
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt(III) catalyzed C-8 selective C–H and C–O coupling of quinoline N-oxide with internal alkynes via C–H activation and oxygen atom transfer†
Nagaraju
Barsu
a,
Malay
Sen
a,
J. Richard
Premkumar
b and
Basker
Sundararaju
*a
aFine Chemical Laboratory, Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur, UP, India. E-mail: basker@iitk.ac.in
bCenter for Molecular Modelling, Indian Institute of Chemical Technology, Hyderabad – 500 067, India
First published on 23rd November 2015
Abstract
An efficient, scalable, atom-economical, regio-selective air stable Cp*Co(III) catalyzed C–H and C–O coupling via a C–H activation/oxygen atom transfer reaction of quinoline N-oxide and an internal alkyne is reported. Such a catalytic transformation is witnessed for the first time with a cobalt catalyst and using N-oxide as a traceless directing group, in contrast to the existing literature. The developed synthetic methodology is straightforward and possesses various functional group tolerances, including heterocycles.
Quinolines are versatile heterocycles that are widely used as ‘parental’ compounds to integrate molecules with medicinal benefits, natural products and agrochemicals with applications in materials chemistry.1–3 Site selective functionalizations of quinoline at various positions are important, but the majority of the literature deals with C–2(H) bond functionalization.4 The elegant work by Chang5 and Sawamura6 showed that the nitrogen in quinoline can be used as a directing group for C-8 selective functionalization. Chang and others further developed a system based on quinoline-N-oxide for C-8 selective functionalization, where oxygen is used as a traceless directing group for various C–C and C–X bond formations using group VII noble metal catalysts such as Rh(III) and Ir(III).7 Since the first report by Fagnou8k, and later development by others, of the utilization of N–O/N–N as a directing group as well as an internal oxidant for C–H bond functionalization, examples have been reported based on this redox-neutral concept as it does not require any external oxidant and produces only environmentally friendly side products (ROH, H2O, etc.).8 Very recently, Li9 