (NBOs), HOMO–LUMO energy gap, mapped molecular electrostatic potential (MEP) surface and density of state (DOS) were also performed. The results show that the presence of a solvent lowers the HOMO and LUMO energy level and increases or decreases the HOMO–LUMO energy gap depending on the chemical system. Different nucleophile and electrophile sites were detected in the molecular electrostatic maps. The softer investigated biosensors were found in more polar media. The highest reorganization energies for the nanobiosensor resulted in water media. The Trp/Pd/SWCNT presents high stability with considerable values of charge transfer, stabilization energies and the energy bond gap in polar medium which confirm both in vitro and in vivo biosensing applications.
1. Introduction
Nanotechnology plays an important role in the development of sensors and biosensors.1–3 Nanomaterials improve the sensitivity and performance of biosensors. There has been an explosion of research due to their characteristics for in vivo and in vitro applications.4 The basic characteristics of nanomaterial (NMs) based biosensors are the linearity, selectivity, sensitivity and response time so they can markedly improve biomolecular detection. Since carbon nanotubes (CNTs) were discovered by Iijima5 and the first use of CNT as a sensor demonstrated by Kong et al.6 CNTs have been the focus of research groups in biosensors,7 diagnostics,8 environmental monitoring9,10 and drug delivery.11 CNTs are a tubular cylindrical arrangement of carbon atoms with a diameter of 1–5 nm. These quasi one dimensional NMs offer unique properties due to long edge/basal plane ratio, higher sensitivity and faster electron transfer kinetics. Single-walled carbon nanotubes (SWCNTs) are at least ten times stronger and six times lighter than steel. SWCNTs with excellent chemical and physical stability display bioelectrocatalytic properties. SWCNTs are suitable component in electrochemical sensors due to their high capacity of charge transfer between heterogeneous phases.12 Their molecular recognition implication is a result of their highly sensitive electric conductivity to change in their chemical environment.13 The solvent environment plays an incredible role on the properties of biosensors. The usage of an appropriate solvent and their effect on the electronic properties of biosensors can lead to higher efficiency.14,15 Solvents can be classified into two categories, polar and non-polar. One of the most common way to define and measure of the solvent polarity is the dielectric constant. Solvents with a dielectric constant of less than 5 are considered to be non-polar. High dielectric constant solvents more than 25 usually have polar functional groups and often high dipole moment. In this work we investigate the solvent effects on the electronic structure properties of the novel tryptophan/Pd doped SWCNTs nanobiosensor that we have introduced previously by the DFT/B3LYP model which exhibits excellent performance on the geometries of organic complexes.16 The polarized continuum model (PCM), which mainly used in this work, is used for the calculation of molecular energies, structure and properties in different medium in DFT approach.17–19 The PCM defines the cavities as envelopes of spheres centered on atoms or atomic groups.20
2. Computational details
Gaussian 09 program package21 have been used to calculate geometry optimization and solvent effects. Density functional calculations with Beck's three parameter hybrid method (B3)22 using correlation functional of Lee, Yang, Parr23 (B3LYP) level have been performed. Different basis sets were tested and all geometry full optimization have been performed with hybrid density functional B3LYP/6-31G(d) and DGDZVP extra basis set for Pd atom as showed good ability in study of long range interactions. The absence of imaginary frequency at the same level verified the optimized structures correspond to the energy minima. Five solvent, water (ε = 80.1), DMSO (ε = 46.7), ethanol (ε = 24.5), acetone (20.7) and carbon tetrachloride (ε = 2.24) were selected to study the solvent effects on titled parameters using Tomasi's polarized continuum (PCM) model. Adsorption energies in solvent were calculated according to eqn (1).| | |
Eads = ETry/Pd/SWCNT − (EPd/SWCNT + ETry)
| (1) |
were ETry/Pd/SWCNT is the total energy of Pd/SWCNT with Try molecule and EPd/SWCNT and are ETry the total energy of Pd/SWCNT and Trp molecule in relax geometry respectively. The noncovalent interaction of Try molecule with Pd/SWCNT were considered via different initial configuration i.e. amine, (complex 1) and carboxyl sits (complex 2) through the perpendicular direction to SWCNT to reduce the unfavorable interactions.
The natural bond orbital (NBO)24 was carried out to quantify the charge transfer between Try and Pd/SWCNT at the same level. AIM 2000 package was employed to deep understanding of the nature of interactions in different solvent medium via the Bader's quantum theory of atom in molecules (QTAIM).25 The sensitivity of investigated bionanosensor model was further analyzed by the calculation of density of stats (DOS).
In order to investigate the surrounding medium and the hydrophobic–hydrophilic behavior of nanobiosensor, the solvation energy of all geometries calculated from the total energies at the same level of theory according to the eqn (2).
| | |
Estab = Ein solvent − Ein gas
| (2) |
DFT-based chemical reactivity and stability descriptors which are electronic chemical potential (μ), chemical hardness (η), chemical softness (S) and electrophilicity (ω) were calculated as defined in eqn (3)–(6) according to Koopmans theorem:
| |
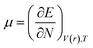 | (3) |
| |
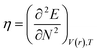 | (4) |
where
μ is chemical potential,
η is chemical hardness,
S is global softness and
ω is electrophilicity index.
