 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Secondary organic aerosol origin in an urban environment: influence of biogenic and fuel combustion precursors†
M. C.
Minguillón
*a,
N.
Pérez
a,
N.
Marchand
b,
A.
Bertrand
b,
B.
Temime-Roussel
b,
K.
Agrios
c,
S.
Szidat
c,
B.
van Drooge
a,
A.
Sylvestre
b,
A.
Alastuey
a,
C.
Reche
a,
A.
Ripoll
a,
E.
Marco
a,
J. O.
Grimalt
a and
X.
Querol
a
aInstitute of Environmental Assessment and Water Research(IDAEA), CSIC, 08034 Barcelona, Spain. E-mail: mariacruz.minguillon@idaea.csic.es
bAix Marseille Université, CNRS, LCE UMR 7376, 13331 Marseille, France
cDepartment of Chemistry and Biochemistry & Oeschger Centre for Climate Change Research, University of Bern, 3012 Bern, Switzerland
First published on 8th December 2015
Abstract
Source contributions of organic aerosol (OA) are still not fully understood, especially in terms of quantitative distinction between secondary OA formed from anthropogenic precursors vs. that formed from natural precursors. In order to investigate the OA origin, a field campaign was carried out in Barcelona in summer 2013, including two periods characterized by low and high traffic conditions. Volatile organic compound (VOC) concentrations were higher during the second period, especially aromatic hydrocarbons related to traffic emissions, which showed a marked daily cycle peaking during traffic rush hours, similarly to black carbon (BC) concentrations. Biogenic VOC (BVOC) concentrations showed only minor changes from the low to the high traffic period, and their intra-day variability was related to temperature and solar radiation cycles, although a decrease was observed for monoterpenes during the day. The organic carbon (OC) concentrations increased from the first to the second period, and the fraction of non-fossil OC as determined by 14C analysis increased from 43% to 54% of the total OC. The combination of 14C analysis and Aerosol Chemical Speciation Monitor (ACSM) OA source apportionment showed that the fossil OC was mainly secondary (>70%) except for the last sample, when the fossil secondary OC only represented 51% of the total fossil OC. The fraction of non-fossil secondary OC increased from 37% of total secondary OC for the first sample to 60% for the last sample. This enhanced formation of non-fossil secondary OA (SOA) could be attributed to the reaction of BVOC precursors with NOx emitted from road traffic (or from its nocturnal derivative nitrate that enhances night-time semi-volatile oxygenated OA (SV-OOA)), since NO2 concentrations increased from 19 to 42 μg m−3 from the first to the last sample.
1 Introduction
Carbonaceous aerosols make up a large proportion of ambient PM10, PM2.5 and PM1 (atmospheric particulate matter finer than 10, 2.5 and 1 μm, respectively). Worldwide tropospheric submicron PM may be from 20 to 90% organic aerosol (OA).1 OA, also named organic matter (OM), may account for 15–26% of both PM10 and PM2.5 in Europe, with the highest loads being recorded at urban and traffic sites.2 For European regional background sites OM + EC (elemental carbon) ranged from 27% to 43% of PM2.5.3 OM concentration ranged from 2 to 4 μg m−3, 3 to 7 μg m−3 and 3 to 7 μg m−3 in PM2.5 for remote-rural, urban background, and traffic and industrial hotspots in Spain, respectively (approx. 25–30%, 20–40% and 30–40% of PM2.5).4In spite of these relatively high proportions in ambient PM, source contributions of OA are still not fully understood5–7 due to the mixed source origins (anthropogenic or biogenic OA) and the atmospheric processes that cause the formation of OA. Moreover, OA may be primary (POA), emitted as solid/liquid PM from both anthropogenic or biogenic sources, or secondary (SOA), formed in the atmosphere from volatile organic compounds (VOCs), which can have an anthropogenic or biogenic origin (AVOCs or BVOCs).7,8 Furthermore, in urban or industrial areas, the high concentrations of specific anthropogenic pollutants (e.g. NOx, SO2 and O3) may enhance the formation of secondary organic aerosols from biogenic precursors (BSOA), giving rise to the so-called enhanced BSOA (eBSOA),7 which, despite its biogenic origin, would be anthropogenically driven. The interaction between the BVOCs–O3–NOx–SOA systems is very complex.9 Thus, several studies have reported higher yields of eBSOA formation from isoprene in the presence of relatively high concentrations of NOx, although this SOA formation becomes lower for very high NOx concentrations.7,10 For the highly reactive sesquiterpenes the interaction with NOx seems also to increase the SOA yields, probably due to the low volatility of the sesquiterpene nitrates.7 Conversely, for monoterpenes increasing NOx/VOC ratios have shown to reduce the BSOA formation from ozonolysis of α-pinene, and highly volatile N-containing organic species are preferentially formed under these conditions.11 Other atmospheric species different to NOx, OH radical and O3 may have a large impact on BSOA formation. Thus, a higher BSOA yield from isoprene can occur under acidic conditions caused by the occurrence of sulphuric acid.12 VOCs play a very relevant role in sulphuric acid related nucleation events and in the growth of newly formed particles.13 In any case, one of the major BSOA formation pathways, especially under high NOx and O3 concentrations, seems to be related to the nocturnal oxidation of VOCs by the nitrate radical (a product of the NO2 + O3 interaction) reaction.7 Thus, both organo-sulphates and organo-nitrates may constitute an important fraction of the SOA due to the prevalence of the above formation pathways.14 Hence, it is expected that OA concentrations in urban environments will increase compared with those from rural areas due to (a) the enlarged anthropogenic POA and VOC emissions leading to anthropogenic SOA, but also to (b) eBSOA formation due to the interaction of BVOCs with other anthropogenic components.
The city of Barcelona, NE Spain, has one of the highest car densities in Europe, with diesel vehicles making up around 47% of the fleet.15 In spite of this adverse fact, PMx concentrations have decreased in the last decade,16 going down from about 40 μg m−3 to 22–30 μg m−3 PM10.
Urban background OA accounted for 20% of the PM10 and >30% of the PM2.5 and PM1 in 2003–2007.17 The SOA contribution to OA, calculated with the EC tracer method,18 was 36–40%. Hence, OA, including SOA, is a major component of PMx in the city of Barcelona and, in general, in regional and urban environments in Europe.
Prior studies carried out at an urban background site during the DAURE campaign in March 2009 using a combination of HR-ToF-AMS and 14C analysis of OC and EC reported a proportion of non-fossil OC of 60% of the total OC measured in PM1, while 57% of the SOA was non-fossil.19,20 This non-fossil SOA could be BSOA or eBSOA. Relatively strong correlations were found between dicarboxylic acids and other oxidation products from BVOCs, and the oxygenated OA (OOA) loading in the urban background of Barcelona.21 Similar results concerning the high non-fossil OA contribution were obtained in Zurich,22 Marseille,23 and Birmingham,24 among other locations.
In order to investigate the origin of OA in Barcelona, with special interest in determining the relative contributions from POA and SOA, and the anthropogenic and the biogenic fractions, a measurement campaign was devised in summer 2013, including the last weeks of August and the beginning of September, coinciding with a period of high insolation and high BVOC emissions. An important proportion of Barcelona's population takes holidays in August, thus traffic flow is substantially reduced compared with the rest of the year. Hence, the campaign comprised a low road traffic period and continued when the traffic resumed its usual flow. Through the combination of several analytical tools, such as offline 14C analysis of OC, offline PM inorganic and organic chemical composition determination, online VOC measurements (using PTR-ToF-MS), online organic aerosol concentration and types/sources determination (using aerosol chemical speciation monitor (ACSM)), online black carbon (BC) measurements, and online NO, NO2, O3, CO, SO2, PM10, PM2.5 and PM1 concentration measurements, we investigated why and how road traffic emissions influence the concentrations and origin of OA in the city.
2 Experimental
An intensive monitoring campaign at the urban background of Barcelona was carried out from 16 August to 6 September 2013. Two periods are distinguished: a summer holiday period with low traffic intensity (16–25 August) named the low traffic (LT) period, and a typical urban traffic emissions period (28 August–6 September), named the high traffic (HT) period, both characterized by similar environmental conditions and high insolation.Measurements were carried out at the urban background site Palau Reial (PR; 41°23′14′′ N, 02°06′56′′E, 78 m a.s.l., Fig. S1†), located 200 m from one of the main traffic avenues of the city (traffic density of 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 vehicles per working day). The main source of atmospheric PMx is road traffic, although contributions from industry, regional secondary atmospheric pollutants, construction, and shipping are also relevant.17,25 Atmospheric dynamics are driven by the breeze regime, with a NW wind component during the night and the development of breezes during the day turning progressively from SE to SW direction, with gradually increasing wind speeds reaching maximum levels around noon.26
000 vehicles per working day). The main source of atmospheric PMx is road traffic, although contributions from industry, regional secondary atmospheric pollutants, construction, and shipping are also relevant.17,25 Atmospheric dynamics are driven by the breeze regime, with a NW wind component during the night and the development of breezes during the day turning progressively from SE to SW direction, with gradually increasing wind speeds reaching maximum levels around noon.26
2.1 On line measurements of VOCs and other gaseous pollutants
Measurements of VOCs were performed with a Proton-Transfer Reaction Time of Flight Mass Spectrometer (PTR-ToF-MS, Ionicon PTR TOF 8000, Ionicon Analytik, Innsbruck, Austria).27,28 The instrument was operated in standard conditions, with a reaction chamber pressure fixed at 2.05 mbar, drift tube voltage at 500 V and drift tube temperature at 323 K corresponding to an electric field strength applied to the drift tube (E) to buffer gas density (N) ratio of 125Td (1Td = 10−17 V cm2). Mass spectra were recorded up to m/z 500 with a time resolution of 1 minute. Instrumental background signal was measured regularly by passing the sample flow through a catalytic converter that efficiently removes VOCs (platinum wool heated at 350 °C). VOC mixing ratios are expressed in parts per billion by volume (ppbv):29| [VOC H+] = [H3O+]0[VOC]kt |
Real-time concentrations of NO and NO2 (Chemiluminescence, SIR S-5012), CO (Ecotech, GFC, EC 9830), O3 (UV absorption, SIR S-5014), and SO2 (Pulsed Fluorescence, Thermo Scientific Model 43C) were measured and supplied by the Department of Environment of the Autonomous Government of Catalonia.
