
Photo-induced room temperature phosphorescence and thermally activated photochromism based on thianthrene derivatives†
Nan
Li
b,
Yunsheng
Wang
 *a and
Zhen
Li
*abc
*a and
Zhen
Li
*abc
aJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China. E-mail: wangys_18@tju.edu.cn; lizhen@whu.edu.cn
bInstitute of Molecular Aggregation Science, Tianjin University, Tianjin, 300072, China
cHubei Key Lab on Organic and Polymeric Opto-Electronic Materials, Department of Chemistry, Wuhan University, Wuhan, 430072, China
First published on 8th July 2024
Abstract
In the field of smart materials, designing organic compounds that exhibit both photo-induced room temperature phosphorescence (RTP) and photochromism presents significant challenges. Despite this, the broad potential applications of such materials continue to attract substantial interest from the scientific community. Here, we engineered three thianthrene derivatives by modifying the π-conjugation, creating the polymer-doped systems TN-2Me-p, TN-PhNap-p, and TN-2Nap-p, each with both photo-induced RTP and reversible, reusable photochromic properties. Notably, the poly(methyl methacrylate) film doped with TN-2Nap (TN-2Nap-p), a molecule featuring two naphthyl groups, displayed remarkable thermally erasable photochromism with an RTP lifetime of 431 ms. In contrast, the TN-2Me-doped film, with its naphthyl groups replaced by methyl groups, showed no photochromism under UV exposure but exhibited an increased photoluminescent quantum yield from 3.67% to 13.92%, and an emission lifetime of 132.58 ms. Strikingly, the photochromic response of TN-2Nap-p showed a significant temperature dependency due to radical generation. At room temperature, the radical lifetime was 272.44 s, but decreased 125-fold to 1.76 s at 80 °C. No photochromic activity was observed in liquid nitrogen. Utilizing the distinctive properties of photoinduced RTP and/or thermally activated photochromism, these materials have been successfully applied in mask-based graphic and text writing. Their high quantum yields, exceptional processability, and flexibility also highlight their potential for integration into flexible display technologies.
 Yunsheng Wang | Dr Yunsheng Wang earned his PhD in Chemistry from Tianjin University in 2022 under the guidance of Professor Ben Zhong Tang and Professor Zhen Li. He is currently a Postdoctoral Researcher at the Joint School of National University of Singapore and Tianjin University in Fuzhou and a recipient of the Postdoctoral Fellowship Program of CPSF. Dr Wang's research focuses on the unique role of back electron transfer (BET) in organic luminescent materials, particularly in organic high-temperature thermoluminescent (TL) materials. Additionally, his interests include the energy storage mechanisms in long-lived metastable states and the controlled release of stored energy, especially in the form of photons. |
Introduction
In the evolving domain of smart materials, the synthesis and functional integration of organic compounds that exhibit photo-responsive properties, specifically photo-induced room temperature phosphorescence (RTP) and photochromism, represent a frontier of significant scientific interest and technological potential.1–5 Here, ‘photo-induced RTP’ refers to the significant enhancement of RTP properties under continuous UV irradiation, which facilitates the removal of molecular oxygen as a quencher, thereby allowing the RTP to grow stronger. This specific activation process distinguishes our findings from general RTP behavior observed under standard conditions. These materials are crucial for applications spanning from advanced display to security and data storage solutions, prompting a vigorous exploration of their underlying photophysical mechanisms and practical implementations.4–8Historically, the exploration of RTP materials has focused on harnessing and manipulating triplet excited states, pivotal for achieving long-lived luminescence at ambient conditions.1–4,9–12 The process benefits from strategies like heavy atom facilitation and rigid matrix encapsulation, which could enhance intersystem crossing (ISC) and suppress non-radiative decay, overcoming the challenges of spin-forbidden singlet-to-triplet transitions.9–13 Despite notable advances in RTP material development, achieving high quantum efficiency and long phosphorescence lifetimes without resorting to cryogenic temperatures or heavy atom doping remains a big challenge.9–15 On a parallel trajectory, materials exhibiting photochromism with the reversible transformation of molecular structure upon light exposure in a color change have garnered attention for their dynamic optical switching capabilities.4–8 However, integrating photochromism with phosphorescence to create multifunctional smart materials has encountered substantial barriers, primarily due to the competing dynamics of radical formation and energy transfer processes within the same molecular frameworks.4–8,13–19
The synthesis of materials that concurrently demonstrate both photo-induced RTP and reversible photochromism not only expands the functional repertoire of smart materials but also introduces unique opportunities for precise optical modulation. Herein, we report the design and synthesis of three novel thianthrene derivatives of TN-2Me, TN-PhNap and TN-2Nap obtained through strategic π-conjugation modifications. These derivatives are embedded in a polymethyl methacrylate (PMMA) matrix, resulting in polymer-doped systems (0.03 mmol of emitter molecules per 1 g of PMMA) that showcase an intricate balance of photo-induced RTP and dynamic photochromic behavior (Fig. 1). Notably, TN-2Nap with its dual naphthyl groups and TN-2Me, where naphthyl is replaced by methyl, illustrated how subtle structural variations can profoundly influence the photophysical outcomes, highlighting the critical role of molecular design in tailoring the photo-responsive characteristics. This study demonstrated the significant temperature dependency of these photo-responsive behaviors. TN-2Nap-p showed no photochromic activity in liquid nitrogen, whereas at elevated temperatures, the materials exhibited rapid changes in its photochromic and phosphorescent properties. The integration of such dual-functionality paves the way for developing advanced optical materials capable of high-resolution information encryption, multi-level anti-counterfeiting, and erasable graphic/text writing.
