Novel, electrolyte solutions comprising fully inorganic salts with high anodic stability for rechargeable magnesium batteries†
Robert E.
Doe
*a,
Ruoban
Han
a,
Jaehee
Hwang
a,
Andrew J.
Gmitter
a,
Ivgeni
Shterenberg
b,
Hyun Deog
Yoo
b,
Nir
Pour
b and
Doron
Aurbach
b
aPellion Technologies, STE 105, 625 Mount Auburn St., Cambridge, MA, USA. E-mail: rob@pelliontech.com
bDepartment of Chemistry, Bar-Ilan University, Ramat-Gan, Israel 92100
First published on 8th November 2013
Abstract
Herein the first inorganic magnesium salt solution capable of highly reversible magnesium electrodeposition is presented. Synthesized by acid–base reaction of MgCl2 and Lewis acidic compounds such as AlCl3, this salt class demonstrates upwards of 99% Coulombic efficiency, deposition overpotential of <200 mV, and anodic stability of 3.1 V.
More than ever there is demand for energy storage devices with substantially higher energy density than lithium ion (Li-ion) batteries will be capable of delivering. One increasingly attractive avenue to achieving such energy density has been to replace monovalent Li+ with a multivalent ion such as Mg2+ in order to shuttle twice the charge between electrode materials that are site limited, but not electron limited. Furthermore, previous efforts provide strong evidence for the prospect that Mg chemistry is capable of providing ultrahigh volumetric energy density obtained by employing an Mg metal anode (3833 A h l−1).1 That said, there are many limitations with implementing Mg as a viable anode for secondary batteries. One major issue is the reductive nature of Mg metal towards a variety of common electrolyte components (e.g., organic solvents) and impurities (e.g., water, oxygen, organics). In general, unintended reduction of electrolyte components precipitates as insoluble products that passivate the Mg electrode surface and ‘block’ transport of both ions and electrons. This characteristic is distinctly different from Li metal and Li-ion anodes which form passivating layers with a wide variety of electrolyte solution components that generally provide sufficient Li-ion transport between the solid and solution state. Consequently, the nature of Mg-ion mobility dictates which salts and solvents are compatible with a magnesium anode. Ideally all electrolyte components are thermodynamically stable, but in principle they must be at least kinetically stable in the sense that they do not form a stable, transport blocking film, or soluble reaction products that result in complete irreversible consumption of the electrolyte in contact with magnesium.
Nearly 100 years ago organometallic Grignard-containing solutions were first shown to meet the above requirements.2 To date, organometallic Mg-ion electrolyte solutions are the only class of solutions to repeatedly demonstrate facile, highly reversible Mg electrodeposition although many other Mg salts have been examined, including those akin to that used in Li-ion (i.e., inorganic salts and the wide variety of polar, aprotic solvents).3,4 The general trend among the wide variety of Grignard-based solutions demonstrated thus far is that solutions based purely upon Grignard compounds exhibit low conductivity and low anodic stability (<1 V), which makes them impractical for use in Mg-ion batteries. However the concept that complexing organometallic species will increase Mg-ion conductivity and anodic stability while mitigating chemical reactivity was first shown about 25 years ago.5 Subsequently it took 10 more years of effort to further that idea, and to identify a proper combination of electrolyte solution and cathode in order to demonstrate the first prototype Mg battery.1 In this prototype the electrolyte solution consists of complex products represented as (Mg(AlCl4−nRn)2) wherein R represents an organic moiety (e.g., alkyl, aryl, etc.) resulting from an acid base reaction between R2Mg Lewis base and a Lewis acid (e.g., AlCl2Et, AlCl3, BPh3).6 A plethora of similar electrolyte solutions followed along with many attempts to enable reversible Mg electrodeposition from inorganic salts akin to Li-ion, however none of the latter class has clearly and repeatedly demonstrated facile, highly reversible Mg electrodeposition.3,4 Commercialization of Mg-ion batteries employing electrolytes based upon organometallic reagents such as Grignard compounds would be challenging. Virtually all organometallic species of the alkalis, earth alkalis and aluminium are highly unstable in the presence of air and water and many are classified as pyrophoric. In addition, organometallic species are easily oxidized species, and generally limit the electrochemical stability window of the solution and dominate the chemical reactivity of the solution in contact with various electrode materials and other cell components. Hence, there is real need to enhance the space of possible electrolytes for Mg batteries.
