
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zipper-like molecular packing of donor–acceptor conjugated co-oligomers based on perylenediimide†
Laure
Biniek
a,
Pierre-Olivier
Schwartz
b,
Elena
Zaborova
c,
Benoît
Heinrich
b,
Nicolas
Leclerc
c,
Stéphane
Méry
b and
Martin
Brinkmann
*a
aInstitut Charles Sadron, CNRS, Université de Strasbourg, 23 rue du Loess, BP 84047, 67034 Strasbourg Cedex2, France. E-mail: martin.brinkmann@ics-cnrs.unistra.fr
bInstitut de Physique et de Chimie des Matériaux de Strasbourg, Université de Strasbourg, CNRS UMR 7504, 23 rue du Loess, BP 43, 67034 Strasbourg Cedex2, France
cICPEES, Université de Strasbourg, CNRS UMR 7515, ECPM, 25 rue Becquerel, 67087 Strasbourg Cedex2, France
First published on 18th February 2015
Abstract
The molecular packing of a new class of perylene diimide-based acceptor–donor (A–D) co-oligomers has been investigated by combining electron diffraction and X-ray scattering methods for AD dyads and an ADA triad structure. The AD and ADA compounds form highly ordered lamellar mesophases with well-defined donor and acceptor domains. To determine the structure of the co-oligomers, highly oriented films with different orientations were prepared. Both flat-on and edge-on orientations of the lamellae were obtained by using two different alignment methods. High temperature rubbing leads to edge-on oriented lamellae with the long molecular axis of the co-oligomer oriented almost parallel to the rubbing direction. Instead, on oriented substrates of poly(tetrafluoroethylene) (PTFE), flat-on lying lamellae with the π-stacking direction oriented parallel to the PTFE chains are obtained. The structural data gathered by low dose selected area electron diffraction and high resolution transmission electron microscopy are used to establish a structural model of an AD dyad. Driven by the strong π-stacking of the PDI core, both AD and ADA co-oligomers form two similar self-assembled lamellar structures with an original zipper-like organization of the PDI blocks.
I. Introduction
In the last decade, composite materials associating electron-donor (D) and electron-accepting (A) π-conjugated units have gained an important place in plastic electronics.1–3 Research on these systems is mainly motivated by applications such as organic photovoltaics (OPV) or ambipolar charge transport. Of high interest is the possibility to create a bulk heterojunction between donor and acceptor molecules or polymers for organic photovoltaic applications.4,5 Several teams have shown that a well-controlled nanostructuring of donor and acceptor domains can be obtained by the design of self-assembling block copolymers6 or co-oligomers.7 Many of these systems are based on acceptor groups of fullerene C61 or C71.7d–g However, in the past decade, perylene diimide (PDI) has become an interesting alternative acceptor molecule to C61. Since absorption properties are essential for photovoltaic conversion, PDI has been more and more integrated into macromolecular and molecular systems.8 Many PDI-based architectures have been synthesized and interesting properties e.g. n-type transport9 and OPV activity were demonstrated.10,11 PDI is a conjugated macrocycle with remarkable electron accepting properties, chemical stability, dye properties and can moreover be functionalized in various positions (bay and imide), giving a handle on electronic and optical properties. Various groups have recently reported the design and electronic properties of donor–acceptor co-oligomers based on PDI rather than block copolymers. Geng et al. showed that PDI-based AD dyads form well defined lamellar mesophases with a perfect phase separation at a sub-10 nm length scale between the PDI acceptor and the donor block, as is required for an efficient bulk heterojunction in OPV.11Recently we reported the synthesis and physico-chemical properties of new co-oligomer architectures including ADA and DAD triads of interest for OPV applications.12 Contrary to AD and ADA co-oligomers that form lamellar mesophases with long range order, the DAD triads were found to be essentially amorphous. The absence of long range order of DAD triads was explained by the marked discrepancy between molecular areas of D and A segments. Since the molecular area of D is about twice the area of A, the packing of AD and ADA implies a double-layer organization with layers of overlapping PDI blocks (see Fig. 1). Such a stacking mode is not possible for DAD because of the presence of the sterically demanding alkyl side-chains on the donor blocks. Based on the available powder diffraction data, two different packing schemes were proposed (see Fig. 1) but could not be discriminated.
 | ||
| Fig. 1 Left: molecular structures of the AD and ADA co-oligomers used in this study. Right: schematic view of the two structural models proposed for the self-assembled lamellar structure of AD dyads.12 Donor blocks shown in dark blue are in an upper layer, whereas light-blue ones correspond to the layer beneath. | ||
Many of these self-assembling co-oligomers cannot be obtained as macroscopic single crystals suitable for classical X-ray structure determination. This is even more of a problem as the self-assembled systems form mesophases rather than crystalline phases. It is therefore necessary to resort to alternative approaches to construct realistic structural models of such co-oligomeric systems in order to understand the impact of packing of donor and acceptor moieties on electronic properties of the systems. The classical approach is to grow oriented fibers, that yield however only limited structural information.13 In this study, we use a different methodology that combines low dose electron diffraction, high resolution Transmission Electron Microscopy (HR-TEM) and X-ray diffraction data on highly oriented thin films. This approach was successful in the case of regioregular poly(3-hexylthiophene) and poly(9,9′-dioctyl-fluorene) thin films prepared by directional epitaxial crystallization.14–16 It is applied here to the AD0 dyad composed of a PDI acceptor block and a donor block made of thiophene, benzothiadiazole and fluorene units (see Fig. 1). The difficult single-crystal growth is circumvented by producing highly oriented films of the polymers or molecular materials that can potentially yield single-crystal like electron diffraction patterns used further for the structural modeling. In the present case, alignment of the lamellar mesophases was obtained by using high-temperature rubbing and epitaxy on friction-transferred poly(tetrafluoroethylene) (PTFE). On the one hand, high-T rubbing produces very high levels of both orientation and crystallinity in conjugated semi-conducting polymers e.g. poly(3-alkylthiophene)s and poly(thienothiophene)s as well as in alternating co-polymers.17–20 On the other hand, epitaxy on oriented PTFE is a simple means to grow highly oriented films of molecular semi-conductors such as acenes, phthalocyanines or columnar mesophases of larger macrocycles such as hexabenzocoronene.21,22 This study presents successively, (i) the growth of oriented films of the co-oligomers by high-temperature rubbing and by epitaxy on friction-transferred poly(tetrafluoroethylene) (PTFE), (ii) the evidence of a zipper-like packing of the co-oligomers by HR-TEM and (iii) the structural determination using ED and GIXD data.