2.2 Online aerosol measurements
Ultrafine particle number size distributions within the range 15–660 nm (N15–660, 5 min time resolution) were measured with a Scanning Mobility Particle Sizer (SMPS), composed by a Differential Mobility Analyser (DMA, TSI 3081) coupled with a condensation particle counter (CPC, TSI 3772). Equivalent BC mass concentrations31 were measured by a multi-angle absorption photometer (MAAP, model 5012, Thermo ESM Andersen Instruments, 1 min time resolution) with a PM10 inlet. Semi-hourly PM10, PM2.5, and PM1 concentrations were measured by an optical particle counter (GRIMM 180) and corrected with simultaneous 24 hour gravimetric samples.An Aerosol Chemical Speciation Monitor (Aerodyne Research Inc., ACSM)32 was used to measure non-refractory submicron aerosol species (organic aerosol, nitrate, sulphate, ammonium and chloride) with a time resolution of 15 minutes. Mass calibration of the ACSM was based on determining the instrument response factor (RF) using ammonium nitrate calibration aerosol.32 Relative Ionization Efficiency (RIE) for ammonium was directly determined from the ammonium nitrate calibration (5.61). Sulphate RIE was determined by doing the aforementioned calibration exercise with ammonium sulphate monodisperse aerosol, and resulted in a value of 1.38. The default RIE for organic aerosol (1.4)33 has been used. The data were acquired using Aerodyne Research software ACSM_DAQ_v1438 and processed with the ACSM data analysis software version 1.5.3.0 (Aerodyne Research Inc.) written in Igor Pro (WaveMetrics, Inc., Lake Oswego, OR, USA). A correction for the instrument performance limitations was applied based on the inlet pressure and N2 signal. The aerosol mass concentrations were then corrected for particle collection efficiency (CE) following the Middlebrook approach.34 Comparison with co-located measurements (real-time PM1, and particle size distribution determined by an SMPS) allowed the identification of three periods with different correlation slopes. The ACSM data were then corrected according to these intercomparisons.
2.3 Offline aerosol analyses
PM2.5 samples were collected in 8 h time periods (0:00 h–8:00 h, 8:00 h–16:00 h and 16:00 h–0:00 h UTC) on quartz filters using high volume samplers (30 m3 h−1).A complete chemical characterization was performed.35 Briefly, an acid digestion of half filter was carried out for each sample to yield solutions for the subsequent analysis of major and trace elements by Inductively Coupled Atomic Emission Spectroscopy (ICP-AES) and Mass Spectrometry (ICP-MS). Water leachates from a quarter of the filter were used to determine soluble ion concentrations by Ion Chromatography and NH4+ concentrations by a selective electrode. OC and EC concentrations were determined from 1.5 cm2 filter sections by means of a thermal-optical carbon analyser (Sunset, EUSAAR_2 protocol).36
Molecular organic species were determined in the same filter samples after extraction by a mixture of methanol and dichloromethane (1:1 v/v), derivation of esters to TMS-esters and subsequently detection and quantification in a gas-chromatograph coupled to a mass-spectrometer (GC-MS) in full-scan mode.21 Hence, several organic species in a wide range of polarity were analyzed, including levoglucosan, hopanes, and formation products of VOC oxidation, such as dicarboxylic acid, poly acids, and polyols.
Additional 24 h PM2.5 samples were collected using high volume samplers and used for 14C analysis on the OC fraction. The separation of the OC and collection as CO2 was carried out according to the Swiss_4S method.37 The 14C isotopic ratio (fraction of modern carbon, fM) of each sample was determined using an Accelerator Mass Spectrometer MICADAS (MIni CArbon Dating System) at the Laboratory for the Analysis of Radiocarbon with AMS (LARA)38 and corrected for the OC field blank.
2.4 Supporting measurements
Meteorological variables (atmospheric pressure, wind components, solar radiation, temperature and relative humidity) were recorded at a meteorological station located 300 m from our sampling site and provided by the Faculty of Physics from the Universitat de Barcelona. Road traffic intensity data from the Diagonal Avenue were supplied by the Council of Barcelona (Servei de Mobilitat).2.5 Source apportionment to the organic aerosol
The source apportionment to the organic aerosol was investigated by applying Positive Matrix Factorization (PMF)39 using the Multilinear Engine (ME-2)40 to the ACSM organic mass spectra, using the toolkit SoFi (Source Finder) version 4.8.41 The dataset used comprised from 25 July to 19 September 2013. The ME-2 allows the user to introduce a priori information about sources e.g. using the a-value approach. Thus the user may ‘constrain’ a factor by introducing its source profile and determining the extent to which the output profile can differ from the one input to the model (the a-value). Only m/z ≤ 120 were used given that those >120 have lower signal to noise and they account for a minor fraction of the total signal. Details on the different solutions are given in the results section.The source apportionment of OC was also assessed based on the 14C isotopic ratios (fM). The determination of the contribution from different sources to the total OC is done knowing their fM. The fM for fossil sources is zero. The fM of OC from non-fossil sources (fMOCnf) depends on the contribution from the different non-fossil sources, mainly biomass burning and biogenic sources, and it has to be defined in advance,22 although subsequent iterations could be applied if necessary.19 For the present study, the fMOCnf was calculated assuming a 10/90 share between biomass burning and biogenic source, in agreement with other studies42 and based on the ACSM results from this study. The fMOC for biomass burning emissions is 1.063, corresponding to emissions from the burning of 25-year-old trees harvested in 2011 as determined with a tree-growth model,43 and the fM for biogenic sources is 1.023, corresponding to the year 2013.44 The resulting fMOCnf is 1.027. OC was then separated into fossil OC (OCf) and non-fossil OC (OCnf). OCf was attributed to POA and SOA from fossil fuel combustion. OCnf may have different origins, such as biogenic SOA (bio), biomass burning (bb), cooking, biofuel combustion, brake lining dust, natural rubber in tire dust, and others.45
3 Results and discussion
3.1 General conditions
Similar meteorological patterns were recorded during the LT period (means of 25 °C, 64% RH, and insolation of 239 W m−2) and the HT period (23 °C, 59% RH, 230 W m−2), which were separated by coincidence by a rainy inter-period (26 to 27 August, Monday and Tuesday), characterized by a clean atmosphere, low insolation and temperature and high humidity (Fig. S2†). However, in the HT period lower wind speed and higher pressure were recorded (1.7 vs. 2.1 m s−1, and 1019 vs. 1016 hPa) as a consequence of an anticyclonic episode leading to atmospheric stagnation.The HYSPLIT back-trajectory analysis evidenced that both periods were characterized by regional recirculation of air masses, with relatively short air mass transport distances, although in the LT period a North-western direction prevailed, whereas in the HT period this short transport had a European origin (Fig. S3 and S4†).
Mean traffic flow at the Diagonal Avenue increased +37% from the LT to the HT period. The rush traffic hours were more clearly identified during the HT period at 6 and 16–18 UTC, as well as the maximum traffic intensity/wind speed ratio at 6 UTC and 19 UTC (Fig. S5†).
The separation of the LT and HT periods was done based on these results. Hence, in the subsequent sections the influence of traffic emissions on PM composition and source contribution, with a special emphasis on OC, will be investigated.
3.2 PMx, N and gases concentrations
PM10, PM2.5 and PM1 concentrations increased in the HT vs. the LT period +18, +24 and +38%, respectively (Table 1). This increase is due to the more pronounced increase during traffic rush hours and the development of the high pressure episode, reaching the maximum PMx on Tuesday 3 September (Fig. 1), one day before the maximum pressure.| LT | HT | Difference (%) | ||
|---|---|---|---|---|
| Meteorogical conditions and traffic | ||||
| T | (°C) | 25 | 23 | −6 |
| RH | (%) | 64 | 59 | −8 |
| SR | (W m−2) | 239 | 230 | −4 |
| WS | (m s−1) | 2.1 | 1.7 | −20 |
| P | (hPa) | 1016 | 1019 | <1 |
| Traffic | (Vehicles per h) | 1663 | 2271 | 37 |
|
||||
| Gases | ||||
| SO2 | (μg m−3) | 2.8 | 3 | 7 |
| NO | (μg m−3) | 3 | 5 | 79 |
| NO2 | (μg m−3) | 16 | 29 | 81 |
| O3 | (μg m−3) | 80 | 85 | 6 |
| CO | (μg m−3) | 333 | 495 | 48 |
|
||||
| Aerosol | ||||
| PM10 | (μg m−3) | 20 | 24 | 18 |
| PM2.5 | (μg m−3) | 13 | 16 | 24 |
| PM1 | (μg m−3) | 10 | 14 | 38 |
| N15 | (# cm−3) | 7741 | 9822 | 27 |
| N15–20 | (# cm−3) | 1227 | 1493 | 22 |
| N20–100 | (# cm−3) | 4610 | 6511 | 41 |
| N100–500 | (# cm−3) | 1180 | 1756 | 49 |
 | ||
| Fig. 1 Time series of gases and selected aerosol parameters along the campaign. The LT and HT periods are shown. Date format is dd/mm/yyyy. | ||
Particle number concentrations increased +22% from the LT to the HT period for the N15–20 (the nucleation mode), and +41% and +49% for the N20–100 and N100–500 (Table 1). N15–20 in the LT period showed intensive midday maxima, attributed to nucleation burst by prior studies in Barcelona,17,46–48 whereas these midday nucleation burst maxima were lower in the HT period (Fig. 1).