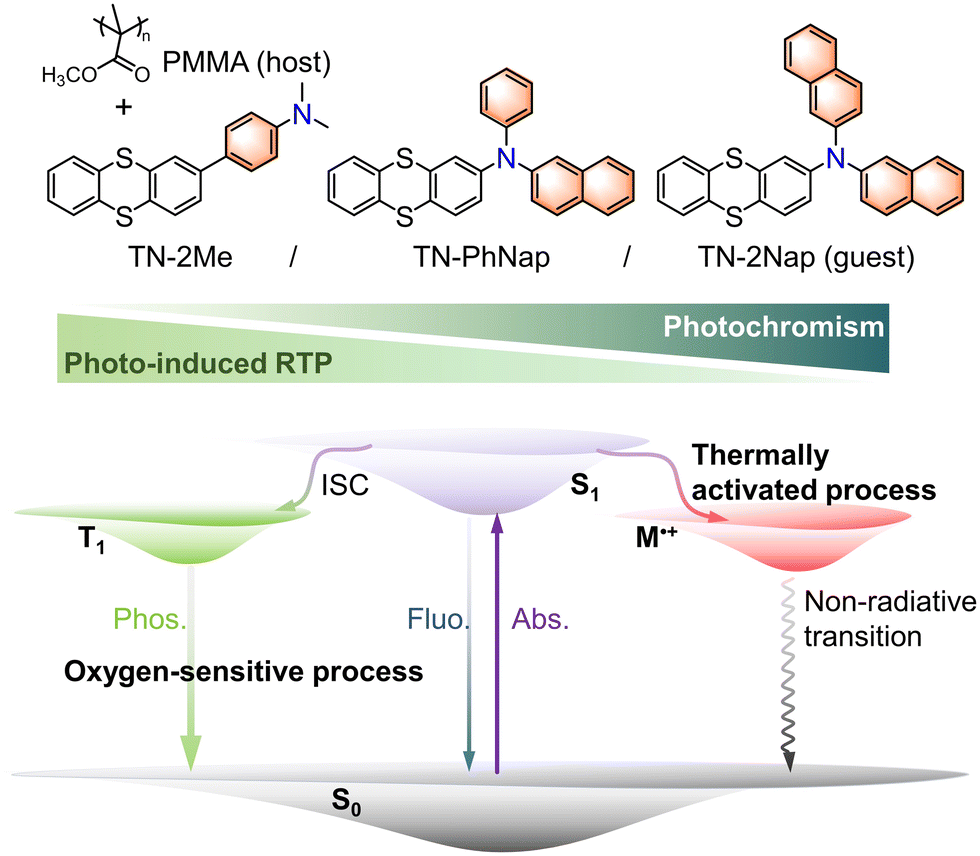 | ||
| Fig. 1 Schematic representation of the structures of thianthrene derivatives and the different photo-responsive behaviors in the PMMA films (T1, triplet state; Sn, singlet state; Phos, phosphorescence; Fluo, fluorescence; Abs, absorption; M+˙, cationic radical). | ||
Results and discussion
Synthesis
TN-2Me, TN-PhNap and TN-2Nap are readily available through Suzuki–Miyaura coupling and Buchwald–Hartwig coupling reactions. The synthetic route is described in Scheme S1 (ESI†). The molecular structures and purity of the three compounds have been fully confirmed by 1H NMR, 13C NMR, high-resolution mass spectrometry (HRMS), and high-performance liquid chromatography (HPLC).Photophysical properties
The photophysical behaviors of these luminogens were investigated in 1 × 10−5 M dichloromethane (DCM) (Fig. S1, ESI†). At room temperature, TN-2Me, TN-PhNap, and TN-2Nap exhibited steady-state spectra with single emission peaks. However, a new emission band ranging from 490 nm to 680 nm emerged in TN-2Me solution at 77 K. The comparison with delayed spectra identified this band as phosphorescence, corresponding to radiative transitions of triplet excitons. In contrast, for TN-PhNap and TN-2Nap solutions, no significant new bands appeared in the steady-state spectra across temperatures from 77 K to 298 K. Nevertheless, their corresponding delayed spectra revealed strong phosphorescence, similar to TN-2Me in frozen solution. When comparing steady-state spectra, TN-2Me exhibited the strongest phosphorescence, which correlated with its shortest afterglow lifetime of 208.03 ms among the three molecules. The weaker emission from TN-PhNap and TN-2Nap resulted in longer lifetimes at low temperatures, of 857.73 ms and 676.05 ms respectively, which were over three and four times longer than that of TN-2Me, likely due to their weaker spin–orbit coupling (SOC).9–11The phosphorescence observed in frozen solutions suggested that the three molecules could achieve room temperature phosphorescence if non-radiative decays were sufficiently suppressed. To realize this goal, we considered embedding the luminophores within a polymer matrix to provide a rigid environment that suppressed non-radiative transitions (Fig. 1). Compared to their behavior in solution at 77 K, the doped films (TN-2Me-p, TN-PhNap-p, TN-2Nap-p) exhibited similar photoluminescence (PL) spectra (Fig. 2). Notably, TN-2Me-p displayed a significant photo-induced phosphorescence phenomenon, with its quantum yield increasing from 3.67% to 13.92% after UV irradiation (Fig. 2f). For such polymer-doped systems, the depletion of oxygen under continuous UV irradiation was commonly considered the cause of the photo-induced RTP effect (Fig. S2–S4, ESI†). Initially, triplet excitons were easily quenched by oxygen in the film, resulting in only weak phosphorescence.20 Upon sustained UV irradiation, a significant depletion of triplet oxygen occurred, allowing for the observation of brighter phosphorescence emissions.20,21 The RTP lifetime were significantly enhanced, reaching 132.58 ms, 477.73 ms, and 431.78 ms respectively (Fig. 2e). The prolonged lifetimes for TN-PhNap and TN-2Nap were attributed to the weaker spin–orbit coupling between the singlet and triplet states within the molecules (Fig. 3a). Additionally, TN-PhNap exhibits the minimal overlap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), that help stabilize the triplet excitons, thereby extending the phosphorescence lifetime. In contrast, the significant overlap between HOMO and LUMO in TN-2Me, the effective intersystem crossing (ISC) channel and stronger spin–orbit coupling promoted the decay of the triplet exciton, consistent with its efficient but short-lived phosphorescence (Fig. 2e and f).