Herein we present for the first time an inorganic Mg-ion electrolyte capable of highly reversible Mg electrodeposition, and a window of electrochemical stability a near 3 V Mg-ion battery at room temperature.7 The salt solution comprises the reaction products of MgCl2 and AlCl3 and occurs in a variety of solvents, including ethereal solutions such as tetrahydrofuran (THF), 1,2-dimethoxyethane (monoglyme, DME) and higher glymes such as tetraglyme. Our preliminary findings indicate that the inorganic Magnesium Aluminium Chloride complex (i.e., MACC) forms as a result of an acid–base reaction in a similar manner to that reported for the transmetalation in the formation of the Grignard-based complexes. In a general fashion:
| mMgCl2 + nAlCl3 ⇆ MgmAlnCl[(2*m)+(3*n)] | (1) |
The principle of the above reaction is a simple and direct route to circumventing the limitations of the solutions containing organometallic moieties; however it is quite counterintuitive that it should occur in practice.9 This is owing to the low solubility of MgCl2 in a number of solvents and also the fact that MgCl2 is relatively chemically inert as it does not dissociate in solutions based upon aprotic organic solvents to an appreciable extent. Consequently a solution containing only MgCl2 will have very low to no measurable conductivity, and enable no Mg deposition, dissolution, or intercalation. In contrast, Fig. 1 depicts a typical cyclic voltammogram of a 0.25 M Mg salt solution depicted in eqn (1) as carried out in DME. The reductive peak displaying maximum current density near −1 V is due to the deposition of Mg metal while the oxidative peak with maximum current density at about 0.3 V is attributed to the subsequent electrochemical dissolution of the Mg metal. The electrochemical window obtained with this system exceeds 3.1 V vs. Mg metal. It is clearly evident from the cyclic voltammogram that the process of magnesium deposition and dissolution is highly reversible. Similarly, Fig. 2 shows the results of preliminary microcoulometry experiments for a cell containing a Cu working electrode and an Mg counter electrode and MACC 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in THF. About 2.5 μm of Mg were initially plated onto the working electrode surface