26
3. Result and discussion
3.1. Optimized configurations
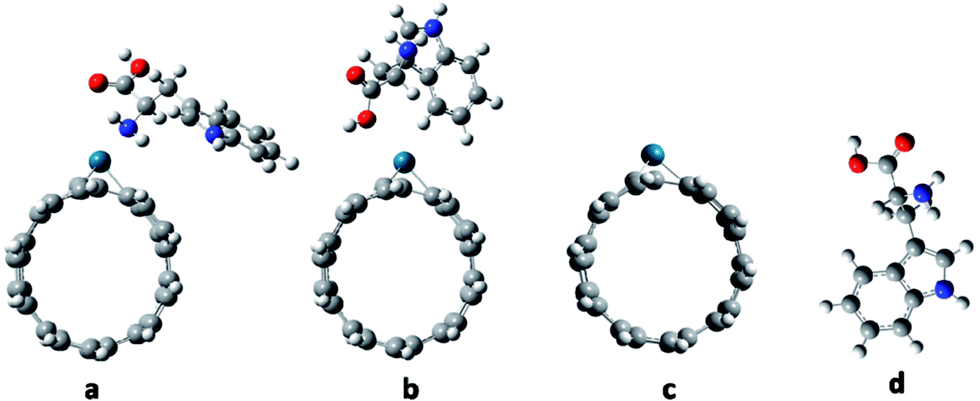
The molecular geometries of Try/Pd-SWCNT were optimized in vacuum without symmetry constrain and then in different solvents at B3LYP/6-31G(d) level. Fig. 1 shows the optimized geometry structures of titled nanobiosensor complexes and its components. It is worth mentioning that the results show that the proposed recognition element, aromatic amino acids, maintains its stability and orientation in different solvents that is an important limiting factor in the use of the biomolecules in sensing elements so they could also be excellent molecular templates. To evaluate the biosensing strength, adsorption energy as one of the most important parameters were investigated. To relate the bionanosensor applications to the real case, high polar water and DMSO solvents, ethanol and acetone with moderate polarity and non-polar carbon tetrachloride were selected. The calculated data indicates that the total energy of the complexes are lower than that in the gas phase and the total energies shift to lower values with the increasing of the electric permittivity of the solvent. The electric field of the solvent will interact with the complexes and lead to stabilization. The values of the adsorption energies (Eads), reorganization energies (EReorg) and dipole moments of Try/Pd-SWCNT in the five mentioned medium for all complexes with DFT/6-31G(d) calculations are given in Table 1. The magnitude of ground state electronic dipole moment of the systems is increased in more polar media due to the inductive solvent polarization effects in the polar solvents. The maximum adsorption energies for the complexes were found in the nonpolar solvent and the maximum of the reorganization energy were found in more polar solvent due to the hydrophobic nature of the nanobiosensor.
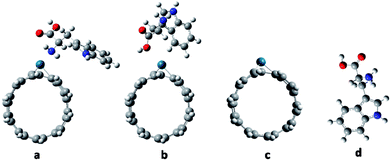 |
| | Fig. 1 The optimized geometry structures of Try-Pd/SWCNT nanobiosensor complexes and its components: (a) complex 1 (b) complex 2 (c) Pd/SWCNT and (d) Try. | |
Table 1 The calculated values of the adsorption energies (Eads in kcal mol−1), reorganization energies (EReorg in kcal mol−1) and dipole moments of Try-Pd/SWCNT nanobiosensor complexes and its components in the five mentioned medium
| Media |
Compound |
Eads |
EReorg |
Dipole moment |
| Water |
Pd/SWCNT |
— |
−418.585 |
9.036 |
| Try |
— |
−8.758 |
4.166 |
| Complex 1 |
−17.087 |
−24.727 |
13.877 |
| Complex 2 |
−2.796 |