CO, NO2 and NO average concentrations increased from 333 μg m−3, 16 μg m−3 and 3 μg m−3, respectively, for the LT period to 495 μg m−3, 29 μg m−3 and 5 μg m−3 for the HT period (+48, +81 and +79%, respectively, Table 1). O3 concentrations were 80 μg m−3 (LT) and 85 μg m−3 (HT). SO2 concentrations did not show clear variations between LT and HT periods (average around 3 μg m−3), attributed to the main influence of shipping emissions, which are not dependent on the period, and resulting in a midday maximum coinciding with the maximum sea breeze intensity.
3.3 Volatile organic compounds
With the exception of formic acid, isoprene, methyl vinyl ketone (MVK) + MACR, and monoterpenes, all VOCs presented higher mixing ratios during the HT period. Aromatic hydrocarbons related to traffic emissions showed the highest differences between the LT and HT periods, increasing from +78% to +85% (C2, C3 and C4-alkylbenzenes), +63% (benzene), and +52% (toluene). VOCs were dominated by methanol (2.7 ppbv and 3.1 ppbv as averages for the LT and HT periods, respectively), acetone (2.2 ppbv and 2.7 ppbv), and acetic acid (0.9 ppbv and 1.1 ppbv). Isoprene average mixing ratios were 0.16 ppbv and 0.12 ppbv for the LT and HT periods, respectively, in the range measured during the winter DAURE campaign in Barcelona,49 although mixing ratios of benzene and toluene were slightly lower than those registered in DAURE; and higher for acetone, acetic acid, or MVK (Table 2).| m/z | Chemical formula (protonated ions) | Most probable VOCs | k rates (10−9 cm3 s−1) | LT (ppbv) | HT (ppbv) | Difference (%) |
|---|---|---|---|---|---|---|
| 33.033 | (CH4O)H+ | Methanol | 2.22 | 2.7 | 3.1 | 14 |
| 47.012 | (CH2O2)H+ | Formic acid | 1.99 | 0.3 | 0.1 | −46 |
| 59.049 | (C3H6O)H+ | Acetone | 3.32 | 2.2 | 2.7 | 21 |
| 61.028 | (C2H4O2)H+ | Acetic acid | 2.25 | 0.9 | 1.1 | 19 |
| 69.069 | (C5H8)H+ | Isoprene | 1.96 | 0.16 | 0.12 | −22 |
| 71.049 | (C4H6O)H+ | MVK + MACR | 3.39 | 0.03 | 0.03 | −10 |
| 73.065 | (C4H8O)H+ | MEK | 3.28 | 0.06 | 0.08 | 34 |
| 79.054 | (C6H6)H+ | Benzene | 1.9 | 0.03 | 0.05 | 63 |
| 93.069 | (C7H8)H+ | Toluene | 2.08 | 0.13 | 0.20 | 52 |
| 107.085 | (C8H10)H+ | C2-Alkylbenzene | 2.27 | 0.07 | 0.13 | 78 |
| 121.101 | (C9H12)H+ | C3-Alkylbenzene | 2.45 | 0.02 | 0.04 | 84 |
| 135.117 | (C10H14)H+ | C4-Alkylbenzene | 2.4 | 0.002 | 0.004 | 85 |
| 137.132 | (C10H16)H+ | Monoterpenes | 2.37 | 0.017 | 0.014 | −16 |
The traffic-related VOCs showed a marked daily cycle during the HT period, peaking during traffic rush hours and following the same daily pattern as BC (Fig. 2).
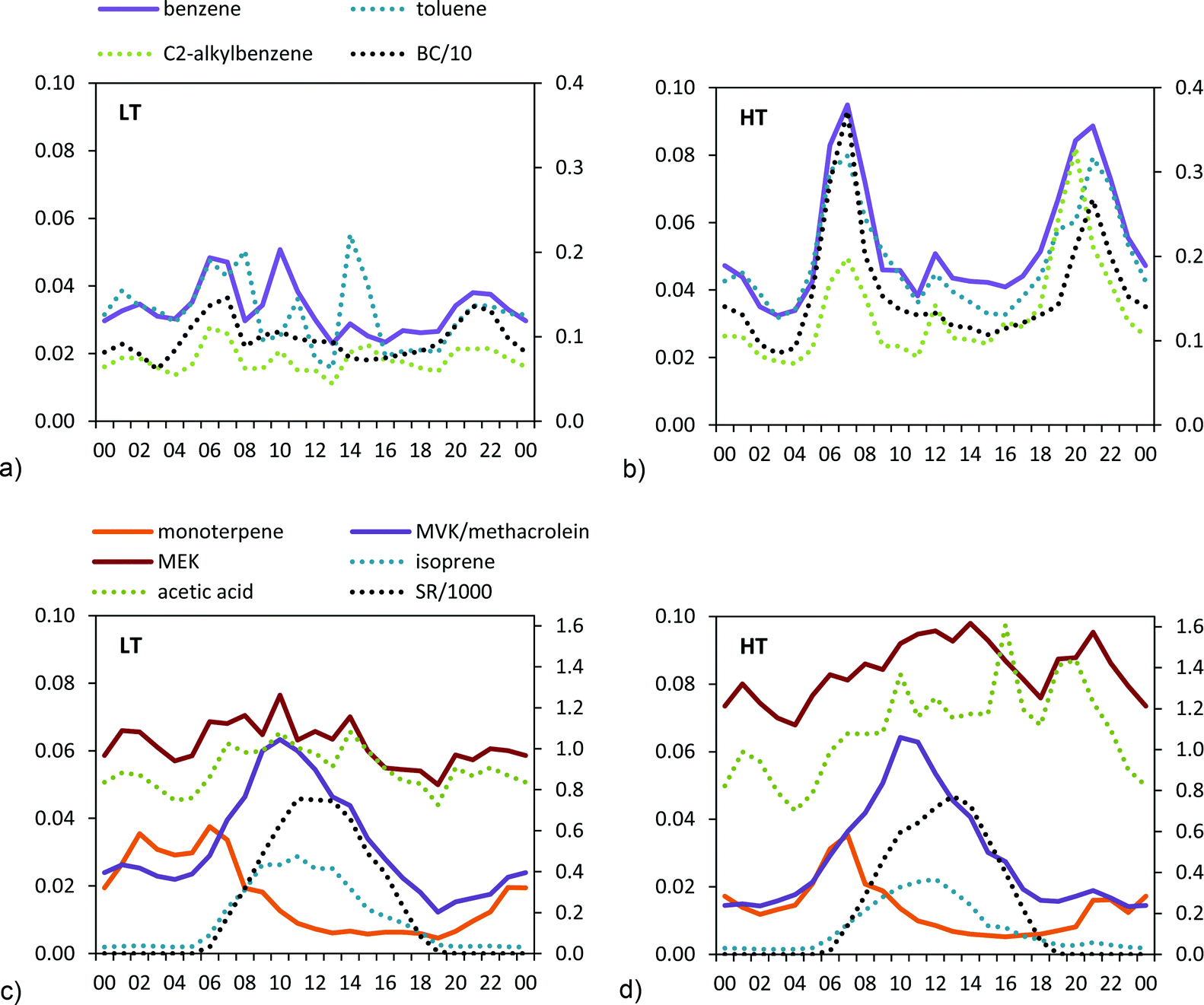 | ||
| Fig. 2 Average daily cycle of the mixing ratios (ppbv) of selected VOCs (a and b: traffic related VOCs; c and d: biogenic VOCs) during the LT and HT periods. BC concentrations in μg m−3 divided by 10 are also plotted. Solar radiation (SR) in W m−2 divided by 1000 is also plotted. In each of the plots, the full lines correspond to the left axis and the dotted lines correspond to the right axis. The time is UTC. | ||
Daily variability of the biogenic VOCs is related to temperature and solar radiation cycles (Fig. 2). Monoterpenes showed a marked decrease during the day (Fig. 2), attributed to degradation by O3.50
Other VOCs, such as methanol or acetone increased between +14% to +21% in the HT period (Table 2). These compounds did not show a clear diurnal pattern, although higher mixing ratios were recorded during the daytime (Fig. S6†). The higher mixing ratios measured during the HT period are probably related to the higher oxidation of BVOCs and AVOCs.