and 20% of that was cycle 50 times at 0.5 mA cm−2. The Coulombic efficiency of this process to a final stripping potential of 1 V is 98.8%. Despite the initial activation overpotential the cycling occurred over 50 cycles with an average cell overpotential of <200 mV. The preliminary values are similar to, but slightly less than Grignard-based reagents, so further comparative study is necessary to understand what performance differences are intrinsic to the solutions.10
:1 in THF. About 2.5 μm of Mg were initially plated onto the working electrode surface and 20% of that was cycle 50 times at 0.5 mA cm−2. The Coulombic efficiency of this process to a final stripping potential of 1 V is 98.8%. Despite the initial activation overpotential the cycling occurred over 50 cycles with an average cell overpotential of <200 mV. The preliminary values are similar to, but slightly less than Grignard-based reagents, so further comparative study is necessary to understand what performance differences are intrinsic to the solutions.10
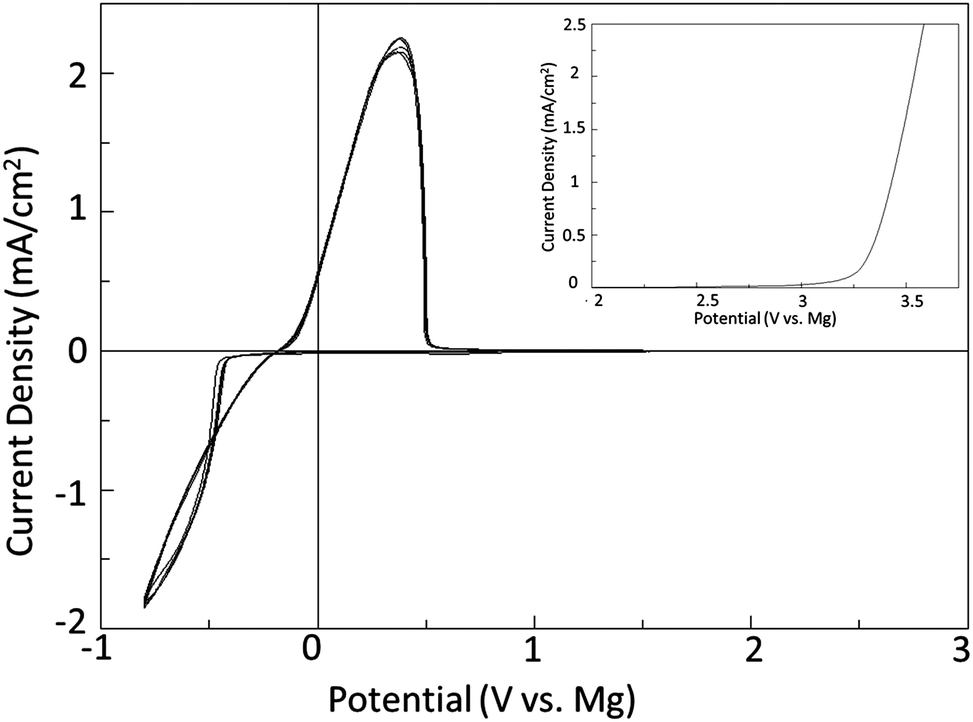 | ||
| Fig. 1 Depicts the cyclic voltammogram and linear sweep voltammetry (insert) of 0.25 M MACC 2:1 solution in DME. The working electrode is Pt while the counter and reference electrodes are Mg metal. Measurements are obtained at 25 mV s−1 and ambient conditions. | ||
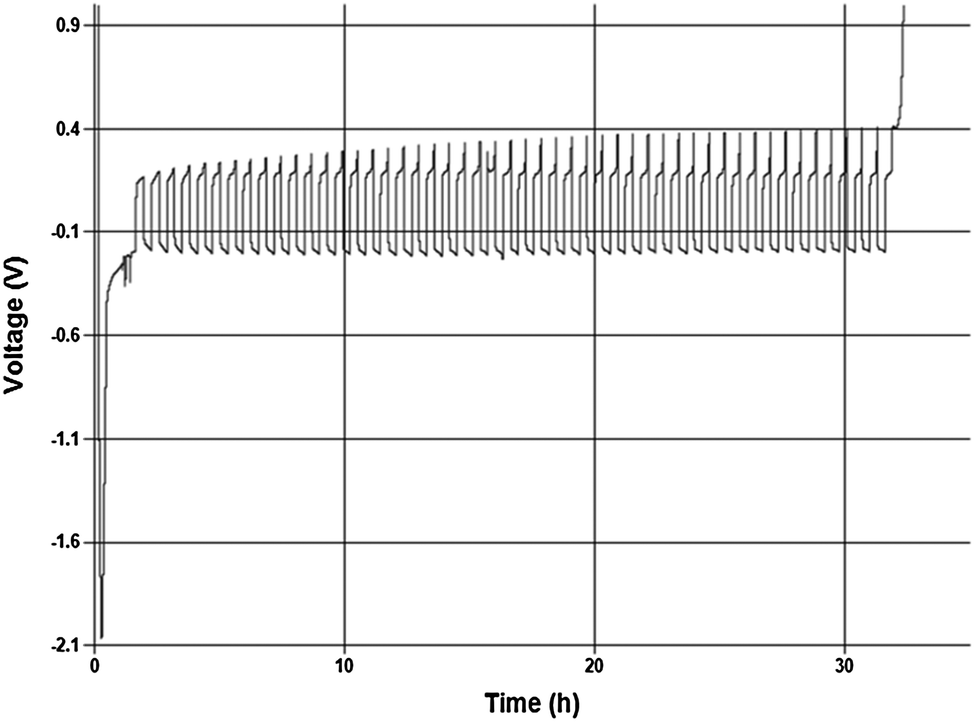 | ||
| Fig. 2 Displays a graph of the potential response resulting during chronopotentiometry experiments carried out with 0.25 M MACC 2:1 solution in THF. The test utilize a two electrode cell (Cu vs. Mg) and a current density of 0.5 mA cm−2. 20% of an initial deposit of about 2.5 micron Mg was cycled 50 times at ambient conditions. | ||
In a similar experiment several microns of Mg were deposited on a Pt working electrode in order to observe the morphology as compared to Grignard-based complexes such as the all-phenyl complex.11Fig. 3A and B are scanning electron micrographs showing uniform, evenly distributed micron-sized particles similar to that observed in the Grignard-based solutions (Fig. 3C depicts a similar sample of magnesium plated from APC solution under the same conditions).10 The EDS elemental mapping of the sample in micrographs 3A and 3B confirms about 