
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel co-crystals of the nutraceutical sinapic acid†
A. S.
Sinha
a,
U. B.
Rao Khandavilli
a,
E. L.
O’Connor
a,
B. J.
Deadman
a,
A. R.
Maguire
b and
S. E.
Lawrence
*a
aDepartment of Chemistry, Analytical and Biological Chemistry Research Facility, Synthesis and Solid State Pharmaceutical Centre, University College Cork, Cork, Ireland. E-mail: simon.lawrence@ucc.ie
bDepartment of Chemistry and School of Pharmacy, Analytical and Biological Chemistry Research Facility, Synthesis and Solid State Pharmaceutical Centre, University College Cork, Cork, Ireland
First published on 29th May 2015
Abstract
Sinapic acid (SA) is a nutraceutical with known anti-oxidant, anti-microbial, anti-inflammatory, anti-cancer, and anti-anxiety properties. Novel co-crystals of SA were prepared with co-formers belonging to the category of GRAS [isonicotinic acid (INC), nicotinamide (NIA)], non-GRAS [4-pyridinecarbonitrile (PYC)], and active pharmaceutical ingredients (APIs) [6-propyl-2-thiouracil (PTU)] list of compounds. Structural study based on the X-ray crystal structures revealed the intermolecular hydrogen-bonded interactions and molecular packing. The crystal structure of sinapic acid shows the anticipated acid–acid homodimer along with discrete hydrogen bonds between the acid carbonyl and the phenolic moiety. The robust acid–acid homodimer appears to be very stable and is retained in the structures of two co-crystals (SA·NIA and SA·PYC). In these cases, co-crystallization occurs via intermolecular phenol O–H⋯Naromatic hydrogen bonds between the co-formers. In the SA·PTU·2MeCN co-crystal the acid–acid homodimer gives way to the anticipated acid–amide heterodimer, with the phenolic moiety of SA hydrogen-bonded to acetonitrile. Attempts at obtaining the desolvated co-crystal led to lattice breakdown, thus highlighting the importance of acetonitrile in the formation of the co-crystal. Among the co-crystals examined, SA·INC (5 weeks), SA·NIA (8 weeks) and SA·PYC (5 weeks) were found to be stable under accelerated humidity conditions (40 °C, 75% RH), whereas SA·PTU·2MeCN decomposed after one week into individual components due to solvent loss.
Introduction
‘Crystal engineering’, a term initially introduced by Pepinsky1 and developed by Schmidt,2 has been defined as: “the understanding of intermolecular interactions in the context of crystal packing and the utilisation of such understanding in the design of new solids with desired physical and chemical properties.”3 The bulk properties of materials are directly affected by the assembly of molecules in the solid state,4 hence an important goal is the controlled assembly/ordering of molecules in the solid state.Co-crystallization is an important part of crystal engineering and can be defined as the coming together of two or more neutral molecules in a crystalline lattice under ambient conditions utilising intermolecular interactions such as hydrogen bonding, halogen bonding and π–π bonding.5 It is a viable and effective method for improving the key bulk properties of a compound such as solubility,6 stability7 and bioavailability.8 Pharmaceutical co-crystallization has gained particular attention within the past decade and is rapidly emerging as a valuable tool in the synthesis of new drugs with controlled physicochemical properties.9 Co-crystallization has been used in the agrochemical industry,10 and in the manufacture of explosives.11
The term nutraceutical was first introduced by Stephen Felice in the late eighties,12 and refers to compounds that are found in food or food products and can potentially aid in the treatment or even the prevention of diseases.13 Polyphenols are a common class of nutraceuticals found in fruits and vegetables which exhibit disease preventing capabilities and have been utilised as anti-oxidants, free radical scavengers and in cosmetic formulations.14,15 Although this valuable class of compounds possess obvious health benefits, some nutraceuticals are not physicochemically efficient due to poor stability, solubility and bioavailability.15 Thus, it is important to alter these key bulk properties of nutraceuticals in order to develop medicinally active forms that could prove valuable to the pharmaceutical industry.15