| LT | HT | Difference (%) | Difference of component/OA (%) | ||
|---|---|---|---|---|---|
| Chemical composition (μg m −3 ) | |||||
| BC | 1.0 | 1.6 | 62 | 6 | |
| Sulphate | 2.4 | 3.8 | 60 | 1 | |
| Nitrate | 0.5 | 1.0 | 99 | 26 | |
| Ammonium | 1.1 | 1.8 | 72 | 9 | |
| OA | 6.8 | 10.8 | 58 | — | |
| OA types | HOA | 0.6 | 1.1 | 79 | 14 |
| COA | 1.2 | 1.9 | 62 | 3 | |
| LV-OOA | 2.5 | 3.5 | 40 | −11 | |
| SV-OOA | 2.6 | 4.4 | 69 | 7 | |
| Organic compounds (ng m −3 ) | |||||
| Diacids (urban SOA) | Succinic acid | 16.8 | 21.3 | 27 | −19 |
| Glutaric acid | 2.9 | 3.7 | 29 | −17 | |
| Adipic acid | 0.9 | 1.2 | 25 | −20 | |
| Pimelic acid | 0.8 | 0.8 | 4 | −34 | |
| Azelaic acid | 1.8 | 3.0 | 65 | 6 | |
| Phthalic acid | 3.9 | 5.2 | 32 | −15 | |
| Pinene SOA | cis-Pinonic acid | 12.1 | 42.6 | 251 | 125 |
| Malic acid | 28.8 | 53.0 | 84 | 18 | |
| 3-Hydroxyglutaric acid | 4.8 | 8.6 | 80 | 16 | |
| MBTCA | 4.2 | 10.0 | 135 | 51 | |
| Isoprene SOA | 2-Methylglyceric acid | 23.3 | 32.3 | 39 | −11 |
| C5-Alkene triols | 28.0 | 37.7 | 35 | −14 | |
| 2-Methylthreitol | 19.9 | 19.4 | −2 | −37 | |
| 2-Methylerythritol | 67.5 | 57.6 | −15 | −45 | |
| Urban POA | Norhopane | 0.5 | 0.9 | 85 | 19 |
| Hopane | 0.5 | 0.7 | 45 | −7 | |
| Fluorathene | 0.11 | 0.33 | 217 | 103 | |
| Pyrene | 0.12 | 0.42 | 249 | 124 | |
| Benz[a]anthracene + chrysene | 0.17 | 0.32 | 84 | 18 | |
3.4 PM chemical composition
The PM1 relative composition was very similar during both the LT and HT periods (Fig. 3). It was dominated by OA (=OM) (59% and 57%, respectively), followed by sulphate (20%), ammonium (9% and 10%), nitrate (4% and 5%) and EC (8%). Table 3 shows the average concentration of the different PM components for the LT and HT periods, as well as the difference between both periods. To better assess the variation in the concentrations of the different PM components, a normalized concentration (relative to OA) was used, and the difference in these normalized concentrations is also shown in Table 3.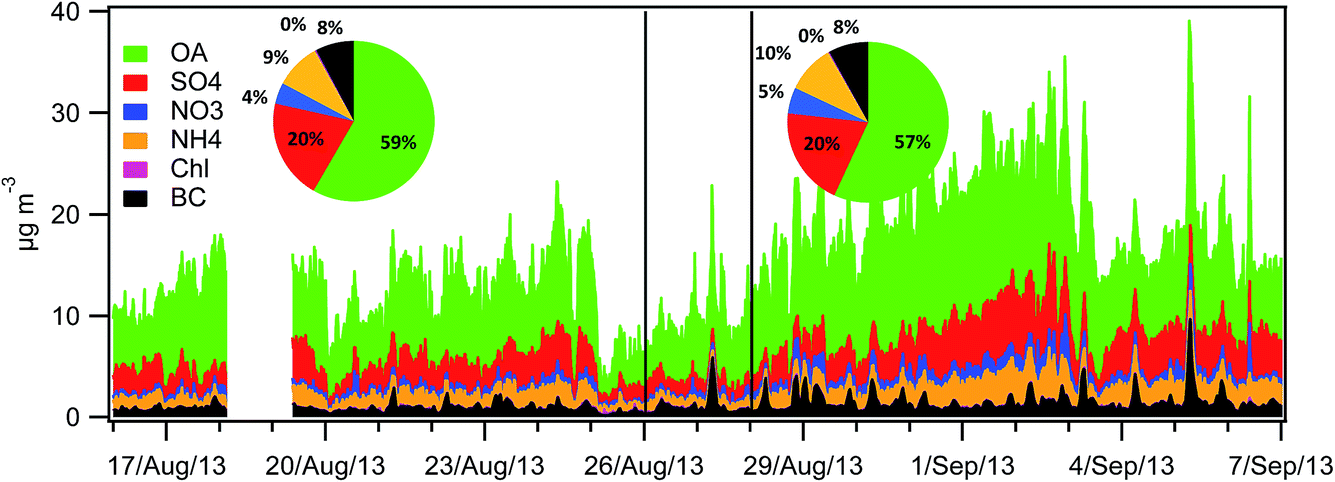 | ||
| Fig. 3 Stacked OA, sulphate, nitrate, ammonium, chloride and BC concentrations in PM1 (15 min time resolution) and average composition for the LT and HT periods. | ||
Average BC concentrations increased from 1.0 μg m−3 in the LT period to 1.6 μg m−3 in the HT period. BC concentrations were more driven by the traffic cycles than by the anticyclone development during the HT period (Fig. 2 and 3). Thus, maximum BC levels during traffic rush hours reached 4 μg m−3 on most days of the HT period (and up to 10 μg m−3), whereas during the LT period these rarely exceeded 1 μg m−3, and the daily pattern was less marked or even missing (Fig. 2 and 3).
OA concentrations increased +58% from the LT to the HT period. Nitrate and ammonium concentrations increased +99% and +72%, respectively, comparable to the BC concentrations (+62%). In terms of relative concentrations (with respect to OA), the increase was +26% and +9%. Part of this increase may be the result of the higher NOx (+80%) and NH3 exhaust traffic emissions in the HT period, resulting in the formation of ammonium nitrate. Nevertheless, and given that the increase for nitrate is higher than that for ammonium, part of the nitrate could potentially be attributed to the presence of organonitrates in the HT period. The sulphate concentrations increased +60%, similarly to the reference OA concentration. The higher sulphate concentration could be related to an increase in the sulphate formation at a regional scale (note that SO2 concentrations only increased +7% in Barcelona, Table 1) or to the accumulation of pollutants over the Barcelona area. The intra-day variation of the different components varied from day to day, resulting in average daily patterns that were relatively flat for sulphate and ammonium (Fig. 4). Nitrate and OA concentrations partially followed the daily pattern observed for BC (Fig. 4), especially during the HT period.
 | ||
| Fig. 4 Average daily pattern of OA (right axis), sulphate, nitrate, ammonium and BC concentrations (left axis) for the LT and HT periods. The time corresponds to the end of the hourly slot. | ||
Some mineral dust tracer concentrations, such as Al and Fe, increased in the HT period +60% and +18%, whereas Ca decreased by −27%, yielding a mineral dust increase of only +12% during the HT.
Sb and Sn concentrations, tracers of non-exhaust traffic emissions (mainly from brake pads),51 increased in the HT period +78% and +79% (Fig. 5), similarly to BC concentrations, hence confirming their road traffic origin. Conversely concentrations of sea salt tracers such as Na decreased by −13%.
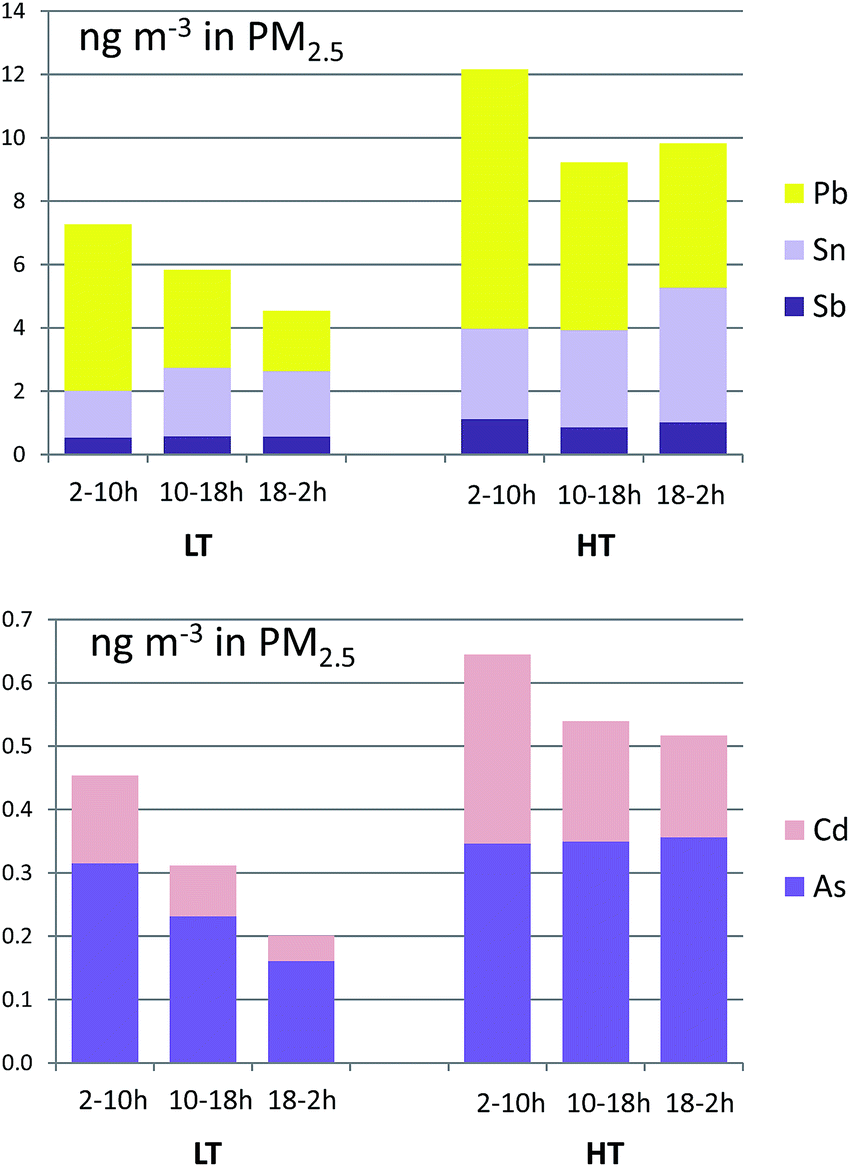 | ||
| Fig. 5 Eight-hour averages of concentrations of selected trace elements in PM2.5 during the LT and HT periods. Time is local time (UTC+2). | ||
Also, Pb and Cd concentrations, industrial tracers, increased between +49% and +151% during the HT period (Fig. 5), probably due to an increase in industrial activity.