and Chang10 have independently developed a Rh(III) catalytic system to advance this redox-neutral process for C-8 selective functionalization of quinoline via sequential C–H activation/oxygen atom transfer (OAT) with internal alkynes. Though many new catalytic C–H bond functionalizations have been exemplified in the last decade using relatively expensive noble metal catalysts,11 the challenge still remains to effect the same or new transformations by utilizing relatively cheap, abundant first row transition metal catalysts.12 The continuing theme of our research efforts to develop C–H bond functionalizations using first row transition metal catalysts13 has led us to investigate the novel reaction of the C(8)–H bond functionalization of quinoline. Herein, we report the Cp*Co(III)14 catalyzed C(8)–H bond functionalization of quinoline with internal alkynes, in the absence of additives and using quinoline-N-oxide as a traceless directing group.15 This elegant methodology has many advantages; as it does not require any directing groups, external oxidants or deprotection of N-oxide, it provides 100% atom efficiency (Scheme 1). We have also shown that the Co(III) catalyst efficiency and reactivity for the C(8)–H bond functionalization of quinoline are better than those for Ir(III) and Rh(III) catalysts.9,10
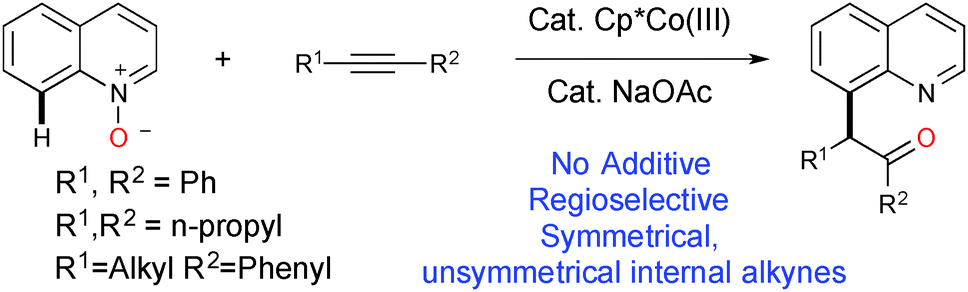 | ||
| Scheme 1 Co(III) catalyzed C–H activation/oxygen atom transfer process. | ||
A summary of the results for the optimization is presented in Table S1 (ESI†).16 We began our investigation using quinoline-N-oxide 1a, as a model substrate, and diphenylacetylene 2a in the presence of Cp*Co(CO)I2 (10 mol%) and NaOAc (20 mol%) in dioxane at 80 °C for 24 h; this did not give the expected product 3a. A solvent dependency study for the same reaction was carried out (Table S1, entries 2–8, ESI†) and we found that the reaction proceeded more efficiently in TFE (trifluoroethanol) compared to other solvents, with an isolated yield for 3a of 80% (Table S1, entry 2, ESI†). An improved yield for 3a was obtained when we employed NaOPiv (Table S1, entry 14, ESI†) compared to other carboxylates (Table S1, entries 9–14, ESI†). There was no significant improvement in the