−29.257 |
13.480 |
| DMSO |
Pd/SWCNT |
— |
−418.007 |
8.829 |
| Try |
— |
−9.220 |
4.149 |
| Complex 1 |
−16.571 |
−24.095 |
13.734 |
| Complex 2 |
−2.161 |
−28.506 |
13.325 |
| Ethanol |
Pd/SWCNT |
— |
−416.838 |
8.404 |
| Try |
— |
−8.900 |
4.115 |
| Complex 1 |
−16.794 |
−22.829 |
13.447 |
| Complex 2 |
−2.095 |
−26.950 |
12.723 |
| Acetone |
Pd/SWCNT |
— |
−416.350 |
8.233 |
| Try |
— |
−8.758 |
4.098 |
| Complex 1 |
−16.893 |
−22.299 |
13.329 |
| Complex 2 |
−2.130 |
−26.356 |
12.740 |
| Carbon tetrachloride |
Pd/SWCNT |
— |
−409.456 |
5.843 |
| Try |
— |
−6.392 |
3.849 |
| Complex 1 |
−18.747 |
−14.892 |
11.660 |
| Complex 2 |
−2.650 |
−17.615 |
10.640 |
3.2. Electronic structures
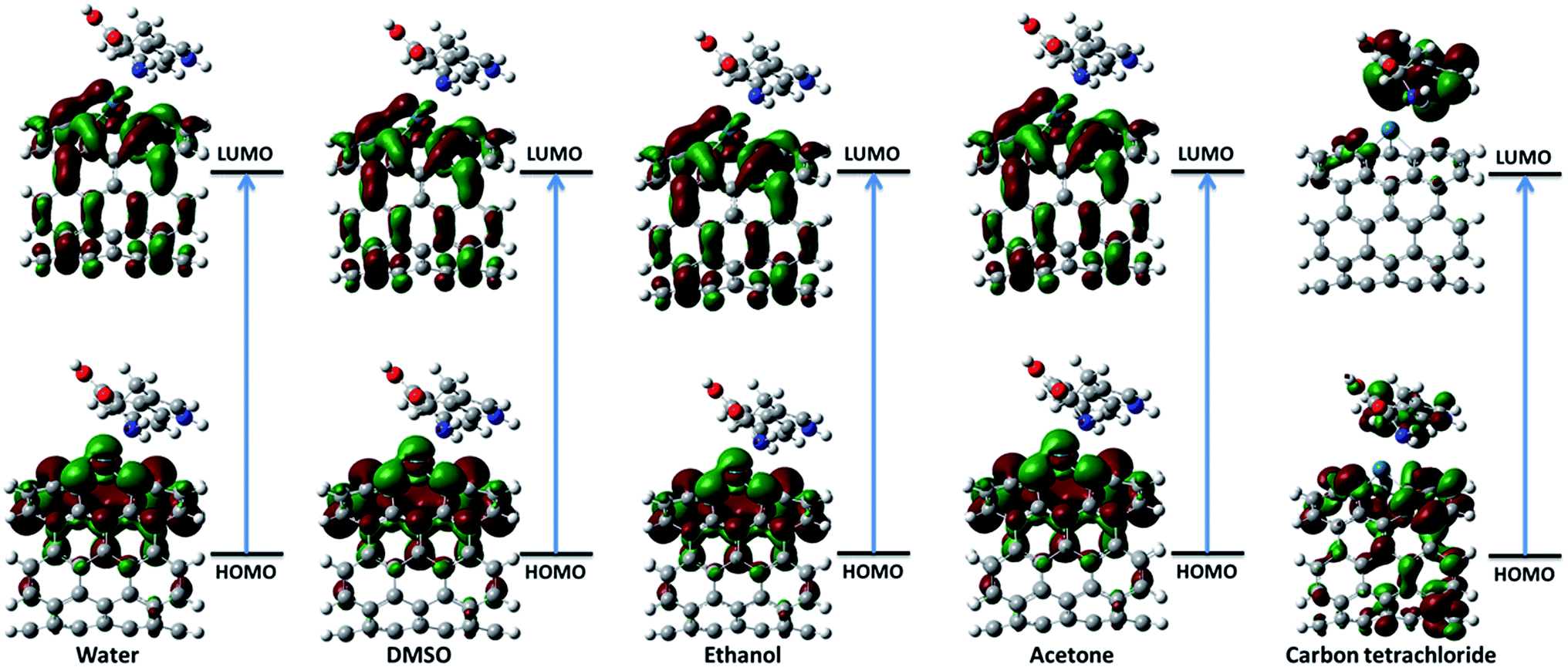
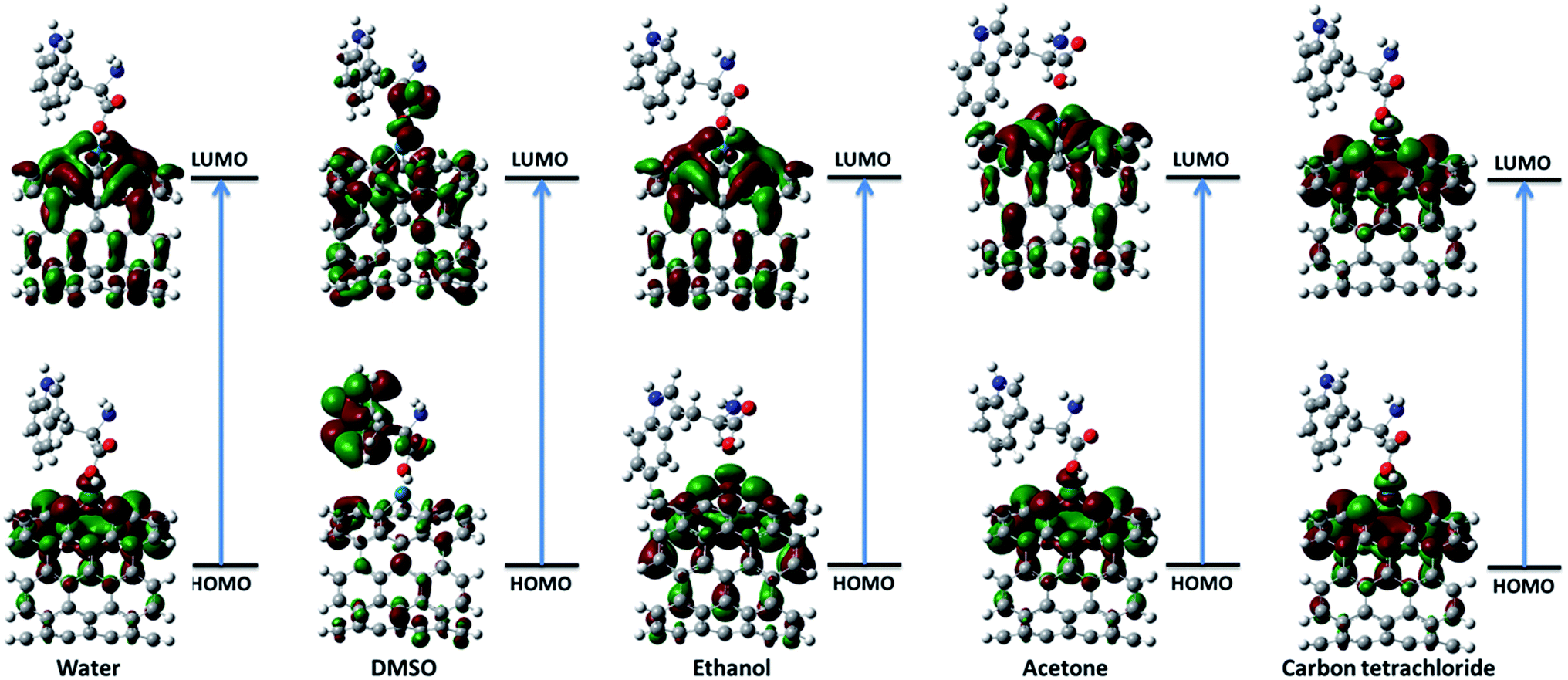
3.2.1. Frontier molecular orbital theory. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are the most important frontier molecular orbitals (FMOs) which play crucial role in the chemical stability and reactivity of molecules. The HOMO and the LUMO represent the ability to donate and accept an electron respectively. The chemical reactivity, chemical hardness and softness concept and optical polarizability determine by the energy gap between HOMO and LUMO.27 In the present study, the HOMO and LUMO energies are predicted at B3LYP method with 6-31G(d) basis set. The distribution and energy level of HOMO and LUMO orbitals in different medium are presented in Fig. 2 and 3 and Table 2 respectively. The positive and negative phase is represented in red and green respectively. The atoms occupied by more densities of HOMO and LUMO should have strong ability to detach and gain electron respectively. The DFT-based reactivity descriptors are listed in Table 2. The reactivity of active sites of biosensor can be elucidated by computation using global chemical reactivity indices. As can be seen in Table 2, the HOMO and LUMO energies shift to lower values in more polar media. Its worth mentioning that the larger the HOMO–LUMO gap, the harder and more stable/less reactive the system. On the basis of energy gap, global hardness and electronic chemical potential of complex 1 of nanobiosensor in different media can be arranged as follow:
| Water < DMSO < ethanol < acetone < carbon tetrachloride |
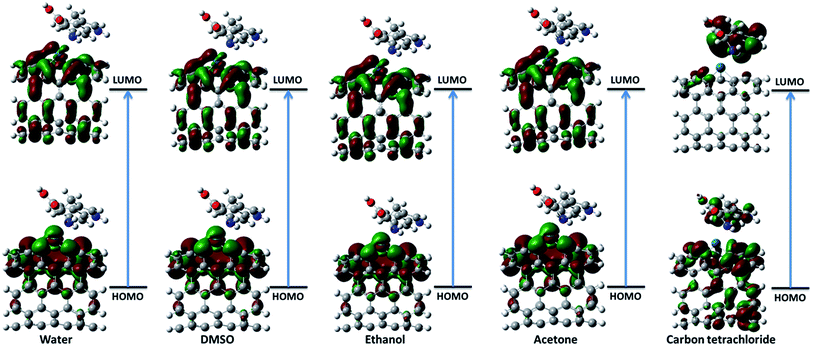 |
| | Fig. 2 The distribution of HOMO and LUMO orbitals of complex 1 in different solvents. | |
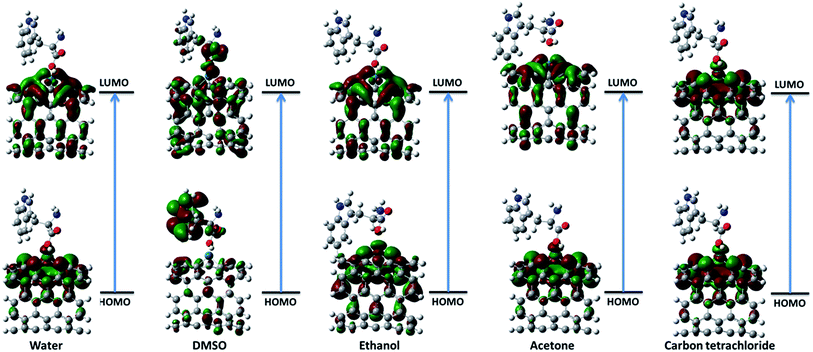 |
