 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCathodic photocurrent generation from zinc-substituted cytochrome b562 assemblies immobilized on an apocytochrome b562-modified gold electrode
Akira Onoda*,
Yasuaki Kakikura and
Takashi Hayashi*
Department of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: onoda@chem.eng.osaka-u.ac.jp; thayashi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7930; Tel: +81-6-6879-7928
First published on 16th August 2013
Abstract
Hierarchical assemblies of the noteworthy photoactive cytochrome b562 reconstituted with zinc protoporphyrin IX covalently linked with the protein surface were constructed on a gold electrode modified with an apoprotein of cytochrome b562. The integrated photoactive hemoproteins were characterized by electrochemical impedance and quartz crystal microbalance analyses. The protein-immobilized electrode exhibits enhanced photocurrent generation relative to the one having a Zn-substituted hemoprotein monolayer.
Introduction
Creation of an electron-transfer interface between a redox-active protein and electrode is a key technological step toward further development of biosensors and photoelectric conversion biodevices.1 To achieve an efficient electrochemical communication using a protein, many methodologies have been developed including protein immobilization which is mediated by noncovalent interactions, such as an electrostatic interaction with self-assembled monolayers (SAMs) on electrodes,2 an affinity interaction with tag peptides3 and a specific protein–cofactor interaction.4 A further strategy to enhance the electrochemical communication is the accumulation of proteins on the electrode. The immobilization of proteins as multilayers has been reported using electrostatic interaction,5 hydrophobic interaction,6 interaction with polyelectrolytes,7 and protein–nanoparticle interaction.8Among the redox-active proteins, hemoproteins seem to be promising components for the construction of functional protein-immobilized electrodes due to their remarkable functions, such as electron transfer, catalysis, and sensing. Hemoprotein-immobilized electrodes have thus become of wide interest to evaluate their electrochemical behaviors9 and/or generate a functional system, such as a biocatalyst,10 a biosensor,11 and a biofuel cell.12
We previously described the preparation of linear hemoprotein self-assemblies via a specific heme–heme pocket interaction, in which a heme moiety is externally attached to the surface of the H63C single mutant of cytochrome b562 (CYT)13 or the A125C single mutant of myoglobin.14 The supramolecular self-assembling systems of proteins15 also allowed us to immobilize these assemblies on an electrode surface. We thus recently reported the construction of assemblies of photoactive hemoproteins connected with the photosensitizing cofactor, zinc protoporphyrin IX (ZnPP) derivatives,2a,16 on a gold electrode and their enhanced electrochemical communication relative to a monolayer of the zinc-substituted protein.17 The relationship between the function and the protein-assembly orientation on the electrode surface will be important. In this paper, we thus demonstrate the construction of assemblies of photoactive hemoproteins connected with ZnPP, in which each protein immobilized in an opposite orientation to the electrode, to investigate the effect of orientation (Scheme 1). The characterization and the photoelectrochemical behaviors of the photoactive hemoprotein assemblies are described.
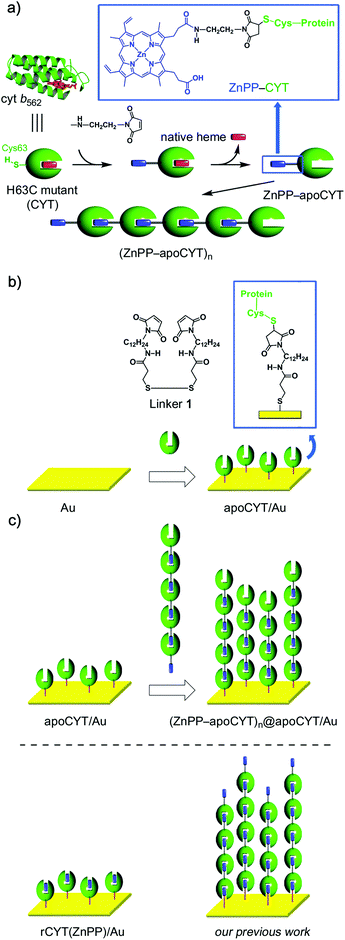 | ||
| Scheme 1 (a) Supramolecular Zn-substituted cyt b562 assemblies.18 Native heme (in red), ZnPP (in blue), and protein matrices (in green). (b) Preparation of an apoCYT-modified gold electrode. (c) Immobilization of the Zn-substituted cyt b562 assemblies on the apoCYT-modified electrode. | ||
Results and discussion