formation of 3a from changing other reaction parameters such as temperature, catalyst loading or ionization with silver salt (Table S1, entries 15–17, ESI†). Further, control experiments revealed that both the [Co] catalyst and NaOPiv are mandatory for a smooth reaction with a high yield of 3a (Table S1, entries 18 and 19, ESI†). The reactions were ineffective when they were performed at 50 °C or ambient temperature (Table S1, entries 20–22, ESI†). The transformation of 1a with alkyne 2a does not proceed forward when using other cobalt sources of Co(III) or Co(II) such as Co(acac)3 or CoCl2 (Table S1, entries 23 and 24, ESI†). Replacing the Co(III) catalyst (in our optimized conditions) with Rh(III)/Ir(III)/Ru(II) resulted in a poor yield or no reaction occurring (Table S1, entries 26 and 27, ESI†). However, a reported catalytic system based on Rh(III) gave a good yield under different conditions;9,10 but the reaction was inefficient when aliphatic alkynes were employed as the substrate under the reported Rh(III) catalytic conditions.
Our further screening revealed that a high yield of 3a was obtained when the alkyne 2a was used as the limiting factor (Table S1, entries 28 and 29, ESI†).
With the best optimized conditions in hand, we further extended the scope of the reaction by exercising various substituted quinoline-N-oxides with 2a to access diverse functionalized quinoline derivatives (Table 1). For example, substituent groups such as bromo, aryl, heteroaryl, alkenyl, alkynyl or unsaturated esters on the C(3)-position of the heteroarene are amenable and produce a good yield (3b–i). Compound 3c was further confirmed using X-ray crystallography.17 Sensitive functional groups, such as ketones and nitro groups, remain intact even after the C–H activation and OAT process (3j–k). Mono- and disubstituted quinoline-N-oxides, at various positions, gave good yields (3l–q). Substituents at the C-7 position suffer from only respectable yields, which is likely due to steric and electronic reasons (3r). The reaction is not limited only to quinoline and can be extended to other heterocycles, in good yields (3s–t).