3.5 Organic aerosol sources
Different source apportionment solutions were investigated. The 3-factor solution consisted of a hydrocarbon-like OA (HOA), a semi-volatile oxidized OA (SV-OOA) and a low-volatility oxidized OA (LV-OOA). The investigation of the residuals for this solution led to the identification of a diurnal pattern for the residuals of m/z 55. Hence, a cooking OA (COA) source was constrained based on a COA profile identified for Barcelona in March 2009.52 The solutions for a-values from 0.025 to 0.2 in steps of 0.025 were investigated, and a final a-value of 0.1 was chosen based on the significance of the source profiles found and their correlation with literature profiles, the correlation of the factor contributions with external tracers, and the significance of the time variation (especially the daily patterns) (Section S3 of the ESI†). The contribution of biomass burning is expected to be low based on previous studies carried out in July 200919 and based on the low signal at m/z 60 registered in the present study, which is below the background threshold53 for more than 95% of the data (Fig. S10†). The possible contribution of an isoprene epoxydiol derived OA factor (IEPOX-OA)54 was investigated, but the solutions with a high number of factors resulted in a split of the HOA and/or the SV-OOA sources but a lack of a clear IEPOX-OA. More details on the solution are given in the ESI (Section S3†).The final solution includes four organic sources: HOA, COA, SV-OOA, and LV-OOA (Fig. 6 and 7), which compares well with other mass spectral profiles from literature, and specifically with those identified for Barcelona in March 2009 (Fig. S9†).52 The HOA is attributed to road traffic and its intra-day variation clearly follows the traffic rush hour times, although it is clearer in the HT period than in the LT period, similarly to the BC variation (Fig. 8). Its contribution was 0.6 and 1.1 μg m−3 for the LT and HT periods (9% and 10% of the total OA), respectively. The HOA/BC ratio ranged from 0.4 to 0.9, which is in agreement with ratios found close to road traffic emission sources (Fig. S11†).55 The average daily pattern for COA shows an increase in the morning lasting until lunch time and an increase at dinner time (Fig. 8), although it may not be as well-defined as would be expected. This average daily pattern may indicate that this factor has some influence on sources other than COA, such as some road traffic influence, although it is expected to be low, given that the mass spectral profile has been constrained according to the COA profile found specifically for Barcelona in a previous study. It contributed 1.2 and 1.9 μg m−3 for the LT and HT periods, respectively, which implies an increase of +62%, similar to the bulk OA increase. The SV-OOA is attributed to recently formed SOA and its variation does not show a constant daily pattern throughout the study period, hence resulting in an average daily pattern not representative of any of the days. It may have an influence of the prevailing wind condition, since the SV-OOA contribution under NW winds was 4.0 μg m−3, while it was only 3.1 μg m−3 for other wind directions, whereas the difference in concentrations of other components depending on the wind direction is lower. The SV-OOA concentrations moderately correlate with nitrate concentrations (R2 = 0.43). This correlation may be due to the similar gas-particle partitioning of both nitrate and SV-OOA, or it could also be attributed to the formation of organo-nitrates from the reaction of biogenic precursors with nitrate.7,56,57 The fraction of SV-OOA with respect to total OA was similar for the LT and the HT periods (38% and 40%, respectively). The LV-OOA is attributed to aged SOA, hence reflecting the SOA contribution that can be in the atmosphere for a longer time. It is moderately correlated with sulphate concentrations (R2 = 0.41). Although the absolute contribution of LV-OOA increased from the LT to the HT period, its relative contribution to total OA was similar (37% and 33%, respectively). The total OOA absolute increase (+2.8 μg m−3, divided into +1.0 μg m−3 of LV-OOA and +1.8 μg m−3 of SV-OOA) is about 6 times that for HOA or BC.
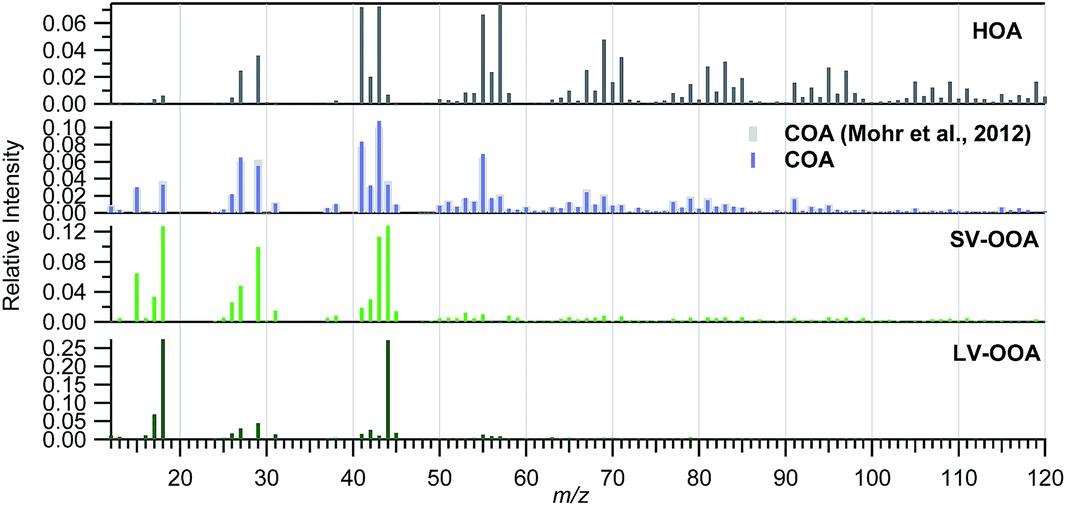 | ||
| Fig. 6 Mass spectral profiles of the organic sources identified. In light grey is the profile used to constrain the COA factor. | ||
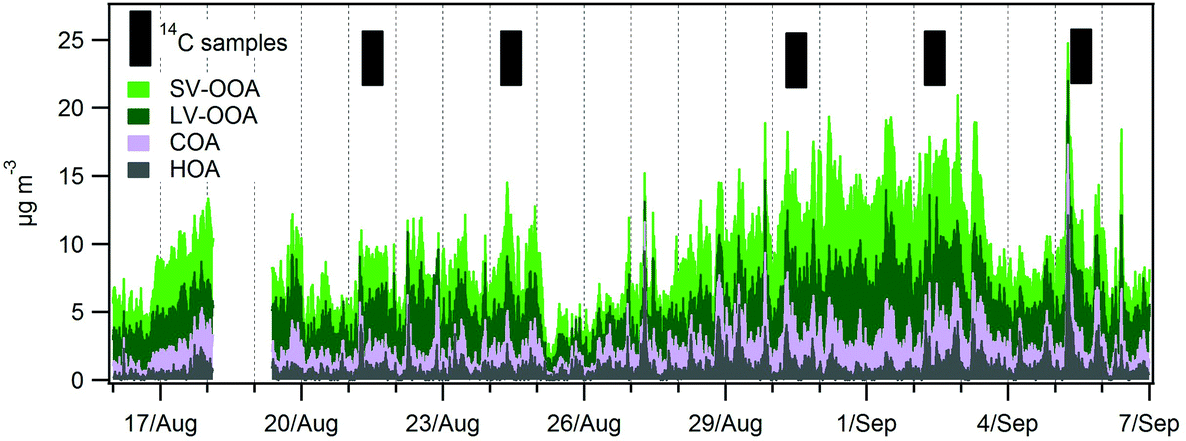 | ||
| Fig. 7 Stacked HOA, COA, LV-OOA and SV-OOA. Selected days for 14C analysis are indicated. | ||
 | ||
| Fig. 8 Average daily pattern of HOA, COA, LV-OOA, SV-OOA and BC concentrations for the LT and HT periods. Error bars correspond to one standard deviation, only shown for some data points for clarity. The time corresponds to the end of the hourly slot. | ||
3.6 Fossil vs. non-fossil OC combined with ME2-ACSM sources
A total of 5 samples were analysed for 14C in OC (Fig. 7). An increase in the fraction of non-fossil OC from the beginning of the campaign towards the end, from 43% to 54% of the total OC, was recorded (Fig. S12†). The absolute concentrations of both the fossil OC and the non-fossil OC increased from the LT to the HT period.Further information can be gained when combining 14C and ACSM OA source apportionment results.19,58 Relative contributions to OC determined by the 14C and ME2-ACSM techniques are shown in Fig. 9. Fractional contributions are compared to avoid differences due to absolute concentration inaccuracies in either method. The OA source contributions from ME2-ACSM were converted into OC source contributions using the following OA-to-OC ratios: 1.21 for HOA, 1.42 for COA, 2.14 for LV-OOA and 1.61 for SV-OOA, as determined in previous studies for Barcelona.52 The sources identified by ME2-ACSM were divided into fossil and non-fossil. HOC was included in the fossil sources and COC was included in the non-fossil sources. As LV-OOC and SV-OOC are thought to be a surrogate for total secondary OC and may originate from both fossil and non-fossil VOC precursors, these two factors were divided into fossil and non-fossil so that the resulting OCf/OCnf ratio from ME2-ACSM sources equals that from the 14C method. The fossil OC was found to be mainly secondary (>70%) except for the last sample, where the fossil SOC only represented 51% of the total fossil OC (Fig. 10). This means that the amount of fossil SOC formed depended on the primary fossil OC for the first four samples, whereas it was influenced by something different for the last sample, in which the fossil SOC formation was lower with respect to the primary fossil OC. This last day resembles the results found in March 2009, when the fossil OC was found to be 53% secondary.19 On the other hand, when analysing the SOC origin, it is observed that the fraction of non-fossil SOC increased from 37% for the first sample to 60% for the last sample (Fig. 10). This enhanced formation of non-fossil SOC could be attributed to the reaction of BVOC precursors with NOx emitted from road traffic (or from its nocturnal derivative NO3 that enhances night-time SVOOA),59 since NO2 concentrations increased from 19 to 42 μg m−3 from the first to the last sample.