To anchor the protein oligomers on the gold electrode via the ZnPP–heme pocket interaction, a linker between a gold electrode and an apoprotein of the H63C mutant (apoCYT) was first prepared as follows: One of the terminal amino groups of 1,12-diaminododecane was protected with t-Boc and then the free amino group was converted to a maleimide moiety. After the deprotection of t-Boc, the obtained 1-(12-aminododecyl)-1H-pyrrole-2,5-dione precursor was then coupled with 3,3′-dithiobis(propionic acid) to yield linker 1. The gold electrode was immersed in a mixed solution of mercaptopropionic acid (100 mM) and 1 (20 mM) to form the self-assembled monolayer (SAM) (Scheme 1b). The maleimide-modified electrode was immersed in the protein solution of apoCYT, and the unbound proteins in the solution were thoroughly washed away to provide an apoCYT-modified gold electrode (apoCYT/Au). The electrochemical impedance spectroscopy (EIS) indicates that the proteins are immobilized by a covalent linkage between the Cys residue of apoCYT and the maleimide on SAM (vide infra).Next, according to our previous report, we prepared the photoactive protein oligomer, (ZnPP–apoCYT)n, as shown in Scheme 1a.17b Coupling of the apoprotein on the gold electrode and noncovalent immobilization of (ZnPP–apoCYT)n were evaluated by the EIS measurement, which provides a sensitive detection of protein binding derived from the resistance changes at the electrode–solution interface. The measurements in Fig. 1 were performed in a buffered solution containing 5 mM K4[Fe(CN)6]/K3[Fe(CN)6]. The charge-transfer resistance RCT for the maleimide-modified gold electrode was ca. 0.5 × 104 Ω, whereas an increased RCT value (1.3 × 104 Ω) was observed after ZnPP-reconstituted CYT (rCYT(ZnPP)) was immobilized on the maleimide-modified electrode via the maleimide–Cys covalent linkage (rCYT(ZnPP)/Au). Furthermore, much larger resistance values, 2.5 × 104 Ω, were obtained when (ZnPP–apoCYT)n was immobilized on apoCYT/Au, indicating the construction of the integrated assemblies of the Zn-substituted CYT on the gold surface via noncovalent ZnPP–heme pocket interaction ((ZnPP–apoCYT)n@apoCYT/Au).
![Alternative-current impedance spectra of (ZnPP–apoCYT)n@apoCYT/Au (■), rCYT(ZnPP)/Au (□), and maleimide-modified gold electrode (○) obtained in a 100 mM MOPS (pH 7.0) buffer containing 2.5 mM K3[Fe(CN)6] and 2.5 mM K4[Fe(CN)6]. Fitted data are shown as solid lines. The biased potential was +0.10 V (vs. Ag|AgCl). The frequency was from 100 kHz to 0.01 Hz and the amplitude was 10.0 mV. The inset shows the equivalent circuit applied to the data fitting.](/image/article/2013/DT/c3dt51469b/c3dt51469b-f1.gif) | ||
| Fig. 1 Alternative-current impedance spectra of (ZnPP–apoCYT)n@apoCYT/Au (■), rCYT(ZnPP)/Au (□), and maleimide-modified gold electrode (○) obtained in a 100 mM MOPS (pH 7.0) buffer containing 2.5 mM K3[Fe(CN)6] and 2.5 mM K4[Fe(CN)6]. Fitted data are shown as solid lines. The biased potential was +0.10 V (vs. Ag|AgCl). The frequency was from 100 kHz to 0.01 Hz and the amplitude was 10.0 mV. The inset shows the equivalent circuit applied to the data fitting. | ||
To evaluate the degree of interprotein assemblies on the (ZnPP–apoCYT)n@apoCYT/Au electrode, QCM measurements were conducted (Fig. 2). Upon the addition of zinc-substituted wild type cytochrome b562 (rCYTWT(ZnPP)) without cysteine residue on the surface to the maleimide-modified electrode, only a slight frequency decrease was observed. In contrast, upon the addition of rCYT(ZnPP) with Cys, the frequency of the maleimide-immobilized QCM decreased by ca. 180 Hz. This value is almost saturated upon the addition of excess rCYT(ZnPP), showing that the the rCYT(ZnPP) protein occupies the electrode surface via the covalent linkage. Furthermore, a significant frequency decrease upon the addition of (ZnPP–apoCYT)n (700 Hz) was observed, supporting the accumulation of ZnPP–apoCYT units on the electrode. Such a prominent frequency decrease was not observed when (ZnPP–apoCYT)n was added to the bare gold electrode. The average number (n) of the oligomer formation within the assemblies was estimated to be 3.8. The limited number of the assembly may stem from the non-specific interaction between the cofactors and the surface of the proteins, which were immobilized in a tightly packed fashion on the electrode surface.