 | ||
| Fig. 2 Normalized steady-state PL spectra of TN-2Me-p, TN-PhNap-p, and TN-2Nap-p (a) before and (b) after 365 nm UV irradiation (500 μW cm−2). (c) Normalized delayed PL of TN-2Me-p, TN-PhNap-p, and TN-2Nap-p after 365 nm UV irradiation (500 μW cm−2). (d) Fluorescence decay profiles of TN-2Me-p (λem = 405 nm), TN-PhNap-p (λem = 405 nm), and TN-2Nap-p (λem = 420 nm). (e) PL intensity decay curves of TN-2Me-p, TN-PhNap-p, and TN-2Nap-p (λem = 520 nm). (f) Quantum yield (Φ) of TN-2Me-p, TN-PhNap-p, and TN-2Nap-p before and after 365 nm UV irradiation (500 μW cm−2). | ||
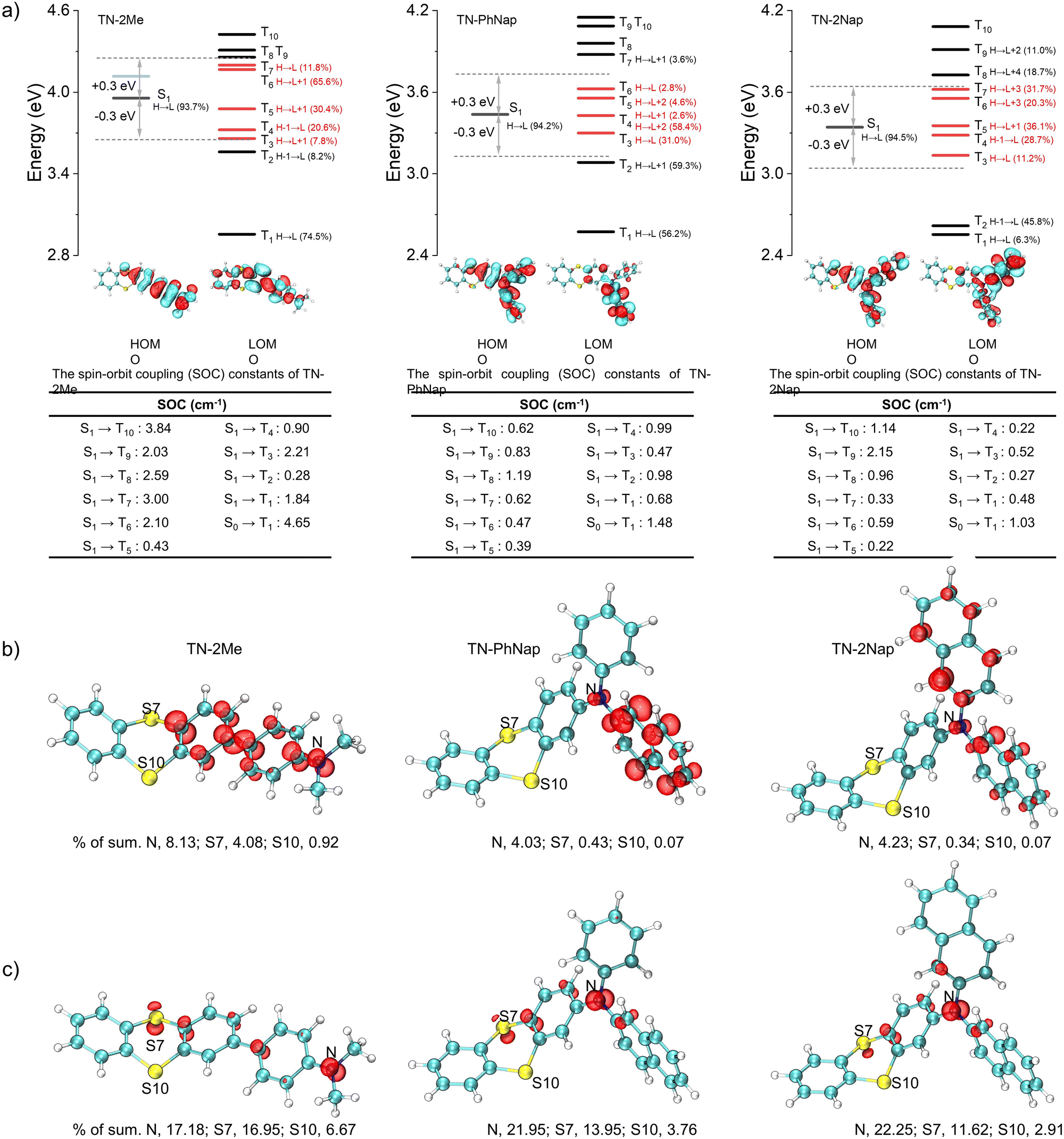 | ||
| Fig. 3 (a) Energy level diagrams and possible ISC channels from excited singlet state (S1) to excited triplet states (Tn) for TN-2Me, TN-PhNap, TN-2Nap and the corresponding spin–orbit coupling (SOC) constants. (b) Odd electron density distribution of TN-2Me, TN-PhNap and TN-2Nap with the contour value of 0.001 a.u. (c) Iso-surface of spin density for the cationic radical TN-2Me+˙, TN-PhNap+˙ and TN-2Nap+˙. | ||
Interestingly, after exposure to UV light, the TN-2Me-p, TN-2Nap-p and TN-PhNap-p films exhibited photochromic phenomena (Fig. S5, ESI†), which was particularly pronounced in TN-2Nap-p (Fig. 4). After UV light irradiation, the film changed from colorless to dark green and it can revert back to its colorless and transparent state upon turning off the UV light and leaving it at ambient for about 1200 s (Fig. 4). The afterglow decay properties were unaffected by the color change and remained stable after 20 cycles (Fig. S6, ESI†). The absorption spectra of the films could accurately describe the process of this reversible change. As shown in Fig. 4a, exposing the films to a UV light generated new absorption peaks of varying intensities. This also corresponded to the observed depth of color change of three films (Fig. S5 and S7, ESI†). Taking TN-2Nap-p as an example, after 20 s of continuous irradiation, a new peak centered around 640 nm gradually increased, and the color of TN-2Nap-p deepened. This indicated that the photo-induced color change was due to the formation of cationic radicals under UV light irradiation,17,22–25 as similar long-wavelength absorption bands have been found in other triphenylamine-based cationic radicals (Fig. S5 and S7, ESI†).