| a Reaction conditions: all reactions were performed under an Ar atm using 1 (0.36 mmol), 2a (0.3 mmol), [Cp*Co(CO)I2] (0.03 mmol) and NaOPiv (0.06 mmol) in CF3CH2OH (1 mL) at 100 °C (oil bath temperature) for 24 h. |
|---|
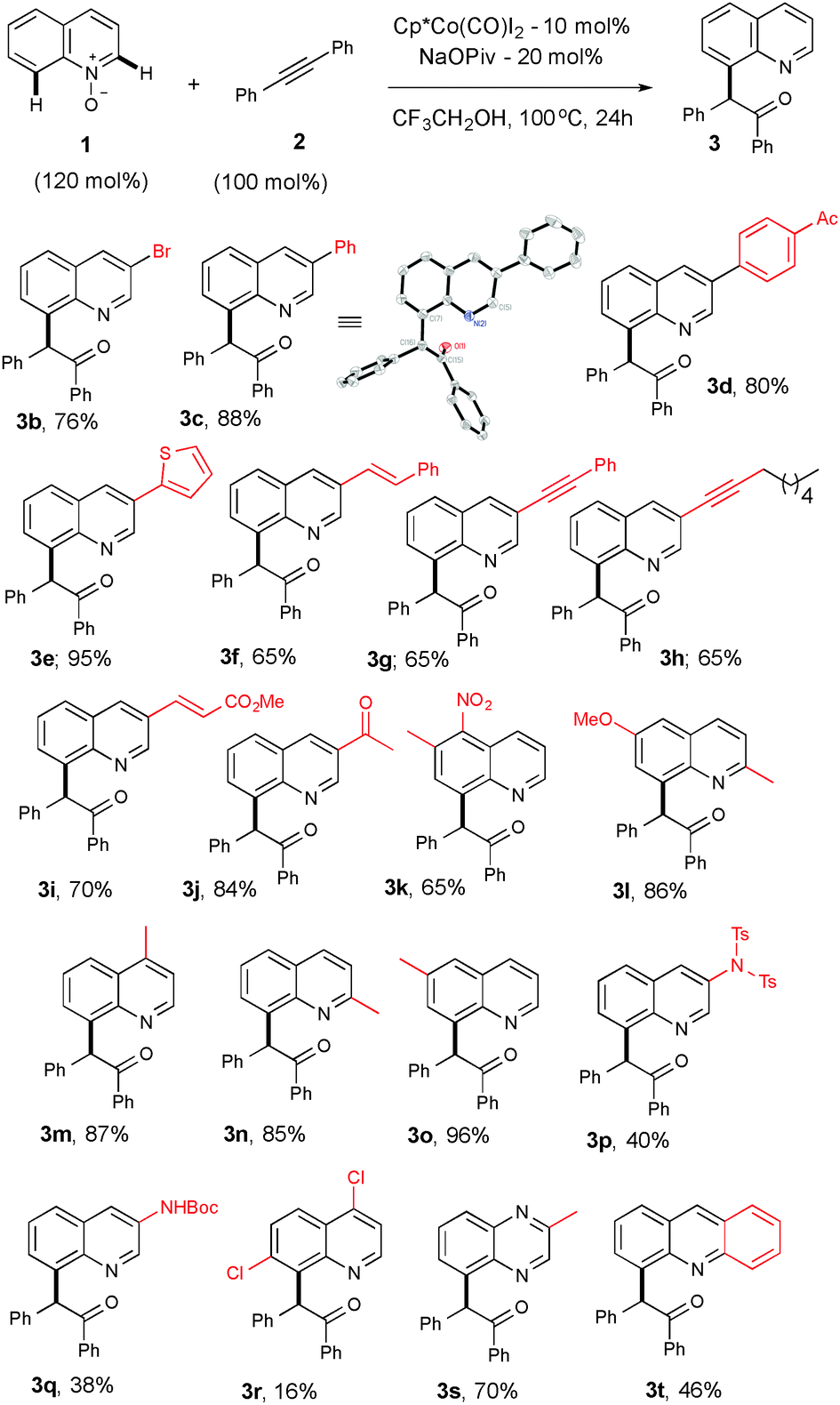
|
A gram-scale reaction was performed with the objective of evaluating the feasibility of this catalytic process for 1a (1.068 g, 6 mmol) and 2a (0.957 g, 6.5 mmol) using 5 mol% of [Cp*Co(CO)I2] and 15 mol% of NaOPiv in trifluoroethanol at 100 °C for 36 h, which produced 3a in 94% (1.839 g) yield (Scheme S1, ESI†). To further demonstrate the generality and practicality of this sequential C–H activation/OAT reaction, we examined an array of alkynes 2 (Table 2). A wide range of symmetrical alkynes could readily undergo the reaction with quinoline-N-oxide 1a with a good to high reactivity and selectivity (4a–d). When a symmetrical aliphatic alkyne such as 4-octyne was employed under the optimized reaction conditions, the reaction gave a 70% yield of 4e with Co(III), whereas the Ir(III) catalyst failed to promote such a reaction and Rh(III) gave a 15% yield under our optimized conditions. On the other hand, the reported Rh(III) catalytic system did not provide any product formation with 4-octyne (4e, Table 2). Thus, this emphasizes the unique reactivity of Co(III) over the other group 9 metals and its importance and applicability with a wide variety of internal alkynes. This change in the order of reactivity of Co(III) vs. Rh(III) for 4-octyne can be explained through charge transfer stabilization energy (Table S3, ESI†).16
| a Reaction conditions: all reactions were performed under an Ar atm using 1 (0.3 mmol), 2a (0.45 mmol), [Cp*Co(CO)I2] (0.03 mmol) and NaOPiv (0.06 mmol) in CF3CH2OH (1 mL) at 100 °C (oil bath temperature) for 24 h. b No reaction was obtained with the reported Rh(III) catalytic system as in ref. 9 and 10. |
|---|
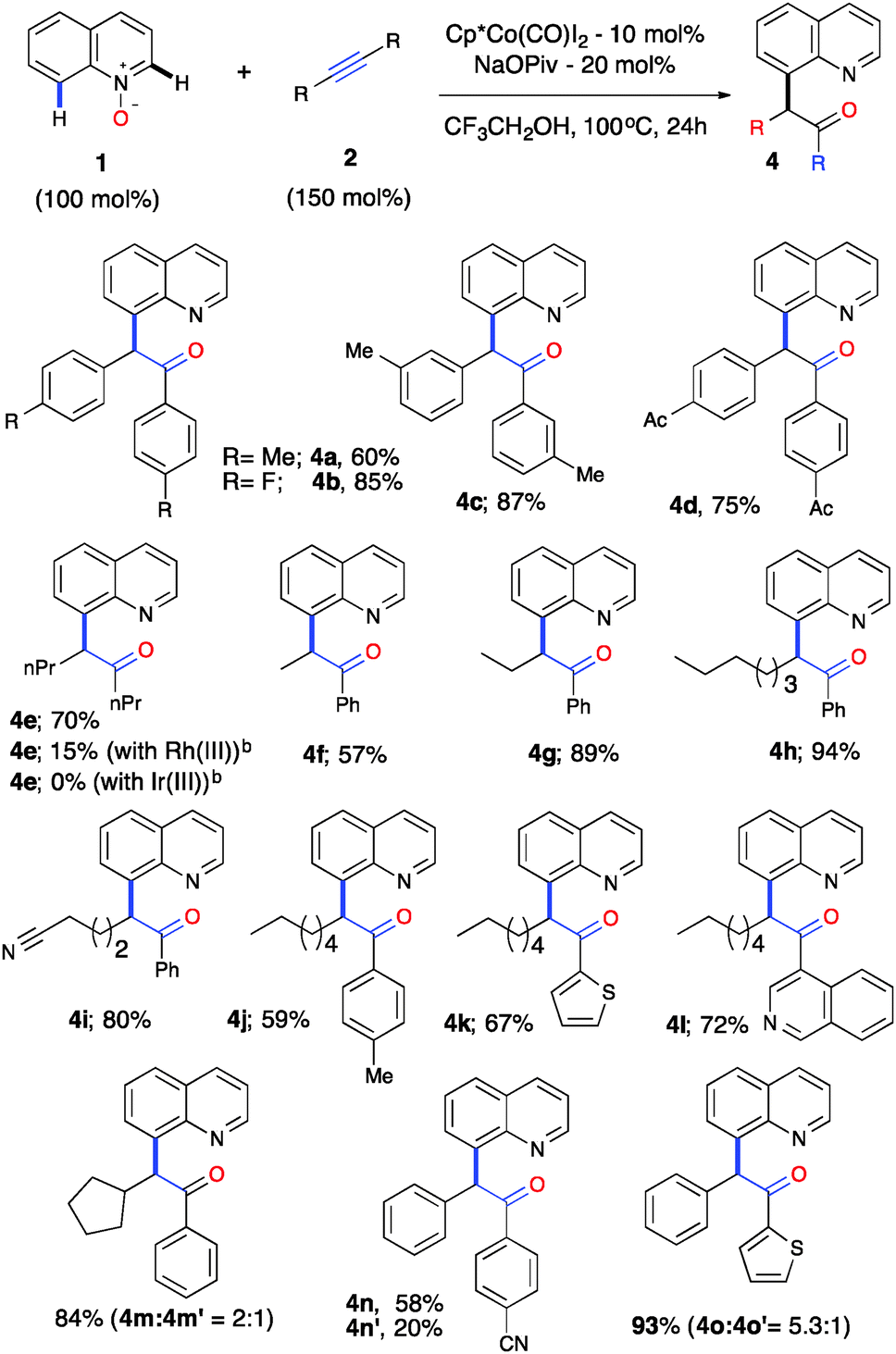
|
To further evaluate the regioselectivity, a number of unsymmetrical internal alkynes were investigated from the arsenal (Table 2, 4f–o). To our delight, all of the unsymmetrical internal alkynes reacted with 1a smoothly to provide a single regioisomer with excellent control over regioselectivity in good yield (4f–m). Electronically biased unsymmetrical biarylacetylene