| | Fig. 3 The distribution of HOMO and LUMO orbitals of complex 2 in different solvents. | |
Table 2 The highest occupied molecular orbital (HOMO) energy, the lowest unoccupied molecular orbitals (LUMO) energy, energy gap and DFT-based reactivity descriptors
| Media |
Compound |
EHOMO |
ELUMO |
Eg |
μ |
η |
S |
ω |
| Water |
Pd/SWCNT |
−4.0765 |
−2.4782 |
1.5983 |
−3.2773 |
0.7991 |
0.6257 |
6.7203 |
| Try |
−5.5385 |
−0.3386 |
5.1998 |
−2.9386 |
2.5999 |
0.1923 |
1.6607 |
| Complex 1 |
−4.0177 |
−2.3993 |
1.6184 |
−3.2085 |
0.8092 |
0.6179 |
6.3609 |
| Complex 2 |
−4.0335 |
−2.4880 |
1.5455 |
−3.2607 |
0.7728 |
0.6470 |
6.8796 |
| DMSO |
Pd/SWCNT |
−4.0727 |
−2.4717 |
1.6010 |
−3.2722 |
0.8005 |
0.6246 |
6.6877 |
| Try |
−5.5401 |
−0.3424 |
5.1976 |
−2.9413 |
2.5988 |
0.1924 |
1.6644 |
| Complex 1 |
−4.0079 |
−2.3863 |
1.6217 |
−3.1971 |
0.8108 |
0.6167 |
6.3030 |
| Complex 2 |
−4.0270 |
−2.4834 |
1.5436 |
−3.2552 |
0.7718 |
0.6478 |
6.8645 |
| Ethanol |
Pd/SWCNT |
−4.0680 |
−2.4621 |
1.6059 |
−3.2651 |
0.8029 |
0.6227 |
6.6386 |
| Try |
−5.5390 |
−0.3397 |
5.1993 |
−2.9394 |
2.5996 |
0.1923 |
1.6617 |
| Complex 1 |
−3.9908 |
−2.3667 |
1.6241 |
−3.1787 |
0.8121 |
0.6157 |
6.2214 |
| Complex 2 |
−4.0248 |
−2.1208 |
1.9040 |
−3.0728 |
0.9520 |
0.5252 |
4.9590 |
| Acetone |
Pd/SWCNT |
−4.0653 |
−2.4581 |
1.6072 |
−3.2617 |
0.8036 |
0.6222 |
6.6191 |
| Try |
−5.5385 |
−0.3386 |
5.1998 |
−2.9386 |
2.5999 |
0.1923 |
1.6607 |
| Complex 1 |
−3.9843 |
−2.3593 |
1.6249 |
−3.1718 |
0.8125 |
0.6154 |
6.1912 |
| Complex 2 |
−4.0150 |
−2.4771 |
1.5379 |
−3.2460 |
0.7689 |
0.6502 |
6.8515 |
| Carbon tetrachloride |
Pd/SWCNT |
−4.0735 |
−2.4488 |
1.6247 |
−3.2611 |
0.8123 |
0.6155 |
6.5460 |
| Try |
−5.5319 |
−0.3223 |
5.2096 |
−2.9271 |
2.6048 |
0.1920 |
1.6447 |
| Complex 1 |
−3.9192 |
−2.2794 |
1.6399 |
−3.0993 |
0.8199 |
0.6098 |
5.8575 |
| Complex 2 |
−3.9720 |
−2.4651 |
1.5069 |
−3.2186 |
0.7534 |
0.6636 |
6.8746 |
The measure of energy lowering due to maximal electron flow between HOMO (donor) and LUMO (acceptor) represent the electrophilicity. The electrophilicity index of nanobiosensor increase by the increase of solvent dielectric constant of the media.
Except for ethanol media, in the complex 2, the greater the electronic chemical potential, the less stable or more reactive is the nanobiosensor. Results demonstrate that water media represents a good opportunity to apply the more stable complex of the investigated nanobiosensor in the biological environment with higher electron affinity and reactivity.
