
Ligand-customized colloidal quantum dots for high-performance optoelectronic devices†
Hang
Xia‡
a,
Huicheng
Hu‡
b,
Ya
Wang
a,
Mengxuan
Yu
a,
Mohan
Yuan
a,
Ji
Yang
b,
Liang
Gao
abce,
Jianbing
Zhang
 cde,
Jiang
Tang
abce and
Xinzheng
Lan
*abce
cde,
Jiang
Tang
abce and
Xinzheng
Lan
*abce
aSchool of Optical and Electronic Information (OEI), Huazhong University of Science and Technology (HUST), Wuhan, Hubei 430074, People's Republic of China. E-mail: xinzhlan@mail.hust.edu.cn
bWuhan National Laboratory for Optoelectronics (WNLO), Huazhong University of Science and Technology (HUST), Wuhan, Hubei 430074, People's Republic of China
cOptics Valley Laboratory, Wuhan, Hubei 430074, People's Republic of China
dSchool of Integrated Circuit, Huazhong University of Science and Technology, Wuhan, Hubei 430074, People's Republic of China
eWenzhou Advanced Manufacturing Technology Research Institute of Huazhong University of Science and Technology, Wenzhou, Zhejiang 325035, People's Republic of China
First published on 29th May 2024
Abstract
Colloidal quantum dots (CQDs) have attracted considerable attention owing to their potential applications in low-cost and high-performance optoelectronic devices. Achieving electronic-grade CQD materials requires precise control over their semiconducting properties via surface management. Herein, we developed a new surface modification strategy that allows for enhanced control over both the type and the amount of ligands by decoupling colloidal-stabilization and property-engineering ligands. This enables the fine control of the electronic properties of CQD materials. Using PbS CQDs, we showed improved passivation and suppressed band tail states. The resultant solar cells exhibited a higher power conversion efficiency of 13.3% compared to the 10.4% efficiency of the control device, and photodetectors showed a detectivity (D*) of 1.8 × 1012 Jones, which is a more than four-fold improvement in comparison to the control devices. Moreover, the versatility of this approach was evaluated in other CQD systems, including emissive CdSe/ZnCdSe/ZnSe core/shell and infrared HgTe CQDs. The high photoluminescence quantum yield of CdSe/ZnCdSe/ZnSe core/shell CQDs was well preserved in polar solvents following a solution ligand exchange process, and tunable doping of HgTe CQDs was achieved. Overall, this ligand-customization approach verifies the significance of fine control of ligands towards the development of electronic-grade CQD materials, which might have implications for other QD materials.
 Xinzheng Lan | Xinzheng Lan received his PhD from Hefei University of Technology in 2015. Later, he worked on his postdoctoral research in the University of Toronto and the University of Chicago from 2014 to 2019. He is currently a full professor in the School of Optical and Electronic Information at the Huazhong University of Science and Technology. His current research interests include semiconductor materials and their optoelectronic devices such as photodetectors, light-emitting diodes, and solar cells. |
Introduction
Colloidal quantum dots (CQDs) are solution-processed semiconductor nanocrystals.1,2 Owing to quantum confinement, they offer size-tunable band gaps independent of their compositions, which is peculiar of QDs.3,4 The excellent solution processability renders them compatible with a wide range of substrates, allowing for monolithic integration with Si electronics. These advantages have boosted extensive research efforts, and a range of optoelectronic devices based on CQD materials have been reported.5–10Similar to bulk materials, the semiconductor properties of CQDs are central to high-performance devices. Early efforts were focused on the fine control of colloidal synthetic dynamics that produces monodispersed CQDs with sharp excitonic absorption, an important determinant evaluating their band edge properties due to energetic disorder.11–13 This is much similar to compositional tuning typically applied in engineering band gaps in bulk semiconductors.14,15 Surface management is another big challenge in achieving electronic-grade materials, particularly in the nanoscale regimes where the surface does matter.16–18
Solid-state ligand exchange (SSLE) was first developed to reduce inter-dot distances,2,19–21 resulting in improved electronic coupling. This is typically achieved by applying short-chain ligands (such as amine-, thiol- or halide-containing molecules) to replace long-chain ligand-capped CQD films. The undesired film shrinkage and nonuniform ligand exchange dynamics, however, curtail film quality and induce energetic disorder. Solution-phase ligand exchange (SPLE) was later explored where ionic ligands enable electrostatic stabilization in polar solvents rather than steric hindrance stabilization in nonpolar solvents.7,22–25 QD films with a thickness of the order of a few hundred nanometers can be readily assembled via a single-step spin-coating process. The resulting QD films show high quality in terms of structural uniformity and film compactness. More importantly, a homogeneous colloidal solution also translated into QD solids with more flattened energy landscape that greatly suppresses undesirable charge trapping and recombination and finally contributes to improved charge collection.7 The process is currently widely employed for the best-performing electronic and optoelectronic devices such as LEDs, solar cells and photodetectors.26–31 The cascade surface modification of colloidal quantum dot inks enables efficient bulk homojunction photovoltaics.32 The advantages of solution-phase ligand exchange over traditional solid-state ligand exchange are however compromised by restricted ligands available. Only those ligands that are able to offer electrostatic stabilization of the target CQDs in certain polar solvents meet the prerequisites, allowing solution-phase ligand exchange. In addition, the properties of CQD solids are bounded by the available ligands, and fine tuning of the semiconducting properties remains a challenge.
In this work, we aimed to address this issue and improve the semiconductor properties of CQDs by employing a two-step ligand engineering approach. Using infrared PbS CQDs as a model system, we developed a new functional-ligand-customized (FLC) approach that first employs colloidal-stabilization ligands of 2-mercaptoethanol (ME), promoting ligand exchange, and subsequently adds specific functional ligands, finely tuning the electronic properties. Using a combination of infrared CQDs including PbS and HgTe, and highly emissive CdSe/ZnCdSe/ZnSe core/shell CQDs, we demonstrate that our new approach is able to finely tune the semiconductor properties including passivation, doping and band tails. These improvements allow us to achieve solar cells and photodetectors with greatly improved performance, compared with those prepared via a traditional process.