The choice of the co-former (co-crystal former) is important in pharmaceutical co-crystals and usually they are chosen from lists such as Generally Regarded As Safe (GRAS)16 and Everything Added to Food in the United States (EAFUS).17 An obvious choice for the class of compounds that can be used to extend the above lists of co-formers are nutraceuticals, which can act as good co-formers due to the variable co-crystal-forming functional groups present on their backbone.15 Also, their potential medicinal benefits along with their ready availability mean that nutraceuticals are attractive co-formers for pharmaceutical co-crystallization.15,18 Apart from the above, the three most important benefits of utilising nutraceuticals as co-formers for pharmaceutical co-crystallization are: (a) nutraceuticals that are bioactive can be co-administered with other bioactive drugs, thus targeting the synergy between the two components; (b) active pharmaceutical ingredients (APIs) with poor physicochemical properties can be co-crystallized with nutraceuticals in an attempt to synthesise new crystalline forms with improved physicochemical benefits; (c) anti-oxidant behavior of nutraceuticals can be used to impart stability to APIs that are prone to oxidation, or to reduce the incidence of diseases related to oxidative stress in the physiological state.
Sinapic acid is a widespread nutraceutical in the plant kingdom and is commonly found in fruits, vegetables, cereal grains, oilseed crops, and some spices and medicinal plants (Fig. 1).19 It can be classed as a phenolic acid and is known for its anti-oxidant,20 anti-microbial,21 anti-inflammatory,22 anti-cancer,23 and anti-anxiety activity.24 Indeed, it is a superior anti-oxidant than ferulic acid,25 which is already used as a natural anti-oxidant in foods, beverages, and cosmetics.26 4-Vinylsyringol (Fig. 1), a decarboxylation product of sinapic acid, is a potent anti-oxidative and anti-mutagenic agent.27 Sinapine (sinapoyl choline) is an acetylcholinesterase inhibitor with biological applications in the treatment of various diseases (Fig. 1).28
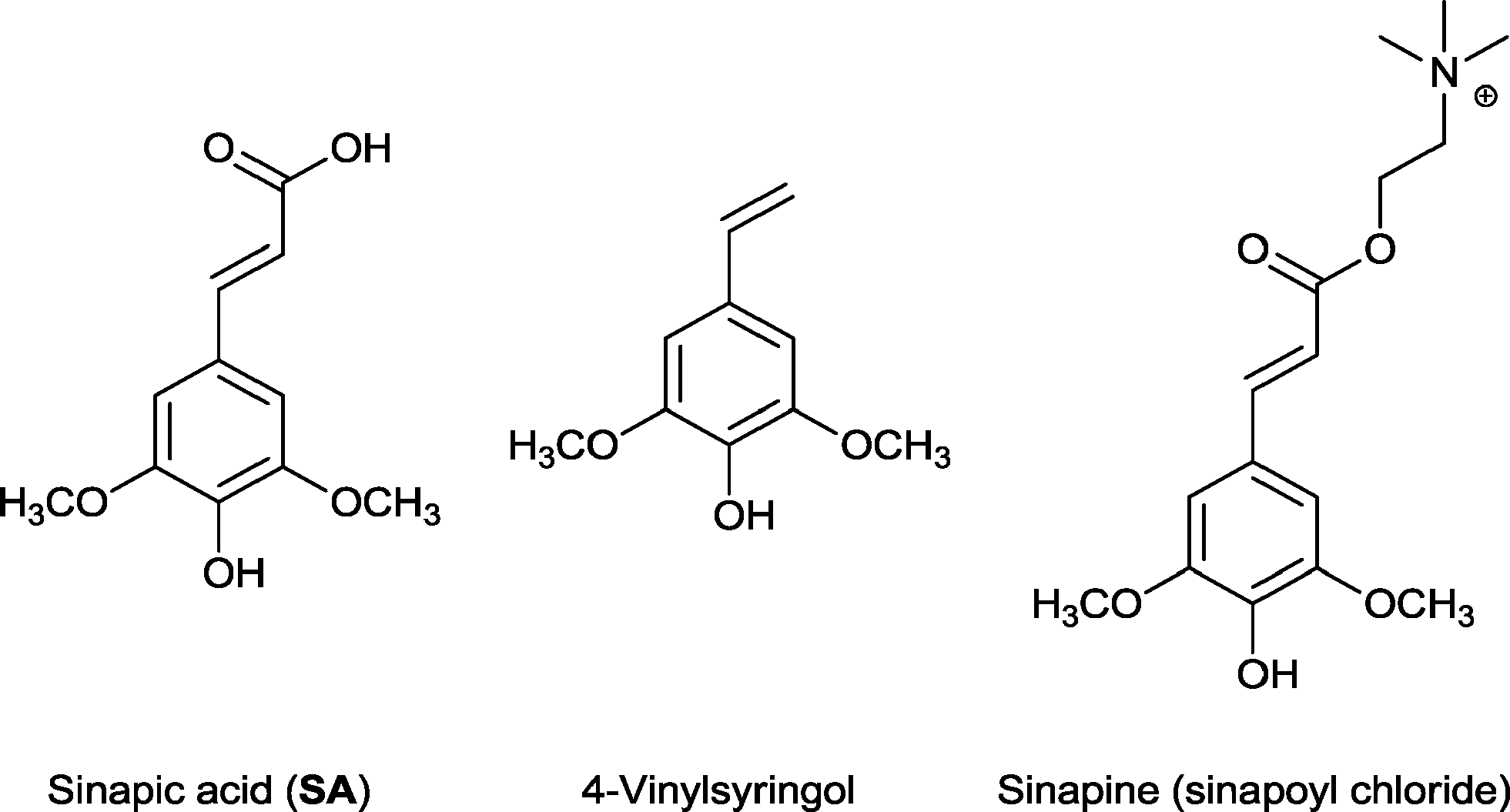 | ||
| Fig. 1 Structure of sinapic acid and its bioactive derivatives. | ||
Pharmacokinetic studies conducted on sinapic acid and its derivatives have highlighted the role of these phenolic acids in the human body.29–31 Sinapic acid is transported in blood due to its ability to bind with serum albumin via hydrogen bonds and hydrophobic interactions.29 In a study on non-processed cereal, the maximum recorded sinapic acid level in the plasma was found to be 40 nM with a bioavailability of 3%.30 Another study on cranberry juice intake by humans reported the plasma-sinapic acid level to be 1.5 μg mL−1.31 However, its poor solubility and, in turn, poor bioavailability means there are, to date, no pharmaceutically viable formulations of sinapic acid.
The solid-state structure of sinapic acid has not been described. Furthermore, there are no known co-crystals of sinapic acid, and the only reported crystal structure of SA is a methanol solvate.32 An understanding of the hydrogen bond behaviour of the functional groups on sinapic acid in the solid state is a key step in the selection of appropriate co-formers towards the design of sinapic acid co-crystals. The solid-state chemistry of phenols and acids, the two hydrogen-bonding functional groups present in sinapic acid, is well known.33,34 Acids usually form co-crystals via hydrogen bonding with functional groups including carboxylic acids, amides and pyridines.34 Phenols can act as both a hydrogen bond donor and an acceptor. Thus, they can hydrogen bond with acceptors such as pyridines and the oxygen atom of various carbonyl functional groups, as well as with donors including alcohols and other phenols.33 The calculated molecular electrostatic potentials (MEPs) of the carboxylic acid (+246 kJ mol−1) and the phenolic (+218 kJ mol−1) moieties on sinapic acid also highlight the hydrogen-bond donor potential of sinapic acid.15 Therefore, we anticipated that sinapic acid would form a range of intermolecular interactions such as acid–acid, acid–amide, acid–Naromatic, phenol–Naromatic and phenol–carbonyl hydrogen bonds, as illustrated in Fig. 2.15 Furthermore, any unsatisfied donors or acceptors can potentially hydrogen bond with the five hydrogen-bond acceptors (carboxylic, phenolic and methoxy oxygen atoms) in sinapic acid that are also capable of a myriad of supramolecular interactions, thus giving rise to various possibilities for co-crystallization.
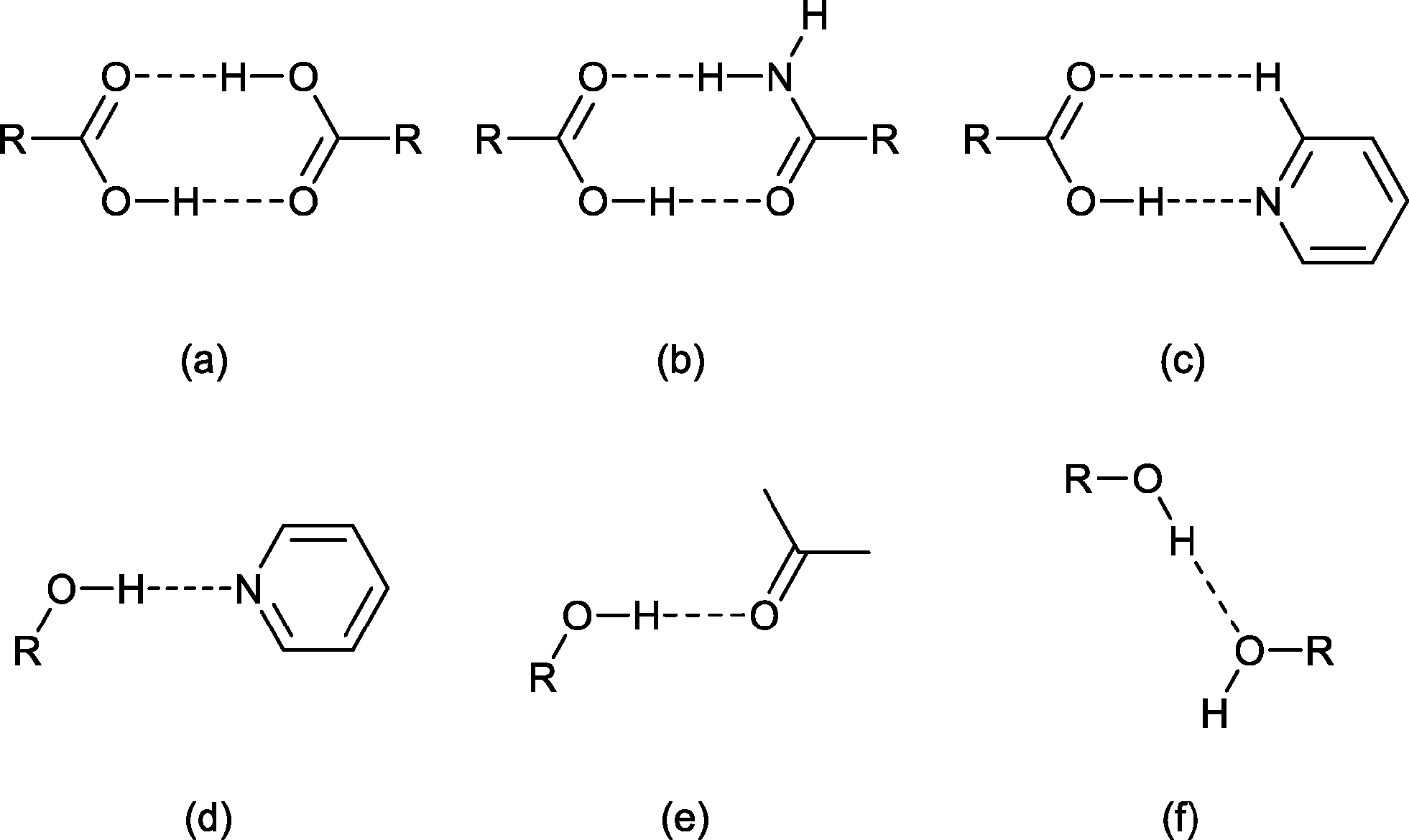 | ||
| Fig. 2 Common hydrogen-bonding motifs involving acids (a–c) and phenols (d–f). | ||
Accordingly, based on knowledge of hydrogen bond compatibility, calculated MEPs of sinapic acid and the expected motifs, we investigated the co-crystallization of sinapic acid with 21 co-formers containing three hydrogen-bond compatible functional groups, namely carboxylic acids (Fig. 2a and e), amides (Fig. 2b and e), and pyridines (Fig. 2c and d) (detailed list provided in ESI†). These co-formers were chosen because they possess relatively high aqueous solubilities. Additionally, we also investigated the co-crystallization of sinapic acid with four model APIs containing the carboxylic acid and secondary amide moieties (Fig. 2a–b, e), with the aim of exploring the possibility of co-administered drugs. The structural and stability data of sinapic acid and its co-crystals are reported herein.