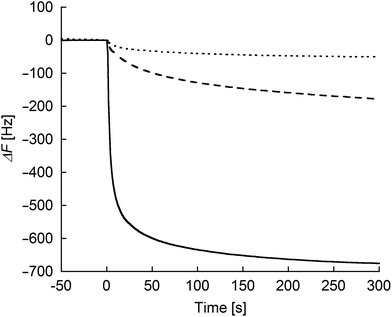 | ||
| Fig. 2 Frequency changes in response to the addition of the proteins: rCYTwt(ZnPP) (dotted line) and rCYT(ZnPP) (dashed line) to the maleimide-modified Au electrode, respectively, and (ZnPP–apoCYT)n (solid line) to the apoCYT/Au electrode. Samples were added into 500 μL of a buffered solution (100 mM KPi (pH 7.0)) in the QCM cell at 25 °C (final concentration of the sample was 4 μM). | ||
Photocurrent measurements of (ZnPP–apoCYT)n@apoCYT/Au employed as a working electrode were carried out to investigate the properties of the hierarchical protein assemblies containing the photosensitizer. Fig. 3 shows photocurrent response patterns of the prepared electrodes during on/off cycles of white light in the presence of methylviologen (MV2+) as an electron acceptor. We observed the clear rise and fall of the cathodic current under the bias potential of −200 mV (vs. Ag|AgCl). When the electrode covalently immobilized with Zn-substituted CYT via Cys63 was irradiated with monochromatic light at 420 nm (0.043 mW), we observed a photocurrent response of 18 nA cm−2, which was slightly increased relative to that found in the monomer-immobilized via ZnPP in our previous report (13 nA cm−2). This suggests that the two Tyr residues which position between the heme pocket and Cys63 would be possibly involved in the electron transfer pathway from the electrode to ZnPP (Fig. 4). In addition, the photocurrents of (ZnPP–apoCYT)n@apoCYT/Au were almost doubled (38 nA cm−2), indicating that more densely layered assembly of the zinc-substituted proteins on the electrode surface enhances the photocurrent generation. Although the four layers of the protein including three layers of the photosensitizing moieties were assembled on average, the photocurrents increased only twice. This suggests that the proteins more distant from the electrode surface would be less efficient to generate photocurrents. We also found that our previously reported Zn-substituted cytochrome b562, in which the zinc cofactor is located directly on the electrode, is more effective (60 nA cm−2) than the assembly here in photocurrent generation (Fig. 3). Hence a smaller assembly (3.8 units) was formed relative to the previously reported system (7.0 units).
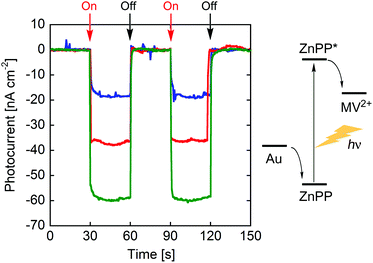 | ||
| Fig. 3 Photocurrent response patterns of (ZnPP–apoCYT)n@apoCYT/Au (in red), rCYT(ZnPP)/Au (in blue), and our previously reported hemoprotein assembly (in green)17b during on/off cycles of white light. The bias potential was −200 mV (vs. Ag|AgCl). | ||
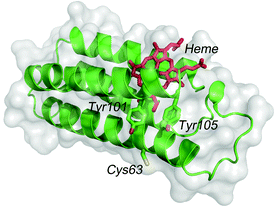 | ||
| Fig. 4 The structure of cytb562 showing Tyr101 and Tyr105 residues between heme and Cys63. | ||
Conclusions
In conclusion, Zn-substituted hemoprotein assemblies via a specific noncovalent ZnPP–heme pocket interaction were constructed on the apocytochrome b562-immobilized electrode, which was characterized by EIS and QCM measurements. The present work demonstrates that the fabricated supramolecular assemblies of a photoactive electron transfer protein in the orientation opposite to our previously reported system also generate cathodic photocurrents. Efforts to expand the attractive features of hemoprotein assemblies with a controlled orientation by this strategy to photoelectric conversion are now in progress.Experimental
Instruments