 | ||
| Fig. 4 In situ monitoring of absorption spectra and absorbance decay curves of TN-2Nap-p during continuous exposure to 365 nm UV light (500 μW cm−2) and subsequent recovery in air after the UV source is turned off, recorded at temperatures of 25 °C, 50 °C, and 60 °C (a)–(f). Time-dependent decay of EPR signals for three doped materials before and after photoactivation: (g) TN-2Me-p, (h) TN-PhNap-p, (i) TN-2Nap-p. | ||
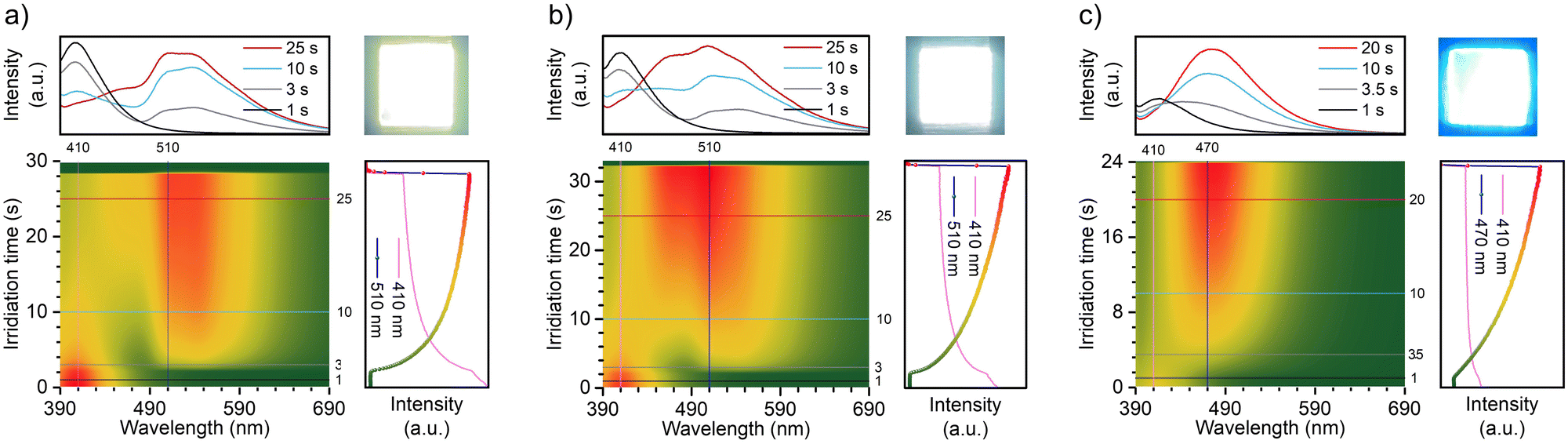
Based on the analysis of the electron paramagnetic resonance (EPR) results (Fig. 4g–i), this photochromism was caused by photo-generated cation radicals, and the photo-generated radicals of the three molecules exhibited different decay characteristics. To clearly demonstrate these color change and recovery processes, Fig. 4b and Fig. S7 (ESI†) present scatter plots of the intensity change over time at the wavelength of the new absorption peak. After 15, 10, and 20 s of irradiation, respectively, the TN-2Me-p, TN-2Nap-p, and TN-PhNap-p showed a continuous increase in the intensity of their absorption peaks, evidencing the ongoing generation of radicals. The rate of radical formation in TN-PhNap-p was initially fast, but it showed a linear decrease during the subsequent 3.5 to 10 s of continuous irradiation (Fig. S7d, ESI†), indicating poor photostability of the radicals. Upon turning off the UV lamp, an exponential decay of radicals was observed, with TN-2Me-p and TN-PhNap-p returning to their original state within 10 s, and TN-2Nap-p within 1200 s. To describe the decay kinetics of this process, referring to the fitting method of phosphorescence lifetime,9,10 τr was used to describe the decay lifetime of the radicals, which was 0.56 s, 18.28 s, and 272.44 s, respectively.