Results and discussion
We first used PbS CQDs (with an exciton peak at ∼940 nm) as a model system to investigate the process of ligand exchange. The PbS CQDs were synthesized based on the previous report (Fig. S1, ESI†). Fig. 1 depicts the schematic of the FLC process (Fig. 1b) in comparison with the traditional SPLE process (Fig. 1a) (Fig. S2, ESI†). For the SPLE process, halides (usually lead iodide) were employed to boost the ligand exchange process. Following an antisolvent purification process, halide-capped PbS CQDs can be well dispersed in hybrid solvents of dimethylformamide (DMF) and butylamine (BTA). The resulting PbS CQD ink is ready for film deposition. The process is widely employed in current high-performance PbS optoelectronic devices. However, it remains a challenge to achieve precise control over the amount of the ligands. For our new FLC approach, 2-mercaptoethanol (ME), which has been extensively investigated in the previous studies due to the strong bonding between the surface metal sites and the SH group,32–34 was first employed to displace native oleic acid of PbS CQDs via a nonpolar single-phase process in a toluene solvent. Following the purification process, the resulting PbS QD solids are redispersed in DMF: BTA hybrid solvents with the addition of functional ligands to achieve excellent colloidal stabilization (Fig. S3, ESI†). Due to the large steric hindrance of ME compared to the atomic ligand, this process facilitates the introduction of functional ligands (such as PbI2, NH4I) to the CQD surfaces. This can be achieved by pre-dissolving functional ligands in the hybrid solvents where the amount of functional ligands can be readily tuned by varying their concentrations. Compared with the SPLC process, the FLC strategy decouples colloidal stabilization and property engineering using hybrid ligands and, therefore, allows for the control of both the type and the concentration of functional ligands in the hybrid solvent. | ||
| Fig. 1 Schematic illustration of ligand exchange through (a) SPLE and (b) FLC processes. | ||
Fourier-transform infrared (FTIR) spectra were used to monitor ligand exchange using an FLC approach (Fig. 2a). Following the ligand exchange with ME, the vibrations of COO− (around 1536 cm−1) and CH (centered at 2871 and 2927 cm−1) are significantly reduced. Meanwhile, the –OH vibrations at 3200–3600 cm−1 see a dramatic increase in the intensity, which are assigned to the –OH groups of ME.35 These results manifest the ligand displacement of oleic acid (OA) by ME.36–38 To demonstrate the successful modification of the CQD surface by functional ligands, we employed X-ray photoelectron spectroscopy (XPS) to analyze the surface composition of the PbS-ME (without functional ligands) and PbS–PbI2 (with PbI2 as the functional ligands) films. Compared with the PbS-ME film, the XPS I 3d spectra revealed a new peak corresponding to the I− of the PbS–PbI2 film, evidencing the reprogramming of the CQD surface by I− (Fig. S4a, ESI†). Meanwhile, the presence of C–S bonds evidenced from the XPS S 2p spectra (Fig. S4b, ESI†) suggests that ME ligands remain bonded to the surface of the CQDs.32 This finding aligns consistently with the results obtained from the XPS spectra (Fig. S4c, ESI†) of the sample fabricated by the double ligand exchange with both ME and PbI2.
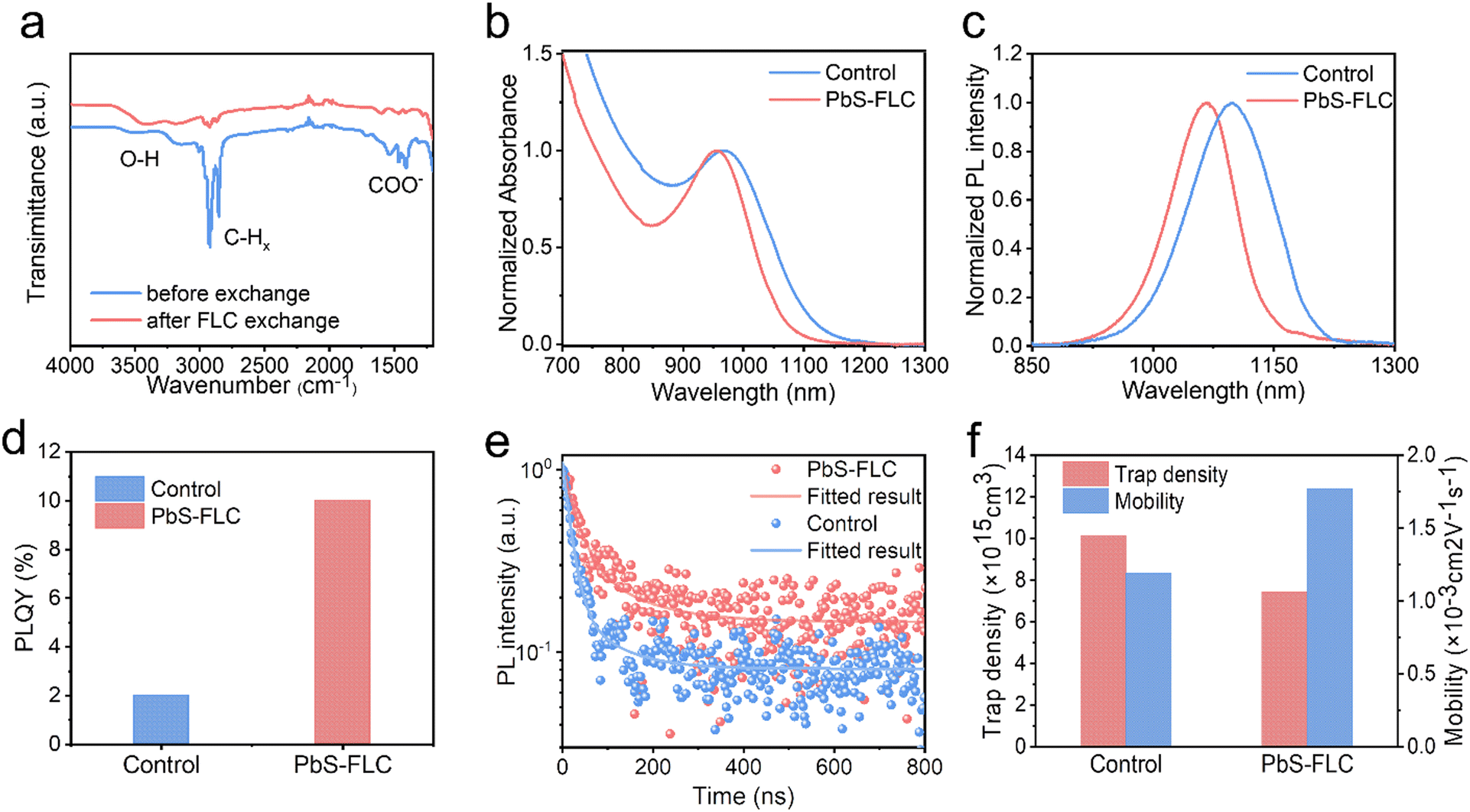 | ||
| Fig. 2 (a) FTIR spectra of PbS films before and after FLC exchange. (b) Normalized optical absorption and (c) PL spectra of the PbS-FLC and control ink. (d) PLQY measurement of PbS-FLC and control inks. (e) Time-resolved PL decay of PbS-FLC and control solid films. (f) Extracted average tarp density and mobility of PbS-FLC and control film from SCLC. | ||
To study the effect of the type of functional ligands on the optoelectronic properties of CQDs, a variety of ligands including PbI2, NH4I, CdI2, and CsI were used to fabricate CQD inks. The absorbance spectra (Fig. S5, ESI†) suggest that compared with the PbS-ME ink, all exciton peaks of PbS inks with the addition of ligands exhibit blue shifts in their excitonic peaks. In the case of FLC without additives, the redshift and broadening of the excitonic peak following the ligand exchange may be attributed to a combination of incomplete ligand coverage and the presence of thiolates.39 With the introduction of smaller atomic ligands such as halides (PbI2, NH4I, CdI2, and CsI) the coverage of surface ligands increased, providing improved colloidal stability and defect passivation. Consequently, the excitonic absorption peak shows a blue shift, and more importantly, presents a narrower half-width at half-maximum (HWHM), an important determinant evaluating the energetic disorder of CQDs.