1H NMR spectra (400 MHz) were recorded using a Bruker DPX400 NMR spectrometer, and chemical shifts are reported in ppm relative to the residual solvent resonances. ESI-TOF MS analyses were performed using a Bruker Daltonics micrOTOF. UV-Vis spectra were taken using a Shimadzu UV-3150 or UV-2550 spectrophotometer equipped with a thermostated cell holder. The pH values were monitored using an F-52 Horiba pH meter. Quartz-crystal microbalance (QCM) measurements were performed using AFFINIX QNμ (Initium, Japan). Electrochemical measurements were made with a potentiostat (CompactStat, Ivium Technologies) using a platinum wire as a counter electrode and standard Ag|AgCl as a reference electrode. The electrolyte was 100 mM MOPS buffer (pH 7.0) containing 5 mM K4[Fe(CN)6]/K3[Fe(CN)6].Materials
Zinc protoporphyrin IX with maleimide at the propionate side chain,17b mono-N-Boc protected 1,12-dodecane diamine13c and 3,3′-dithiobis(propionic acid) were prepared according to a previous report.19 Other reagents and chemicals were purchased and used as received. The H63C mutant of cytochrome b562 (CYT) was expressed in E. coli and purified as previously reported.13c,20 Zinc protoporphyrin IX was obtained by a conventional procedure. Apo forms of proteins (apoCYT and apoCYTwt), the reconstituted proteins with zinc protoporphyrin IX (rCYT(ZnPP) and rCYTwt(ZnPP)), and the photoactive protein oligomer (ZnPP–apoCYT)n were prepared according to the methods of previous reports with modification.16b,17bSynthesis
Preparation of modified gold electrodes
A gold-coated quartz slide (AuroSheet(111), Tanaka Kikinzoku Kogyo K.K., Japan) with (111) surface (>80%) was used as an electrode. An electrochemically cleaned gold electrode was modified by soaking in 300 μL of a DMSO solution of mercaptopropionic acid (100 mM) and 1 (20 mM) to form a self-assembled monolayer. After incubation for 6 h at room temperature, the electrode was thoroughly rinsed with water and then immersed in a 100 μM protein solution (apoCYT, rCYT(ZnPP)) in 200 μL of 10 mM Tris-HCl buffer (pH 7.0) to give the electrode modified with each protein.Quartz-crystal microbalance (QCM) measurements
QCM measurements were performed using a cell equipped with a 27 MHz QCM plate oscillating at the fundamental frequency. The maleimide- or apoCYT-immobilized QCM cell was filled with 500 μL of a buffered solution (10 mM KPi, pH 7.0), and the time course frequency changes were observed on the addition of the protein solution (4 μM (ZnPP–apoCYT)n, rCYT(ZnPP), or rCYTwt (ZnPP)).Photocurrent measurements
Photocurrent measurements were performed in a typical three-electrode configuration connected to a potentiostat as reported previously.17b A 50 mM Tris-HCl (pH 7.5) containing methylviologen (1 mM) was used. The maximum current with clear photoresponses was obtained under the bias potential of −200 mV.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research ((B), KAKENHI 17350085 and Innovative Areas “Coordination Programming”, area 2107, KAKENHI 22108518, 24108726) from MEXT and the Japan Society for the Promotion of Science (JSPS). Y.K. acknowledges support from the Global COE Program of Osaka University and from JSPS. A.O. acknowledges support from the Frontier Research Base for Global Young Researchers, Osaka University, on the Program of MEXT, and from the Ogasawara Foundation. T.H. acknowledges support from the Asahi Grass Foundation.Notes and references
- (a) I. Willner and E. Katz, Bioelectronics, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) L. Fruk, C. H. Kuo, E. Torres and C. M. Niemeyer, Angew. Chem., Int. Ed., 2009, 48, 1550–1574 CrossRef CAS PubMed; (c) L.-Y. Cheng, Y.-T. Long, H.-B. Kraatz and H. Tian, Chem. Sci., 2011, 2, 1515–1518 RSC; (d) N. Terasaki, N. Yamamoto, T. Hiraga, Y. Yamanoi, T. Yonezawa, H. Nishihara, T. Ohmori, M. Sakai, M. Fujii, A. Tohri, M. Iwai, Y. Inoue, S. Yoneyama, M. Minakata and I. Enami, Angew. Chem., Int. Ed., 2009, 48, 1585–1587 CrossRef CAS PubMed; (e) M. Miyachi, Y. Yamanoi, Y. Shibata, H. Matsumoto, K. Nakazato, M. Konno, K. Ito, Y. Inoue and H. Nishihara, Chem. Commun., 2010, 46, 2557–2559 RSC.