II. Results and discussion
A. Orientation of AD0 co-oligomers
To build a structural model of AD0, highly oriented films were prepared using high-T rubbing and epitaxy on friction-transferred PTFE. The detailed procedures used to prepare and characterize the structure of the films are given in the ESI.†Let us first consider the results obtained on rubbed films of the co-oligomers. For both the AD0 and the AD1 dyads, UV-vis spectroscopy and POM (see Fig. S1 and S2, ESI†) show that high-T rubbing aligns the co-oligomer molecules parallel to the rubbing direction but TEM does not reveal any ordered structures (see Fig. S3a, ESI†). As demonstrated nicely by in situ TEM experiments (see Fig. S3b, ESI†), an additional post-rubbing thermal annealing at T ≥ 160 °C is necessary to induce the growth of an oriented lamellar mesophase of AD1 with the co-oligomer molecules lying essentially in the plane of the substrate and almost parallel to the rubbing direction R. Large-scale orientation of the lamellar mesophases is indicated by both the ED patterns shown in Fig. 2a and the GIXD patterns (see also Fig. S4 and S5, ESI†). In rubbed AD0 films, the crystalline lamellae are oriented normal to the substrate plane (so-called standing lamellae) and perpendicular to the rubbing direction (the indexation of the rather complex ED patterns is discussed in the ESI†). The carefull analysis of the ED pattern indicates that two populations of standing lamellae coexist, they are made of either “edge-on” or “face-on” molecules relative to the substrate plane (see the schematic illustration in Fig. 2e and f). Characteristic fingerprints of these two populations of standing lamellae are also observed in the corresponding GIXD patterns (see Fig. S5, ESI†).
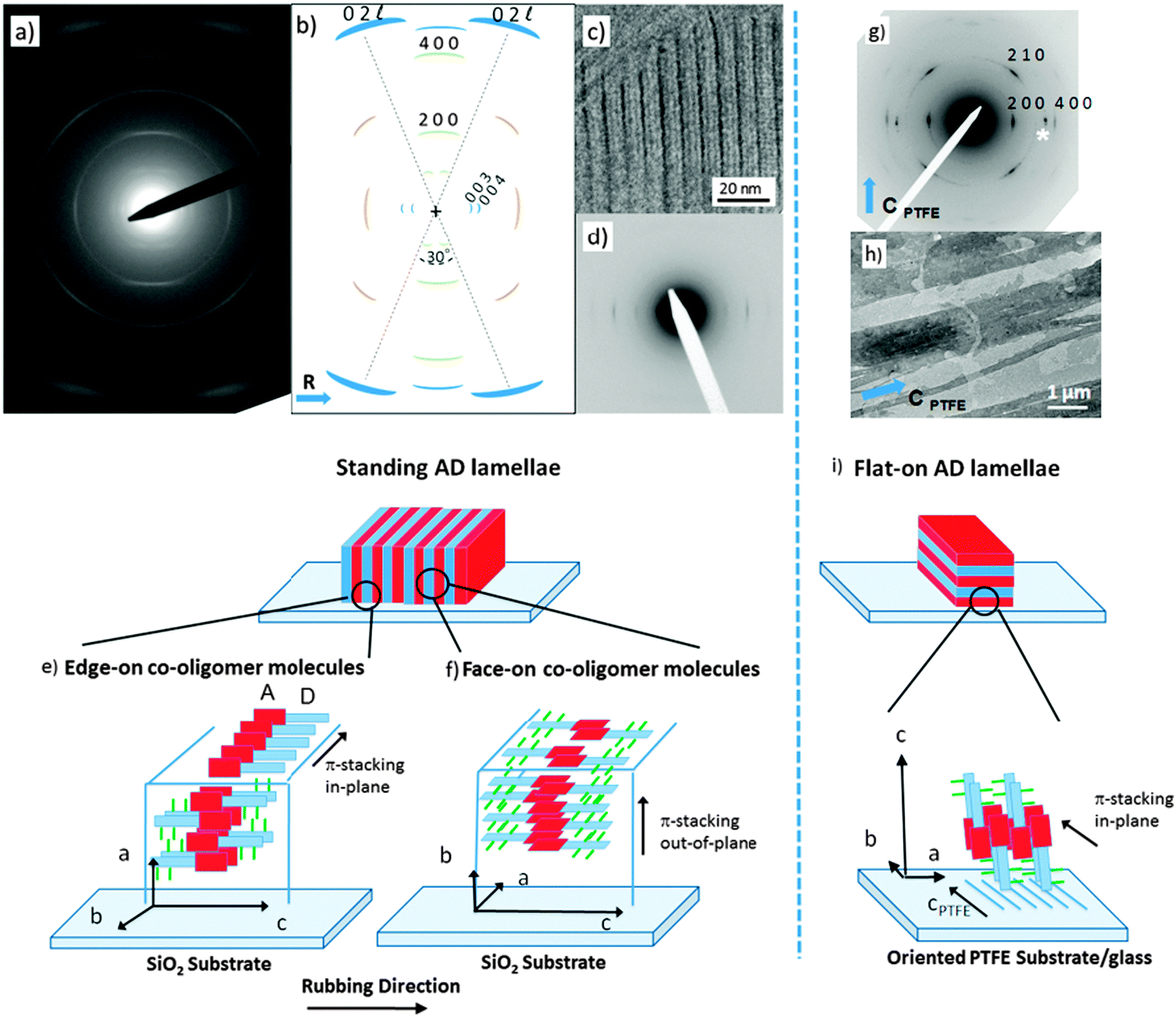 | ||
| Fig. 2 (a) ED pattern of a rubbed and annealed AD0 thin film, (b) schematic representation of the ED pattern, (c) BF-TEM showing the lamellar structure of AD0; (d) low angle diffraction pattern; (e) and (f) schematic illustrations of the orientation of the co-oligomer molecules in the standing lamellae seen in the BF image in (c); (g) SAED pattern of AD0 on oriented PTFE; (h) BF-TEM image of the flat-lying lamellae on the PTFE fibers. The rubbing direction and the polymer chain direction of PTFE are indicated by a blue arrow; (i) schematic representation of the flat-on orientations of the crystalline lamellae. The acceptor PDI group is shown in red, the donor block in blue and the lateral alkyl chains in green. | ||