Since PbI2 is mostly employed for the passivation of PbS CQDs, here we focus on the use of PbI2 as the functional ligand and show the advantages of our FLC approach over the traditional SPLC method. We compared PbS–PbI2 (termed PbS-FLC) CQDs with the control prepared by the traditional SPLE method.7Fig. 2b and c show the normalized absorbance and photoluminescence (PL) spectra of the CQD inks. The HWHM in the absorbance spectra, full-width at half-maximum (FWHM) in the PL spectra and Stokes shift are shown in Fig. S6a (ESI†). The PbS-FLC film exhibits a HWHM of 81 nm, an FWHM of 104 nm, and a Stokes shift of 136 nm, which are much smaller than those of the control sample (HWHM: 103 nm, FWHM: 128 nm, and Stokes shift: 151 nm). The results suggest that the FLC strategy is able to engineer the electronic structures of PbS CQDs. Photoluminescence quantum yield (PLQY) measurements were then performed to further evaluate the passivation of PbS-FLC. The PLQY of PbS-FLC CQDs is 10%, a fivefold increase in comparison with the control CQDs (Fig. 2d). The improved emissive properties suggested improved passivation of the PbS-FLC CQD inks compared to the traditional SPLE approach. Moreover, as shown Fig. S6b and S7 (ESI†), these property improvements of PbS CQDs are well preserved in their final solid-state film, which indicates suppressed energetic disorder in the film state.
To show the energetic disorder in the CQD film, we acquired the Urbach energy by fitting the relation curves between the absorption coefficient of the band tail state and the photon energy, as shown in Fig. S8 (ESI†). The Urbach energy (30 meV) of the PbS-FLC film is smaller than that of the control (42 meV), which indicates that the PbS-FLC film has fewer band tail or defect states. Time-resolved PL (TRPL) measurements were performed to determine the carrier lifetime of CQD films. As shown in Fig. 2e, a bi-exponential decay was used to fit the curves and extract the carrier lifetime. The average carrier lifetime for radiative recombination in the PbS-FLC film and the control film is 29 and 17 ns, respectively, which indicates improved defect passivation in the PbS-FLC CQD film.
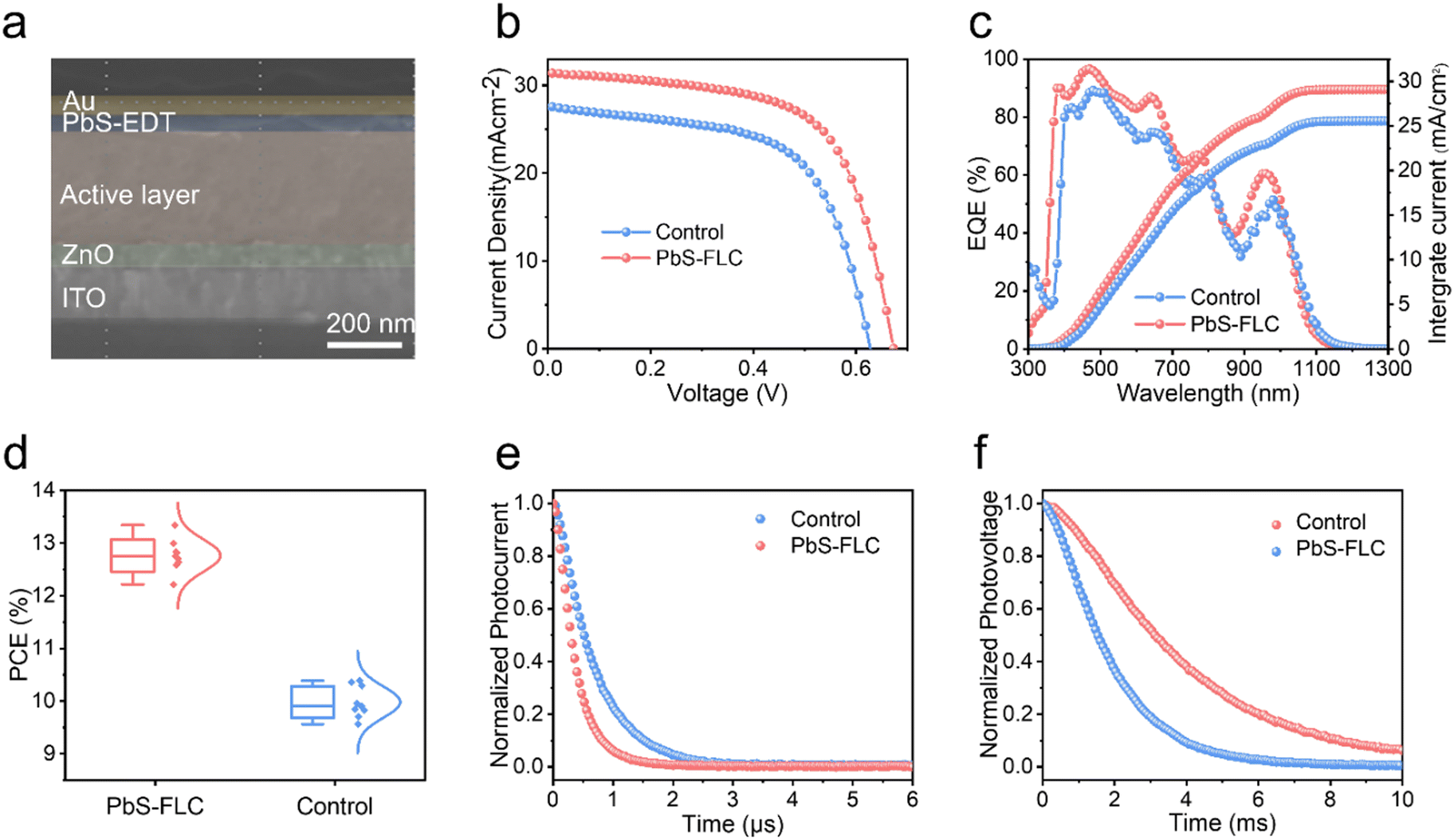
Space charge-limited current (SCLC) trap-filling measurements of hole-only devices (ITO/PbS-EDT/PbS-CQDs/PbS-EDT/Au) were performed to quantify the trap density and transporting properties (Fig. 2f and Fig. S9, ESI†).27,40,41 The trap-filling region can be identified by a significant increase in current injection at a voltage (>VTFL), where all the traps are filled, then trap density can be identified using eqn (1):
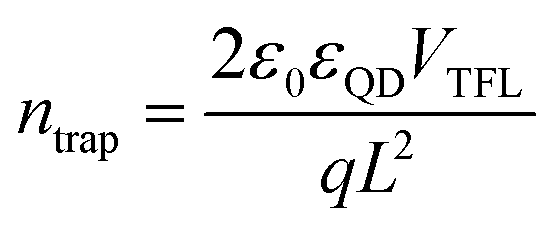 | (1) |
We employed ultrafast transient absorption to analyze the carrier kinetics in PbS CQD films. A low pump power (27 μJ cm−2) was applied to minimize the Auger recombination, thereby favoring a high signal-to-noise ratio. The spectro-temporal transient absorption (TA) maps are presented in Fig. 3a and b. Detailed analysis in Fig. 3c reveals a distinctive red shift of approximately 6.1 meV for the PbS-FLC film, in comparison with the control film that exhibits a more pronounced red shift of 18.2 meV. The observed reduction in energy funneling within the PbS-FLC film substantiates a more flattened energy landscape, which will help improve charge extraction in QD devices. Additionally, the whole TA spectra of PbS-FLC exhibited a slower pump–probe decay behavior than that of the control film. As depicted in Fig. 3d, the average lifetime (τ) of ∼8 ns for the PbS-FLC film was obtained by fitting using a biexponential function, in contrast to that of 4.1 ns for the control film. Such differences can be attributed to more trap states in the control film.
 | ||
| Fig. 3 Spectro-temporal transient absorption maps for (a) control and (b) PbS-FLC films. The shifts of peak position for transient bleach over time for PbS-FLC and control films (c). TA decay of PbS-FLC and control films (d). Representative transient bleach data plotted as normalized absorbance changes ΔT/T for (e) control and (f) PbS-FLC films at different pump fluences. | ||
Meantime, we checked the excitation kinetics of thin film states under high luminous fluxes to extract nonradiative and radiative components (Fig. 3e and f). The pump-power-dependent TA kinetics of PbS-FCL and the control films were well fitted using a biexponential equation, and the corresponding parameters obtained from the fitting are summarized in Table S1 (ESI†). The fast decay – assigned to the nonradiative process component – was extracted in the TA decay curves. In contrast, the slow decay can be attributed to the combination of the inter-dot charge transfer and intrinsic radiative recombination.43 As shown in Table S1 (ESI†), as the pump power increases, the fast component (RA1) content in both films continuously increase, accompanied by a slow decay rate. Notably, the PbS-FLC film shows suppressed non-radiative recombination (∼10% lower) independent of power intensities, in comparison with the control film.