- (a) Y. Tokita, J. Shimura, H. Nakajima, Y. Goto and Y. Watanabe, J. Am. Chem. Soc., 2008, 130, 5302–5310 CrossRef CAS PubMed; (b) Y. Tokita, S. Yamada, W. Luo, Y. Goto, N. Bouley-Ford, H. Nakajima and Y. Watanabe, Angew. Chem., Int. Ed., 2011, 50, 11663–11666 CrossRef CAS PubMed; (c) K. Nakano, T. Yoshitake, Y. Yamashita and E. F. Bowden, Langmuir, 2007, 23, 6270–6275 CrossRef CAS PubMed.
- (a) C. Ley, D. Holtmann, K. M. Mangold and J. Schrader, Colloids Surf., B, 2011, 88, 539–551 CrossRef CAS PubMed; (b) N. Terasaki, M. Iwai, N. Yamamoto, T. Hiraga, S. Yamada and Y. Inoue, Thin Solid Films, 2008, 516, 2553–2557 CrossRef CAS PubMed.
- (a) Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS PubMed; (b) M. Zayats, E. Katz, R. Baron and I. Willner, J. Am. Chem. Soc., 2005, 127, 12400–12406 CrossRef CAS PubMed; (c) A. Das and M. H. Hecht, J. Inorg. Biochem., 2007, 101, 1820–1826 CrossRef CAS PubMed; (d) H. Zimmermann, A. Lindgren, W. Schuhmann and L. Gorton, Chem.–Eur. J., 2000, 6, 592–599 CrossRef CAS; (e) L. H. Guo, G. McLendon, H. Razafitrimo and Y. L. Gao, J. Mater. Chem., 1996, 6, 369–374 RSC.
- R. Dronov, D. G. Kurth, H. Möhwald, R. Spricigo, S. Leimkühler, U. Wollenberger, K. V. Rajagopalan, F. W. Scheller and F. Lisdat, J. Am. Chem. Soc., 2008, 130, 1122–1123 CrossRef CAS PubMed.
- P. N. Ciesielski, C. J. Faulkner, M. T. Irwin, J. M. Gregory, N. H. Tolk, D. E. Cliffel and G. K. Jennings, Adv. Funct. Mater., 2010, 20, 4048–4054 CrossRef CAS.
- F. Lisdat, R. Dronov, H. Möhwald, F. W. Scheller and D. G. Kurth, Chem. Commun., 2009, 46, 274–283 RSC.
- L. Frolov, O. Wilner, C. Carmeli and I. Carmeli, Adv. Mater., 2008, 20, 263–266 CrossRef CAS.
- (a) K. Kobayashi, M. Shimizu, T. Nagamune, H. Sasabe, Y. Fang and W. Knoll, Bull. Chem. Soc. Jpn., 2002, 75, 1707–1713 CrossRef CAS; (b) D. H. Murgida and P. Hildebrandt, Phys. Chem. Chem. Phys., 2005, 7, 3773–3784 RSC; (c) A. E. F. Nassar, W. S. Willis and J. F. Rusling, Anal. Chem., 1995, 67, 2388–2392 CrossRef; (d) I. Taniguchi, K. Watanabe, M. Tominaga and F. M. Hawkridge, J. Electroanal. Chem., 1992, 333, 331–338 CrossRef CAS; (e) J. Xu, F. Shang, J. H. T. Luong, K. M. Razeeb and J. D. Glennon, Biosens. Bioelectron., 2010, 25, 1313–1318 CrossRef CAS PubMed.
- (a) E. E. Ferapontova, Electroanalysis, 2004, 16, 1101–1112 CrossRef CAS; (b) F. W. Scheller, U. Wollenberger, C. Lei, W. Jin, B. Ge, C. Lehmann, F. Lisdat and V. Fridman, Rev. Mol. Biotech., 2002, 82, 411–424 CrossRef CAS.