Notably, the solid powders of the three molecules did not exhibit the aforementioned photochromic and long afterglow properties (Fig. S8 and S9, ESI†). However, in the doped films based on polystyrene (PS) matrix (TN-2Me-ps, TN-PhNap-ps and TN-2Nap-ps), similar photophysical behaviors to the PMMA doped system were observed, but with a weaker degree of photochromism and faster fading (Fig. S10–S12, ESI†). Meanwhile, the RTP decay rate was faster in the PS-doped film compared to the PMMA-doped system (Fig. S13, ESI†).
Differences in the photophysical behaviors of the three molecules may arise from distinct reorganization energies and spin electronic properties. For TN-2Nap, the transition from its neutral state to a radical cation involved a change in the thianthrene ring's dihedral angle from 130.91° to 142.8°, accompanied by a relatively low reorganization energy of 0.20 eV (Fig. S14, ESI†). This facilitated the conversion of the molecule from the excited state to a cationic radical through structural relaxation.26 In contrast, TN-2Me required overcoming a higher energy barrier, evidenced by a reorganization energy of 0.39 eV. During this process, the dihedral angle of the S-heterocycle shifted from 129.92° to 148.79° with an alteration of 18.87°, resulting in minimal radical formation under illumination. Also, the reorganization energy of TN-PhNap was relatively small, and thus a noticeable photochromic phenomenon was observed. However, as shown in Fig. 3b and c, the unpaired electron of TN-2Nap+˙ was predominantly located on the nitrogen atom, accounting for as high as 22.25% (Tables S1–S6, ESI†), which contributed to the high stability of TN-2Nap+˙. In contrast, for TN-2Me+˙ and TN-PhNap+˙, the percentages are 17.18% and 21.95%, respectively, indicating a slightly lower radical stability compared to TN-2Nap+˙, which is reflected in their shorter radical decay lifetime. Structurally, the π-conjugation in TN-2Me, TN-PhNap, and TN-2Nap was systematically expanded by incorporating zero, one, and two naphthyl groups, respectively. This progressive extension was believed to enhance radical stability by enlarging the π-conjugated system, which formed a protective barrier around the nitrogen centers, insulating them from environmental quenching.17,23–25
In addition to manipulating radical stability through molecular structural adjustments, we have discovered that temperature variations could effectively modulate both the generation and decay rates of radicals, thus controlling the kinetics of photochromism. As illustrated in Fig. 4 and Fig. S15–S17 (ESI†), the photochromic process of TN-2Nap-p at 25 °C, 50 °C, 60 °C, and 80 °C was monitored in situ, yielding corresponding UV-visible absorption spectra. It was observed that the decay lifetime of radicals decreased rapidly with increasing temperature, from 272.44 s at room temperature to 23.9 s, 2.57 s, and 1.76 s, respectively (Fig. 4 and Fig. S15, ESI†). This trend was visually demonstrated in Videos S1–S4 (ESI†). When photochromic patterns were immersed in hot water (60 °C, 80 °C water), the blue pattern at the higher temperature was erased very quickly. Notably, both patterns were created by photomask on the same TN-2Nap-p film loaded on a quartz substrate. Similarly, the TN-PhNap-p film also exhibited an accelerated decay rate of radicals upon heating (Fig. S16, ESI†). Regarding the excitation process, an increase in temperature markedly accelerated the rate of color change in the materials. For instance, in TN-2Nap-p, the absorption peak at ∼640 nm increased rapidly with rising temperatures compared to the first 10 s of continuous UV exposure (Fig. 4). Conversely, at sufficiently low temperatures, the photochromic properties were completely absent. As demonstrated in Video S3 (ESI†), upon UV excitation in liquid nitrogen, TN-2Nap-p displayed only intense prolonged afterglow, with no observable color change in the film. The results demonstrated that photochromism is a thermally activated process. Raising the temperature promoted structural rearrangement in excited-state, facilitating radical generation. This behavior could be explained by the Marcus rate equation,27 indicating that an increase in temperature enhanced both the electron transfer and non-radiative decay, thus accelerating both photochromism and fading. Furthermore, TN-2Nap-p in a nitrogen atmosphere also exhibited significant photochromism after continuous irradiation, with rapid color fading at 80 °C (Fig. S18 and S19, ESI†). This further demonstrated that the radical decay kinetics are controlled by heat. However, it is noteworthy that inducing photochromism in TN-2Nap-p films in the presence of oxygen was more difficult compared to those in air or nitrogen. Additionally, the slight discoloration observed in oxygen was less persistent than in air or nitrogen. This indicates that oxygen also has a significant quenching effect on photogenerated radicals.
As previously noted, the longer duration and stronger afterglow observed at low temperatures suggested an enhanced release of energy via radiative transitions of triplet excitons. At liquid-nitrogen temperature, molecular freezing inhibited radical generation, making phosphorescence the predominant energy release mechanism in the excitation evolution pathways. As demonstrated in Video S3 (ESI†), the afterglow of TN-2Nap in liquid nitrogen lasted over one minute, far exceeding the 2.2 s observed at room temperature. With increasing temperatures above ambient, the afterglow duration shortened, and both radical generation and deactivation accelerated, indicating a dominance of non-radiative energy dissipation pathways, as characterized by molecular thermal vibrations. This change was validated in the variable temperature PL spectra (Fig. 5 and Fig. S20, S21, ESI†). These spectra revealed a decrease in afterglow relative to fluorescence with increasing temperature, indicating greater dissipation of excited-state energy via non-radiative pathways. Notably, for TN-2Me-p, a temperature rise led to a significant red shift in fluorescence, altering the emission color from yellow-green at room temperature to sky blue at 100 °C (Fig. S22, ESI†). This shift was likely due to changes in the predominant molecular conformation of TN-2Me.
 | ||
| Fig. 5 The in situ monitoring of changed steady-state PL spectra of TN-2Me-p with different temperature (a) 25 °C, (b) 60 °C, (c) 100 °C (PL spectra were collected by QE65 Pro. mode: high-speed scanning). | ||
Through the study of the aforementioned photophysical properties and combined with theoretical calculations, we were able to clearly delineate the excitation state evolution process of these molecules. The molecular structures of their corresponding cationic radicals were optimized. The study found that the cationic radicals possessed more planar conformations, which favored the stability of the radicals (Fig. S14, ESI†). By analyzing the excitation process, the molecule was excited from the ground state to the singlet state (S0), after which there were four possible pathways: first, directly returning to the ground state and emitting fluorescence; second, losing an electron to form a cationic radical and then transitioning back to the ground state; third, transitioning to the triplet state (T1) through the intersystem crossing (ISC) process, then returning to S0 and emitting phosphorescence; fourth, undergoing other non-radiative transitions. Therefore, such photochromism was a result of competition among multiple potential pathways, including the ISC process. From the computational results, it was observed that TN-2Me had a stronger ISC channel and a larger spin–orbit coupling constant between T1 and S0 (Fig. 3), hence its phosphorescence emission predominated, allowing only very weak photochromic phenomena to be observed. The opposite was true for TN-PhNap and TN-2Nap, consistent with the experimental results that the quantum yields of TN-PhNap and TN-2Nap actually decreased after irradiation.
With their intriguing and reversible photo-induced phosphorescence and photochromic characteristics, these materials offered a non-contact optical information writing tool that could also serve for information encryption and anti-counterfeiting purposes (Videos S5–S7, ESI†). As demonstrated in Fig. 6, the films allowed for the easy writing of graphic and textual information via UV irradiation through a mask. This photo-induced writing technology provided a high resolution capable of millimeter-scale precision (Fig. S23 and S24, ESI†), ensuring that the content was clear and legible, which was crucial for display applications. The flexibility of films was a key trait that enabled integration with bendable and foldable display technologies, broadening its potential applications (Fig. 6c). The photoinduced RTP patterns gradually disappeared as oxygen slowly rediffused into the film, giving it repeatable write/erase characteristics. Meanwhile, photochromic patterns can be quickly erased by raising the temperature, allowing new information to be written again. The high quantum yield ensures that the information was discernible in well-lit environments, reinforcing its practicality. Meanwhile, by utilizing the thermally activated reversible on/off switching of photochromic properties in the material, rapid writing and erasing of information are possible. As shown in Fig. 6d and e, different Loong patterns were sequentially written three times on the same film, each capable of being quickly erased at high temperatures. To better demonstrate this process, hot water at 60 °C and 80 °C was used to erase the information, further showcasing the exceptional stability of the material (Fig. 6e and Videos S1, S2 and S8, ESI†). In brief, the exceptional flexibility, resolution, and reversible optical recording capabilities made these films an ideal medium for state-of-the-art display systems.
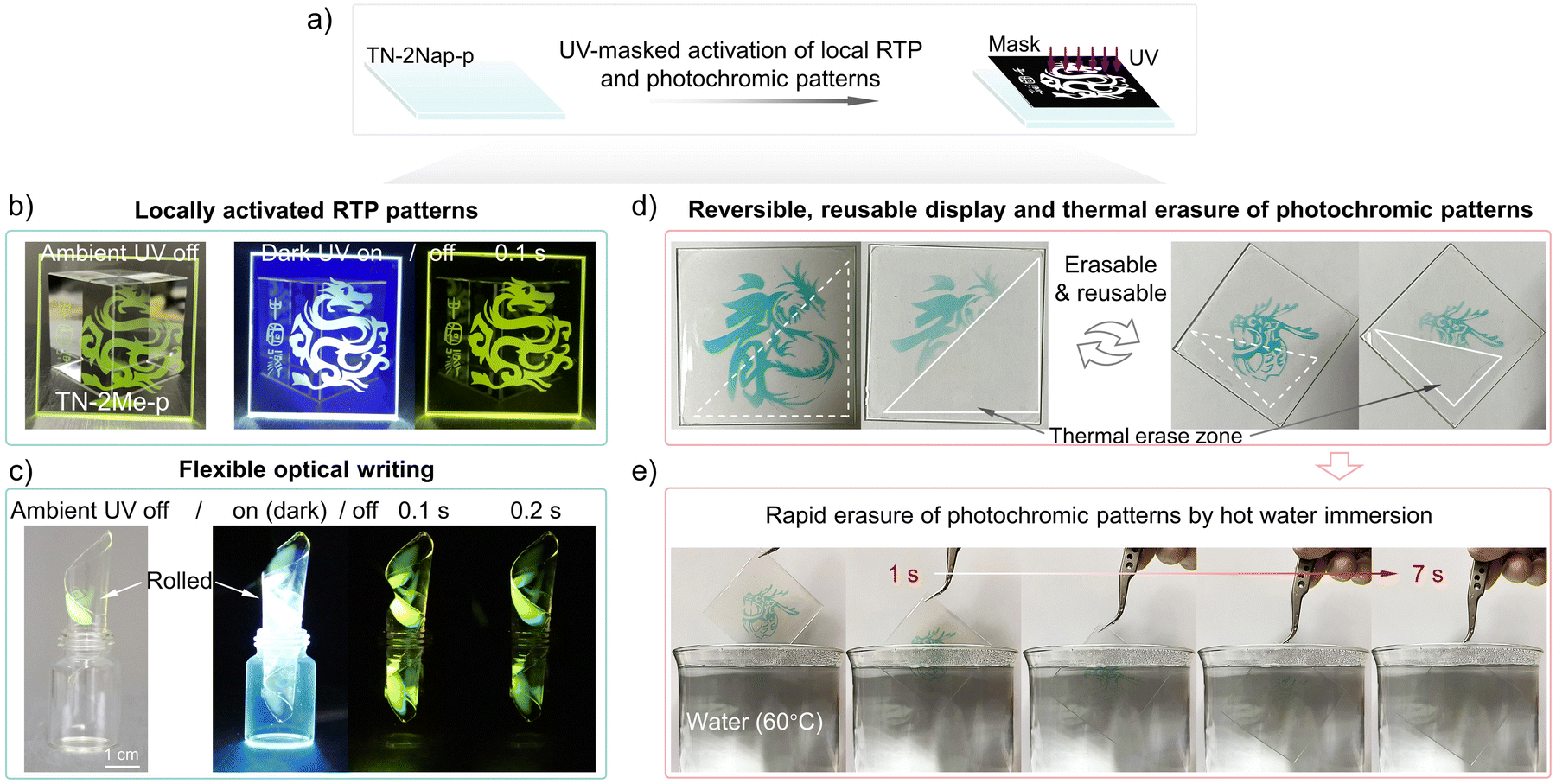 | ||
| Fig. 6 (a) Schematic and outputs of localized activation of the RTP and photochromic patterns by UV light through masks, showing both phosphorescence and color change responses. (b) Photographs of the afterglow patterns during the day and at night. The film (TN-2Me-p) was prepared by drop coating on quartz substrate. (c) Photo-induced RTP from a film curled into a cylinder ∼1.0 cm in diameter, obtained by peeling the film from a quartz substrate as shown in (b). (d) Photochromic patterns at room temperature on the same film (TN-2Nap-p), which disappear after about 20 minutes but can be rapidly erased by localized heating (white triangular area). (e) After re-exposure to light, a ‘Loong head’ pattern is created and then immersed in 60 °C hot water, erasing the pattern within ∼7 s. (film size, 50 mm × 50 mm × 0.7 mm). | ||
Conclusion
In this study, three thianthrene derivatives, TN-2Me, TN-2Nap, and TN-PhNap, were successfully synthesized and fully characterized. When doped into PMMA, these materials displayed diverse photo-responsive behaviors under continuous irradiation, including reversible photochromism and photoinduced RTP. This is attributed to the competition between ISC transitions and radical formation during the excitation process, which is closely associated with their structural attributes, especially the reorganization energy and the extent of π-conjugation. TN-2Nap-p exhibits a pronounced photochromic effect, primarily due to its low reorganization energy and extensive π-conjugation, which collectively facilitate the photo-induced transitions and stabilize radicals. Moreover, the radical stability observed in TN-2Nap, which shows a dramatic reduction in decay rate from over four minutes at 25 °C to under two seconds at 80 °C, highlights the capacity for precise temperature-based control of photochromic responses. These findings are pivotal for the development of advanced photonic materials capable of functioning across a broad temperature spectrum, thereby addressing a wider array of scientific and technological challenges. The advancements in RTP and photochromism not only enhance our understanding but also expand the potential applications of these materials in fields such as flexible displays and encrypted data storage, offering robust solutions for future optical and materials science applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y.-S. W and Z. Li spearheaded the implementation of the project; L. N., Y.-S. W., and Z. L. wrote and edited the paper; N. L. was primarily responsible for the experiments and then measured and analyzed the data; all authors contributed to the data analysis, discussion and revision of the manuscript; The computational work was conducted by Y.-S. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (NSFC 22305172), China Postdoctoral Science Foundation (2023M732586 and 2024T170640), Postdoctoral Fellowship Program of CPSF (GZB20230509).References
- G. Baryshnikov, B. Minaev and H. Ågren, Theory and calculation of the phosphorescence phenomenon, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS PubMed
.