80 atomic percent Mg, but also significant Al and Cl, at about 13 and 9 atomic percent, respectively (after removing the background signal due to Pt substrate and O uptake during sample transfer). Although further study is required, it is anticipated that the Mg/Al ratio can be tuned with improved understanding of the role of solution composition and plating conditions has in regard to MACC solutions. Thus far we find that reasonable conductivity (i.e., ∼2 mS cm−1 at 25 °C) and electrodeposition can be obtained from solutions wherein the MgCl2:AlCl3 ratio ranges from about 1:2 to 3:1 and the Mg molarity is between about 0.1 M and 0.5 M, depending upon the solvent of choice.7
 | ||
| Fig. 3 Displays SEM micrographs of about 5 μm thick Mg resulting from galvanostatic plating of Mg onto a Pt working electrode at 1 mA cm−2. 0.25 M MACC 2:1 solution in DME is shown at lower and higher magnification in 3A and 3B, respectively. 3C results from 0.25 M APC 2:1 under the same conditions. | ||
While the evidence presented above demonstrates that this inorganic salt solution is capable of highly reversible Mg electrodeposition it is also important to determine whether any issues would be present when intercalating Mg2+ ions into electrode materials at potentials positive of Mg metal. A model compound to explore this characteristic is the so-called Chevrel phase, Mo6S8. Fig. 4 depicts a demonstration of Mg intercalation and removal under galvanostatic conditions in 0.25 M MACC 3:2 in DME for the first time. A typical capacity response of about 80 mA h g−1 is observed over eight charge–discharge cycles at a rate of C/15. The cell voltages comparable to those observed when cycling in Grignard-based Mg complex salt solutions. An additional 15 cycles indicates minimal capacity fade upon increasing the rate to C/2.
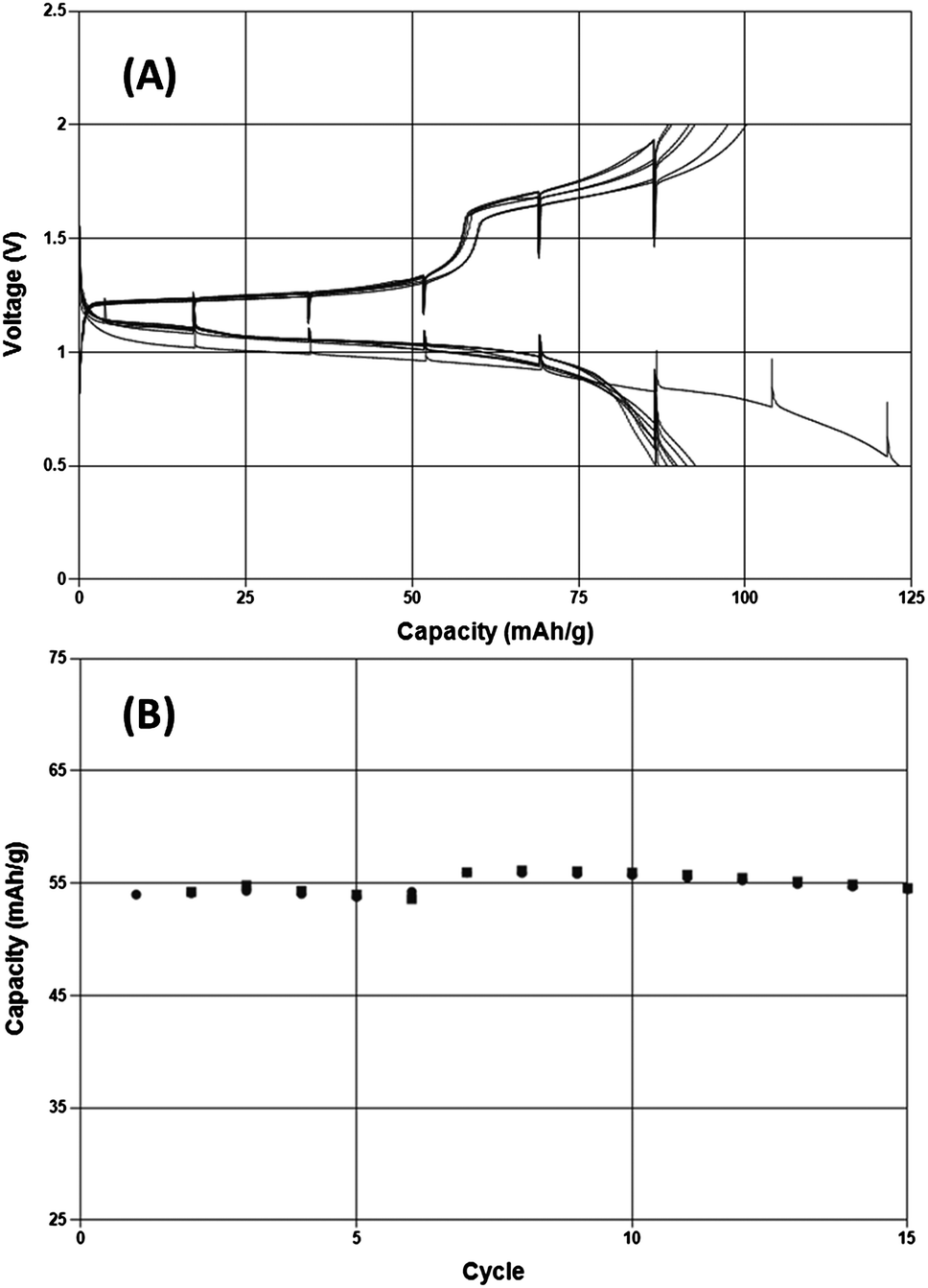 | ||
| Fig. 4 Galvanostatic cycling of Mo6S8 in 0.25 M MACC 3:2 in DME taken at C/15 for several cycles, shown as a voltage profile in (A) and followed by C/2 for an additional 15 cycles depicted as charge and discharge capacity vs. cycle number (B). Mg serves as the negative electrode. | ||