gave a mixture of regioselective products, including heteroarylacetylene, in good yields (4n–o). Further, biarylacetophenone 3a was put forth for cascade C–H bond functionalization using the reported procedure18 and gave a tetracyclic compound as a single diastereomer in moderate yield (Scheme S2, ESI†). To understand the high reactivity and selectivity of the C–H activation/OAT process, we conducted a series of preliminary experiments in order to understand the mechanism. Deuterium-labelled experiments were carried out under the optimized reaction conditions to examine the incorporation of deuterium on the C(8)–H position of quinoline N-oxide (Scheme S3a–c, ESI†). It was revealed that no deuterium was detected on the C(8) position, with or without 2a, in the presence of Co(III), this suggests that the C–H bond cleavage could be irreversible in nature. To confirm this further, competitive experiments were conducted with electronically variant alkynes and quinoline N-oxides, which resulted in no selectivity (Scheme S3d and e, ESI†). C(8)-alkenylquinoline-N-oxide 5 was exposed to our reaction conditions which resulted in no formation of the oxygen transfer product, suggesting that the C–H activation and OAT are cascade processes in nature, ruling out the possibility of a C(8)-alkenyl intermediate (Scheme S3f, ESI†).
Based on our preliminary studies and the relevant reports on Rh(III),19 a plausible mechanism has been drawn (Scheme 2). Cationic complex I undergoes an irreversible cyclometallation followed by alkyne coordination and migratory insertion between the Co and C atoms, leading to the alkenyl cobaltacyclic intermediate V. It further undergoes reductive elimination to the Co(I) intermediate VI, which will further proceed via oxidative addition between the N and O atoms, leading to intermediate VII. This then undergoes protodemetallation leading to C-8 functionalized quinoline 3/4 and the regeneration of the active catalyst. The proposed mechanism is further supported by DFT calculations performed using the Gaussian 09 program and the DFT method B3LYP.16
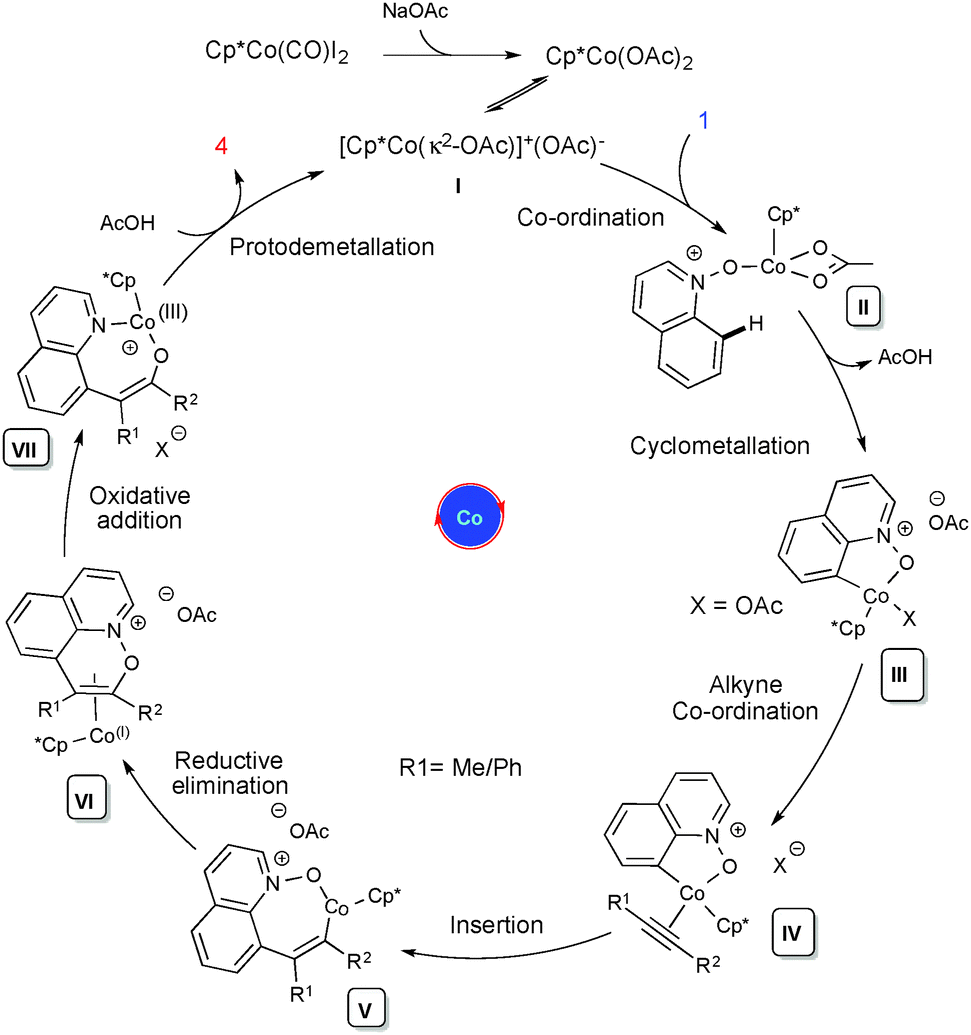 | ||
| Scheme 2 Proposed mechanism. | ||