We then pursued to show the advantages of our new FLC approach in building high-performance devices. We examined the excitonic peaks of PbS CQD inks prepared by varying the concentrations of the functional ligand PbI2 (Fig. S10, ESI†). With the increase in PbI2 content, the surface of PbS QDs will be capped by more PbI2, which will increase the inter-dot distance and induce the blueshift of the peak position.44
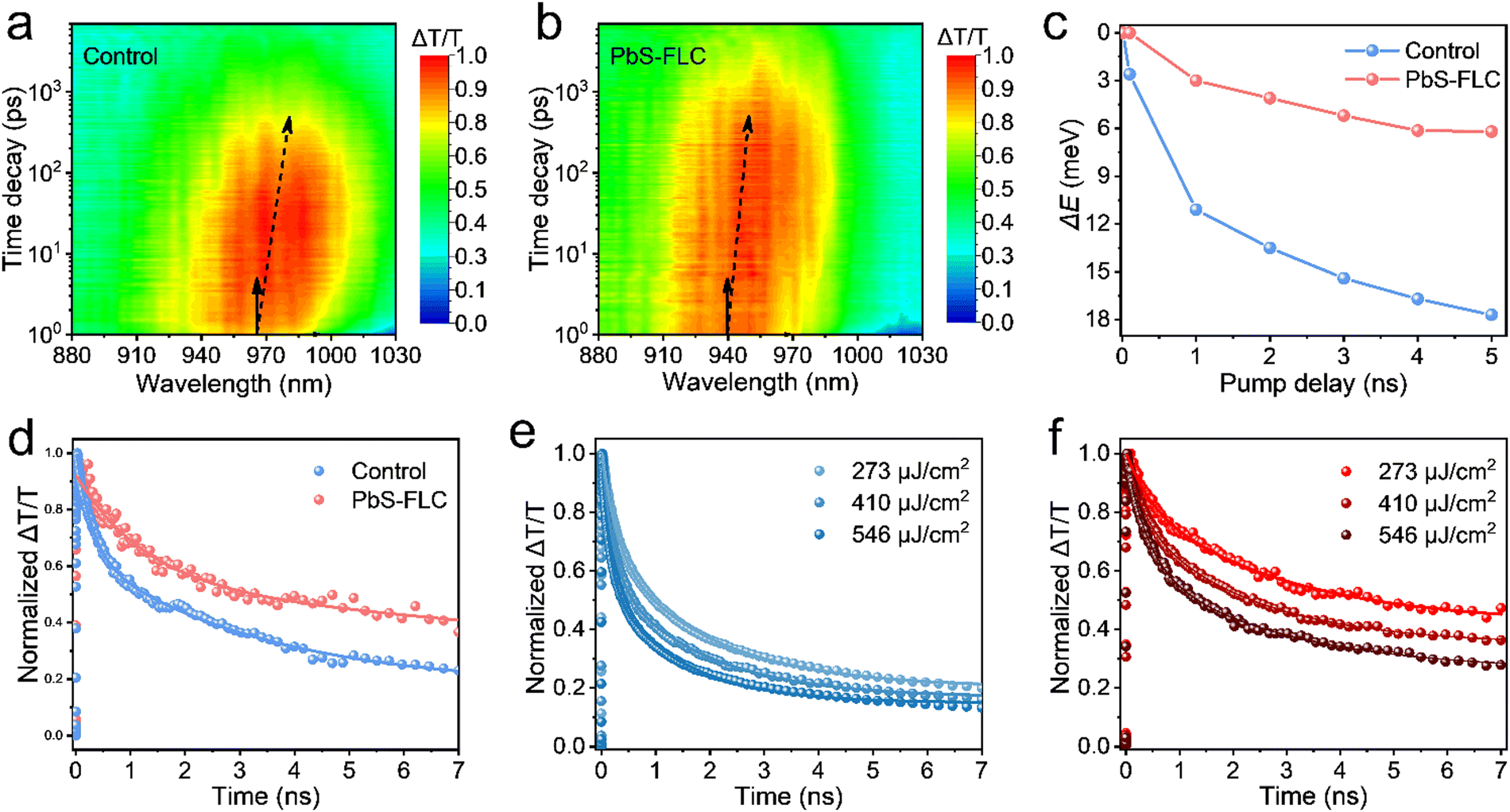
However, the presence of excess PbI2 curtails the colloidal stability and leads to uncontrollable and undesirable agglomeration, resulting in the redshift of the peak position. Meanwhile, to show the advantages of our new FLC approach in constructing high-performance devices, we fabricated the PbS CQD solar cells with the device architecture of ITO/ZnO/PbS-inks/PbS-EDT/Au. The ZnO layer serves as the electron transport layer, while PbS-EDT (CQD treated with 1,2-ethanedithiol) functions as the hole transport layer. The cross-sectional scanning electron microscopic (SEM) image reveals a dense CQD active layer with a thickness of approximately 400 nm (Fig. 4a). The device performance is summarized in Fig. S11 (ESI†). The device performance first increases proportionally with the increase in the amount of PbI2. We propose that the introduction of the functional ligand PbI2 facilitates improved surface passivation of PbS QDs. However, the presence of excess PbI2 leads to a sharp decline in the device performance. This is attributed to the decreased colloidal stability of PbS CQDs, which compromises the film quality and leads to reduced device performance. These findings suggest that an optimized PbI2 concentration (30 mg mL−1) is required in this system. The best current–voltage (J–V) curves are shown in Fig. 4b. The best-performing device with the PbS-FLC film achieved a higher AM1.5 PCE of 13.3% (with a Voc of 0.672 V, a Jsc of 31.4 mA cm−2, and a fill factor (FF) of 63.2%) compared to 10.4% (with a Voc of 0.628 V, a Jsc of 26.7 mA cm−2, and a (FF) of 60.4%) for the control device, which is on a par with the best performance reported in the literature (Table S2, ESI†). Compared to previous cascade surface modification methods,32 our approach allows fine tuning of the ligands, which enables enhanced tuning of the electronic properties of CQD solids. The increment in the open-circuit voltage (Voc) of the PbS-FLC device may benefit from the flattened energy landscape and enhanced surface passivation, as indicated by the TA and PLQY results.