3.2.2. Natural bond orbital analysis. In the natural bond orbital (NBO) analysis, the highest possible percentage of the electron densities were included to provide the possible natural Lewis structure of the wave function. So the information of inter and intera-molecular interactions in both filled and virtual orbital spaces could be accurately obtained. The donor–acceptor interactions have been evaluated by the second-order Fock matrix. The stabilization energy, E(2), associated with the delocalization of donor (i) to acceptor (j) is estimated as| |
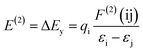 | (7) |
where qi, εi, εj and F(ij) are the donor orbital occupancy, diagonal elements and the off diagonal NBO Fock matrix elements respectively.28 The maximum stabilization energies of the intramolecular interactions have been distributed to LP(N) to LP*(Pd) and σ*(C–Pd) interactions in complex 1 and LP(O) to LP*(Pd) interactions in complex 2 in all investigated media. Calculated results show that the higher the E(2) the more intensive is the interaction between occupied Lewis-type NBO orbitals and unoccupied non-Lewis NBO orbitals. Therefore the polar solvent increase the energy of transitions. Results were tabulated in Table 3.
Table 3 The donor–acceptor interactions and the second-order perturbation energy (E(2) in kcal mol−1) of investigated complexes in 5 different solvents
| Compound |
Donor NBO |
Acceptor NBO |
E(2) water |
E(2) DMSO |
E(2) ethanol |
E(2) acetone |
E(2) carbon tetrachloride |
| Complex 1 |
σN111–H116 |
LP*(4)Pd70 |
5.46 |
|
— |
— |
— |
| σN111–H116 |
LP*(5)Pd70 |
— |
4.46 |
4.34 |
— |
— |
| σN111–H116 |
LP*(6)Pd70 |
— |
|
— |
4.3 |
3.39 |
| σN111–H116 |
LP*(7)Pd70 |
— |
|
— |
— |
3.51 |
| σN111–H117 |
LP*(4)Pd70 |
4.69 |
|
— |
— |
— |
| σN111–H117 |
LP*(5)Pd70 |
— |
4.19 |
4.02 |
— |
— |
| σN111–H117 |
LP*(6)Pd70 |
— |
|
— |
3.97 |
— |
| σN111–H117 |
LP*(7)Pd70 |
— |
|
— |
— |
— |
| LP(1) N111 |
LP*(4)Pd70 |
28.22 |
14.89 |
15 |
— |
— |
| LP(1) N111 |
LP*(5)Pd70 |
8.97 |
18.38 |
17.63 |
15.04 |
15.7 |
| LP(1) N111 |
LP*(6)Pd70 |
— |
8.35 |
9.26 |
17.42 |
12.75 |
| LP(1) N111 |
LP*(7)Pd70 |
— |
|
— |
9.52 |
15.05 |
| LP(1) N115 |
σ*C49–Pd70 |
16.05 |
12.34 |
12.14 |
11.73 |
10.71 |
| Complex 2 |
LP(1) O114 |
LP*(4)Pd70 |
16.3 |
16.53 |
13.48 |
15.68 |
15.06 |
| LP(2) O114 |
LP*(4)Pd70 |
3.18 |
3.18 |
3.95 |
3.94 |
3.57 |
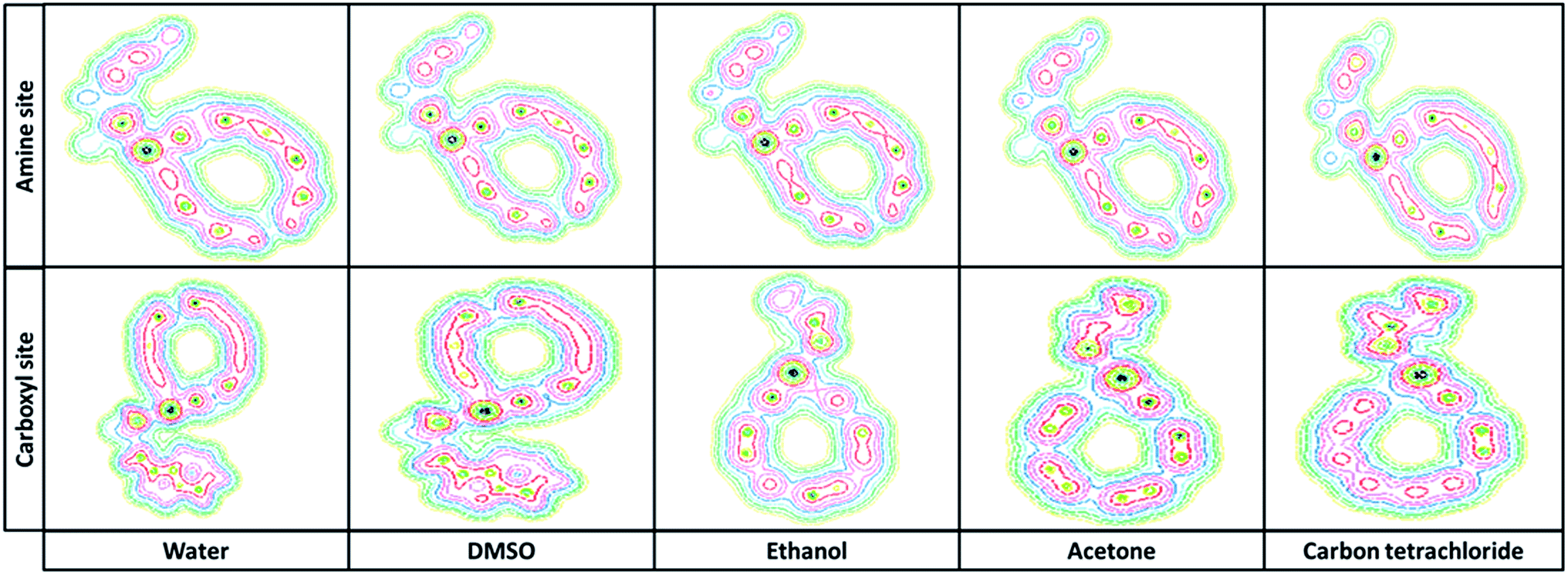
3.2.3. Quantum theory atoms in molecules. The Bader “atoms in molecules” theory is based on the analysis of the properties of the electron density (ρBCP) and Laplacian of electro density (∇2ρBCP) at the bond critical points (BCP). In covalent interactions the electronic charge is concentrated in the internuclear region and the sign of the Laplacian is negative (∇2ρBCP < 0). For closed shell interactions with the positive sign Laplacian (∇2ρBCP > 0), the electronic charge is depleted in the interatomic surface.29–31 The topological parameters derived from electron density properties of titled nanobiosensor showed that N⋯Pd and O⋯Pd bondings have high ρBCP and positive ∇2ρBCP which these properties are typically for closed-shell interactions. Based on topological parameters reported in Table 4, the shorter distance at Pd⋯N and Pd⋯O and greater electron density, ρPd⋯N and ρPd⋯O are observed. Fig. 4 shows the contour map of Try/Pd-SWCNT obtained from the ab initio calculations.