Experimental
Materials and methods
The compounds utilised in this study were obtained from Sigma Aldrich and used as received. Solvents were obtained from commercial sources and distilled before use. Melting points were determined using an Electrothermal IA9100 melting point apparatus.Screening involving mechanical grinding was employed with subsequent crystallization by slow evaporation from solution in all cases, even where grinding and IR indicated co-crystallization had not occurred.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (0.3 mmol) of the sinapic acid and co-formers. Mechanical grinding experiments were conducted in a Retsch MM400 Mixer mill, equipped with two stainless steel 5 mL grinding jars and one 2.5 mm stainless steel grinding ball per jar. The mill was operated at a rate of 30 Hz for 30 min.
:1 ratio (0.3 mmol) of the sinapic acid and co-formers. Mechanical grinding experiments were conducted in a Retsch MM400 Mixer mill, equipped with two stainless steel 5 mL grinding jars and one 2.5 mm stainless steel grinding ball per jar. The mill was operated at a rate of 30 Hz for 30 min.
Solution crystallization
Based on solubility considerations of both sinapic acid and the co-formers, four solvents (acetone, acetonitrile, ethanol and methanol) were selected for solution crystallization studies.Crystallography
Single crystal X-ray data were collected on a Bruker APEX II DUO diffractometer at room temperature or 100 K using graphite monochromatised Mo Kα (λ = 0.7107 Å) radiation or on a Bruker X2S at room temperature using graphite monochromatised Mo Kα (λ = 0.7107 Å) radiation. The structures were solved using direct methods and refined on F2. Analysis was undertaken with the SHELX suite of programs35 and diagrams prepared with Mercury 3.3.36 All non-hydrogen atoms were located and refined with anisotropic thermal parameters. Hydrogen atoms were either placed in calculated positions or they were located and refined with isotropic thermal parameters. The detailed crystallographic data and structure refinement parameters for these compounds are summarised in Table 1.| SA | SA·NIA | SA·PYC | SA·PTU·2MeCN | |
|---|---|---|---|---|
| Formula | C11H12O5 | C11H12O5·C6H6N2O | C11H12O5·C6H4N2 | C11H12O5·C7H10N2OS·2(C2H3N) |
| MW | 224.21 | 346.33 | 328.32 | 476.54 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Triclinic |
| Space group, Z | P21/n, 4 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , 2 , 2 |
P21/c, 8 |
P, 2 |
| a, Å | 4.7531(7) | 4.9109(11) | 14.893(3) | 8.2922(8) |
| b, Å | 15.683(2) | 9.1732(18) | 31.878(6) | 10.5857(10) |
| c, Å | 14.170(2) | 18.814(4) | 6.5992(14) | 14.7818(15) |
| α, deg | 90 | 99.563(7) | 90 | 98.274(2) |
| β, deg | 90.383(3) | 94.601(7) | 95.192(4) | 94.315(2) |
| γ, deg | 90 | 90.531(7) | 90 | 105.620(2) |
| V, Å3 | 1056.2(3) | 832.9(3) | 3120.2(11) | 1227.7(2) |
| D c, g cm−3 | 1.410 | 1.381 | 1.398 | 1.289 |
| μ, mm−1 | 0.112 | 0.106 | 0.104 | 0.175 |
| 2θ range, deg | 1.94–26.41 | 3.31–26.28 | 1.51–26.79 | 1.40–26.34 |
| T, K | 296.(2) | 300.(2) | 100.(2) | 100.(2) |
| Total reflns. | 14863 |
8751 | 69717 |
16145 |
| Unique reflns. | 2159 | 3230 | 6650 | 4920 |
| R int | 0.0415 | 0.0572 | 0.0689 | 0.0260 |
| Obs. reflns., I > 2σ(I) | 1546 | 1746 | 4854 | 4229 |
| No. parameters | 155 | 240 | 449 | 319 |
| No. restraints | 0 | 11 | 4 | 10 |
| R 1 [I > 2σ(I)] | 0.0392 | 0.0536 | 0.0652 | 0.0393 |
| wR2 [all data] | 0.1148 | 0.1322 | 0.1683 | 0.1089 |
| S | 1.301 | 0.995 | 1.155 | 1.103 |
| ρ max, e Å−3 | 0.156 | 0.166 | 0.348 | 0.589 |
| ρ min, e Å−3 | −0.159 | −0.191 | −0.339 | −0.374 |
Physical form stability studies
200 mg of each solid was taken in 8 dram vials and the physical stability was evaluated at 40 ± 0.5 °C in 75% relative humidity. The vials of each co-crystal were subjected to the above accelerated stability conditions for 3 days and weekly intervals from 1 week to 8 weeks. PXRD and high performance liquid chromatography (HPLC) techniques were used to monitor the stability of the co-crystals.Results and discussion
Co-crystallization of sinapic acid has been attempted with 21 different co-formers which belong to the GRAS16 and EAFUS lists,17 and non-GRAS compounds in order to develop novel crystalline materials of this nutraceutical. Of the 21 co-formers examined, three successful co-crystals (SA·INC, SA·NIA and SA·PYC) were identified. Isonicotinic acid (INC) and nicotinamide (NIA) are GRAS compounds, and 4-pyridinecarbonitrile (PYC) is a non-GRAS co-former (Fig. 3). | ||
| Fig. 3 Structure of the four successful co-formers. | ||
Finally, four model APIs were also selected for co-crystallization with sinapic acid to investigate the potential of co-administered drugs. One successful co-crystal of SA with the API 6-propyl-2-thiouracil (PTU) was isolated as an acetonitrile solvate, and was fully characterised by solid-state techniques. PTU is an important drug used to treat hyperthyroidism including Graves' disease,37 and any new crystalline forms of this API have the potential to lead to new pharmaceutical formulations.
Infrared (IR) spectroscopy
The two main hydrogen bonding functional groups in sinapic acid are the phenolic and acid moieties. The characteristic IR carbonyl stretch (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) shows up in the region of 1650–1730 cm−1 depending on the nature of the carbonyl group (acid/amide/aldehyde/ester). The carbonyl stretch of carboxylic acids generally appear between 1650–1700 cm−1, whereas phenols are known to absorb strongly in the region of 3400–3700 cm−1.38 Since the phenolic stretches are generally broad, and also coincide with the acid O–H stretch of SA, along with the co-former acid O–H (INC) and amide N–H (NIA) stretches, it is difficult to monitor these bands for any changes. Hence, the SA carbonyl stretch is a better indicator of hydrogen bonding in these co-crystals. The important carbonyl stretches in SA and its co-crystals are given in Table 2.
O) shows up in the region of 1650–1730 cm−1 depending on the nature of the carbonyl group (acid/amide/aldehyde/ester). The carbonyl stretch of carboxylic acids generally appear between 1650–1700 cm−1, whereas phenols are known to absorb strongly in the region of 3400–3700 cm−1.38 Since the phenolic stretches are generally broad, and also coincide with the acid O–H stretch of SA, along with the co-former acid O–H (INC) and amide N–H (NIA) stretches, it is difficult to monitor these bands for any changes. Hence, the SA carbonyl stretch is a better indicator of hydrogen bonding in these co-crystals. The important carbonyl stretches in SA and its co-crystals are given in Table 2.
Co-crystal formation is indicated by blue shifts in the carbonyl stretch of sinapic acid, which is reflected in the longer O–H⋯OC hydrogen bond distances in the cocrystals compared to sinapic acid. Blue-shifts of hydrogen bonds upon co-crystallization are not expected, however, computational investigations into this behaviour exist.39 In the case of SA·INC, the carbonyl stretch of SA is unidentifiable, but the overall shifts in the different bands in the IR spectrum are clearly indicative of the formation of a new solid phase. Another clear indicator of co-crystal formation is the presence of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch due to the nitrile functional group at 2230 cm−1 for SA·PYC, 2253 and 2262 cm−1 for SA·PTU·2MeCN (Fig. 4).
N stretch due to the nitrile functional group at 2230 cm−1 for SA·PYC, 2253 and 2262 cm−1 for SA·PTU·2MeCN (Fig. 4).
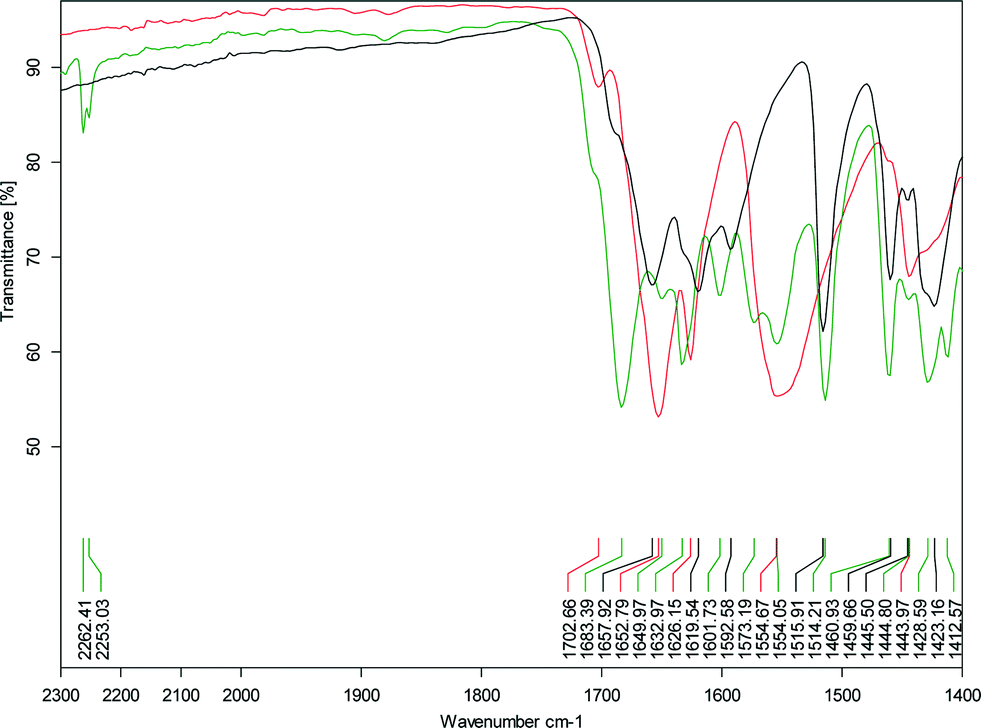 | ||
| Fig. 4 Expanded view of the IR spectrum of SA·PTU·2MeCN showing the region from 1400–2300 cm−1 (black – SA; red – PTU; green – SA·PTU·2MeCN). | ||
Structural analysis
Sinapic acid by itself forms the anticipated acid–acid homodimer [Fig. 5 (blue)]. The phenol OH moieties are forming discrete hydrogen bonds with the oxygen atom of the carbonyl group [Fig. 5 (orange)], thus forming a two-dimensional sheet. No polymorphs of sinapic acid were found using three solvents: acetone, acetonitrile and ethanol, while the methanol solvate obtained matched the previous report.32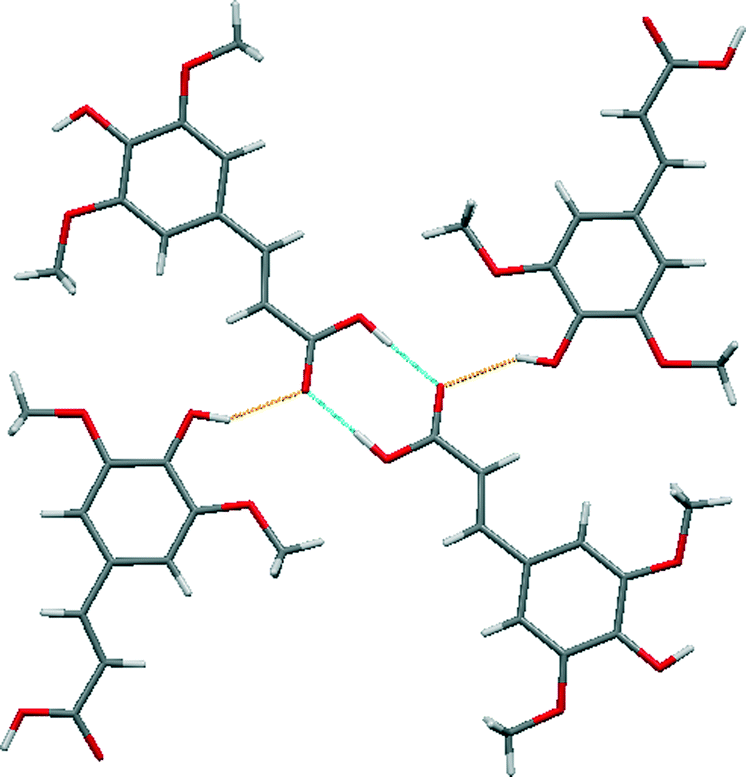 | ||
| Fig. 5 A section of the crystal structure of SA highlighting the key hydrogen bonding interactions. | ||
A comparison of the structure of sinapic acid with the reported structure of its methanol solvate (WAMFOG) reveals that the acid–acid homodimer motif is common to both crystal structures [Fig. 6 (blue)], with the exception being the R44(8) tetramer [Fig. 6 (orange)], which is formed by hydrogen bonds between two molecules of methanol and two phenolic moieties on adjacent sinapic acid molecules.32,40 This observation is in accordance with the data from the CSD on carboxylic acids,41 where in the absence of competing hydrogen bond acceptors, the carboxylic acid moiety will preferentially form the homodimer. Likewise, the phenolic substituent exhibits the common motifs associated with phenols,33i.e. O–H⋯OC acid heterosynthon in SA, and phenol O–H⋯O alcohol homosynthon in the methanol solvate.
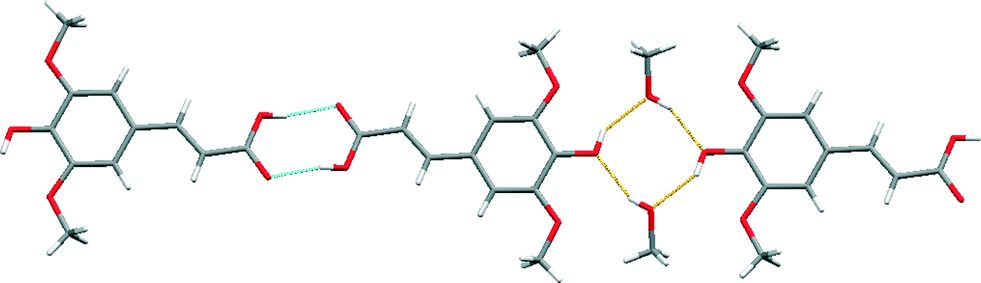 | ||
| Fig. 6 The reported crystal structure of the methanol solvate of sinapic acid (WAMFOG).32 | ||
The robust acid–acid homodimer in SA is retained in SA·NIA, with co-crystallization occurring via sinapic acid O–H⋯Naromatic hydrogen bonds (Fig. 7). Essentially, there are four different hydrogen bonds in the structure of SA·NIA: (a) acid–acid homodimer between two neighbouring sinapic acid molecules [Fig. 7 (blue)]; (b) discrete phenol O–H⋯Naromatic hydrogen bonds between SA and NIA responsible for extending the architecture [Fig. 7 (orange)]; (c) bifurcated amide N–H⋯O methoxy/phenol hydrogen bonds which extends the structure in two dimensions [Fig. 7 (magenta)]; and (d) anti-amide N–H⋯OC amide C(4) chains responsible for interlinking adjacent layers [Fig. 7 (black) and 8]. Also, prevalent in the structure of this co-crystal is the R44(22) tetramer formed by two sinapic acid and two nicotinamide molecules.
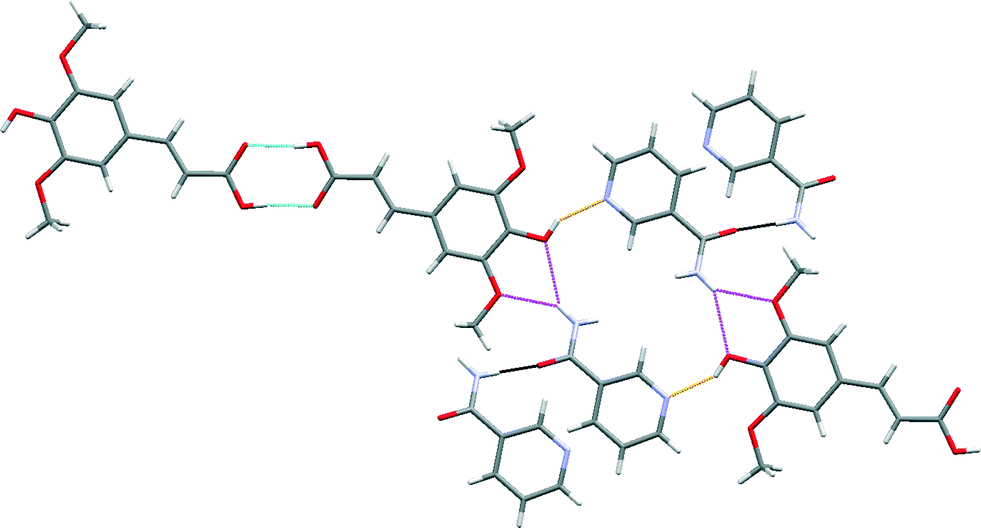 | ||
| Fig. 7 A section of the crystal structure of SA·NIA highlighting the key hydrogen bonding interactions. | ||
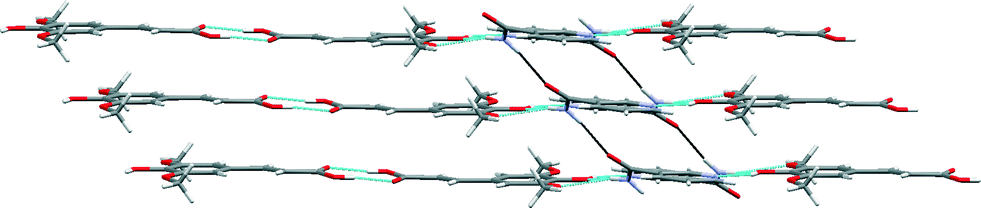 | ||
| Fig. 8 Amide N–H⋯OC amide C(4) chains (black) responsible for interlinking adjacent layers in the SA·NIA co-crystal. | ||
The crystal structure of sinapic acid–4-pyridylcarbonitrile (SA·PYC) co-crystal displays the same two key hydrogen bonds as SA·NIA, i.e. the robust acid–acid homodimer [Fig. 9 (blue)] and the phenol O–H⋯Naromatic hydrogen bonds [Fig. 9 (orange)].
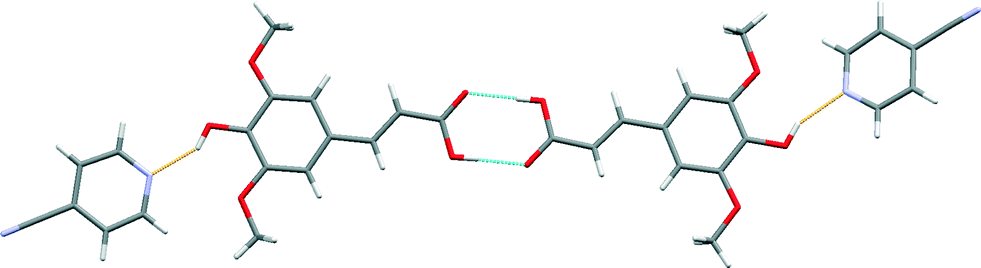 | ||
| Fig. 9 A section of the crystal structure of SA·PYC highlighting the key hydrogen bonding interactions. | ||
The carboxylic acid (+246 kJ mol−1) moiety on sinapic acid is a stronger hydrogen bond donor than the phenolic (+218 kJ mol−1) moiety based on its molecular electrostatic potentials (MEPs).15 The presence of the electron donating methoxy groups in the ortho position on the aromatic ring are responsible for decreasing the calculated MEP values of the phenolic moiety. Thus, it is expected that the carboxylic acid moiety would form co-crystals utilizing the range of heteromeric hydrogen bonds available, such as acid–acid, acid–amide and acid–Naromatic.34 Despite this, surprisingly, in the structures of the co-crystals SA·NIA and SA·PYC, the best donor (carboxylic acid) is not hydrogen-bonded to the best acceptor (pyridine nitrogen atom).42 The acid–acid homodimer is retained and co-crystallization occurs via phenol O–H⋯Naromatic hydrogen bonds. This has also been observed in the co-crystals of ferulic acid,43 a similar hydroxycinnamic acid, where the acid–acid dimer is retained, and no hydrogen bonds are observed between the best donor (carboxylic acid) and the best acceptor (pyridine nitrogen atom).
The crystal structure of SA·PTU·2MeCN displays three primary hydrogen-bonded interactions: (a) SA acid–amide (secondary) PTU R22(8) heterodimer [Fig. 10 (blue)]; (b) PTU thioamide–thioamide R22(8) homodimer between two adjacent PTU molecules (homodimer is capped on both ends by the heterodimer) [Fig. 10 (magenta)]; and (c) SA phenol O–H⋯N acetonitrile discrete hydrogen-bonds [Fig. 10 (orange)]. Furthermore, the hydrogen-bonded network is completed by structure-stabilising weak hydrogen bonds between the methyl hydrogen atom of acetonitrile and the methoxy oxygen atom of sinapic acid [Fig. 10 (black)] (C31–H31B⋯O12 2.28 Å, C31⋯O12 3.21 Å, C31–H31B⋯O12 158.7°). This hydrogen-bonded network resembles 1-D zig-zag chains, and adjacent chains are close-packed to give an overall supramolecular architecture resembling 2-D sheets. The acetonitrile molecules occupy voids in the sheets with weak C–H⋯N hydrogen bonds with adjacent molecules of SA and PTU (Fig. 11).
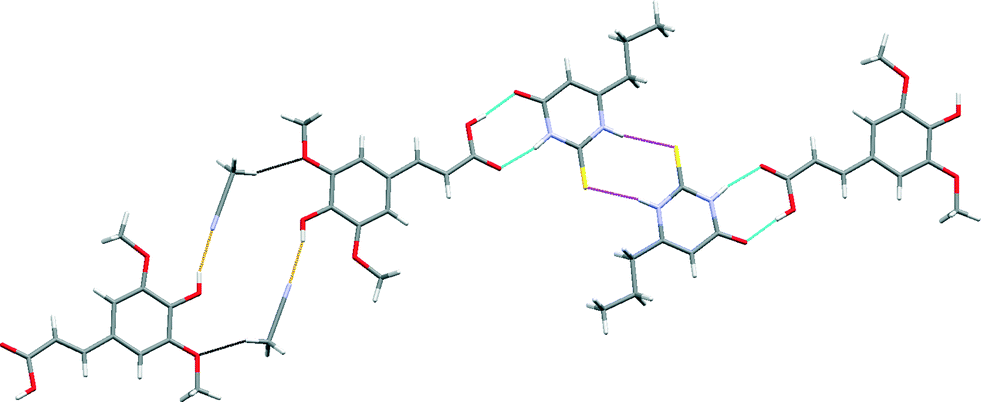 | ||
| Fig. 10 A section of the crystal structure of SA·PTU·2MeCN highlighting the key hydrogen bonding interactions. | ||
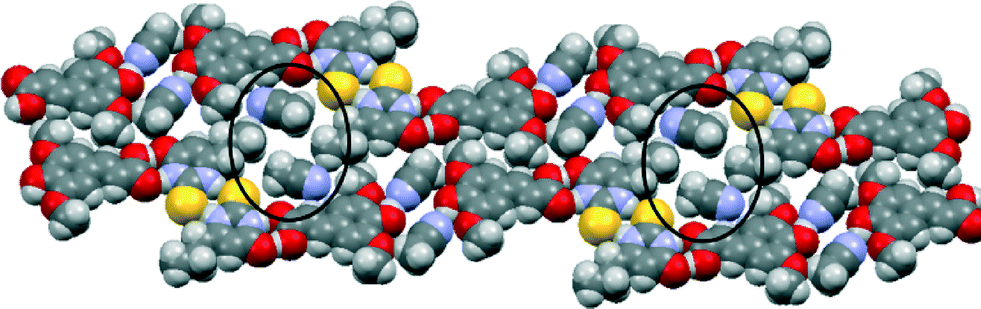 | ||
| Fig. 11 Space-filling model of the crystal structure of SA·PTU·2MeCN highlighting the voids occupied by acetonitrile molecules. | ||
A comparison of the structure of SA·PTU·2MeCN with the reported structure of PTU reveals that the thioamide–thioamide R22(8) homodimer motif is common to both crystal structures.44 A notable difference is the secondary amide–amide homodimer motif in PTU, which is unsurprisingly replaced by the preferred secondary amide–acid heterodimer in SA·PTU·2MeCN.
Acetonitrile molecules are required for the formation of this co-crystal since all other attempts to grow single crystals of a co-crystal of SA with PTUvia solution crystallization proved unsuccessful. DSC analysis of the powder obtained from a grinding experiment of an equimolar mixture of SA and PTU reveals the formation of a material with the same melting point (170 °C) as observed in the DSC analysis of SA·PTU·2MeCN (see ESI†). Based on this knowledge, it may be theorised that for 6-propyl-2-thiouracil to co-crystallize with sinapic acid, the unsatisfied hydrogen-bond donor (phenolic moiety of SA) must be satisfied by a suitable hydrogen bond acceptor (acetonitrile nitrogen atom). Failing this, the two components would not come together in the crystal lattice. Hence, perhaps due to the size and shape of acetonitrile molecules, and the presence of a hydrogen bond acceptor in the form of the nitrile nitrogen atom, it was an ideal candidate to facilitate co-crystallization.
Thermal stability
The thermal stability and phase transitions of APIs are important in the study of their physicochemical properties. The DSC data for the co-crystals SA·INC, SA·NIA and SA·PYC show single endotherm peaks, which indicates that these co-crystals are thermally stable up to 224, 175 and 143 °C respectively, with the melting point of the co-crystals lying in-between that of the respective co-formers in all cases. Also, single endotherms in co-crystals SA·INC, SA·NIA and SA·PYC indicate that there is no decomposition prior to melting. Evolution of acetonitrile from SA·PTU·2MeCN occurred between 52–82 °C and the second endotherm for the melting point of the desolvated material was observed at 170 °C (Fig. 12). TGA data also support the equimolar stoichiometry in SA·PTU·2MeCN (Fig. 13). The powder X-ray diffraction pattern of the desolvated material clearly shows that the crystal lattice of SA·PTU·2MeCN falls apart, degrading to a physical mixture of the two components (Fig. 14). This further highlights the importance of the acetonitrile molecules in the formation of SA·PTU·2MeCN.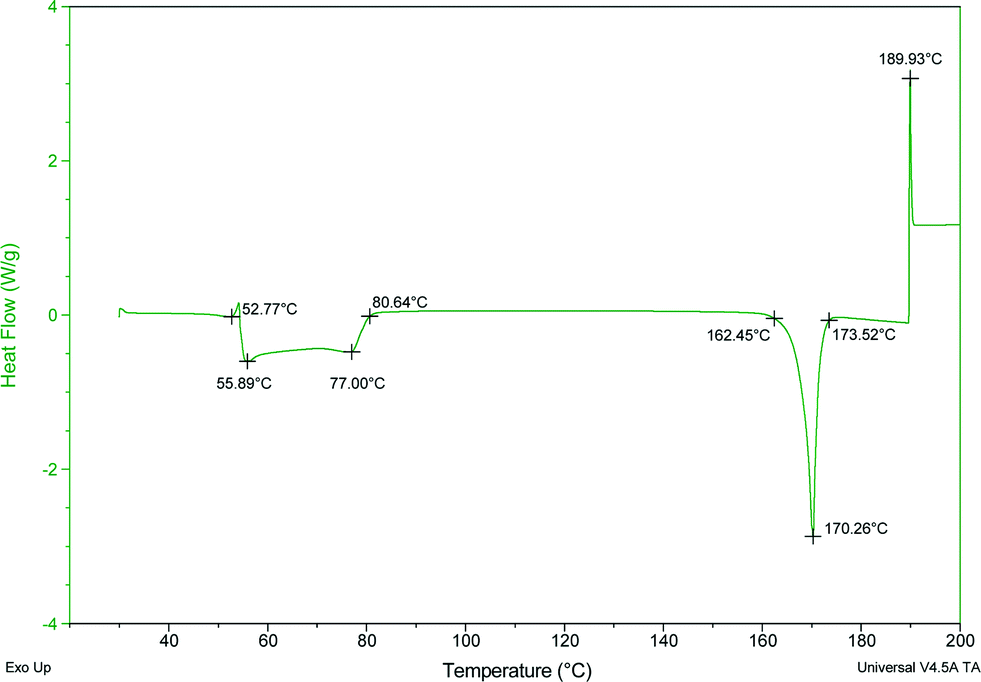 | ||
| Fig. 12 DSC spectrum of SA·PTU·2MeCN showing the loss of the acetonitrile molecules. | ||
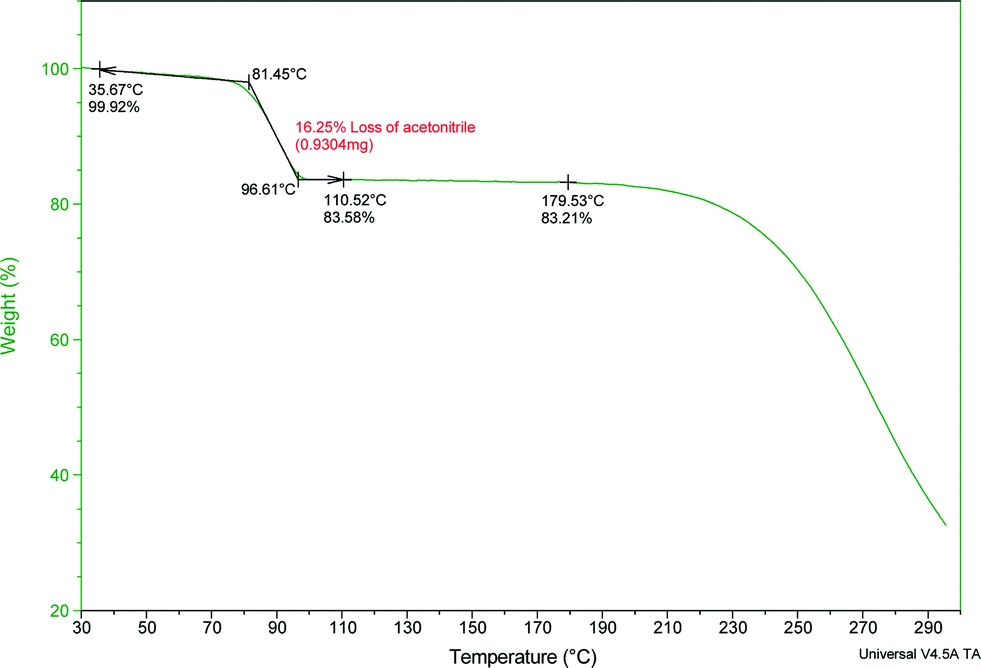 | ||
| Fig. 13 TGA spectrum of SA·PTU·2MeCN showing the loss of the acetonitrile molecules. | ||
 | ||
| Fig. 14 PXRD pattern of SA·PTU·2MeCN (blue) compared with desolvated sample (black), sinapic acid (green), and 6-propyl-2-thiouracil (red). | ||
Physical form stability
An important characteristic of new pharmaceutical formulations that should be determined is the stability of the material with respect to changes in relative humidity and temperature. This knowledge aids in the storage and transportation of APIs without affecting drug activity. Sinapic acid is stable at accelerated ICH conditions of 40 °C and 75% RH for up to 8 weeks as determined by PXRD and HPLC (Table 3). Three of the co-crystals, SA·INC, SA·NIA and SA·PYC, are stable under the same conditions for 5, 8 and 5 weeks, respectively, as determined by PXRD and HPLC, while SA·PTU·2MeCN is stable for only one week. This is indicated in the PXRD pattern by the appearance of peaks due to the co-former and loss of peaks associated with the co-crystal (see ESI†). HPLC analyses of the dissolved co-crystals displayed only co-former peaks at 250, 280 and 320 nm, which indicates that dissociation of the co-crystals is the major degradative pathway operating under these ICH conditions. We have also examined the equilibrium solubility of the co-crystals and did not observe any improvement in the solubility of sinapic acid in the co-crystals.| Compound | 1 week | 2 week | 3 week | 4 week | 5 week | 6 week | 7 week | 8 week |
|---|---|---|---|---|---|---|---|---|
| √ = stable; X = unstable. | ||||||||
| SA | √ | √ | √ | √ | √ | √ | √ | √ |
| SA·INC | √ | √ | √ | √ | √ | X | X | X |
| SA·NIA | √ | √ | √ | √ | √ | √ | √ | √ |
| SA·PYC | √ | √ | √ | √ | √ | X | X | X |
| SA·PTU·2MeCN | √ | X | X | X | X | X | X | X |
Conclusions
The co-crystallization of sinapic acid was attempted with a total of 25 different co-formers, 21 of which were simple model compounds (ten carboxylic acids, eight amides, three pyridines) and four were APIs. Four co-formers were successful and formed co-crystals which were isolated and characterised (one carboxylic acid, one amide, one pyridine and one API), and three co-crystals were structurally characterised by X-ray diffraction. The model compounds that were successful all had a pyridyl functional group present, although this did not guarantee success as three compounds containing this group (isonicotinamide, 4-hydroxypridine and 4,4′-bipyridine) did not form a co-crystal with sinapic acid. In the two model compounds that were structurally characterised, the sinapic acid forms acid–acid homodimers with the other co-formers joining these homodimers together via hydrogen bonding. It is only in the case of the acetonitrile solvate of sinapic acid with 6-propyl-2-thiouracil that acid–amide heterodimers between the two components are observed, and the presence of acetonitrile is necessary for the co-crystal to form. Thus, it appears that the sinapic acid homodimer is not easily replaced by acid–acid or acid–amide heterodimers. Participation of the phenol group is an important consideration when considering co-crystallization of sinapic acid, with phenol O–H⋯N hydrogen bonds consistently displayed in all the co-crystals structurally characterised.All co-crystals show good stability towards accelerated ICH humidity conditions with the exception of the solvate, which decomposes after one week with the loss of solvent. The co-crystal of sinapic acid with nicotinamide shows high stability and, thus, is an ideal candidate for exploring pharmaceutical formulations of sinapic acid.
Acknowledgements
This publication has emanated from research conducted with the financial support of Science Foundation Ireland under grant numbers 12/RC/2275 and 05/PICA/B802/EC07, and UCC 2013 Strategic Research Fund.Notes and references
- R. Pepinsky, Phys. Rev., 1955, 100, 971 CAS.
- G. M. Schmidt, Pure Appl. Chem., 1971, 27, 647 CrossRef CAS.
- G. R. Desiraju, Crystal Engineering: The design of Organic Solids, Elsevier, Amsterdam, 1989 Search PubMed.
- C. B. Aakeröy, B. M. T. Scott, M. M. Smith, J. F. Urbina and J. Desper, Inorg. Chem., 2009, 48, 4052 CrossRef CAS PubMed; L. Garzon-Tovar, A. Duarte-Ruiz and K. Wurst, Inorg. Chem. Commun., 2013, 32, 64 CrossRef PubMed; T. Mes, S. Cantekin, D. W. R. Balkenende, M. M. M. Frissen, M. A. J. Gillissen, B. F. M. De Waal, I. K. Voets, E. W. Meijer and A. R. A. Palmans, Chem. – Eur. J., 2013, 19, 8642 CrossRef PubMed.
- C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439 RSC.
- P. Sanphui, S. Tothadi, S. Ganguly and G. R. Desiraju, Mol. Pharmaceutics, 2013, 10, 4687 CrossRef CAS PubMed; D. J. Good and N. Rodríguez-Hornedo, Cryst. Growth Des., 2009, 5, 2252 Search PubMed; C. B. Aakeröy, S. Forbes and J. Desper, J. Am. Chem. Soc., 2009, 131, 17048 CrossRef PubMed; K. Shiraki, N. Takata, R. Takano, Y. Hayashi and K. Terada, Pharmacol. Res., 2008, 25, 2581 CrossRef PubMed.
- A. V. Trask, W. D. Samuel Motherwell and W. Jones, Int. J. Pharm., 2006, 320, 114 CrossRef CAS PubMed; N. Rodríguez-Hornedo, S. J. Nehm and A. Jayasankar, Cocrystals: Design, Properties, and Formation Mechanisms, in Encyclopedia of Pharmaceutical Technology, ed. J. Swarbrick, Informa Healthcare, USA, 3rd edn, 2006 Search PubMed; A. V. Trask, W. D. Samuel Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013 Search PubMed.
- M. L. Cheney, N. Shan, E. R. Healy, M. Hanna, L. Wojtas, M. J. Zaworotko, V. Sava, S. Song and J. R. Sanchez-Ramos, Cryst. Growth Des., 2010, 10, 394 Search PubMed; M. B. Hickey, M. L. Peterson, L. A. Scoppettuolo, S. L. Morrisette, A. Vetter, H. Guzmán, J. F. Remenar, Z. Zhang, M. D. Tawa, S. Haley, M. J. Zaworotko and Ö. Almarsson, Eur. J. Pharm. Biopharm., 2007, 67, 112 CrossRef CAS PubMed; D. P. McNamara, S. L. Childs, J. Giordano, A. Iarriccio, J. Cassidy, M. S. Shet, R. Mannion and A. Park, Pharmacol. Res., 2006, 23, 1888 CrossRef PubMed.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. Swift, R. Thaimattam, T. S. Thakur, R. K. Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147 Search PubMed; N. Blagden, S. J. Coles and D. J. Berry, CrystEngComm, 2014, 16, 5753 RSC; J. W. Steed, Trends Pharmacol. Sci., 2013, 34, 185 CrossRef CAS PubMed; H. G. Brittain, J. Pharmacol. Sci., 2013, 102, 311 CrossRef PubMed.
- E. Nauha, Crystalline forms of selected agrochemical actives: design and synthesis of co-crystals, Department of Chemistry, University of Jyväskylä, Research report No. 151, 2012 Search PubMed.
- O. Bolton, L. R. Simke, P. F. Pagoria and A. J. Matzger, Cryst. Growth Des., 2012, 12, 4311 Search PubMed; K. B. Landenberger, O. Bolton and A. J. Matzger, Angew. Chem., Int. Ed., 2013, 52, 6468 CrossRef CAS PubMed; J. Evers, I. Gospodinov, M. Joas, T. M. Klapötke and J. Stierstorfer, Inorg. Chem., 2014, 53, 11749 CrossRef PubMed.