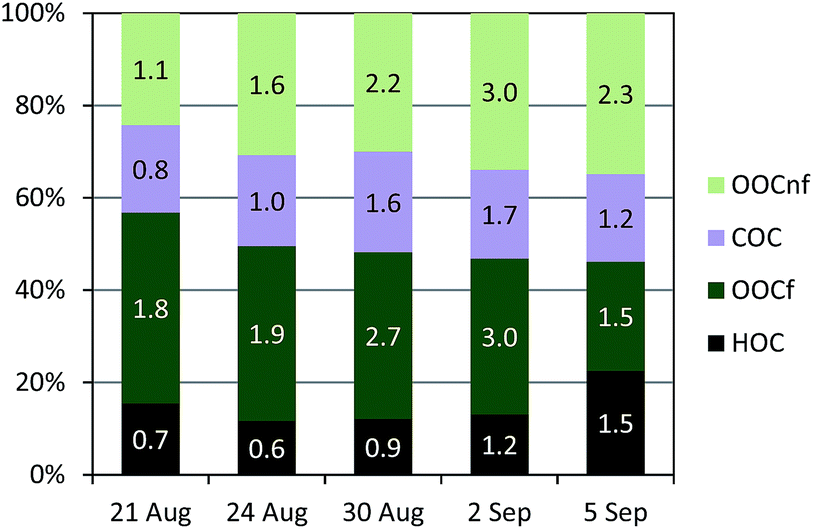 | ||
| Fig. 9 Relative source apportionment to OC using the 14C technique and ME2-ACSM. Numbers indicate the absolute concentrations in μg m−3. HOC: hydrocarbon-like organic carbon; OOC: oxygenated organic carbon (secondary OC); COC: cooking organic carbon. | ||
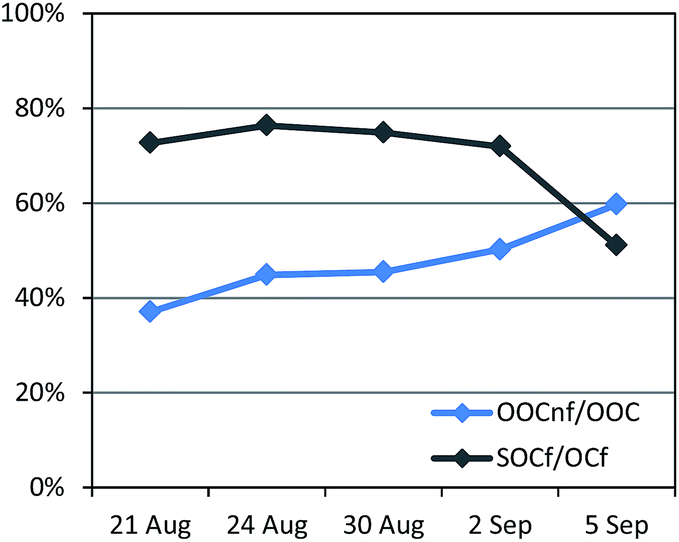 | ||
| Fig. 10 Percentage of non-fossil secondary OC with respect to total secondary OC (represented by the oxygenated organic carbon (OOC)), and percentage of secondary fossil OC with respect to total fossil OC. | ||
3.7 Organic compounds and relation with OA sources
The urban POA related organic compound concentrations increased from +45% to +249% in the HT period (Table 3). In relative terms, when considering their normalized concentrations with respect to total OA, they increased from +18% to +124%, except for the hopanes, attributed to their reactivity.60 PAHs showed substantial correlations (p < 0.05) with typical traffic tracers, such as BC (0.57 < R2 < 0.81) and NO2 (0.50 < R2 < 0.71), as well as the HOA contribution (the R2 correlation coefficient of HOA and fluoranthene and pyrene was 0.68 and 0.79, respectively). Dicarboxylic acids, such as succinic acid, glutaric acid and azelaic acid, as well as poly- and aromatic acids, such as malic acid and phthalic acid are SOA-species in the absence of primary biomass burning emissions.61,62 The very low levoglucosan concentrations (<1 ng m−3) and the low signal at m/z 60 found by the ACSM suggest that these acids are mainly SOA. Their concentrations increased between +20% and +50% from LT to HT, although their normalized concentrations with respect to OA decreased from the LT to the HT period. The compounds related to SOA originated from isoprene showed a decrease of −11% to −45% in their relative concentrations. On the other hand, the compounds related to pinene SOA showed an increase in their normalized concentrations (+16% to +125%).The precursors of the SOA species may be anthropogenic or biogenic. Thus, phthalic acid can be related to oxidation of naphthalene (i.e. anthropogenic fossil fuel combustion emissions), while azelaic acid can be related to ozonolysis of (particulate) oleic acid (e.g. an anthropogenic non-fossil carbon source from cooking). The increase in the normalized concentrations of some aged oxidation products of biogenic VOCs, such as 3-hydroxyglutaric acid and 3-methyl-1,2,3-butanetricarboxylic acid (MBTCA) in the case of α-pinene oxidation,63,64 may reflect the enhanced production of BSOA at higher traffic intensities in the metropolitan area, as observed in other studies.7,14,65 On the other hand, 2-methylglyceric acid, from oxidation of isoprene,66,67 showed a decrease of −11% in its normalized concentration. While 2-methylglyceric acid is favoured in a high NOx regime, 2-methylthreitol and 2-methylerythritol are preferentially formed under low NOx conditions,65 which explains the larger decrease in the HT period of normalized concentrations of 2-methylthreitol and 2-methylerythritol (−37% and −45%) compared to 2-methylglyceric acid (−11%).
Substantial correlations (p < 0.05) were observed among the different SOA species, as well as with the ME2-ACSM OA sources (Fig. 11), suggesting heterogeneous reactions and mixing of these compounds in the urban airshed. The long-chained dicarboxylic azelaic acid showed a strong correlation (R2 = 0.61) with the COA component, suggesting that cooking activities could be involved, in agreement with a previous study in the urban area of Barcelona,62 although the correlation between pimelic acid and COA was weaker (R2 = 0.21). The short-chained dicarboxylic acids, i.e. succinic, glutaric and adipic acids, showed strong or relatively strong correlations with the LV-OOA component (R2 = 0.72, 0.65 and 0.50, respectively), reflecting aged-SOA. These correlations were also observed for poly-acids, such as malic acid, 3-hydroxyglutaric acid, MBTCA and 2-methylglyceric acid (R2 = 0.49, 0.41, 0.66 and 0.49, respectively). Nevertheless, in contrast to the former compounds these latter compounds also showed substantial correlations (p < 0.05) with the SV-OOA component (0.43 < R2 < 0.71), supporting the idea of enhanced BSOA production at higher traffic intensities. The concentrations of the isoprene SOA products did not correlate to any other variable, including the ME2-ACSM OA sources. It is unlikely that these compounds are readily transformed to other compounds, due to their relatively high concentrations, although part may be transformed to organosulphates in urban atmospheres.67,68 The decrease in their normalized concentrations from the LT to the HT period could be attributed to their formation in the region under relatively low NOx conditions (LT period) and their transport to the urban area.68
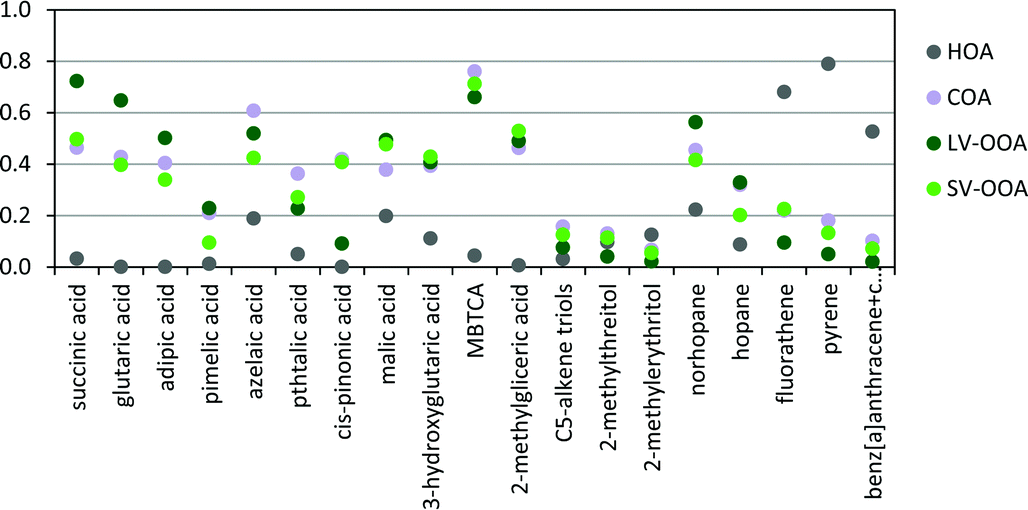 | ||
| Fig. 11 Squared Pearson correlation coefficients (R2) between organic tracer compounds and OA sources. | ||
4 Final discussion and conclusions
The increase of traffic related organic species is in agreement with the increases observed for inorganic traffic indicators as well as the HOA. Moreover, an increase in OOA determined by ME2-ACSM from the LT to the HT period was simultaneously recorded for almost all analyzed SOA species, including those from BSOA. These findings may implicate an enhanced formation of BSOA in the urban airshed under higher traffic intensities (average NOx concentrations were 20 μg m−3 for the LT period and 36 μg m−3 for the HT period). This is further suggested by the higher concentrations of non-fossil SOC found for the samples in the HT period. However, part of these SOA species and other possible oxidation products (including those from isoprene oxidation), may have been formed outside of the city and subsequently transported. This would mean that they were formed under conditions that were similar for both the LT and HT periods, in spite of the increase of traffic intensities in the HT period.Maximum concentrations of C4–C6 dicarboxylic acid and phthalic acid were recorded during the daytime, similarly to the LV-OOA component. On the other hand, the products of pinene oxidation, cis-pinonic (a first generation product), 3-hydroxyglutaric acid and MBTCA (further oxidation products), and malic acid are sometimes registered in higher concentrations in the evening and night-time samples.
To further investigate how the different species, components, and parameters relate to each other, a factor analysis was applied to a combined dataset including: VOC-factors (obtained from the processing of the VOC dataset), ACSM data (inorganic compounds and OA sources), and meteorological and online air quality parameters. It resulted in the identification of 5 factors, which accounted for 72% of the variance:
- Factor 1: this reflects mainly the road traffic influence, with high factor loadings for the VOC traffic factor, N20–500, BC, NO, NO2, HOA and traffic/wind speed.
- Factor 2: this reflects mainly the biogenic VOC influence, with high factor loadings for the VOC biogenic factor, temperature, and the solar radiation.
- Factor 3: this reflects the components with the higher influence on the PM mass, like sulphate, nitrate, ammonium, COA, LV-OOA and SV-OOA.
- Factor 4: this reflects the fine particles, grouping the different particle number size ranges up to 500 nm.
- Factor 5: this reflects the more oxidized components, like LV-OOA and ozone.