Table 4 Topological (in a.u.) and geometrical (in Å) parameters of important interaction between Try and Pd/SWCNT in 5 different solvents
| Media |
Complex 1 |
Complex 2 |
| ρPd⋯N |
∇2ρPd⋯N |
rPd⋯N |
ρPd⋯O |
∇2ρPd⋯O |
rPd⋯N |
| Water |
0.0598 |
0.2534 |
2.3039 |
0.0252 |
0.0929 |
2.6058 |
| DMSO |
0.0596 |
0.2527 |
2.3050 |
0.0254 |
0.0941 |
2.6010 |
| Ethanol |
0.0594 |
0.2513 |
2.3070 |
0.0242 |
0.0862 |
2.6327 |
| Acetone |
0.0593 |
0.2507 |
2.3078 |
0.0267 |
0.0992 |
2.5818 |
| Carbon tetrachloride |
0.0583 |
0.2449 |
2.3162 |
0.0256 |
0.0922 |
2.6085 |
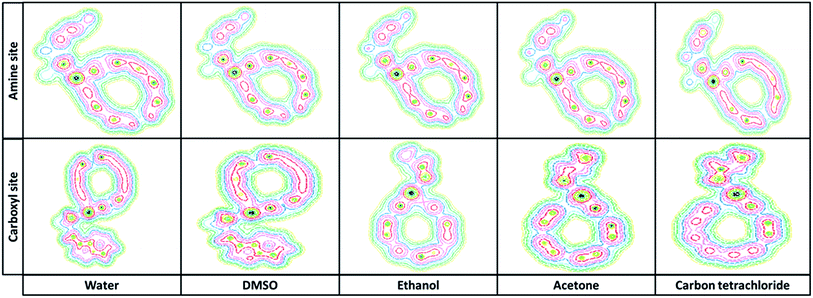 |
| | Fig. 4 The contour map of Try-Pd/SWCNT obtained from the DFT calculations in different solvents. | |
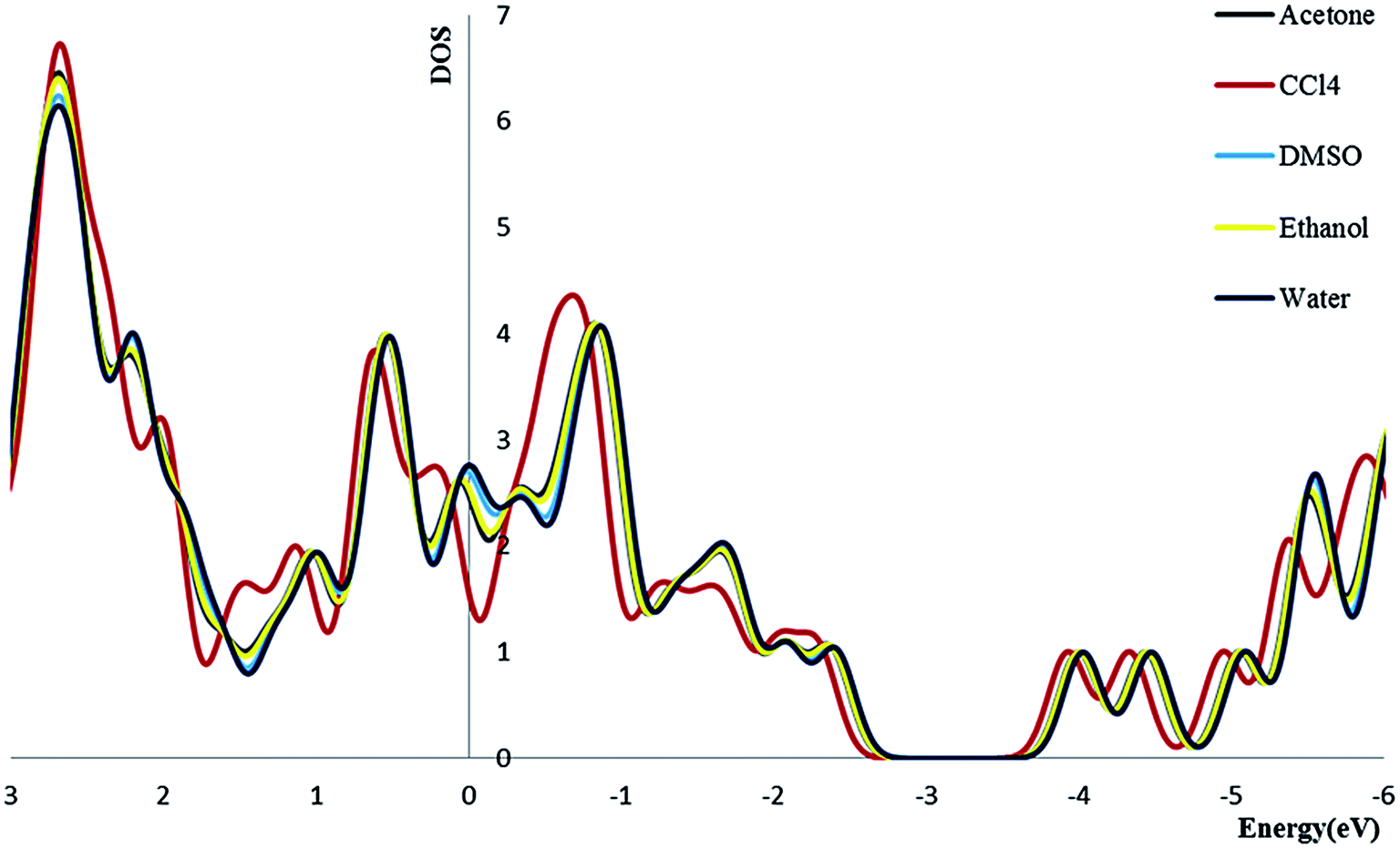
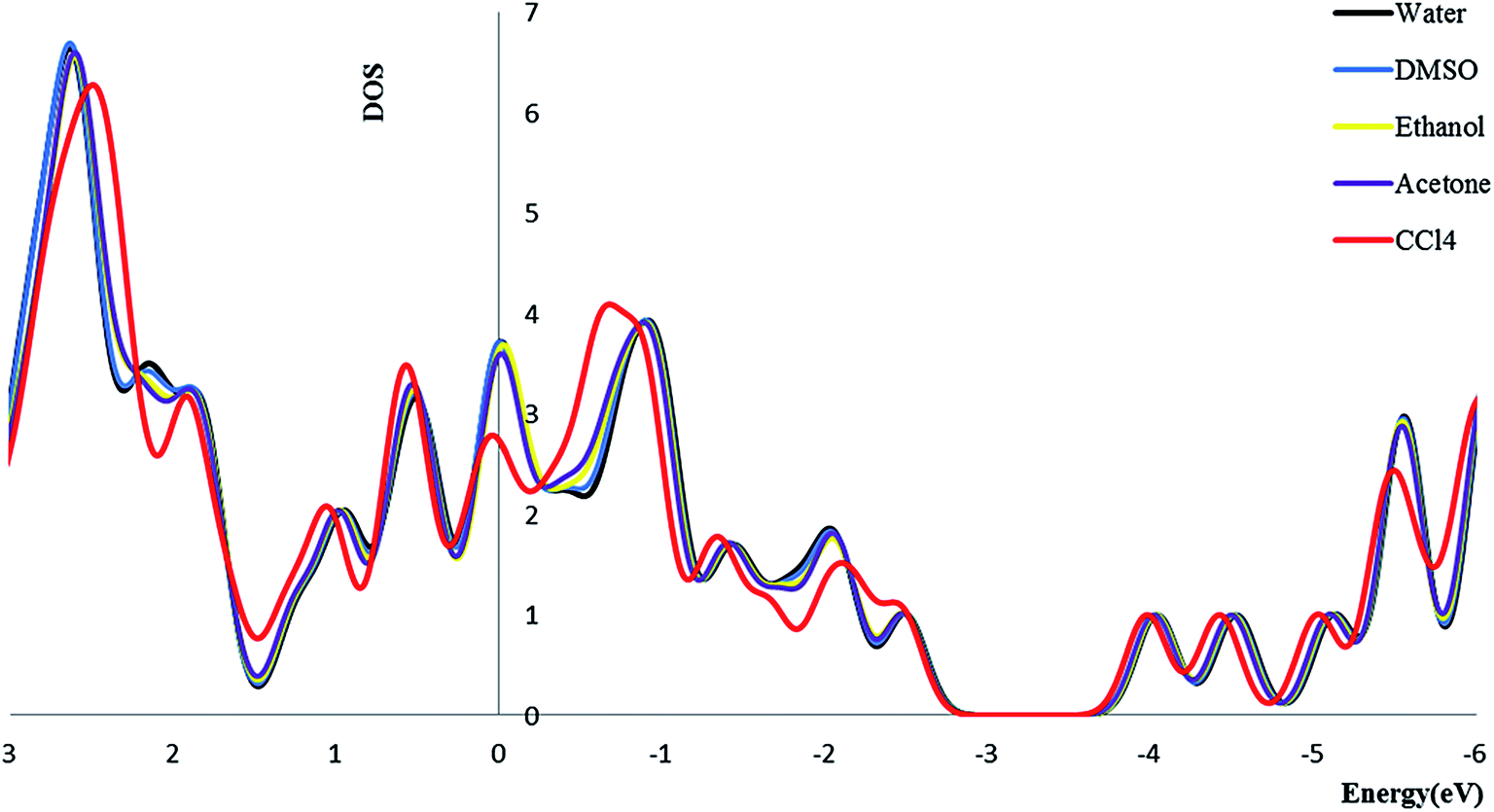
3.2.4. Density of state. We calculate the electronic densities of state for the Try/Pd-SWCNT nanobiosensor in different media. Results show that by increasing the dielectric constant of the solvents, the Fermi energy increases in both titled complexes. We can find that the Eg decrease for complex 1 and increase for complex 2 (exception in ethanol media) in more polar media. The DOSs increase around the Fermi level slightly which indicates the enhance of the conductance. Its also could be seen that a slightly right move happens to the DOS around the Fermi level. Also a noticeable DOS peak appears below the Fermi level for the complex 2 plots. Fig. 5 and 6 show the comparison of the total DOS for complex 1 and 2 in different media. As can be seen the DOSs for polar solvents are almost the same.