 | ||
| Fig. 4 (a) Cross-sectional SEM image of the device architecture. (b) J–V curves of FLC and control devices. (c) External quantum efficiency (EQE) curves and integrated JSC of the 940 nm PbS CQD solar cells. (d) Distribution of PCE values. Transient photocurrent (e) and photovoltage (f) decay spectroscopy of the FLC and control devices. | ||
External quantum efficiency (EQE) measurements (Fig. 4c) confirmed a high integrated Jsc value of the PbS-FLC device (29.07 mA cm−2) compared with the control device (25.55 mA cm−2), which is consistent with that obtained from the J–V curves. Fig. 4d shows the statistical data that further verify the performance enhancement via an FLC approach. Control experiments rule out the possibility that the performance improvement mainly came from the introduction of ME ligands (Fig. S12, ESI†). Instead, it evidences the importance of the amount of functional ligands in the final QD assembly for the best device performance. We further evaluated the stability of unencapsulated PbS-FLC devices, which retained over 90% of their original efficiency after 30 days of storage in air (Fig. S13, ESI†), showing great potential in the photovoltaic applications. Transient photocurrent (TPC) (Fig. 4e) and transient photovoltage (TPV) (Fig. 4f) were then employed to investigate the dynamics of charge carriers of these two kinds of devices.45,46 The PbS-FLC devices exhibited a shorter carrier extraction time (τTPC) of 0.37 μs and a longer recombination time (τTPV) of 4.10 ms. In contrast, the τTPC and τTPV values of the control devices are 0.71 μs and 2.02 ms, respectively. The results indicated that the FLC strategy can enhance charge carrier extraction and reduce recombination rates in the resulting devices, which agrees well with the results from SCLC.
In addition to solar cells, PbS CQDs are also promising optoelectronic materials for infrared photodetectors. To demonstrate the advantages of our FLC-processed PbS CQDs as photodetectors, we selected PbS CQDs with an exciton peak at 1250 nm, which is beyond the response range of the traditional Si detectors. The TEM images of PbS CQDs are exhibited in Fig. S1 (ESI†). Similar to the above-mentioned results, the PbS-FLC films exhibited a narrower exciton peak with a smaller FWHM of ∼89 meV, while for the control PbS CQD films, an FWHM of ∼174 meV was shown (Fig. S14, ESI†). This verifies that our FLC strategy can be applied to PbS CQDs with different sizes. The J–V curves of the PbS-FLC and control devices were measured under 1300 nm LED illumination of 470 μW cm−2 (Fig. 5a). The dark current density of the PbS-FLC device at −0.5 V is 3.67 × 102 nA cm−2, which is more than an order of magnitude lower than that of the control device (1.0 × 104 nA cm−2). We argue that the increase in carrier lifetime suppresses the thermally excited generation currents at the reverse bias, which is the main reason for the decrease in dark currents. The higher responsivity is also observed from the PbS-FLC device. The photocurrent is 280 μA cm−2 at −0.1 V, in comparison with 226 μA cm−2 of the control device at the same applied voltage bias. The dark J–V curves were fitted based on a diode circuit model to explore the underlying mechanisms of the performance enhancement (Table S3 and Fig. S15, ESI†). The fitting curves consist of the diode current, ohmic leakage current, and non-ohmic leakage current as in eqn (2):47,48
| Jd = J0 {exp[q(V − Jd × Rs)/ηKBT] − 1} + (V − Jd × Rs)/Rsh + k(V − JdRs)m | (2) |
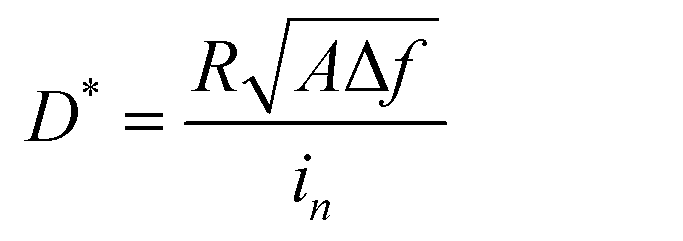 | (3) |
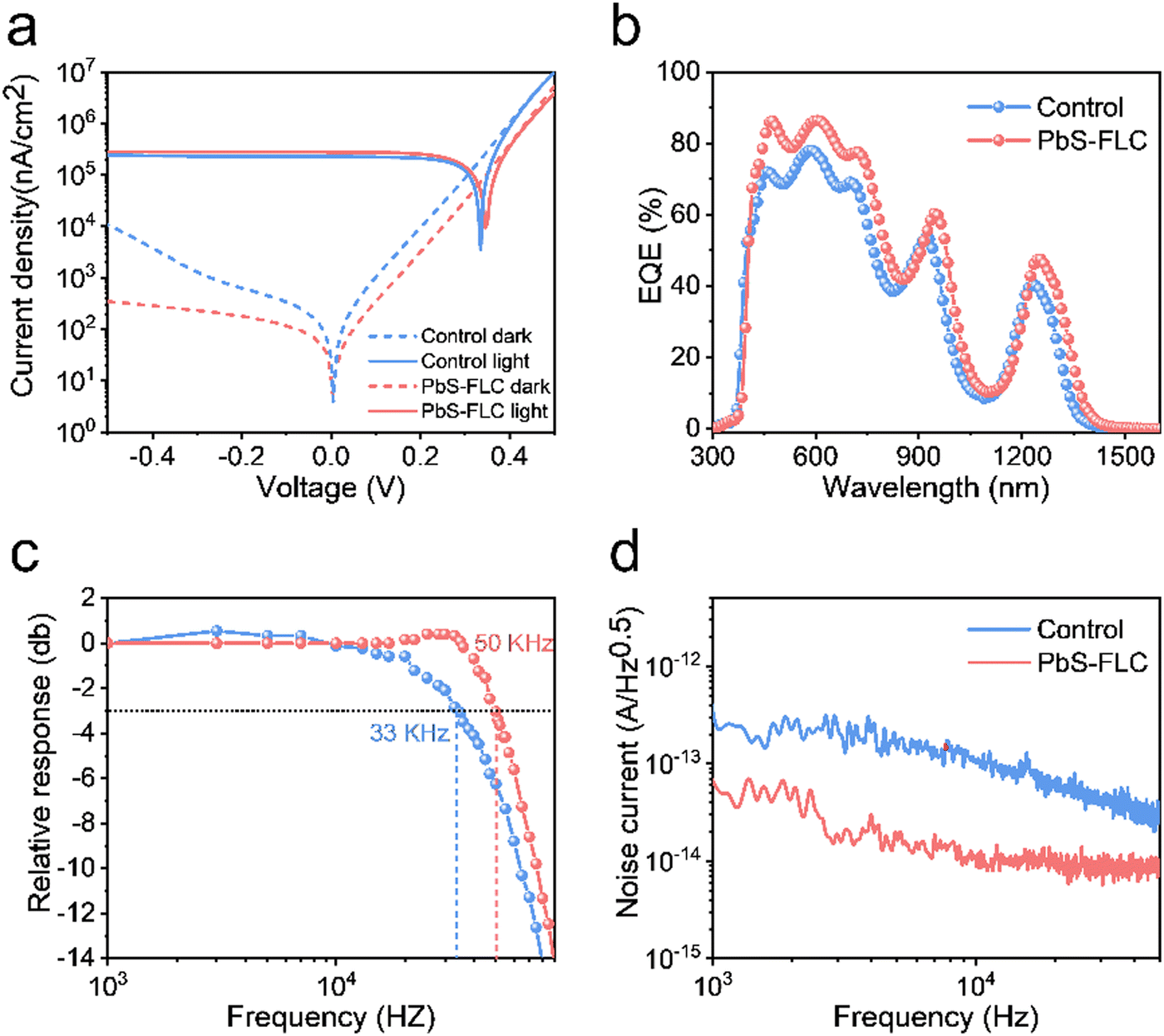 | ||
| Fig. 5 (a) J–V curves of the FLC and control devices measured under dark conditions and 1300 nm light illumination. (b) EQE curves of the 1250 nm PbS CQD devices. (c) −3 dB bandwidth. (d) Noise power spectra of the control and FLC devices. | ||