Second, epitaxied layers on oriented PTFE substrates yield films with standing co-oligomer molecules. The TEM images in Fig. 2h depict the typical terraced morphology of melt-annealed co-oligomer films deposited on oriented PTFE for AD0. Fig. 2h shows mono-layer-thick AD0 lamellae between PTFE fibers and multi-layer thick lamellae on top of PTFE fibers. Accordingly, after cooling from the melt, the co-oligomers crystallize as flat-lying lamellae characterized by a typical terraced morphology corresponding to standing/tilted co-oligomer molecules (Fig. 2g). The selected area electron diffraction pattern of AD0 flat-lying lamellae indicates that the lamellae grow with a (001) contact plane and that the π-stacking direction of the AD0 molecules (the b axis) is parallel to the PTFE chains (cPTFE) (see Fig. 2i). In conjugated materials like pentacene and some phthalocyanines, such an orientation of lamellar crystals was explained by ledge-directed nucleation.21 In that case, oriented aggregates are nucleated by the PTFE fibers and the crystals grow along the PTFE fibers (the fast growth direction usually aligns parallel to the PTFE chain direction). A similar mechanism is probable also in the present case because the π-stacking of all investigated co-oligomers aligns parallel to the PTFE chains.
B. Evidence for a zipper-like packing using low dose HR-TEM
Let us first find an argument to discriminate between the two proposed models in Fig. 1.12 As seen in this figure, the major difference between the two packing modes stems from the arrangement of the AD0 molecules along the a axis. In model 1, the donor groups are strongly π-stacked in (b, c) planes separated by the alkyl side-chains rejected on both sides of the donor backbone along a. As a result, the AD0 molecules form layers of alternating conjugated backbones and layers of alkyl side chains with periodicity a = 1.74 nm. By contrast, for model 2, the donor groups are packed with their conjugated backbones side-by-side and the alkyl side chains are rejected outwards along the a axis, which results in a period 2a = 3.5 nm between layers of conjugated backbones and layers of alkyl side-chains. Moreover, in model 2, the donor groups are not strongly π-stacked because the successive layers of AD0 molecules along the b axis point alternatively along a and −a. Interestingly, these two models can be discriminated based on some HR-TEM images of the co-oligomers. Fig. 3 shows some representative HR-TEM images of the rubbed and annealed films of AD0, AD1 and ADA. As shown previously, the films consist essentially of edge-on oriented lamellae. However, the ED patterns show that within the edge-on lamellae, the stacking of the molecules can occur with the π-stacking either in-plane or along the film normal. As a matter of fact, the low dose HR-TEM images show some areas with a zipper-like self-organization of the ADn and ADA co-oligomers (see Fig. 3). To observe this