- Y. Fan, Q. Li and Z. Li, Afterglow bio-applications by utilizing triplet excited states of organic materials, Sci. China: Chem., 2023, 66, 2930–2940 CrossRef CAS
- J. Wang, Y. Yang, X. Sun, X. Li, L. Zhang and Z. Li, Management of triplet excitons transition: fine regulation of Förster and dexter energy transfer simultaneously, Light: Sci. Appl., 2024, 13, 35 CrossRef CAS PubMed
- B. Wu, X. Xu, Y. Tang, X. Han and G. Wang, Multifunctional optical polymeric films with photochromic, fluorescent, and ultra-long room temperature phosphorescent properties, Adv. Opt. Mater., 2021, 9, 2101266 CrossRef CAS
- J. Zheng, J. Chen, Y. Jin, Y. Wen, Y. Mu, C. Wu, Y. Wang, P. Tong, Z. Li, X. Hou and J. Tang, Photochromism from wavelength-selective colloidal phase segregation, Nature, 2023, 617, 499–506 CrossRef CAS
- H. Nie, J. L. Self, A. S. Kuenstler, R. C. Hayward and J. R. Alaniz, Multiaddressable photochromic architectures: from molecules to materials, Adv. Opt. Mater., 2019, 7, 1900224 CrossRef
- G. K. Gulati, L. K. Gulati and S. Kumar, Recent progress in multi-stimulable photochromic oxazines with their wide-ranging applications, Dyes Pigm., 2021, 192, 109445 CrossRef CAS
- Y. Wakayama, R. Hayakawa, K. Higashiguchi and K. Matsuda, Photochromism for optically functionalized organic field-effect transistors: a comprehensive review, J. Mater. Chem. C, 2020, 8, 10956 RSC
- S. Hirata, Molecular physics of persistent room temperature phosphorescence and long-lived triplet excitons, Appl. Phys. Rev., 2022, 9, 011304 CAS
- W. Zhao, Z. He and B. Tang, Room-temperature phosphorescence from organic aggregates, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS
- Y. Fan, Q. Li and Z. Li, Afterglow bio-applications by utilizing triplet excited states of organic materials, Sci. China: Chem., 2023, 66, 2930–2940 CrossRef CAS
- Z. Li, K. Fu, H. Deng, K. Nie, Z. Pan, Z. Mao, J. Zhao, G. Li, P. Zhu, Z. Chi and R. Sun, Polymeric ultralong organic phosphorescence with excellent humidity and temperature resistance via hydrophobic effect, Aggregate, 2024, 5, e440 CrossRef CAS
- E. Hamzehpoor, C. Ruchlin, Y. Tao, C.-H. Liu, H. M. Titi and D. F. Perepichka, Efficient room-temperature phosphorescence of covalent organic frameworks through covalent halogen doping, Nat. Chem., 2023, 15, 83–90 CrossRef CAS
- J. Zhang, D. Li, W. Li, Y. Wu, X. Mu, C. Liu, K. Fang and Z. Ge, Highly efficient through-space charge transfer TADF molecule employed in TADF- and TADF-sensitized organic light-emitting diodes, Sci. China: Chem., 2024, 67, 1270–1276 CrossRef CAS
- J. H. Zhang, S. Liu, Y. Yuan and Y. L. Chen, Wide-ranging force responsive composites based on 1,2-dioxetane and ZnS as luminescent probes in polyurethanes, Chin. J. Polym. Sci., 2023, 41, 1162–1168 CrossRef
- X. Ping, J. Zhan, Y. Zhu, Y. Wu, C. Hu, J. Pan, C. Yao, J. Zuo, H. Feng and Z. Qian, Photoactivation of solid-state fluorescence through controllable intermolecular [2 + 2] photodimerization, Chem. – Eur. J., 2023, 29, e202301520 CrossRef CAS PubMed
- Y. Yang, J. Wang, D. Li, J. Yang, M. Fang and Z. Li, Tunable photoresponsive behaviors based on triphenylamine derivatives: the pivotal role of π-conjugated structure and corresponding application, Adv. Mater., 2021, 33, 2104002 CrossRef CAS
- L. Zhan, T. Chen, C. Zhong, X. Cao, Y. Zhang, Y. Zou, Z. Bin, J. You, D. Zhang, L. Duan, C. Yang and S. Gong, Luminescent gold(III) exciplexes enable efficient multicolor electroluminescence, Sci. China: Chem., 2023, 66, 3213–3222 CrossRef CAS
- V. A. Dini, D. Genovese, C. Micheletti, N. Zaccheroni, A. Pucci and C. Gualandi, Emission or scattering? Discriminating the origin of responsiveness in AIEgen-doped smart polymers using the TPE dye, Aggregate, 2023, 4, e373 CrossRef CAS
- Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Tang and Z. Li, New phenothiazine derivatives that exhibit photoinduced room-temperature phosphorescence, Adv. Funct. Mater., 2021, 31, 2101719 CrossRef CAS
- C. S. Redondo, P. Kleine, K. Roszeitis, T. Achenbach, M. Kroll, M. Thomschke and S. Reineke, Interplay of fluorescence and phosphorescence in organic biluminescent emitters, J. Phys. Chem. C, 2017, 121, 14946–14953 CrossRef
- S. Amthor, B. Noller and C. Lambert, UV/Vis/NIR spectral properties of triarylamines and their corresponding radical cations, Chem. Phys., 2005, 316, 141–152 CrossRef CAS
- E. Moulin, F. Niess, M. Maaloum, E. Buhler, I. Nyrkova and N. Giuseppone, The hierarchical self-assembly of charge nanocarriers: a highly cooperative process promoted by visible light, Angew. Chem., Int. Ed., 2010, 49, 6974–6978 CrossRef CAS PubMed
- J. J. T. Armao, M. Maaloum, T. Ellis, G. Fuks, M. Rawiso, E. Moulin and N. Giuseppone, Healable supramolecular polymers as organic metals, J. Am. Chem. Soc., 2014, 136, 11382–11388 CrossRef CAS
- J.-X. Hu, X.-F. Jiang, Y.-J. Ma, X.-R. Liu, B.-D. Ge, A. N. Wang, Q. Wei and G.-M. Wang, Optically actuating ultra-stable radicals in a large π-conjugated ligand constructed photochromic complex, Sci. China: Chem., 2021, 64, 432–438 CrossRef CAS
- Y. Wang, W. Zhang, J. Yang, Y. Gong, J. Zhang, M. Fang, Q. H. Yang and Z. Li, The key role of molecular aggregation in rechargeable organic cathodes, Matter, 2022, 5, 4467–4479 CrossRef CAS
- J. T. Blaskovits, K. Lin, R. Fabregat, I. Swiderska, H. Wu and C. Corminboeuf, Is a single conformer sufficient to describe the reorganization energy of amorphous organic transport materials?, J. Phys. Chem. C, 2021, 125, 17355–17362 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc02034k |
| This journal is © The Royal Society of Chemistry 2024 |