- V. Brower, Nat. Biotechnol., 1998, 16, 728 CrossRef CAS PubMed.
- S. H. Zeisel, Science, 1999, 285, 1853 CrossRef CAS; E. K. Kalra, AAPS PharmSci, 2003, 5, e25 CrossRef PubMed.
- A. Munin and F. Edwards-Lévy, Pharmaceutics, 2011, 3, 793 CrossRef CAS PubMed; M. Massaro, E. Scoditti, M. A. Carluccio and R. De Caterina, Cardiovasc. Ther., 2010, 28, e13 CrossRef PubMed; M. Pandey, R. K. Verma and S. A. Saraf, Asian J. Pharm. Clin. Res., 2010, 3, 11 Search PubMed.
- A. S. Sinha, A. R. Maguire and S. E. Lawrence, Cryst. Growth Des., 2015, 15, 984 CAS.
- Generally Regarded as Safe: http://www.cfsan.fda.gov/~rdb/opa gras.html and http://www.cfsan.fda.gov/~dms.grasguid.html.
- Everything Added to Food in the United States: http://www.usc.es/caa/EdulcWeb/EAFUS.pdf.
- B. S. Sekhon, RGUHS J. Pharm. Sci., 2012, 2, 16 Search PubMed.
- N. Nićiforović and H. Abramovič, Compr. Rev. Food Sci. Food Saf., 2014, 13, 34 CrossRef; C. Chen, Sinapic Acid and Its Derivatives as Medicine in Oxidative Stress-Induced Diseases and Aging, Oxid. Med. Cell. Longevity, 706190 Search PubMed , in press.
- F. Natella, M. Nardini, M. Di Felice and C. Scaccini, J. Agric. Food Chem., 1999, 47, 1453 CrossRef CAS PubMed; H. Kikuzaki, M. Hisamoto, K. Hirose, K. Akiyama and H. Taniguchi, J. Agric. Food Chem., 2002, 50, 2161 CrossRef PubMed; Y. Zou, A. R. Kim, J. E. Kim, J. S. Choi and H. Y. Chung, J. Agric. Food Chem., 2002, 50, 5884 CrossRef PubMed.
- S. Tesaki, S. Tanabe, H. Ono, E. Fukushi, J. Kawabata and M. Watanabe, Biosci., Biotechnol., Biochem., 1998, 62, 998 CrossRef CAS; C. E. Maddox, L. M. Laur and L. Tian, Curr. Microbiol., 2010, 60, 53 CrossRef PubMed; C. Engels, A. Schieber and M. G. Gänzle, Eur. Food Res. Technol., 2012, 234, 535 CrossRef.
- K.-J. Yun, D.-J. Koh, S.-H. Kim, S. J. Park, J. H. Ryu, D.-G. Kim, J.-Y. Lee and K.-T. Lee, J. Agric. Food Chem., 2008, 56, 10265 CrossRef PubMed.
- E. A. Hudson, P. A. Dinh, T. Kokubun, M. S. Simmonds and A. Gescher, Cancer Epidemiol., Biomarkers Prev., 2000, 9, 1163 CAS.
- B. H. Yoon, J. W. Jung, J.-J. Lee, Y.-W. Cho, C.-G. Jang, C. Jin, T. H. Oh and J. H. Ryu, Life Sci., 2007, 81, 234 CrossRef CAS PubMed.
- M.-E. Cuvelier, H. Richard and C. Berset, Biosci., Biotechnol., Biochem., 1992, 56, 324 CrossRef CAS.
- R. J. Robbins, J. Agric. Food Chem., 2003, 51, 2866 CrossRef CAS PubMed.
- H. Kuwahara, A. Kanazawa, D. Wakamatu, S. Morimura, K. Kida, T. Akaike and H. Maeda, J. Agric. Food Chem., 2004, 52, 4380 CrossRef CAS PubMed.
- L. He, H. T. Li, S. W. Guo, L. F. Liu, J. B. Qiu, F. Li and B. C. Cai, Zhongguo Zhongyao Zazhi, 2008, 33, 813 Search PubMed; F. Ferreres, F. Fernandes, C. Sousa, P. Valentão, J. A. Pereira and P. B. Andrade, J. Agric. Food Chem., 2009, 57, 8884 CrossRef CAS PubMed.
- O. G. Bountagkidou, S. A. Ordoudi and M. Z. Tsimidou, Food Res. Int., 2010, 43, 2014 CrossRef CAS PubMed; L. Trnková, I. Boušová, L. Ryšánková, P. Vrabcová and J. Dršata, Proc. ECOpole, 2009, 3, 27 Search PubMed.
- S. M. Kern, R. N. Bennett, F. A. Mellon, P. A. Kroon and M.-T. Garcia-Conesa, J. Agric. Food Chem., 2003, 51, 6050 CrossRef CAS PubMed.
- K. Zhang and Y. Zuo, J. Agric. Food Chem., 2004, 52, 222 CrossRef CAS PubMed.
- H. Qian and W. Huang, J. Mol. Struct., 2005, 743, 191 CrossRef CAS PubMed.
- C. Laurence, M. Berthelot and J. Graton, Hydrogen-Bonded Complexes of Phenols, in Phenols, ed. Z. Rappoport, John Wiley & Sons, Ltd., Chichester, U.K., 2003 RSC; O. Ermer and A. Eling, J. Chem. Soc., Perkin Trans. 2, 1994, 925 RSC; F. G. Bordwell, R. J. McCallum and W. N. Olmstead, J. Org. Chem., 1984, 49, 1424 CrossRef CAS.
- D. Hadzi and S. Detoni, Hydrogen bonding in carboxylic acids and derivatives, in Acid Derivatives, ed. S. Patai, John Wiley & Sons, Ltd., Chichester, U.K., 1979, vol. 1 Search PubMed; W. C. Hamilton and J. A. Ibers, Hydrogen Bonding in Solids, ed. W. A. Benjamin, New York, 1968 Search PubMed.
- G. M. Sheldrick, Acta Cryst., 2008, A64, 112 CrossRef PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Cryst., 2008, 41, 466 CrossRef CAS.
- H. Nakamura, J. Y. Noh, K. Itoh, S. Fukata, A. Miyauchi and N. Hamada, J. Clin. Endocrinol. Metab., 2007, 92, 2157 CrossRef CAS PubMed.
- K. Nakamoto, Y. Morimoto and A. E. Martell, J. Phys. Chem., 1962, 66, 346 CrossRef; G. Socrates, Infrared and Raman charateristic group frequencies; tables and charts, John Wiley and Sons, 2004 Search PubMed.
- S. Inagaki, H. Murai and T. Takeuchi, Phys. Chem. Chem. Phys., 2012, 14, 2008 RSC; J. Joseph and E. D. Jemmis, J. Am. Chem. Soc., 2007, 129, 4620 CrossRef CAS PubMed; X. Li, L. Liu and H. B. Schlegel, J. Am. Chem. Soc., 2002, 124, 9639 CrossRef PubMed.
- For a description of graph set notation for classifying hydrogen-bonding patterns, see: J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS PubMed; M. C. Etter, Acc. Chem. Res., 1990, 23, 120 CrossRef.
- T. R. Shattock, K. K. Arora, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2008, 8, 4533 CAS.
- For a description of best donor - best acceptor theory for hydrogen bonding, see: M. C. Etter and G. M. Frankenbach, Chem. Mater., 1989, 1, 10 CrossRef CAS; M. C. Etter, J. Phys. Chem., 1991, 95, 4601 CrossRef.
- B. Swapna, D. Maddileti and A. Nangia, Cryst. Growth Des., 2014, 14, 5991 CAS; Z. Tan, E. Zhu, L. Luo, Z. Lin and R. Yan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o424 Search PubMed.
- F. F. Ferreira, A. C. Trindade, S. G. Antonio and C. O. Paiva-Santos, CrystEngComm, 2011, 13, 5474 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed list of co-formers, IR, PXRD, DSC, TGA, stability data, and additional figures. CCDC 1060811–1060815. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce00777a |
| This journal is © The Royal Society of Chemistry 2015 |