Acknowledgements
This study was partially funded by the Spanish Ministry of Agriculture, Food and the Environment, the European LIFE+ AIRUSE project (LIFE11 ENV/ES/584), the European Union Seventh Framework Programme (FP7/2007-2013) through ACTRIS (grant agreement no 262254), and the Generalitat de Catalunya (AGAUR 2014 SGR33 and DGQA). We thank Gary Salazar (University of Bern) for his support during the 14C analysis. The publication fee was supported by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- Q. Zhang, J. L. Jimenez, M. R. Canagaratna, J. D. Allan, H. Coe, I. Ulbrich, M. R. Alfarra, A. Takami, A. M. Middlebrook, Y. L. Sun, K. Dzepina, E. Dunlea, K. Docherty, P. F. DeCarlo, D. Salcedo, T. Onasch, J. T. Jayne, T. Miyoshi, A. Shimono, S. Hatakeyama, N. Takegawa, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, P. Williams, K. Bower, R. Bahreini, L. Cottrell, R. J. Griffin, J. Rautiainen, J. Y. Sun, Y. M. Zhang and D. R. Worsnop, Geophys. Res. Lett., 2007, 34, L17811 CrossRef.
- J. P. Putaud, R. van Dingenen, A. Alastuey, H. Bauer, W. Birmili, J. Cyrys, H. Flentje, S. Fuzzi, R. Gehrig, H. C. Hansson, R. M. Harrison, H. Herrmann, R. Hitzenberger, C. Hüglin, A. M. Jones, A. Kasper-Giebl, G. Kiss, A. Kousa, T. A. J. Kuhlbusch, G. Löschau, W. Maenhaut, A. Molnar, T. Moreno, J. Pekkanen, C. Perrino, M. Pitz, H. Puxbaum, X. Querol, S. Rodriguez, I. Salma, J. Schwarz, J. Smolik, J. Schneider, G. Spindler, H. ten Brink, J. Tursic, M. Viana, A. Wiedensohler and F. Raes, Atmos. Environ., 2010, 44, 1308–1320 CrossRef CAS.
- X. Querol, A. Alastuey, J. Pey, M. Cusack, N. Pérez, N. Mihalopoulos, C. Theodosi, E. Gerasopoulos, N. Kubilay and M. Koçak, Atmos. Chem. Phys., 2009, 9, 4575–4591 CAS.
- X. Querol, A. Alastuey, M. Viana, T. Moreno, C. Reche, M. C. Minguillón, A. Ripoll, M. Pandolfi, F. Amato, A. Karanasiou, N. Pérez, J. Pey, M. Cusack, R. Vázquez, F. Plana, M. Dall'Osto, J. de La Rosa, A. Sánchez De La Campa, R. Fernández-Camacho, S. Rodríguez, C. Pio, L. Alados-Arboledas, G. Titos, B. Artíñano, P. Salvador, S. García Dos Santos and R. Fernández Patier, Atmos. Chem. Phys., 2013, 13, 6185–6206 CAS.
- R. Volkamer, J. L. Jimenez, F. San Martini, K. Dzepina, Q. Zhang, D. Salcedo, L. T. Molina, D. R. Worsnop and M. J. Molina, Geophys. Res. Lett., 2006, 33 CrossRef CAS.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- C. R. Hoyle, M. Boy, N. M. Donahue, J. L. Fry, M. Glasius, A. Guenther, A. G. Hallar, K. Huff Hartz, M. D. Petters, T. Petäjä, T. Rosenoern and A. P. Sullivan, Atmos. Chem. Phys., 2011, 11, 321–343 CrossRef CAS.
- S. Fuzzi, M. O. Andreae, B. J. Huebert, M. Kulmala, T. C. Bond, M. Boy, S. J. Doherty, A. Guenther, M. Kanakidou, K. Kawamura, V. M. Kerminen, U. Lohmann, L. M. Russell and U. Pöschl, Atmos. Chem. Phys., 2006, 6, 2017–2038 CrossRef CAS.
- S. N. Pandis, S. E. Paulson, J. H. Seinfeld and R. C. Flagan, Atmos. Environ., Part A, 1991, 25, 997–1008 CrossRef.
- J. H. Kroll, N. L. Ng, S. M. Murphy, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 2006, 40, 1869–1877 CrossRef CAS PubMed.
- A. A. Presto, K. E. Huff Hartz and N. M. Donahue, Environ. Sci. Technol., 2005, 39, 7046–7054 CrossRef CAS PubMed.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, A. Kürten, J. M. St. Clair, J. H. Seinfeld and P. O. Wennberg, Science, 2009, 325, 730–733 CrossRef CAS PubMed.
- M. Kulmala and V.-M. Kerminen, Atmos. Res., 2008, 90, 132–150 CrossRef CAS.
- J. D. Surratt, Y. Gómez-González, A. W. H. Chan, R. Vermeylen, M. Shahgholi, T. E. Kleindienst, E. O. Edney, J. H. Offenberg, M. Lewandowski, M. Jaoui, W. Maenhaut, M. Claeys, R. C. Flagan and J. H. Seinfeld, J. Phys. Chem. A, 2008, 112, 8345–8378 CrossRef CAS PubMed.
- DGT, 2013, https://sedeapl.dgt.gob.es/IEST2/menu.do?path=/vehiculos/parque/&file=inebase&type=pcaxis&L=0&js=1.
- X. Querol, A. Alastuey, M. Pandolfi, C. Reche, N. Pérez, M. C. Minguillón, T. Moreno, M. Viana, M. Escudero, A. Orio, M. Pallarés and F. Reina, Sci. Total Environ., 2014, 490, 957–969 CrossRef CAS PubMed.
- N. Pérez, J. Pey, M. Cusack, C. Reche, X. Querol, A. Alastuey and M. Viana, Aerosol Sci. Technol., 2010, 44, 487–499 CrossRef.
- I. Salma, X. Chi and W. Maenhaut, Atmos. Environ., 2004, 38, 27–36 CrossRef CAS.
- M. C. Minguillón, N. Perron, X. Querol, S. Szidat, S. M. Fahrni, A. Alastuey, J. L. Jimenez, C. Mohr, A. M. Ortega, D. A. Day, V. A. Lanz, L. Wacker, C. Reche, M. Cusack, F. Amato, G. Kiss, A. Hoffer, S. Decesari, F. Moretti, R. Hillamo, K. Teinilä, R. Seco, J. Peñuelas, A. Metzger, S. Schallhart, M. Müller, A. Hansel, J. F. Burkhart, U. Baltensperger and A. S. H. Prévôt, Atmos. Chem. Phys., 2011, 11, 12067–12084 Search PubMed.
- M. Pandolfi, X. Querol, A. Alastuey, J. L. Jimenez, O. Jorba, D. Day, A. Ortega, M. J. Cubison, A. Comerõn, M. Sicard, C. Mohr, A. S. H. Prévôt, M. C. Minguillõn, J. Pey, J. M. Baldasano, J. F. Burkhart, R. Seco, J. Peñuelas, B. L. van Drooge, B. Artiñano, C. Di Marco, E. Nemitz, S. Schallhart, A. Metzger, A. Hansel, J. Lorente, S. Ng, J. Jayne and S. Szidat, J. Geophys. Res.: Atmos., 2014, 119, 4978–5010 CAS.
- B. L. van Drooge, M. Cusack, C. Reche, C. Mohr, A. Alastuey, X. Querol, A. S. H. Prevot, D. A. Day, J. L. Jimenez and J. O. Grimalt, Atmos. Environ., 2012, 61, 482–489 CrossRef CAS.
- S. Szidat, T. M. Jenk, H. A. Synal, M. Kalberer, L. Wacker, I. Hajdas, A. Kasper-Giebl and U. Baltensperger, J. Geophys. Res., 2006, 11, D07206 Search PubMed.
- I. El Haddad, B. D'Anna, B. Temime-Roussel, M. Nicolas, A. Boreave, O. Favez, D. Voisin, J. Sciare, C. George, J. L. Jaffrezo, H. Wortham and N. Marchand, Atmos. Chem. Phys., 2013, 13, 7875–7894 CAS.
- M. R. Heal, P. Naysmith, G. T. Cook, S. Xu, T. R. Duran and R. M. Harrison, Atmos. Environ., 2011, 45, 2341–2348 CrossRef CAS.
- F. Amato, M. Pandolfi, A. Escrig, X. Querol, A. Alastuey, J. Pey, N. Perez and P. K. Hopke, Atmos. Environ., 2009, 43, 2770–2780 CrossRef CAS.
- C. Pérez, M. Sicard, O. Jorba, A. Comerón and J. M. Baldasano, Atmos. Environ., 2004, 38, 3983–4000 CrossRef.
- A. Jordan, S. Haidacher, G. Hanel, E. Hartungen, L. Märk, H. Seehauser, R. Schottkowsky, P. Sulzer and T. D. Märk, Int. J. Mass Spectrom., 2009, 286, 122–128 CrossRef CAS.
- M. Graus, M. Müller and A. Hansel, J. Am. Soc. Mass Spectrom., 2010, 21, 1037–1044 CrossRef CAS PubMed.
- W. Lindinger, A. Hansel and A. Jordan, Int. J. Mass Spectrom. Ion Processes, 1998, 173, 191–241 CrossRef CAS.
- L. Cappellin, T. Karl, M. Probst, O. Ismailova, P. M. Winkler, C. Soukoulis, E. Aprea, T. D. Märk, F. Gasperi and F. Biasioli, Environ. Sci. Technol., 2012, 46, 2283–2290 CrossRef CAS PubMed.
- A. Petzold, J. A. Ogren, M. Fiebig, P. Laj, S. M. Li, U. Baltensperger, T. Holzer-Popp, S. Kinne, G. Pappalardo, N. Sugimoto, C. Wehrli, A. Wiedensohler and X. Y. Zhang, Atmos. Chem. Phys., 2013, 13, 8365–8379 CrossRef CAS.
- N. L. Ng, S. C. Herndon, A. Trimborn, M. R. Canagaratna, P. L. Croteau, T. B. Onasch, D. Sueper, D. R. Worsnop, Q. Zhang, Y. L. Sun and J. T. Jayne, Aerosol Sci. Technol., 2011, 45, 770–784 Search PubMed.
- N. L. Ng, M. R. Canagaratna, J. L. Jimenez, Q. Zhang, I. M. Ulbrich and D. R. Worsnop, Environ. Sci. Technol., 2011, 45, 910–916 CrossRef CAS PubMed.
- A. M. Middlebrook, R. Bahreini, J. L. Jimenez and M. R. Canagaratna, Aerosol Sci. Technol., 2012, 46, 258–271 CrossRef CAS.
- X. Querol, A. Alastuey, S. Rodriguez, F. Plana, C. R. Ruiz, N. Cots, G. Massagué and O. Puig, Atmos. Environ., 2001, 35, 6407–6419 CrossRef CAS.
- F. Cavalli, M. Viana, K. E. Yttri, J. Genberg and J. P. Putaud, Atmos. Meas. Tech., 2010, 3, 79–89 CAS.
- Y. L. Zhang, N. Perron, V. G. Ciobanu, P. Zotter, M. C. Minguillón, L. Wacker, A. S. H. Prévôt, U. Baltensperger and S. Szidat, Atmos. Chem. Phys., 2012, 12, 10841–10856 CAS.
- S. Szidat, G. A. Salazar, E. Vogel, M. Battaglia, L. Wacker, H. A. Synal and A. Türler, Radiocarbon, 2014, 56, 561–566 CrossRef CAS.
- P. Paatero and U. Tapper, Environmetrics, 1994, 5, 111–126 CrossRef.
- P. Paatero, Journal of Computational and Graphical Statistics, 1999, 8, 854–888 Search PubMed.
- F. Canonaco, M. Crippa, J. G. Slowik, U. Baltensperger and A. S. H. Prévôt, Atmos. Meas. Tech., 2013, 6, 3649–3661 Search PubMed.
- P. Zotter, I. El-Haddad, Y. Zhang, P. L. Hayes, X. Zhang, Y. H. Lin, L. Wacker, J. Schnelle-Kreis, G. Abbaszade, R. Zimmermann, J. D. Surratt, R. Weber, J. L. Jimenez, S. Szidat, U. Baltensperger and A. S. H. Prévôt, J. Geophys. Res.: Atmos., 2014, 119, 6818–6835 CAS.
- J. Mohn, S. Szidat, J. Fellner, H. Rechberger, R. Quartier, B. Buchmann and L. Emmenegger, Bioresour. Technol., 2008, 99, 6471–6479 CrossRef CAS PubMed.
- I. Levin, T. Naegler, B. Kromer, M. Diehl, R. J. Francey, A. J. Gomez-Pelaez, L. P. Steele, D. Wagenbach, R. Weller and D. E. Worthy, Tellus, Ser. B, 2010, 62, 26–46 CrossRef.
- L. M. Hildemann, D. B. Klinedinst, G. A. Klouda, L. A. Currie and G. R. Cass, Environ. Sci. Technol., 1994, 28, 1565–1576 CrossRef CAS PubMed.
- J. Pey, S. Rodríguez, X. Querol, A. Alastuey, T. Moreno, J. P. Putaud and R. van Dingenen, Atmos. Environ., 2008, 42, 9052–9062 CrossRef CAS.
- M. Brines, M. Dall'Osto, D. C. S. Beddows, R. M. Harrison and X. Querol, Atmos. Chem. Phys., 2014, 14, 2973–2986 CAS.
- M. C. Minguillón, M. Brines, N. Pérez, C. Reche, M. Pandolfi, A. S. Fonseca, F. Amato, A. Alastuey, A. Lyasota, B. Codina, H. K. Lee, H. R. Eun, K. H. Ahn and X. Querol, Atmos. Res., 2015, 164–165, 118–130 CrossRef.
- R. Seco, J. Peñuelas, I. Filella, J. Llusia, S. Schallhart, A. Metzger, M. Müller and A. Hansel, Atmos. Chem. Phys., 2013, 13, 4291–4306 Search PubMed.
- M. Navazo, N. Durana, L. Alonso, M. C. Gómez, J. A. García, J. L. Ilardia, G. Gangoiti and J. Iza, Environ. Monit. Assess., 2008, 136, 53–68 CrossRef CAS PubMed.
- J. Sternbeck, A. Sjödin and K. Andréasson, Atmos. Environ., 2002, 36, 4735–4744 CrossRef CAS.
- C. Mohr, P. F. DeCarlo, M. F. Heringa, R. Chirico, J. G. Slowik, R. Richter, C. Reche, A. Alastuey, X. Querol, R. Seco, J. Peñuelas, J. L. Jiménez, M. Crippa, R. Zimmermann, U. Baltensperger and A. S. H. Prévôt, Atmos. Chem. Phys., 2012, 12, 1649–1665 CAS.
- M. J. Cubison, A. M. Ortega, P. L. Hayes, D. K. Farmer, D. Day, M. J. Lechner, W. H. Brune, E. Apel, G. S. Diskin, J. A. Fisher, H. E. Fuelberg, A. Hecobian, D. J. Knapp, T. Mikoviny, D. Riemer, G. W. Sachse, W. Sessions, R. J. Weber, A. J. Weinheimer, A. Wisthaler and J. L. Jimenez, Atmos. Chem. Phys., 2011, 11, 12049–12064 Search PubMed.
- S. H. Budisulistiorini, M. R. Canagaratna, P. L. Croteau, W. J. Marth, K. Baumann, E. S. Edgerton, S. L. Shaw, E. M. Knipping, D. R. Worsnop, J. T. Jayne, A. Gold and J. D. Surratt, Environ. Sci. Technol., 2013, 47, 5686–5694 CrossRef CAS PubMed.
- H. L. DeWitt, S. Hellebust, B. Temime-Roussel, S. Ravier, L. Polo, V. Jacob, C. Buisson, A. Charron, M. André, A. Pasquier, J. L. Besombes, J. L. Jaffrezo, H. Wortham and N. Marchand, Atmos. Chem. Phys., 2015, 15, 4373–4387 CAS.
- J. L. Fry, D. C. Draper, K. C. Barsanti, J. N. Smith, J. Ortega, P. M. Winkler, M. J. Lawler, S. S. Brown, P. M. Edwards, R. C. Cohen and L. Lee, Environ. Sci. Technol., 2014, 48, 11944–11953 CrossRef CAS PubMed.
- A. I. Darer, N. C. Cole-Filipiak, A. E. O'Connor and M. J. Elrod, Environ. Sci. Technol., 2011, 45, 1895–1902 CrossRef CAS PubMed.
- V. A. Lanz, M. R. Alfarra, U. Baltensperger, B. Buchmann, C. Hueglin, S. Szidat, M. N. Wehrli, L. Wacker, S. Weimer, A. Caseiro, H. Puxbaum and A. S. H. Prevot, Environ. Sci. Technol., 2008, 42, 214–220 CrossRef CAS PubMed.
- L. Xu, H. Guo, C. M. Boyd, M. Klein, A. Bougiatioti, K. M. Cerully, J. R. Hite, G. Isaacman-vanWertz, N. M. Kreisberg, C. Knote, K. Olson, A. Koss, A. H. Goldstein, S. V. Hering, J. de Gouw, K. Baumann, S. H. Lee, A. Nenes, R. J. Weber and N. L. Ng, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 37–42 CrossRef CAS PubMed.
- I. El Haddad, N. Marchand, H. Wortham, C. Piot, J. L. Besombes, J. Cozic, C. Chauvel, A. Armengaud, D. Robin and J. L. Jaffrezo, Atmos. Chem. Phys., 2011, 11, 2039–2058 CAS.
- T. E. Kleindienst, M. Jaoui, M. Lewandowski, J. H. Offenberg and K. S. Docherty, Atmos. Chem. Phys., 2012, 12, 8711–8726 CrossRef CAS.
- M. Alier, B. L. van Drooge, M. Dall'Osto, X. Querol, J. O. Grimalt and R. Tauler, Atmos. Chem. Phys., 2013, 13, 10353–10371 CrossRef CAS.
- M. Claeys, R. Szmigielski, I. Kourtchev, P. van der Veken, R. Vermeylen, W. Maenhaut, M. Jaoui, T. E. Kleindienst, M. Lewandowski, J. H. Offenberg and E. O. Edney, Environ. Sci. Technol., 2007, 41, 1628–1634 CrossRef CAS PubMed.
- R. Szmigielski, J. D. Surratt, Y. Gómez-González, P. van der Veken, I. Kourtchev, R. Vermeylen, F. Blockhuys, M. Jaoui, T. E. Kleindienst, M. Lewandowski, J. H. Offenberg, E. O. Edney, J. H. Seinfeld, W. Maenhaut and M. Claeys, Geophys. Res. Lett., 2007, 34, L24811 CrossRef.
- A. G. Carlton, C. Wiedinmyer and J. H. Kroll, Atmos. Chem. Phys., 2009, 9, 4987–5005 CrossRef CAS.
- M. Claeys, B. Graham, G. Vas, W. Wang, R. Vermeylen, V. Pashynska, J. Cafmeyer, P. Guyon, M. O. Andreae, P. Artaxo and W. Maenhaut, Science, 2004, 303, 1173–1176 CrossRef CAS PubMed.
- J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6640–6645 CrossRef CAS PubMed.
- Q. T. Nguyen, M. K. Christensen, F. Cozzi, A. Zare, A. M. K. Hansen, K. Kristensen, T. E. Tulinius, H. H. Madsen, J. H. Christensen, J. Brandt, A. Massling, J. K. Nøjgaard and M. Glasius, Atmos. Chem. Phys., 2014, 14, 8961–8981 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Sections 1 to 6, with 12 figures. See DOI: 10.1039/c5fd00182j |
| This journal is © The Royal Society of Chemistry 2016 |