zipper-like organization in HR-TEM, it is necessary that the direction of the electron beam coincides exactly with the π-stacking direction (b axis) of the domains in the thin films. Since the dominant population of standing lamellae corresponds to the (100) contact plane (i.e. co-oligomers with an “edge-on” orientation of the molecule), the zipper-like arrangement is seen occasionally as it corresponds to the minority population of lamellae with (010) orientation on the substrate (i.e. co-oligomers with a “face-on” orientation, see Fig. 2f).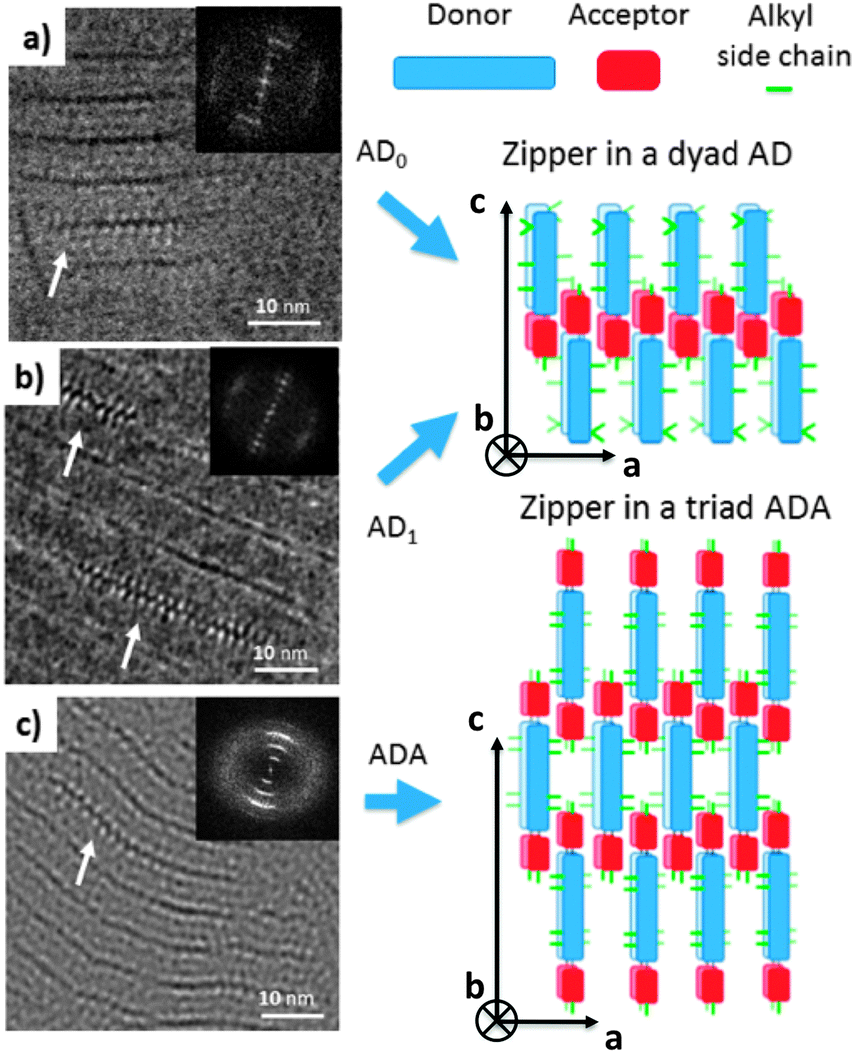 | ||
| Fig. 3 Low dose HR-TEM images of the (a) AD0, (b) AD1 and (c) ADA showing the characteristic zipper-like molecular packing. The white arrows point at the zipper-like arrangement of AD0 co-oligomers in the HR-TEM images. In these zones, the electron beam is parallel to the stacking direction of the PDI blocks (b axis). The insets show the fast Fourier transforms. On the right, a schematic illustration of the zipper-packing of the AD and ADA molecules is shown. It is a top-view of the zip i.e. along the normal to the substrate plane and corresponds to the zones indicates by white arrows in the TEM images. | ||
The contrast in the HR-TEM image of the zipper-like arrangement results from the electronic density difference between the zones of alkyl side-chains (bright) and the conjugated backbone of the co-oligomer (dark grey) (see also the schematic representation in Fig. 3). The important point is that the period along the zip corresponds to a = 1.7 ± 0.1 nm i.e. to what is indeed expected for model 1, whereas a period twice as large is expected for model 2. This result therefore confirms that the molecular packing of Fig. 1a is the most likely. The major novel evidence provided by HR-TEM with respect to the initial model 1 is that the successive columns of π-stacked PDI blocks are not aligned to form a plane of PDI columns as proposed initially in Fig. 1a, but form a zipper-like arrangement as shown schematically in Fig. 3. The shift between successive (b, c) planes in the zipper along the c axis can be estimated from the HR-TEM images: δc = 0.8 ± 0.2 nm. This zipper-like arrangement will be validated further by the structural data gained by ED and GIXD, and its origin will be explained based on the resulting structural model.
The HR-TEM images (Fig. 3c) reveal an additional periodic sub-structure within the lamellae in the form of two additional fringes but with a lower contrast as compared to the dominant dark fringe. This substructure corresponds to a periodic Z contrast modulation inside the lamellae possibly due to the alternation of sulfur-rich/poor zones of the donor block. For AD0, this sub-structure observed by HR-TEM is consistent with the strong intensity of the (003) reflection seen in the FFT and the LAED patterns as it corresponds to a L/3 period. This information will be of much help in the derivation of structural model of AD0.