In conclusion, we have developed a cobalt catalyzed C–H and C–O coupling of quinoline-N-oxide with internal alkynes via a C–H activation/OAT process, which is reported for the first time. We have shown that various substituted quinoline-N-oxide structures can be coupled with symmetrical (diaryl-, dialkylacetylene) and unsymmetrical internal alkynes (alkylarylacetylene) with excellent control over the regioselectivity. DFT calculations showed that regioselective product formation could be achieved with unsymmetrical alkynes due to better conjugation with the alkenyl intermediate and the phenyl group.16 Preliminary studies have revealed that the reaction likely undergoes irreversible cyclometallation. We have also shown that the Co(III) catalyst displayed a better reactivity with aliphatic alkynes compared to its congeners from group 9, metals such as Rh(III) and Ir(III).
Financial support provided by the Indian Institute of Technology Kanpur through an initiation grant (IITK/CHM/20130187) and a DAE-BRNS (BRNS/CHM/2014110) for Young scientist research fund award to BS is gratefully acknowledged. NB and MS acknowledge CSIR for their fellowship. BS wishes to thank Johnson Matthey for their generous support of metal precursors.
Notes and references
- (a) T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles, Wiley-VCH, Weinheim, 2nd edn, 2003, vol. 6, pp. 316–336 CrossRef; (b) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166–187 RSC.
- C. H. McAteer, M. Balasubramanian and R. Murugan, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katrtzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 7, pp. 309–336 Search PubMed.
- C. H. Chen and J. Shi, Coord. Chem. Rev., 1998, 171, 161–174 CrossRef CAS.
- For selected reviews on C–H bond functionalization of quinoline, see: (a) T. Iwai and M. Sawamura, ACS Catal., 2015, 5, 5031–5040 CrossRef CAS; (b) R.-J. Tang, L. Kang and L. Yang, Adv. Synth. Catal., 2015, 357, 2055–2060 CrossRef CAS; (c) D. E. Stephens and O. V. Larionov, Tetrahedron, 2015, 71, 8683–8716 CrossRef CAS PubMed.
- J. Kwak, M. Kim and S. Chang, J. Am. Chem. Soc., 2011, 133, 3780–3783 CrossRef CAS PubMed.
- S. Konishi, S. Kawamorita, T. Iwai, P. G. Steel, T. B. Marder and M. Sawamura, Chem. – Asian J., 2014, 9, 434–438 CrossRef CAS.