- (a) N. Bistolas, U. Wollenberger, C. Jung and F. W. Scheller, Biosens. Bioelectron., 2005, 20, 2408–2423 CrossRef CAS PubMed; (b) Y. Wu and S. Hu, Microchim. Acta, 2007, 159, 1–17 CrossRef CAS.
- (a) G. X. Ma, T. H. Lu and Y. Y. Xia, Bioelectrochemistry, 2007, 71, 180–185 CrossRef CAS PubMed; (b) A. Ramanavicius and A. Ramanaviciene, Fuel Cells, 2009, 9, 25–36 CrossRef CAS.
- (a) H. Kitagishi, Y. Kakikura, H. Yamaguchi, K. Oohora, A. Harada and T. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1271–1274 CrossRef CAS PubMed; (b) H. Kitagishi, K. Oohora and T. Hayashi, Biopolymers, 2009, 91, 194–200 CrossRef CAS PubMed; (c) H. Kitagishi, K. Oohora, H. Yamaguchi, H. Sato, T. Matsuo, A. Harada and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 10326–10327 CrossRef CAS PubMed; (d) A. Onoda, Y. Ueya, T. Sakamoto, T. Uematsu and T. Hayashi, Chem. Commun., 2010, 46, 9107–9109 RSC; (e) A. Onoda, A. Takahashi, K. Oohora, Y. Onuma and T. Hayashi, Chem. Biodiversity., 2012, 9, 1684–1692 CrossRef CAS PubMed; (f) K. Oohora, A. Onoda and T. Hayashi, Chem. Commun., 2012, 48, 11714–11726 RSC.
- (a) K. Oohora, A. Onoda, H. Kitagishi, H. Yamaguchi, A. Harada and T. Hayashi, Chem. Sci., 2011, 2, 1033–1308 RSC; (b) K. Oohora, S. Burazerovic, A. Onoda, Y. M. Wilson, T. R. Ward and T. Hayashi, Angew. Chem., Int. Ed., 2012, 51, 3818–3821 CrossRef CAS PubMed.
- (a) M. M. C. Bastings, T. F. A. de Greef, J. L. J. van Dongen, M. Merkx and E. W. Meijer, Chem. Sci., 2011, 1, 79–88 RSC; (b) J. C. Carlson, S. S. Jena, M. Flenniken, T. F. Chou, R. A. Siegel and C. R. Wagner, J. Am. Chem. Soc., 2006, 128, 7630–7638 CrossRef CAS PubMed; (c) S. Burazerovic, J. Gradinaru, J. Pierron and T. R. Ward, Angew. Chem., Int. Ed., 2007, 46, 5510–5514 CrossRef CAS PubMed; (d) D. A. Uhlenheuer, K. Petkau and L. Brunsveld, Chem. Soc. Rev., 2010, 39, 2817–2826 RSC; (e) H. D. Nguyen, D. T. Dang, J. L. J. van Dongen and L. Brunsveld, Angew. Chem., Int. Ed., 2010, 49, 895–898 CrossRef CAS PubMed.
- (a) E. Topoglidis, C. J. Campbell, E. Palomares and J. R. Durrant, Chem. Commun., 2002, 1518–1519 RSC; (b) S. Takeda, N. Kamiya and T. Nagamune, Biotechnol. Lett., 2004, 26, 121–125 CrossRef CAS; (c) T. Matsuo, A. Asano, T. Ando, Y. Hisaeda and T. Hayashi, Chem. Commun., 2008, 3684–3686 RSC; (d) Y. Hitomi, T. Hayashi, K. Wada, T. Mizutani, Y. Hisaeda and H. Ogoshi, Angew. Chem., Int. Ed., 2001, 40, 1098–1101 CrossRef CAS.
- (a) Y. Kakikura, A. Onoda, E. Kubo, H. Kitagishi, T. Uematsu, S. Kuwabata and T. Hayashi, J. Inorg. Organomet. Polym. Mater., 2013, 23, 172–179 CrossRef CAS; (b) A. Onoda, Y. Kakikura, T. Uematsu, S. Kuwabata and T. Hayashi, Angew. Chem., Int. Ed., 2012, 51, 2628–2631 CrossRef CAS.
- ZnPP contains regioisomers with respect to the substitution position at the two heme-propionate side chains of protoporphyrin IX.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- A. Onoda, T. Himiyama, K. Ohkubo, S. Fukuzumi and T. Hayashi, Chem. Commun., 2012, 48, 8054–8056 RSC.
| This journal is © The Royal Society of Chemistry 2013 |