C. Structural model of AD0 obtained from GIXD and ED data
The GIXD and ED data obtained on the oriented films of AD0 have been used to refine the structure of this co-oligomer beyond the schematic model of the zipper-like arrangement proposed in Fig. 3. The experimental and calculated ED and GIXD patterns with proper indexation are given in Fig. 4 for the refined model of AD0 shown in Fig. 5. The table listing all observed reflections and their indexation is given in the ESI.†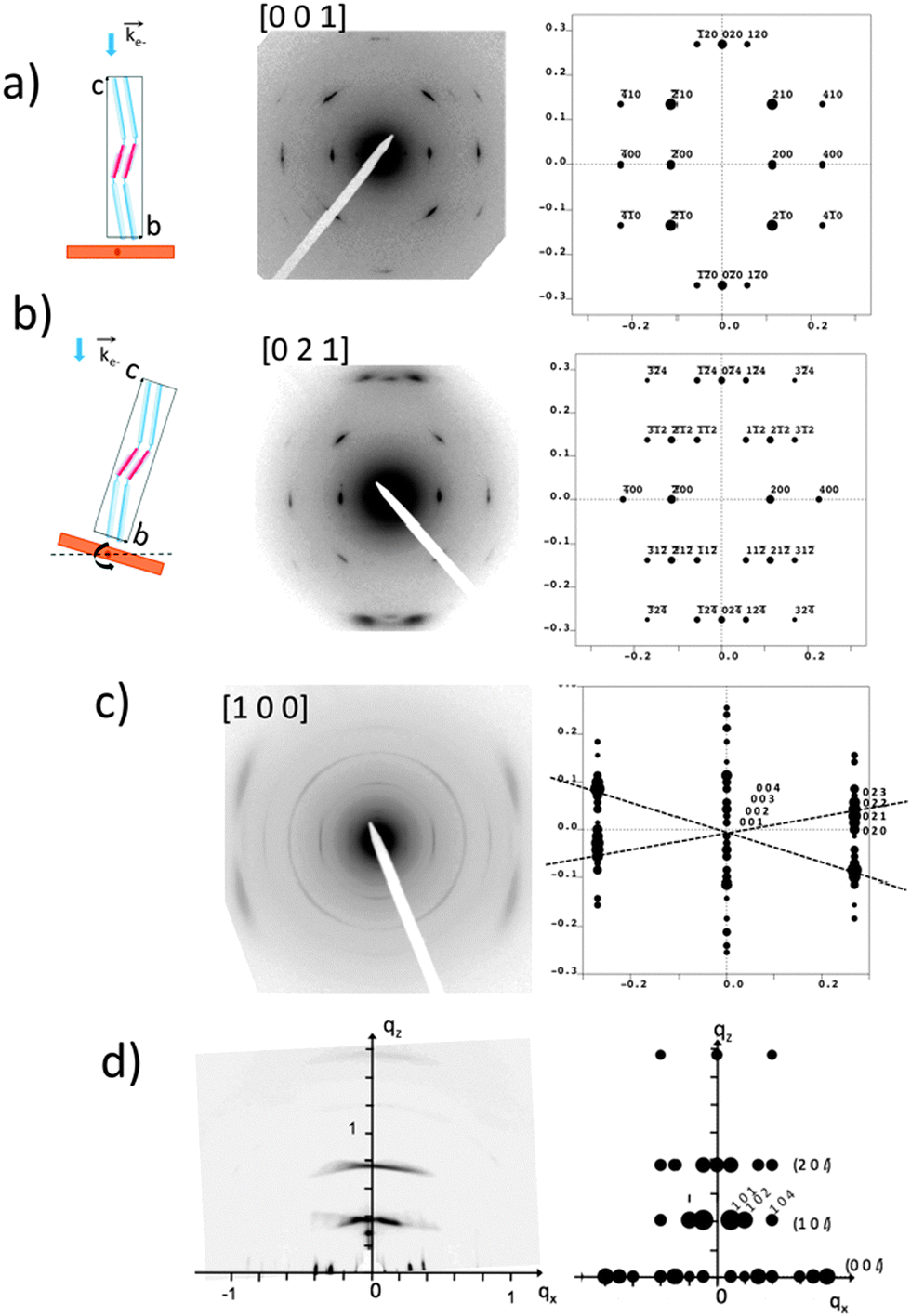 | ||
| Fig. 4 Comparison between experimental and calculated diffraction patterns obtained for the structural model of AD0 shown in Fig. 5. (a) and (b) ED patterns of AD0 films oriented on substrates of PTFE. The pattern in (b) is obtained by tilting the sample around the a* direction by an angle of ±15° (see the schematic illustrations on the left). The patterns (a) and (b) correspond to the [001] and the [021] zones. (c) ED pattern obtained for a rubbed AD0 film after annealing at 190 °C. The experimental pattern highlights the coexistence of three different populations of oriented AD0 lamellae. The dominant population yields a broad set of overlapped (02l) reflections as observed in the calculated ED pattern for the [100] zone. (d) GIXD pattern of an AD0 film oriented by rubbing. The incident beam is oriented perpendicular to the rubbing direction. This pattern is dominated by a population of edge-on oriented co-oligomers with their c axis oriented along the rubbing direction (qx). The calculated X-ray diffraction pattern corresponds to the [010] zone. | ||
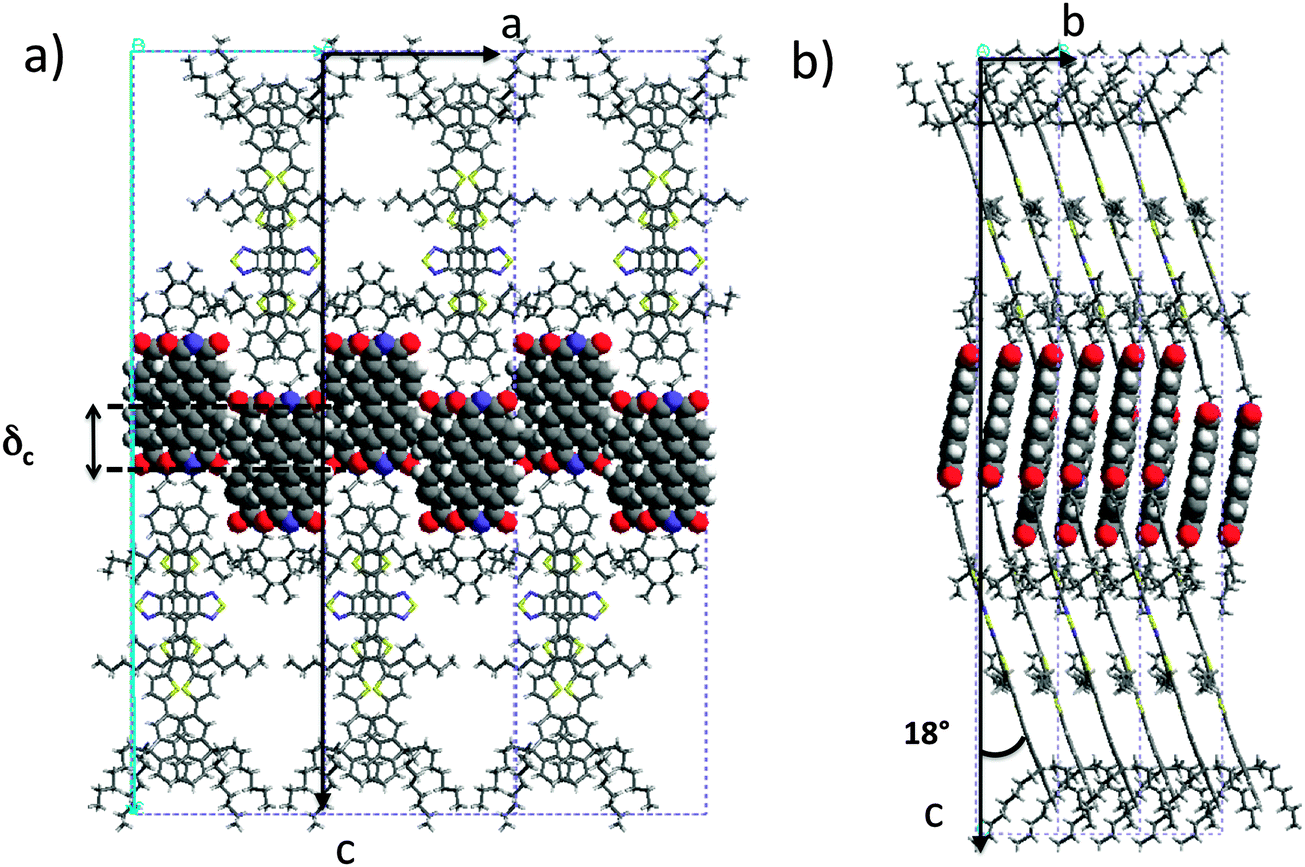 | ||
| Fig. 5 Projections of the refined structural model of AD0 yielding the calculated diffraction patterns in Fig. 4. The projections are along the b axis (a) and along the a axis (b). | ||
AD0 crystallizes in an orthorhombic unit cell: a = 17.47 Å, b = 7.45 Å, c = 70.4 Å and α = β = γ = 90° (Z = 4). Extinction rules can be extracted from the ED and GIXD patterns. The ED pattern in Fig. 4a indicates already the systematic absence of the (0 2n + 1 0) and the (2n + 1 0 0) (n = 0, 1) reflections. Therefore, two selection rules are identified: (h00) with h = 2n and (0k0) with k = 2n. The former condition is consistent with the fact that alkyl side chains of successive donor blocks along b must point alternatively along a and −a. The P21/c space group with unique axis a agrees with the two selection rules and the fact that Z = 4, was chosen for the modeling.
The exact methodology followed in the trial-and-error refinement of the AD0 structure is detailed in the ESI† (see Fig. S6). The model of AD0 yielding the best agreement between experimental and calculated ED patterns along three different zones ([100], [001] and [021]) is shown in Fig. 5 for two characteristic projections along the unit cell axes.
The model accounts for the main features of the experimental patterns obtained by SAED and rotation-tilt. In particular, the presence of the strong (±120) reflections in the pattern corresponding to the [001] zone is very well reproduced. Also, for the [100] zone, the presence of several (02l) reflections forming a quasi-continuum explains the broad reflection seen in the ED pattern of Fig. 2a and the GIXD pattern of Fig. 4d. Accordingly, the structural model is consistent with HR-TEM data and supports a zipper-like packing of the co-oligomers. In addition, the calculated powder X-ray diffraction pattern in Fig. S7 (ESI†) matches well the experimental pattern for AD0. Discrepancies can be accounted for by the fact that the model represents a crystalline structure in which the ethylhexyl and octyl side-chains are given a unique position whereas in the films analyzed by TEM and GIXD, AD0 forms a lamellar mesophase with disorder in the arrangement of the alkyl side-chains as indicated by the broad reflection at 1.5 Å−1 in the XRD patterns.12
Both the PDI and the donor blocks are π-stacked in the structure of AD0, in agreement with results obtained by dark field TEM (see Fig. S8, ESI†). The AD0 molecules are π-stacked in pairs with a strong overlap of both the perylene units and the donor groups. The PDIs form π-stacked columns in a slipped cofacial arrangement: two successive PDIs in a column along b are shifted both along the a and the c axes. In a pair of successive AD0 molecules along b, the lateral alkyl chains of the donor are rejected on either side of the conjugated skeleton. Moreover, head-to-tail stacking of benzothiadiazole units along the b axis leads to strong overlaps between their benzene groups, reminding the situation found for some alternating benzothiadiazole-based copolymers as well as some oligomers.23 Of importance is the fact that the strong π–π overlap between PDI blocks enforces a similar very tight π–π stacking of the donor block with short 3.4–3.5 Å intermolecular distances.
It is worth noting that the terminal ethylhexyl chain of the PDI is packed together with the lateral ethylhexyl chains of the thiophene of the donor block. Successive co-oligomer pairs along the a axis organize in a “head-to-head” arrangement with the donor skeletons pointing alternatively along c and −c and the PDIs located close side-by-side. This organization brings about a regular zipper-like molecular packing. This zipper-like structure is such that the PDI cores in two successive (b, c) planes are shifted along the c axis by δc = 0.7 nm. This shift of every second (b, c) layer leads to the observed intensity-evolution of the (00l) reflections and in particular to the enhanced intensity of the (003) reflection. This value is further in agreement with that obtained from the low dose HR-TEM images.
D. Discussion
The type of zipper-like arrangement derived for AD0, can readily be transposed to the ADA triad, as illustrated in Fig. 3. The two terminal PDI groups on each side of the donor block in ADA can π-stack while preserving an efficient packing of the donor groups and the lateral alkyl chains. Nevertheless and as noted earlier, the ADA co-oligomers form large-scale lamellar mesophases without annealing, indicating a stronger self-assembling than for the AD0 and AD1 dyads. This enhanced ordering ability of ADA derives from the fact that successive lamellae of ADA are strongly interdigitated along the c axis as seen in Fig. 3. The AD dyad lamellae can grow in a sequential mode i.e. lamella after lamella, in a smectic-like system whereas for the ADA triad, interdigitation of the molecules confers a stronger 3D cohesion to the self-assembled structure.
These results demonstrate that beside the strong π-stacking forces of the perylene cores, the lateral interactions between perylenes are also very important to generate 2-dimensional planes of PDI blocks. It would appear that a subtle choice of the alkyl side-chain on the imide position of the PDI unit may give a handle on the π-stacking of PDI units by controlling the shift δc between successive (b, c) layers of π-stacked co-oligomers in the zipper-like structure illustrated in Fig. 3. This is an essential feature of these systems since the precise π-orbital overlaps between both the PDI blocks and the donor blocks controls charge transport properties via the charge transfer integral.25 Accordingly, not only do the alkyl side chains play an essential role in terms of processing by imparting solubility in organic solvents to the conjugated co-oligomers, but they additionally can be rationally chosen in order to fine-tune the self-assembled structures generated in the solid state by the co-oligomers.
III. Conclusion
The most important results of this study deal with the growth, the structure and the orientation of PDI-based dyads and triads. First, high temperature rubbing, originally developed for large-scale alignment of conjugated macromolecules, can equally well align co-oligomers without using any alignment substrate. This is of interest for addressing both structural and charge transport issues in co-oligomer thin films. Second, by using different substrates and orientation methods, major features of the structure of these complex molecular systems have been established by a combination of electron diffraction, grazing incidence-X-ray diffraction and high-resolution TEM. As a major result, an original zipper-like arrangement of both ADn and ADA co-oligomers has been uncovered. This particular organization appears to be driven by two antagonistic self-assembling forces: (i) the strong π-stacking of PDI units into dense PDI sheets and (ii) the grouping of the alkyl side chains of both the PDI and the donor blocks. This organization differs markedly from the previously reported PDI-based structures and illustrates the strong impact of the chemical design on both electronic and self-assembling properties of such donor–acceptor co-oligomers.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work has been supported by the French National Agency (ANR PICASSO Project) and by the European Community via the Interreg IV-A program. (C25, Rhin Solar Project). We thank Pohang Accelerator Laboratory (PAL) for giving us the opportunity to perform the GIWAXS measurements, MEST and POSTECH for supporting these experiments, Dr Tae Joo Shin for adjustments and help, and other colleagues from the 9A U-SAXS beamline for assistance.References
- J. T. Chen and C.-S. Hsu, Polym. Chem., 2011, 2, 2707–2722 RSC.
- H. Liu, J. Xu, Y. Li and Y. Li, Acc. Chem. Res., 2010, 43, 1496–1508 CrossRef CAS PubMed.
- K. M. Coakley and M. D. McGehee, Chem. Mater., 2004, 16, 4533–4542 CrossRef CAS.
- W. Ma, C. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS PubMed.
- (a) B. De Boer, U. Stalmach, C. Melzer, V. V. Krasnikov and G. Hadziioannou, Polymer, 2001, 43, 9097–9109 CrossRef; (b) F. Zhang, M. Svensson, M. R. Andersson, M. Maggini, S. Bucella, E. Menna and O. Ingänas, Adv. Mater., 2001, 13, 1871–1874 CrossRef CAS; (c) S. Miyanishi, Y. Zhang, K. Tajima and K. Hashimoto, Chem. Commun., 2010, 46, 6723–6725 RSC; (d) S. M. Lindner and M. Thelakkat, Macromolecules, 2004, 37, 8832–8835 CrossRef CAS; (e) M. Sommer, S. Huettner and M. Thelakkat, J. Mater. Chem., 2010, 20, 10788–10797 RSC; (f) Q. Zhang, A. Cirpan, T. P. Russel and T. Emrick, Macromolecules, 2009, 42, 1079–1082 CrossRef CAS; (g) M. Sommer, S. M. Lindner and M. Thelakkat, Adv. Funct. Mater., 2007, 17, 1493–1500 CrossRef CAS; (h) R. H. Lohwasser, G. Gupta, P. Kohn, M. Sommer, A. S. Lang, T. Thurn-Albrecht and M. Thelakkat, Macromolecules, 2013, 46, 4403–4410 CrossRef CAS; (i) D. Zeng, I. Tahar-Djebbar, Y. Xiao, F. Kameche, N. Kayunkid, M. Brinkmann, D. Guillon, B. Donnio, D. A. Ivanov, E. Lacaze, D. Kreher, F. Mathevet and A.-J. Attias, Macromolecules, 2014, 47, 1715–1731 CrossRef CAS.
- (a) J.-F. Nierengarten, Sol. Energy Mater. Sol. Cells, 2004, 83, 187–199 CrossRef CAS; (b) J. Roncali, Chem. Soc. Rev., 2005, 34, 483–495 RSC; (c) J. L. Segura, N. Martin and D. M. Guldi, Chem. Soc. Rev., 2005, 34, 31–47 RSC; (d) J.-F. Nierengarten, J.-F. Eckert, J.-F. Nicoud, L. Ouali, V. Krasnikov and G. Hadziioannou, Chem. Commun., 1999, 617–618 RSC; (e) W. S. Li, Y. Yamamoto, T. Fukushima, A. Saeki, T. Seki, S. Tagawa, H. Masunaga, S. Sasaki, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 8886–8887 CrossRef CAS PubMed; (f) T. Nishizawa, K. Tajima and K. Hashimoto, J. Mater. Chem., 2007, 17, 2440–2445 RSC; (g) T. Nishizawa, K. Tajima and K. Hashimoto, Nanotechnology, 2008, 19, 424017 CrossRef PubMed; (h) T. Roland, J. Léonard, G. Ramirez-Hernandez, S. Mery, O. Yurcheko, S. Ludwigs and S. Haacke, Phys. Chem. Chem. Phys., 2012, 14, 273–279 RSC.
- (a) F. Würthner, Chem. Commun., 2004, 1564–1579 RSC; (b) C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed; (c) E. Kozma and M. Catellani, Dyes Pigm., 2013, 98, 160–179 CrossRef CAS.
- (a) H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed; (b) R. Steyrleuthner, M. Schubert, F. Jaiser, J. C. Blakesley, Z. Chen, A. Facchetti and D. Neher, Adv. Mater., 2010, 22, 2799–2803 CrossRef CAS PubMed; (c) M. Schubert, D. Dolfen, J. Frisch, S. Roland, R. Steyrleuthner, B. Stiller, Z. Chen, U. Scherf, N. Koch, A. Facchetti and D. Neher, Adv. Energy Mater., 2012, 2, 369–380 CrossRef CAS.
- (a) X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed; (b) P. E. Hartnett, A. Timalsina, H. S. Matte, S. Ramakrishna, N. Zhou, X. Guo, W. Zhao, A. Facchetti, R. P. H. Chang, M. C. Hersam, M. R. Wasielewski, R. Michael and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 16345–16356 CrossRef CAS PubMed; (c) M. Guide, S. Pla, A. Sharenko, P. Zalar, F. Fernandez-Lazaro, A. Sastre-Santos and T.-Q. Nguyen, Phys. Chem. Chem. Phys., 2013, 15, 18894–18899 RSC; (d) X. Zhang, J. Yao and C. Zhan, Chem. Commun., 2015, 51, 1058 RSC; (e) Y. Lin, Y. Wang, J. Wang, J. Hou, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2014, 26, 5137 CrossRef CAS PubMed.
- (a) L. Bu, X. Guo, B. Yu, Y. Qu, Z. Xie, D. Yan, Y. Geng and F. Wang, J. Am. Chem. Soc., 2009, 131, 13242–13243 CrossRef CAS PubMed; (b) L. Bu, X. Guo, B. Yu, Y. Fu, Y. Qu, Z. Xie, D. Yan, Y. Geng and F. Wang, Polymer, 2011, 52, 4253–4260 CrossRef CAS; (c) Y. Qu, B. Gao, H. Tian, X. Zhang, Y. Wang, Z. Xie, H. Wang, Y. Geng and F. Wang, J. Mater. Chem. A, 2014, 2, 3632–3640 RSC.
- P.-O. Schwartz, E. Zaborova, L. Biniek, B. Heinrich, M. Brinkmann, N. Leclerc and S. Mery, J. Am. Chem. Soc., 2014, 136, 5981–5992 CrossRef CAS PubMed.
- (a) V. Percec, M. Peterca, T. Tadjiev, X. Zeng, G. Ungar, P. Leowanawat, E. Aqad, M. R. Imam, B. M. Rosen, U. Akbey, R. Graf, S. Sekharan, D. Sebastiani, H. W. Spiess, P. A. Heiney and S. D. Hudson, J. Am. Chem. Soc., 2011, 133, 12197–12219 CrossRef CAS PubMed; (b) L. F. Dössel, V. Kamm, I. A. Howard, F. Laquai, W. Pisula, X. Feng, C. Li, M. Takase, T. Kudernac, S. De Feyter and K. Müllen, J. Am. Chem. Soc., 2012, 134, 5876–5886 CrossRef PubMed; (c) V. Percec, H.-J. Sun, P. Leowanawat, M. Peterca, R. Graf, H. W. Spiess, X. Zeng, G. Ungar and P. A. Heiney, J. Am. Chem. Soc., 2013, 135, 4129–4148 CrossRef CAS PubMed.
- M. Brinkmann and J.-C. Wittmann, Adv. Mater., 2006, 18, 860–863 CrossRef CAS.
- N. Kayunkid, S. Uttiya and M. Brinkmann, Macromolecules, 2010, 43, 4961–4967 CrossRef CAS.
- M. Brinkmann, Macromolecules, 2007, 40, 7532–7541 CrossRef CAS.
- (a) L. Biniek, N. Leclerc, T. Heiser, R. Bechara and M. Brinkmann, Macromolecules, 2013, 46, 4014–4023 CrossRef CAS; (b) L. Hartmann, L. Biniek, K. Tremel, N. Kayunkid and M. Brinkmann, Macromol. Rapid Commun., 2014, 35, 9–26 CrossRef PubMed.
- L. Hartmann, K. Tremel, S. Uttiya, E. Crossland, S. Ludwigs, N. Kayunkid, C. Vergnat and M. Brinkmann, Adv. Funct. Mater., 2011, 21, 4047–4057 CrossRef CAS.
- K. Tremel, F. S. U. Fischer, N. Kayunkid, R. DiPietro, A. Kiriy, D. Neher, S. Ludwigs and M. Brinkmann, Adv. Energy Mater., 2014, 4, 1301659 Search PubMed.
- L. Biniek, S. Poujet, D. Djurado, E. Gonthier, K. Tremel, N. Kayunkid, E. Zaborova, N. Crespo-Monteiro, O. Boyron, N. Leclerc, S. Ludwigs and M. Brinkmann, Macromolecules, 2014, 47, 3871–3879 CrossRef CAS.
- (a) M. Brinkmann, S. Graff, C. Straupé, J.-C. Wittmann, C. Chaumont, F. Nuesch, A. Aziz, M. Schaer and L. Zuppiroli, J. Phys. Chem. B, 2003, 107, 10531–10539 CrossRef CAS; (b) M. Brinkmannn, J.-C. Wittmann, M. Barthel, M. Hanack and C. Chaumont, Chem. Mater., 2002, 14, 904–914 CrossRef; (c) J.-F. Moulin, M. Brinkmann, A. Thierry and J.-C. Wittmann, Adv. Mater., 2002, 14, 436–439 CrossRef CAS; (d) D. W. Breiby, H. T. Lemke, P. Hammershøj, J. W. Andreasen and M. M. Nielsen, J. Phys. Chem. C, 2008, 112, 4569–4572 CrossRef CAS.
- O. Bunk, M. M. Nielsen, T. I. Sœlling, A. M. Van De Craats and N. Stuzmann, J. Am. Chem. Soc., 2003, 125, 2252–2258 CrossRef CAS PubMed.
- (a) H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, R. W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS PubMed; (b) D. Niedzialek, V. Lemaur, D. Dudenko, J. Shu, M. R. Hansen, J. W. Andreasen, W. Pisula, K. Müllen, J. Cornil and D. Beljonne, Adv. Mater., 2013, 25, 1939–1947 CrossRef CAS PubMed; (c) C. L. Donley, J. Zaumseil, J. W. Andreasen, M. M. Nielsen, H. Sirringhaus, R. H. Friend and J.-S. Kim, J. Am. Chem. Soc., 2005, 127, 12890–12899 CrossRef CAS PubMed; (d) F. L. Tobiason, L. Huestis, C. Chandler, S. E. Pedersen and P. Peters, J. Heterocycl. Chem., 1973, 10, 773–778 CrossRef CAS; (e) S.-W. Chang, H. Waters, J. Kettle and M. Horie, Org. Electron., 2012, 13, 2967–2974 CrossRef CAS.
- (a) G. Klebe, F. Graser, E. Hädicke and J. Berndt, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 69–77 CrossRef; (b) E. Hädicke and F. Graser, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 189–195 CrossRef; (c) E. Hädicke and F. Graser, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 195–198 CrossRef.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, high-temperature rubbing of co-oligomer films, interpretation of electron diffraction data, polarized UV-vis absorption spectra of AD0 before and after annealing, UV-vis spectra of rubbed and annealed AD1, result of an in situ thermal annealing experiment in the TEM, GIXD 2D intensity maps for rubbed AD0 films, methodology for structural refinement using a trial-and-error approach, calculated and experimental powder diffraction patterns of AD0, dark field TEM on rubbed AD0, AD1 and ADA films, ED patterns and morphology in AD1 and ADA films oriented on PTFE substrates, table of main ED and GIXD reflections with observed dhkl values and indexation for AD0, AD1 and ADA. See DOI: 10.1039/c5tc00221d |
| This journal is © The Royal Society of Chemistry 2015 |