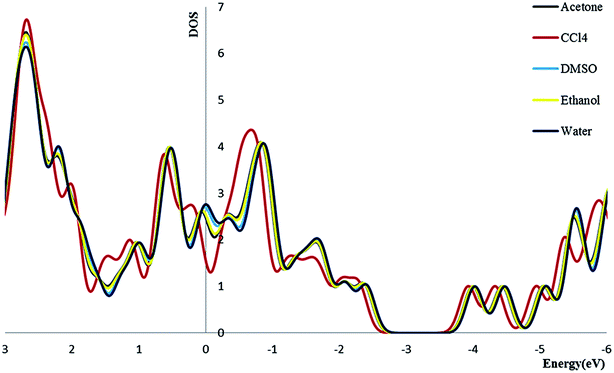 |
| | Fig. 5 The total DOS for complex 1 in different solvents. | |
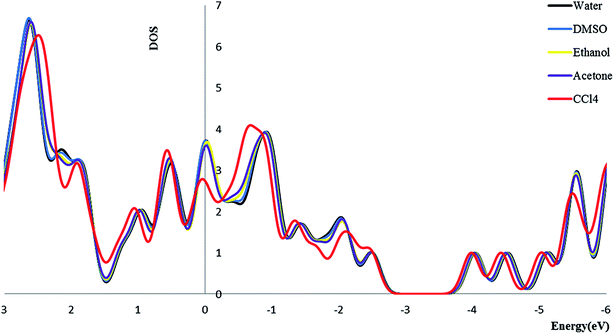 |
| | Fig. 6 The total DOS for complex 2 in different solvents. | |
3.2.5. Molecular potential energy surface. To achieve a deep understanding of the active electrophilic and nucleophilic sites of Try/Pd-SWCNT nanobiosensor, the molecular electrostatic potential (MPE) surfaces were theoretically predicted by DFT at the B3LYP/6-31 G(d)level. Different colors represent the different values of electrostatic potential at the MPE surface. Red, blue and green colors represent the most negative, most positive and zero electrostatic potential respectively.Fig. 7 shows the MPE plots for the titled nanobiosensor. From the MPE it is evident that the negative charge covers the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and the positive region is over the hydrogen bonded to N and O atom. As the ligands and the receptor recognize each other at their molecular surface, the negative region of MPE were related to electrophilic reactivity and the positive region related to nucleophilic reactivity.32
O group and the positive region is over the hydrogen bonded to N and O atom. As the ligands and the receptor recognize each other at their molecular surface, the negative region of MPE were related to electrophilic reactivity and the positive region related to nucleophilic reactivity.32
 |
| | Fig. 7 The molecular electrostatic potential (MPE) surfaces of complex 1 and complex 2 in different solvents. | |
4. Conclusion
The roughly same size of semiconducting single-walled carbon nanotubes as the biological molecules and cell membranes and extreme sensitivity of their environment, gives us a versatile probe to study the biosensing performance of the Try/Pd-SWCNT in 5 solutions. The non-specific solvent effects were described by the PCM model. The solvent effect on the geometry and the electronic structure of the nanobiosensor are presented via DFT/PCM calculation at B3LYP/6-31G(d) level. The obtained results have important implication for the interpretation of nanobiosensor application. The adsorption energies indicate chemisorption of the Try/Pd-SWCNT biosensor in different solvents. In addition the lower HOMO–LUMO energy gap, Eg, for the more stable complex indicated the more reactive and less chemical stable configuration of biosensor in water which emphasis on the chemical biocompatibility and the in vivo application of this novel nanobiosensor.
References
- M. Yoosefian and N. Etminan, RSC Adv., 2015, 5, 31172–31178 RSC.
- M. Yoosefian, Z. Barzgari and J. Yoosefian, Struct. Chem., 2014, 25, 9–19 CrossRef CAS.
- M. Yoosefian, H. Raissi and A. Mola, Sens. Actuators, B, 2015, 212, 55–62 CrossRef CAS.
- S. K. Vashist, A. Venkatesh, K. Mitsakakis, G. Czilwik, G. Roth, F. von Stetten and R. Zengerle, J. Bionanosci., 2012, 2, 115–126 CrossRef.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- J. Kong, N. R. Franklin, C. Zhou, M. G. Chapline, S. Peng, K. Cho and H. Dai, Science, 2000, 287, 622–625 CrossRef CAS PubMed.
- C. Jianrong, M. Yuqing, H. Nongyue, W. Xiaohua and L. Sijiao, Biotechnol. Adv., 2004, 22, 505–518 CrossRef PubMed.
- M. M.-C. Cheng, G. Cuda, Y. L. Bunimovich, M. Gaspari, J. R. Heath, H. D. Hill, C. A. Mirkin, A. J. Nijdam, R. Terracciano and T. Thundat, Curr. Opin. Chem. Biol., 2006, 10, 11–19 CrossRef CAS PubMed.
- C. R. Thomas, S. George, A. M. Horst, Z. Ji, R. J. Miller, J. R. Peralta-Videa, T. Xia, S. Pokhrel, L. Mädler and J. L. Gardea-Torresdey, ACS Nano, 2011, 5, 13–20 CrossRef CAS PubMed.
- M. Yoosefian, M. Zahedi, A. Mola and S. Naserian, Appl. Surf. Sci., 2015, 349, 864–869 CrossRef CAS.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed.
- J. Wildoer, L. Venema, A. Rizler and R. Smalley, Nature, 1998, 391, 6662 Search PubMed.
- S. Peng and K. Cho, Nano Lett., 2003, 3, 513–517 CrossRef CAS.
- N. Etminan, M. Yoosefian, H. Raissi and M. Hakimi, J. Mol. Liq., 2016, 214, 313–318 CrossRef CAS.
- P. Qi, O. Vermesh, M. Grecu, A. Javey, Q. Wang, H. Dai, S. Peng and K. Cho, Nano Lett., 2003, 3, 347–351 CrossRef CAS.
- M. Yoosefian and N. Etminan, Phys. E, 2016, 81, 116–121 CrossRef CAS.
- J. Kohanoff, Electronic structure calculations for solids and molecules: theory and computational methods, Cambridge University Press, 2006 Search PubMed.
- M. Yoosefian, Z. Ansarinik and N. Etminan, J. Mol. Liq., 2016, 213, 115–121 CrossRef CAS.
- M. Yoosefian and A. Mola, J. Mol. Liq., 2015, 209, 526–530 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Chem. Phys., 1997, 107, 3210–3221 CrossRef CAS.
- M. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Inc, Wallingford, CT, 2009, p. 200.
- A. D. Becke, The J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- E. Glendening, A. Reed, J. Carpenter and F. Weinhold, NBO, Version 3.1, Gaussian Inc., Pittsburgh, PA, CT, 2003 Search PubMed.
- R. F. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- R. G. Parr, L. v. Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- B. Kosar and C. Albayrak, Spectrochim. Acta, Part A, 2011, 78, 160–167 CrossRef PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- H. Raissi, A. Khanmohammadi, M. Yoosefian and F. Mollania, Struct. Chem., 2013, 24, 1121–1133 CrossRef CAS.
- H. Raissi, M. Yoosefian, F. Mollania and S. Khoshkhou, Struct. Chem., 2013, 24, 123–137 CrossRef CAS.
- H. Raissi, M. Yoosefian and F. Mollania, Comput. Theor. Chem., 2012, 996, 68–75 CrossRef CAS.
- M. Rocha, A. Di Santo, J. M. Arias, D. M. Gil and A. B. Altabef, Spectrochim. Acta, Part A, 2015, 136, 635–643 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.