To demonstrate the versatility of the FLC strategy, visibly emissive CdSe/ZnCdSe/ZnSe core/shell QDs were further studied. CdSe/ZnCdSe/ZnSe core/shell CQDs were prepared following a procedure previously reported in the literature.49 Our studies indicated that surface-capped OA ligands can be easily replaced by ME and the resulting ME-capped CdSe/ZnCdSe/ZnSe core/shell QDs can be easily dispersed in DMF-BTA hybrid solvents. The photoluminescence quantum yield (PLQY), however, shows a dramatic decrease. The drastic deterioration of the photoluminescence quantum yields can be attributed to the quenching of QDs by thiol ligands.50 In our case, we propose that ME ligands alone are not able to afford the desired defect passivation. Therefore, to regain the highly emissive properties, CdI2 and ZnI2 were used as functional ligands for surface passivation (Fig. 6a). The absorption and PL spectra (Fig. 6b) of FLC CQDs in DMF-BTA solvent show negligible variation, compared with those of native OA-capped CQDs. With the addition of CdI2 and ZnI2, the PLQY loss decreases from 27.1% to 3.9% (CdI2) and 0.4% (ZnI2), respectively (Table S4, ESI†). The recovery of the emissive properties of CdSe/ZnCdSe/ZnSe CQDs using functional ligands verifies the importance of engineering the choice of the ligands. The inorganic ligand-capped emissive QDs are of particular interest in those areas where the thermal management is very important. Note that ZnI2 or CdI2 alone is not able to afford colloidal stabilization of CdSe/ZnCdSe/ZnSe CQDs in polar solvents, supporting the unique advantage of decoupling colloidal stabilization and property engineering in our FLC approach.
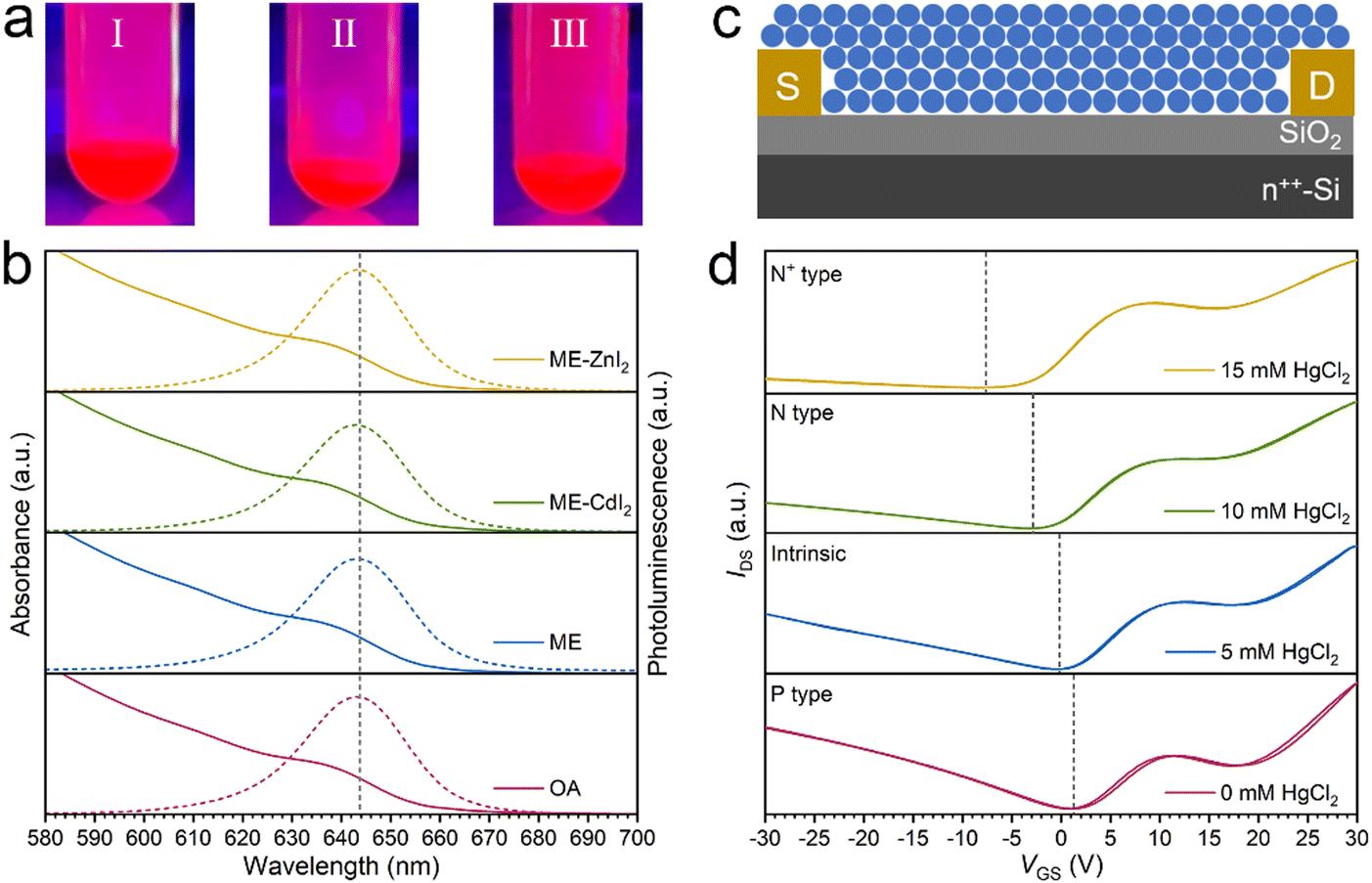 | ||
| Fig. 6 (a) Photographs of the CdSe/ZnCdSe/ZnSe core/shell capped with OA in octane (I), ME-ZnI2 (II) and ME-CdI2 (III) in DMF/BTA. (b) Absorption and PL spectra of CdSe/ZnCdSe/ZnSe core/shell CQDs before and after exchange. (c) Scheme of the FET device architecture. (d) FET transfer curves of HgTe CQDs after exchange. | ||
HgTe CQDs are a new class of infrared-absorbing materials promising low-cost and large-format infrared focal-plane arrays. HgTe CQDs were synthesized following a protocol reported in our previous study.33 With the FLC approach, oleylamine-capped HgTe CQDs can be stabilized by ME in DMF. HgCl2 was selected as the functional ligand engineering the properties of HgTe CQDs. The successful application of our FLC approach in HgTe CQDs was verified by a dynamic light scattering approach (Fig. S17, ESI†). FLC-processed HgTe CQDs with a size of 8 nm and absorption cutoff of 3.75 μm (Fig. S18, ESI†) were then used as building blocks for thin-film field-effect transistor (FET) fabrication (Fig. 6c). In the absence of HgCl2, the HgTe CQD solid shows weak p-type doping. Using HgCl2 as the functional ligand, the doping polarity can be easily switched to n-type and precise control over the amount of HgCl2 allows fine tuning of the doping magnitude (Fig. 6d). Note that an increase in the amount of HgCl2 does not compromise the transporting properties (Table S5, ESI†), indicating the strong interaction between CQDs and their surrounding ligands. This is crucial for the success of our FLC approach. The doping tuning of HgTe CQDs with HgCl2 has been extensively studied in a SPLE process where HgCl2 is added as the ligand for displacement. After ligand exchange, it remains unknown how much HgCl2 was preserved. In contrast, in our FLC approach, HgCl2 was added in the final dispersed solvent such that the amount of HgCl2 in the final QD assembly could be precisely controlled. The ligand-dependent doping characteristics demonstrate that fine tuning of the number of ligands is crucial in engineering the electronic properties of CQD solids.