We have established that it is possible to prepare inorganic Mg salt solutions capable of both highly reversible electrodeposition at the negative electrode and Mg intercalation at the positive electrode. This family of solutions based upon reaction of MgCl2 and Lewis acidic compounds such as AlCl3 in ethereal solvents is capable of providing anodic stability to 3.1 V vs. Mg, conductivity comparable to that of Li-ion electrolytes, and Coulombic efficiency of at least 99%. Consequently we hope this work will spur additional study in the use of the inorganic Magnesium Aluminium Chloride complex and related salt solutions so as to enable the commercialization of high energy density Mg battery technology in the near-term.
The information, data, or work presented herein was funded in part by the Advanced Research Projects Agency – Energy (ARPA-E), U.S. Department of Energy, under Award Number DE-AR0000062 and by Pellion Technologies. The authors wish to thank Dr Yosef Gofer for insightful discussions in the course of this work.
References
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- L. W. Gaddum and H. E. French, J. Am. Chem. Soc., 1927, 49, 1295–1299 CrossRef CAS.
- J. Muldoon, C. B. Bucur, A. G. Oliver, T. Sugimoto, M. Matsui, H. S. Kim, G. D. Allred, J. Zajicek and Y. Kotani, Energy Environ. Sci., 2012, 5, 5941–5950 CAS.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 CAS.
- T. D. Gregory, R. J. Hoffman and R. C. Winterton, J. Electrochem. Soc., 1990, 137, 775–780 CrossRef CAS PubMed.
- D. Aurbach, H. Gizbar, A. Schechter, O. Chusid, H. E. Gottlieb, Y. Gofer and I. Goldberg, J. Electrochem. Soc., 2002, 149, A115–A121 CrossRef CAS PubMed.
- R. E. Doe, R. Han, Y. Gofer, D. Aurbach, N. Pour and I. Sterenberg, Non-Aqueous Electrolyte for Rechargeable Magnesium Ion Cell, International Pat. App., PCT/US2012/071350, 2013 Search PubMed.
- Y. Vestfried, O. Chusid, Y. Goffer, P. Aped and D. Aurbach, Organometallics, 2007, 26, 3130–3137 CrossRef CAS.
- Y. Viestfrid, M. D. Levi, Y. Gofer and D. Aurbach, J. Electroanal. Chem., 2005, 576, 183–195 CrossRef CAS PubMed.
- M. Matsui, J. Power Sources, 2011, 196, 7048–7055 CrossRef CAS PubMed.
- N. Pour, Y. Gofer, D. T. Major and D. Aurbach, J. Am. Chem. Soc., 2011, 133, 6270–6278 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc47896c |
| This journal is © The Royal Society of Chemistry 2014 |