- (a) D. E. Stephens, J. Lakey-Beitia, G. Chavez, C. Ilie, H. D. Arman and O. V. Larionov, Chem. Commun., 2015, 51, 9507–9510 RSC; (b) D. E. Stephens, J. Lakey-Beitia, A. C. Atesin, T. A. Ateşin, G. Chavez, H. D. Arman and O. V. Larionov, ACS Catal., 2015, 5, 167–175 CrossRef CAS PubMed; (c) K. Shin, S.-W. Park and S. Chang, J. Am. Chem. Soc., 2015, 137, 8584–8592 CrossRef CAS PubMed; (d) T. Shibata and Y. Matsuo, Adv. Synth. Catal., 2014, 356, 1516–1520 CrossRef CAS; (e) J. Jeong, P. Patel, H. Hwang and S. Chang, Org. Lett., 2014, 16, 4598–4601 CrossRef CAS PubMed; (f) H. Hwang, J. Kim, J. Jeong and S. Chang, J. Am. Chem. Soc., 2014, 136, 10770–10776 CrossRef CAS PubMed.
- For selected examples of external oxidant free redox-neutral C–H bond functionalization, see: (a) C. V. Suneel Kumar and C. V. Ramana, Org. Lett., 2015, 17, 2870–2873 CrossRef CAS PubMed; (b) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007–1020 CrossRef CAS PubMed and references therein; ; (c) Z. Fan, S. Song, W. Li, K. Geng, Y. Xu, Z.-H. Miao and A. Zhang, Org. Lett., 2015, 17, 310–313 CrossRef CAS PubMed; (d) R. B. Dateer and S. Chang, J. Am. Chem. Soc., 2015, 137, 4908–4911 CrossRef CAS PubMed; (e) F. Yang and L. Ackermann, J. Org. Chem., 2014, 79, 12070–12082 CrossRef CAS PubMed; (f) B. Liu, C. Song, C. Sun, S. Zhou and J. Zhu, J. Am. Chem. Soc., 2013, 135, 16625–16631 CrossRef CAS PubMed; (g) H. Wang, C. Grohmann, C. Nimphius and F. Glorius, J. Am. Chem. Soc., 2012, 134, 19592–19595 CrossRef CAS PubMed; (h) R. K. Chinnagolla, S. Pimparkar and M. Jeganmohan, Org. Lett., 2012, 14, 3032–3035 CrossRef CAS PubMed; (i) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350–2353 CrossRef CAS PubMed; (j) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449–6457 CrossRef CAS PubMed; (k) L. Ackermann and S. Fenner, Org. Lett., 2011, 13, 6548–6551 CrossRef CAS PubMed; (l) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908–6909 CrossRef CAS PubMed.
- X. Zhang, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2014, 53, 10794–10798 CrossRef CAS PubMed.
- U. Sharma, Y. Park and S. Chang, J. Org. Chem., 2014, 79, 9899–9906 CrossRef CAS PubMed.
- Selected recent reviews on C–H bond functionalization: (a) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (b) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308 CrossRef CAS PubMed; (c) L. Ackermann, Org. Process Res. Dev., 2015, 19, 260–269 CrossRef CAS; (d) T. Mesganaw and J. A. Ellman, Org. Process Res. Dev., 2014, 18, 1097–1104 CrossRef CAS PubMed; (e) S. A. Girard, T. Knauber and C. J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (f) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (g) K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CrossRef CAS PubMed; (h) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (i) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (j) K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (k) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (l) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (m) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (n) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (o) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (p) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (q) F. Kakiuchi and T. Kochi, Synthesis, 2008, 3013–3039 CrossRef CAS.
- For selected reviews on C–H bond functionalization using first row transition metal catalysts, see: (a) N. Yoshikai, ChemCatChem, 2015, 7, 732–734 CrossRef CAS; (b) Y. Liang, Y.-F. Liang and N. Jiao, Org. Chem. Front., 2015, 2, 403–415 RSC; (c) T. Hyster, Catal. Lett., 2015, 145, 458–467 CrossRef CAS; (d) X. H. Cai and B. Xie, ARKIVOC, 2015, 184 CAS; (e) I. Bauer and H. J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (f) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed; (g) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS PubMed; (h) L. Ackermann, J. Org. Chem., 2014, 79, 8948–8954 CrossRef CAS PubMed; (i) J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19–30 CrossRef CAS; (j) R. T. Gephart and T. H. Warren, Organometallics, 2012, 31, 7728 CrossRef CAS; (k) N. Yoshikai, Synlett, 2011, 1047–1051 CrossRef CAS; (l) Y. Nakao, Chem. Rec., 2011, 11, 242–251 CrossRef CAS PubMed; (m) E. Nakamura and N. Yoshikai, J. Org. Chem., 2010, 75, 6061–6067 CrossRef CAS PubMed; (n) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087–4109 CAS.
- (a) N. Barsu, D. Kalsi and B. Sundararaju, Chem. – Eur. J., 2015, 21, 9364–9368 CrossRef CAS PubMed; (b) M. Sen, D. Kalsi and B. Sundararaju, Chem. – Eur. J., 2015, 21, 15529–15533 CrossRef CAS PubMed; (c) D. Kalsi and B. Sundararaju, Org. Lett., 2015 DOI:10.1021/acs.orglett.5b03142.
- Selected examples of Cp*Co(III)-catalyzed C–H bond functionalization: (a) D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508–4511 CrossRef CAS PubMed; (b) J. R. Hummel and J. A. Ellman, J. Am. Chem. Soc., 2015, 137, 490–498 CrossRef CAS PubMed; (c) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944–9947 CrossRef CAS PubMed; (d) A. B. Pawar and S. Chang, Org. Lett., 2015, 17, 660–663 CrossRef CAS PubMed; (e) P. Patel and S. Chang, ACS Catal., 2015, 5, 853–858 CrossRef CAS; (f) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 8551–8554 CrossRef CAS PubMed; (g) J. R. Hummel and J. A. Ellman, Org. Lett., 2015, 17, 2400–2403 CrossRef CAS PubMed; (h) T. Gensch, S. Vásquez-Céspedes, D.-G. Yu and F. Glorius, Org. Lett., 2015, 17, 3714–3717 CrossRef CAS PubMed; (i) D.-G. Yu, T. Gensch, F. de Azambuja, S. Vásquez-Céspedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722–17725 CrossRef CAS PubMed; (j) B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Adv. Synth. Catal., 2014, 356, 1491–1495 CrossRef CAS; (k) T. M. Figg, S. Park, J. Park, S. Chang and D. G. Musaev, Organometallics, 2014, 33, 4076–4085 CrossRef CAS; (l) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207–2211 CrossRef CAS PubMed; (m) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Chem. – Eur. J., 2013, 19, 9142–9146 CrossRef CAS PubMed ; also see; (n) L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209–10212 CrossRef CAS PubMed.
- For examples of C-8 selective functionalization of quinoline using N-oxide as a traceless directing group, see: (a) D. E. Stephens, J. Lakey-Beitia, G. Chavez, C. Ilie, H. D. Arman and O. V. Larionov, Chem. Commun., 2015, 51, 9507–9510 RSC; (b) D. E. Stephens, J. Lakey-Beitia, A. C. Atesin, T. A. Ateşin, G. Chavez, H. D. Arman and O. V. Larionov, ACS Catal., 2015, 5, 167–175 CrossRef CAS PubMed; (c) K. Shin, S.-W. Park and S. Chang, J. Am. Chem. Soc., 2015, 137, 8584–8592 CrossRef CAS PubMed; (d) T. Shibata and Y. Matsuo, Adv. Synth. Catal., 2014, 356, 1516–1520 CrossRef CAS; (e) J. Jeong, P. Patel, H. Hwang and S. Chang, Org. Lett., 2014, 16, 4598–4601 CrossRef CAS PubMed; (f) H. Hwang, J. Kim, J. Jeong and S. Chang, J. Am. Chem. Soc., 2014, 136, 10770–10776 CrossRef CAS PubMed.
- See ESI† for more details.
- CCDC 1430321.
- V. P. Mehta, J.-A. García lópez and M. F. Greaney, Angew. Chem., Int. Ed., 2014, 53, 1529–1533 CrossRef CAS PubMed.
- Y. Li, S. Liu, Z. Qi, X. Qi, X. Li and Y. Lan, Chem. – Eur. J., 2015, 21, 10131–10137 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1430321. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc08736h |
| This journal is © The Royal Society of Chemistry 2016 |