Conclusions
In summary, we have developed a novel FLC ligand exchange method that is able to enhance the regulation of the electrical properties of CQD solids, including passivation, doping and band tail characteristics. It was achieved by decoupling the colloidal-stabilization and property-engineering ligands in a two-step process. The universality of our approach is verified using PbS, CdSe/ZnCdSe/ZnSe, and HgTe CQDs. Our results verify the significance of tuning the type and amount of ligands in engineering the properties of CQDs, which is crucial for the construction of high-performance optoelectronic devices. The present limiting factor of our approach is that electronically active ME is dispensable for colloidal stabilization. The advances in colloidal chemistry might enable the use of electronically inert stabilizing ligands instead of ME that will finally allow further engineering of the properties of CQDs. We expect that the ligand engineering strategy presented in this work might have important implications in the construction of other electronic-grade CQD solids.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. U22A2083, 62374068, 61974052), the National Key Research and Development Program of China (No. 2021YFA0715502), Key R & D program of Hubei Province (No. 2021BAA014), the Innovation Project of Optics Valley Laboratory (No. OVL2021BG009, OVL2023ZD002), the Exploration Project of Natural Science Foundation of Zhejiang Province (No. LY23F040005), the Fund from Science, Technology and Innovation Commission of Shenzhen Municipality (No. GJHZ20210705142540010, GJHZ20220913143403007), and the fund for Innovative Research Groups of the Natural Science Foundation of Hubei Province (No. 2020CFA034). The authors thank Prof. Huaibin Shen for kind support of CdSe/ZnCdSe/ZnSe. The authors thank the Analytical and Testing Center of Huazhong University Science and Technology for the material characterizations.Notes and references
- G. Konstantatos, C. Huang, L. Levina, Z. Lu and E. H. Sargent, Adv. Funct. Mater., 2005, 15, 1865–1869 CrossRef CAS
.
- S. A. McDonald, G. Konstantatos, S. Zhang, P. W. Cyr, E. J. D. Klem, L. Levina and E. H. Sargent, Nat. Mater., 2005, 4, 138–142 CrossRef CAS PubMed
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS
- Y. Liu, M. Gibbs, J. Puthussery, S. Gaik, R. Ihly, H. W. Hillhouse and M. Law, Nano Lett., 2010, 10, 1960–1969 CrossRef CAS PubMed
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulović, Nat. Photon., 2013, 7, 13–23 CrossRef CAS
- X. Lan, O. Voznyy, F. P. Garcia de Arquer, M. Liu, J. Xu, A. H. Proppe, G. Walters, F. Fan, H. Tan, M. Liu, Z. Yang, S. Hoogland and E. H. Sargent, Nano Lett., 2016, 16, 4630–4634 CrossRef CAS PubMed
- M. Liu, O. Voznyy, R. Sabatini, F. P. Garcia de Arquer, R. Munir, A. H. Balawi, X. Lan, F. Fan, G. Walters, A. R. Kirmani, S. Hoogland, F. Laquai, A. Amassian and E. H. Sargent, Nat. Mater., 2017, 16, 258–263 CrossRef CAS PubMed
- J.-S. Lee, M. V. Kovalenko, J. Huang, D. S. Chung and D. V. Talapin, Nat. Nanotechnol., 2011, 6, 348–352 CrossRef CAS PubMed
- X. Gong, Z. Yang, G. Walters, R. Comin, Z. Ning, E. Beauregard, V. Adinolfi, O. Voznyy and E. H. Sargent, Nat. Photon., 2016, 10, 253–257 CrossRef CAS
- M. Vafaie, J. Z. Fan, A. Morteza Najarian, O. Ouellette, L. K. Sagar, K. Bertens, B. Sun, F. P. García de Arquer and E. H. Sargent, Matter, 2021, 4, 1042–1053 CrossRef CAS
- M. C. Weidman, M. E. Beck, R. S. Hoffman, F. Prins and W. A. Tisdale, ACS Nano, 2014, 8, 6363–6371 CrossRef CAS PubMed
- Y. Wang, Z. Liu, N. Huo, F. Li, M. Gu, X. Ling, Y. Zhang, K. Lu, L. Han, H. Fang, A. G. Shulga, Y. Xue, S. Zhou, F. Yang, X. Tang, J. Zheng, M. Antonietta Loi, G. Konstantatos and W. Ma, Nat. Commun., 2019, 10, 5136 CrossRef PubMed
- Y. Xia, W. Chen, P. Zhang, S. Liu, K. Wang, X. Yang, H. Tang, L. Lian, J. He, X. Liu, G. Liang, M. Tan, L. Gao, H. Liu, H. Song, D. Zhang, J. Gao, K. Wang, X. Lan, X. Zhang, P. Müller-Buschbaum, J. Tang and J. Zhang, Adv. Funct. Mater., 2020, 30, 2000594 CrossRef CAS
- X. Li, J. Zhang, C. Yue, X. Tang, Z. Gao, Y. Jiang, C. Du, Z. Deng, H. Jia, W. Wang and H. Chen, Sci. Rep., 2022, 12, 7681 CrossRef CAS PubMed
- M.-S. Sang, G.-Q. Xu, H. Qiao and X.-Y. Li, J. Infrared Millim. Waves, 2022, 41, 972 CAS
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 364 CrossRef CAS PubMed
- C. R. Kagan and C. B. Murray, Nat. Nanotechnol., 2015, 10, 1013–1026 CrossRef CAS PubMed
- F. P. García de Arquer, D. V. Talapin, V. I. Klimov, Y. Arakawa, M. Bayer and E. H. Sargent, Science, 2021, 373 Search PubMed
- F. Wu, Y. Zhang, Z. Zhang, G. Li, M. Li, X. Lan, T. Sun and Y. Jiang, Langmuir, 2017, 33, 7124–7129 CrossRef CAS PubMed
- K. Lu, Y. Wang, Z. Liu, L. Han, G. Shi, H. Fang, J. Chen, X. Ye, S. Chen, F. Yang, A. G. Shulga, T. Wu, M. Gu, S. Zhou, J. Fan, M. A. Loi and W. Ma, Adv. Mater., 2018, 30, e1707572 CrossRef PubMed
- C. H. Chuang, P. R. Brown, V. Bulović and M. G. Bawendi, Nat. Mater., 2014, 13, 796–801 CrossRef CAS PubMed
- H. Aqoma and S.-Y. Jang, Energy Environ. Sci., 2018, 11, 1603–1609 RSC
- Z. Ning, Y. Ren, S. Hoogland, O. Voznyy, L. Levina, P. Stadler, X. Lan, D. Zhitomirsky and E. H. Sargent, Adv. Mater., 2012, 24, 6295–6299 CrossRef CAS PubMed
- M. Gu, Y. Wang, F. Yang, K. Lu, Y. Xue, T. Wu, H. Fang, S. Zhou, Y. Zhang, X. Ling, Y. Xu, F. Li, J. Yuan, M. A. Loi, Z. Liu and W. Ma, J. Mater. Chem. A, 2019, 7, 15951–15959 RSC
- J. Z. Fan, N. T. Andersen, M. Biondi, P. Todorovic, B. Sun, O. Ouellette, J. Abed, L. K. Sagar, M. J. Choi, S. Hoogland, F. P. G. de Arquer and E. H. Sargent, Adv. Mater., 2019, 31, e1904304 CrossRef PubMed
- M. Yuan, X. Wang, X. Chen, J. He, K. Li, B. Song, H. Hu, L. Gao, X. Lan, C. Chen and J. Tang, Small, 2022, 18, e2102340 CrossRef PubMed
- X. Yang, J. Yang, M. I. Ullah, Y. Xia, G. Liang, S. Wang, J. Zhang, H.-Y. Hsu, H. Song and J. Tang, ACS Appl. Mater. Interfaces, 2020, 12, 42217–42225 CrossRef CAS PubMed
- M.-Q. Dai and L.-Y. L. Yung, Chem. Mater., 2013, 25, 2193–2201 CrossRef CAS
- S. Yin, C. H. Y. Ho, S. Ding, X. Fu, L. Zhu, J. Gullett, C. Dong and F. So, Chem. Mater., 2022, 34, 5433–5442 CrossRef CAS
- B. K. Jung, H. K. Woo, C. Shin, T. Park, N. Li, K. J. Lee, W. Kim, J. H. Bae, J. P. Ahn, T. N. Ng and S. Oh, J. Adv. Opt. Mater., 2022, 10, 2101611 CrossRef CAS
- K. Qiao, Y. Cao, X. Yang, J. Khan, H. Deng, J. Zhang, U. Farooq, S. Yuan and H. Song, RSC Adv., 2017, 7, 52947–52954 RSC
- M. J. Choi, F. P. Garcia de Arquer, A. H. Proppe, A. Seifitokaldani, J. Choi, J. Kim, S. W. Baek, M. Liu, B. Sun, M. Biondi, B. Scheffel, G. Walters, D. H. Nam, J. W. Jo, O. Ouellette, O. Voznyy, S. Hoogland, S. O. Kelley, Y. S. Jung and E. H. Sargent, Nat. Commun., 2020, 11, 103 CrossRef CAS PubMed
- J. Yang, H. Hu, Y. Lv, M. Yuan, B. Wang, Z. He, S. Chen, Y. Wang, Z. Hu, M. Yu, X. Zhang, J. He, J. Zhang, H. Liu, H.-Y. Hsu, J. Tang, H. Song and X. Lan, Nano Lett., 2022, 22, 3465–3472 CrossRef CAS PubMed
- M. J. Choi, S. W. Baek, S. Lee, M. Biondi, C. Zheng, P. Todorovic, P. Li, S. Hoogland, Z. H. Lu, F. P. G. de Arquer and E. H. Sargent, Adv. Sci., 2020, 7, 2000894 CrossRef CAS PubMed
- A. Shestha, Y. Yin, G. G. Andersson, N. A. Spooner, S. Qiao and S. Dai, Small, 2017, 13, 1602956 CrossRef PubMed
- S. Liu, K. Xiong, K. Wang, G. Liang, M. Y. Li, H. Tang, X. Yang, Z. Huang, L. Lian, M. Tan, K. Wang, L. Gao, H. Song, D. Zhang, J. Gao, X. Lan, J. Tang and J. Zhang, ACS Nano, 2021, 15, 3376–3386 CrossRef CAS PubMed
- S. Wang, C. Bi, A. Portniagin, J. Yuan, J. Ning, X. Xiao, X. Zhang, Y. Y. Li, S. V. Kershaw, J. Tian and A. L. Rogach, ACS Energy Lett, 2020, 5, 2401–2410 CrossRef CAS
- X. Yang, J. Yang, J. Khan, H. Deng, S. Yuan, J. Zhang, Y. Xia, F. Deng, X. Zhou, F. Umar, Z. Jin, H. Song, C. Cheng, M. Sabry and J. Tang, Nano-Micro Lett., 2020, 12, 37 CrossRef CAS PubMed
- C. Giansante, I. Infante, E. Fabiano, R. Grisorio, G. P. Suranna and G. Gigli, J. Am. Chem. Soc., 2015, 137, 1875–1886 CrossRef CAS PubMed
- J. Kwan Kim, J. Hoon Song, H. Choi, S. Jae Baik and S. Jeong, J. Appl. Phys., 2014, 115, 054302 CrossRef
- K. M. Kim, B. J. Choi, Y. C. Shin, S. Choi and C. S. Hwang, Appl. Phys. Lett., 2007, 91, 012907 CrossRef
- M. Liu, F. Che, B. Sun, O. Voznyy, A. Proppe, R. Munir, M. Wei, R. Quintero-Bermudez, L. Hu, S. Hoogland, A. Mandelis, A. Amassian, S. O. Kelley, F. P. García de Arquer and E. H. Sargent, ACS Energy Lett., 2019, 4, 1225–1230 CrossRef CAS
- C. Ding, F. Liu, Y. Zhang, S. Hayase, T. Masuda, R. Wang, Y. Zhou, Y. Yao, Z. Zou and Q. Shen, ACS Energy Lett., 2020, 5, 3224 CrossRef CAS
- M. Liu, S. D. Verma, Z. Zhang, J. Sung and A. Rao, Nano Lett., 2021, 21, 8945–8951 CrossRef CAS PubMed
- W. Ahmad, J. He, Z. Liu, K. Xu, Z. Chen, X. Yang, D. Li, Y. Xia, J. Zhang and C. Chen, Adv. Mater., 2019, 31, e1900593 CrossRef PubMed
- M. Li, S. Chen, X. Zhao, K. Xiong, B. Wang, U. A. Shah, L. Gao, X. Lan, J. Zhang, H. Y. Hsu, J. Tang and H. Song, Small, 2022, 18, e2105495 CrossRef PubMed
- B. L. Williams, S. Smit, B. J. Kniknie, K. J. Bakker, W. Keuning, W. M. M. Kessels, R. E. I. Schropp and M. Creatore, Prog. Photovoltaics Res. Appl., 2015, 23, 1516–1525 CrossRef
- Q. K. Yang, F. Fuchs, J. Schmitz and W. Pletschen, Appl. Phys. Lett., 2002, 81, 4757–4759 CrossRef CAS
- H. Shen, Q. Gao, Y. Zhang, Y. Lin, Q. Lin, Z. Li, L. Chen, Z. Zeng, X. Li, Y. Jia, S. Wang, Z. Du, L. S. Li and Z. Zhang, Nat. Photonics, 2019, 13, 192–197 CrossRef CAS
- W. Lin, Y. Niu, R. Meng, L. Huang, H. Cao, Z. Zhang, H. Qin and X. Peng, Nano Res., 2016, 9, 260–271 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Photo images, absorption spectra, PL spectra, FTIR, XPS, J–V curves, volcano plot of device parameters of PbS QDs solar cells is available in the online version of this article. See DOI: https://doi.org/10.1039/d4tc01182a |
| ‡ Hang Xia and Huicheng Hu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |