
Redox-active covalent organic nanosheets (CONs) as a metal-free electrocatalyst for selective CO2 electro-reduction to the liquid fuel methanol†
Soumitra
Barman
 a,
Anupam
Dey
a,
Faruk Ahamed
Rahimi
a,
Vasudeva Rao
Bakuru
a,
Rohan
Jena
a,
Adrija
Ghosh
b and
Tapas Kumar
Maji
*ab
a,
Anupam
Dey
a,
Faruk Ahamed
Rahimi
a,
Vasudeva Rao
Bakuru
a,
Rohan
Jena
a,
Adrija
Ghosh
b and
Tapas Kumar
Maji
*ab
aMolecular Materials Laboratory, Chemistry and Physics of Materials Unit, School of Advanced Materials (SAMat), Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bangalore-560064, India. E-mail: tmaji@jncasr.ac.in
bNew Chemistry Unit, School of Advanced Materials (SAMat), Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bangalore 560064, India
First published on 24th April 2024
Abstract
In essence, the utilization of renewable energy in the carbon dioxide reduction reaction (CO2RR) holds the potential to transform carbon emissions into valuable chemicals, encompassing a range of hydrocarbons and alcohols. Herein, we have implemented a redox-active triphenylamine (TPA)-based covalent organic nanosheets (CONs) as a metal-free electrocatalyst for CO2 reduction in 0.2 M phosphate buffer (pH-7.2). The reaction produced methanol as the only carbonaceous liquid product reaching a maximum faradaic efficiency (FE) of 51.6% under mild reaction conditions in aqueous medium. Moreover, the corresponding overpotential of the reaction was as low as 210 mV illustrating the superiority of this metal-free electrocatalyst. The mechanistic aspects are supported through experimental in situ RAS-IR (reflection–absorption), in situ Raman study as well as computational study. The superior performance of the electrocatalyst is believed to be due to the presence of redox-active 2-D nanosheets with exposed active sites. This work unlocks a way to produce methanol efficiently by the electrochemical reduction of CO2.
Introduction
Over the last decade, there has been a significant surge of interest in the electrocatalytic CO2 reduction reaction (CO2RR) to value added chemicals and fuels which stems from the high conversion efficiency and selectivity towards desired products.1–4 Electrochemical CO2 reduction can be powered by electricity generated from solar and wind energy, thus intermittent renewable energy can be harnessed for the generation of carbon based fuels and chemicals.4–7 However, the sluggish reaction kinetics limits the CO2 conversion efficiency which prompts the urgency of developing an efficient electrocatalyst. To achieve high energy efficiency and scalability, the reaction must occur rapidly and selectively at low overpotentials. The reduction of CO2 to hydrocarbons and alcohols has the potential to generate a sustainable supply of valuable feedstocks for chemical industries and fuels to meet our energy demands. In this context, advanced electrocatalysts are required to boost the CO2RR with both high conversion efficiency and selectivity.8–12 Nevertheless, metal-based CO2 reduction electrocatalysts exhibit numerous significant drawbacks, including limited selectivity, inadequate durability, susceptibility to gas poisoning, and adverse environmental effects. As a result, there is a strong aspiration to substitute metal-based electrocatalysts with more favourable alternatives such as hetero-atom doped carbonaceous materials, g-C3N4, etc.13–16 These alternatives aim to facilitate cost-effective, yet efficient and stable electrocatalytic activities in terms of overpotential, product selectivity and efficiency.17,18 However, they still face some major issues with the limitation of CO2 adsorption or efficient electron transmission due to the non-porous and undefined structure. In this context, metal-free covalent organic frameworks with porous crystalline structures have been recently explored as a promising platform for the CO2RR with more exposed catalytic active sites.19–25 The tailoring of the COF structure by covalent conjugation of suitable redox active building blocks will help facile electron transfer during the CO2RR process. Furthermore, the rational design of these building blocks with electron rich functional groups would allow the binding of CO2 during catalysis.26–28 In addition, the ultrathin structure endows catalysts with attractive properties, including large surface area, highly exposed electrocatalytic active sites, rapid mass transport, and superior electron transfer. Therefore, the use of ultrathin 2D covalent organic nanosheets (CONs) is an effective strategy to improve electrochemical activity.19,29,30 However, in this growing field, metal free-COFs are rarely reported towards the electrochemical CO2RR and to the best of our knowledge production of methanol as the CO2 reduced product is yet to be reported.31–38Redox active triphenyl amine (TPA) is a well-studied electron donor and acts as a hole transporting moiety.39 TPA shows two reversible redox states based on the formation of radical species, which allow it to switch between different oxidation states. This property makes TPA a versatile building block in various applications, particularly in electrochemistry, for catalysing electron transfer processes.40 TPA based materials including porous organic polymers have been studied for photo and electrocatalytic applications including the HER and ORR.41–43 Here, we envisioned that redox active COF comprised of a triphenyl amine based moiety would significantly influence the electron mobility because of the extended π-conjugation along the 2D network. Building upon this concept, we have successfully employed a triphenylamine-based COF (TPA–TPA) in the form of covalent organic nanosheets (CONs) as a heterogeneous, metal-free electrocatalyst for facilitating the reduction of CO2 in aqueous medium. The highly reducing CONs produced methanol along with a trace amount of CO as carbonaceous products along with H2. The FE reached as high as 51.6%. The electrocatalyst suppresses the thermodynamic barrier with an overpotential as low as 210 mV. These results are indeed superior in terms of metal-free electrocatalysts for methanol production. Additional in situ IR and Raman experiments were carried out to prove the mechanistic pathway along with density functional theory (DFT)-based calculation. This work demonstrated a straightforward strategy towards designing an efficient CO2RR catalyst for liquid fuel production utilizing low cost and high surface area-based porous materials.
Results and discussion
The targeted COF was synthesized under solvothermal conditions through a one-pot condensation reaction with tris(4-aminophenyl)amine (TAPA) and tris(4-formylphenyl)amine (TFPA) following the reported literature and is denoted as TPA–TPA COF (Fig. 1a).44,45 The structure was analyzed using powder X-ray diffraction study in combination with computational simulation. The diffraction pattern of the TPA–TPA COF showed a sharp low-angle diffraction at 2θ of 5.05° corresponding to a (100) reflection (Fig. 1b and c). Additionally, two diffraction peaks at 9.15 and 13.66° were assigned to (200) and (210) reflections, respectively. A slightly broad peak at a value of 2θ of 20.25° was attributed to the (001) reflection caused by π-stacking between the COF layers. These observations are in good agreement with the previously reported literature.44 The successful preparation was confirmed using Fourier transform infrared (FTIR) spectroscopy, and solid-state 13C-NMR spectroscopy. The FTIR spectrum of the as-prepared TPA–TPA COF displayed characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration at 1618 cm−1 corroborating the formation of COF through Schiff-base condensation.
N stretching vibration at 1618 cm−1 corroborating the formation of COF through Schiff-base condensation.
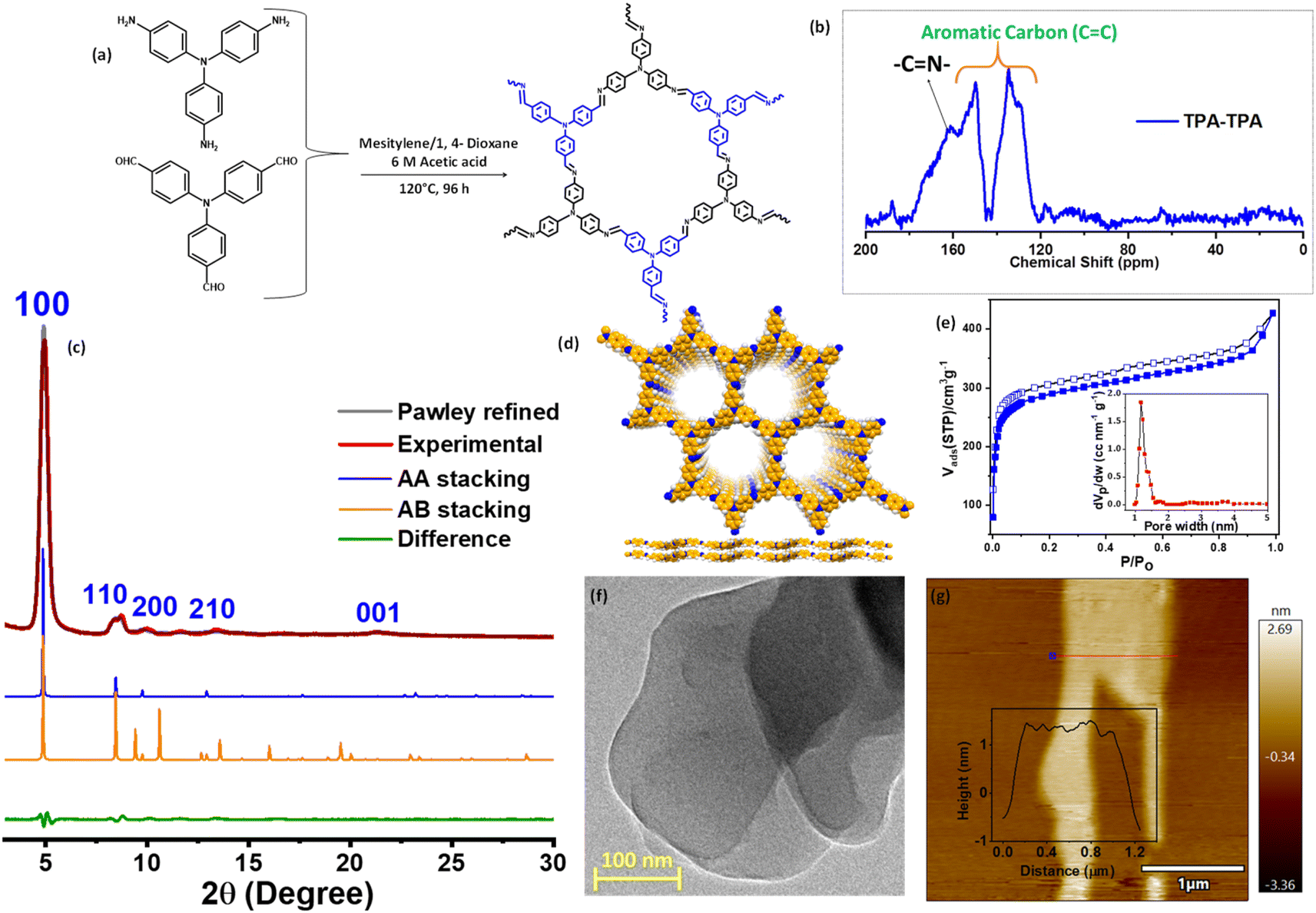 | ||
| Fig. 1 (a) Schematic illustration of the preparation of TPA–TPA COF. (b) Solid state 13C-NMR spectra of TPA–TPA COF. (c) Powder X-ray diffraction pattern of TPA–TPA COF along with the simulated pattern. (d) Optimized extended structure of TPA–TPA COF based on π–π interaction. (e) N2 adsorption isotherm of TPA–TPA COF; inset showing pore size distribution. (f) HRTEM image of TPA–TPA COF. (g) Tapping mode AFM image of the exfoliated COF nanosheets (CONs). | ||
Additionally, the peaks in the range at 1500–1595 and 3032 cm−1 corresponded to the aromatic CC stretching frequency and aromatic C–H stretching frequency of the triaryl monomers, respectively (Fig. S1†). The utilization of solid-state 13C-NMR further confirmed the formation of the TPA–TPA COF. The presence of the peak at around 161 ppm is attributable to the resonances of the imine CN carbon for TPA–TPA COF (Fig. 1d). Additionally, the peaks at 120–150 ppm could be attributed to the aromatic ring. Moreover, thermogravimetric analysis (TGA) revealed that the framework is stable up to 475 °C (Fig. S2†). The permanent porosity of the TPA–TPA COF was established by measuring N2 sorption at 77 K which resembled a type-I adsorption isotherm demonstrating the microporous nature of the polymer. The Brunauer–Emmett–Teller (BET) surface area was calculated to be 1086 m2 g−1 (Fig. 1e). The pore size distribution was analyzed using nonlocal density functional theory (NLDFT) and found to be 1.4 nm (Fig. 1e inset). In the bulk COF, 2D layers are extended through the π–π interactions, which are easily exfoliated into ultrathin 2D covalent organic nanosheets (CONs) via one-step ethanol-assisted 2 h-long ultra-sonication. After exfoliation, the structure of the TPA–TPA COF remains intact in the nanosheets as realized by PXRD measurement (Fig. S3†). The morphology of the as-synthesized TPA–TPA COF was visualized using SEM and high-resolution TEM images (HRTEM). The SEM image displayed uniform arrangements in a layered fashion, whereas, the TEM image displayed layered morphology stacked one upon another layer (Fig. S4† and 1f). Atomic force microscopy (AFM) analysis of the exfoliated COF suggested the formation of CONs with the thickness of 1.5 ± 0.5 nm. Taking into account the interplanar distance of 0.39 nm between adjacent layers, it was concluded that the CONs consisted of ∼4 atomic layers (Fig. 1g). Next, we examined the redox activity of the TPA–TPA COF by an electrochemical study. Cyclic voltammogram (CV) study revealed the redox behavior of the triphenyl moiety present in the COF skeleton. The reversible peak at E1/2 = 0.57 V vs. Ag/Ag+ demonstrated the formation of TPA˙+ species. Another reversible peak which appeared at E1/2 = 1.15 V vs. Ag/Ag+ illustrates the formation of TPA+ species (Fig. S5†).42,46
The electrocatalytic CO2RR performance was carried out in CO2-saturated 0.2 M phosphate buffer (K2HPO4/KH2PO4) (pH-7.2) in a proton-conducting membrane (Nafion-117) separated two-compartment customized gas-tight H-cell with TPA–TPA CONs using Ag/AgCl as the reference electrode and Pt as the counter electrode (Fig. S6†). The CO2 reduction ability was first examined by performing linear sweep voltammetry (LSV) measurements (Fig. 2a). The potential was swept between 0.5 V and −1.2 V vs. RHE with a scan rate of 10 mV s−1. From the linear sweep voltammetry (LSV) curves, the much higher current densities of the TPA–TPA COF in CO2-saturated phosphate-buffer than in Ar-saturated solution indicates the activity in the CO2RR. The Ar-saturated electrolyte showed an increase of current density which was ascribed to the proton reduction with applied cathodic current. In the CO2-saturated electrolyte, enhanced current density was observed. The enhanced current density under the CO2-saturated atmosphere was associated with the catalytic CO2 reduction and H+ reduction. The current density being a function of the kinetics of various electron transfer processes between the reactant and the electrode material, the increased current densities obtained in the CO2-saturated electrolyte compared with that in the Ar-saturated electrolyte indicated the significant contribution of the current due to the CO2 reduction process. Additionally, cyclic voltammetry (CV) measurement showed significant increment of cathodic current from −0.2 V vs. RHE in a CO2 atmosphere (Fig. S7†). Energy efficiency and selectivity was assessed by performing controlled potential electrolysis (CPE) at different applied potentials between −0.18 V and −0.98 V (vs. RHE) in CO2-saturated phosphate buffer (pH-7.2) (Fig. 2b). Comprehensive product analysis using gas chromatography-mass spectrometry (GC-MS) and 1H-NMR spectroscopy (Fig. 2c and S8, S9†) revealed that methanol (CH3OH) was generated as the carbonaceous product along with CO as a minor product. Apart from that, H2 was produced from proton reduction. No other liquid or gaseous products were detected during the CO2RR at different applied potentials. Reduction of CO2 to CH3OH started at a low potential of −0.18 V vs. RHE (Fig. S10†) suggesting an overpotential as low as 210 mV which is significantly lower than that of other reported electrocatalysts forming CH3OH.32,47,48 Faradaic efficiencies (FEs) were calculated for complete product distribution over the entire potential window (Fig. 2d). The maximum FE of 51.6% for CH3OH formation was achieved at the potential of −0.78 V vs. RHE. This corresponds to a turnover frequency (TOF) of 0.04 s−1 with a turnover number (TON) of 252 after 2 h which is comparable with a multistep proton coupled electron transfer process.49 Additionally, we took a deeper look to compare partial current densities at different potentials. The partial current density for CH3OH formation (jMeOH) reached 3.2 mA cm−2 at −0.78 V vs. RHE (Fig. 2e). The Nyquist plots demonstrated much smaller interfacial charge-transfer resistance for the TPA–TPA COF during CO2 reduction, suggesting a favorable faradaic process (Fig. S11†). Apart from the catalytic activity, the durability and capability of repetitive use are key to high-performance electrocatalysis. The robust TPA–TPA CONs have long-term catalytic durability. The chronoamperometric current density and faradaic efficiency versus time was recorded over 12 h (Fig. 2f and S12†). There was no significant loss either in the activity or current density. Notably, both the catalytic efficiency and selectivity showed almost similar values to the pristine one. The remarkable long-term catalytic durability could be attributed to the highly stable π-conjugated structure, which produces high catalytic current densities. Post-electrolysis characterization was performed to test the stability. Post-run powder X-ray diffraction study and HRTEM images displayed an unaltered skeleton as well as morphology of the TPA–TPA-COF (Fig. S13 and S14†). Additionally, the solid-state 13C-NMR spectrum also revealed the intact COF framework after electrocatalysis (Fig. S15†).
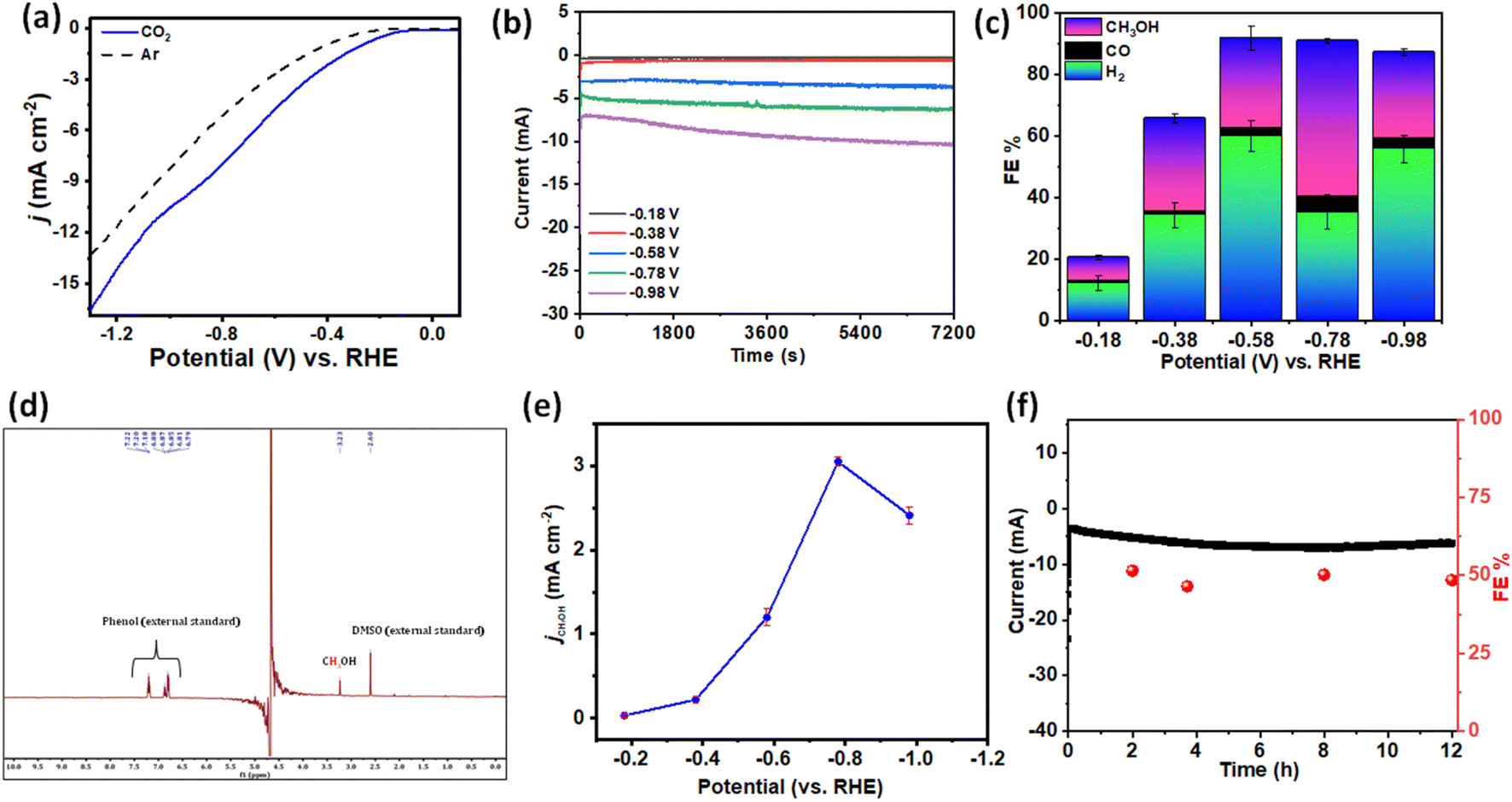 | ||
| Fig. 2 (a) LSV curves showing the activity in Ar (black) and CO2 (blue)-saturated phosphate buffer. (b) Controlled potential electrolysis at different potentials for 2 h. (c) Faradaic efficiencies of CO2 reduction showing product distribution at different potentials. (d) Representative 1H-NMR spectrum showing the formation of methanol at δ = 3.23 ppm using the solvent suppression method. (e) Partial current density plot for CH3OH production. (f) Long-term durability test during the electrocatalytic CO2RR at −0.78 V. | ||
Furthermore, we have also carried out the experiment using isotope-labeled 13CO2 in place of CO2 in the electrolysis over TPA–TPA-COF to trace the source of the carbonaceous products. The product was analyzed using 1H-NMR (Fig. S16†) and GC-MS spectroscopy (Fig. S17†). The NMR signal corresponding to the proton of the 13CH3 group of methanol showed splitting of the singlet peak while 13CO2 was used as the feed gas (Fig. S16†). Additionally, the extracted mass spectra from the GC-MS displayed a base peak of 32 (13CH3O), whereas the standard methanol mass spectra showed a base peak of 31 (12CH3O) further illustrating that methanol as a carbonaceous product originated from CO2 reduction over the TPA–TPA-COF electrocatalyst (Fig. S17†).
To understand the mechanistic pathway of the aforementioned product formation, we have implemented in situ FTIR set-up using an external reflection configuration (Fig. S18†). A Si prism was used bevelled at 60°. A detailed description of the setup can be found in the ESI.† First of all, the spectra collected at different potentials in an Ar atmosphere were used as the background to minimize the effect of proton reduction. Then CO2 was passed as the feeding gas to perform controlled potential electrolysis (CPE). The rapid scan was performed using a synchronization technique to obtain a large number of spectra in a single pulse for 5 min. The potential of −0.78 V vs. RHE was applied to observe the effect of CO2 reduction. As demonstrated in Fig. 3a, the peak at 1450 cm−1 which increased gradually with time corresponded with the C–H bending vibration of –CH3 of methanol, illustrating the presence of methanol near the electrode surface.50 Additionally, the observed peak at 2990 cm−1 was assigned as the C–H stretching vibration of methanol confirming the methanol formation.51 The peak at 1660 cm−1 could be attributed to the formation of COOH*. Furthermore, the electrolysis was carried out at different potentials ranging from −0.58 to −1.18 V vs. RHE. The results demonstrated the production of CH3OH at the corresponding applied potential due to the presence of peaks at ca. 1200, 1450, 1660, 2343 and 2990 cm−1 corresponding to COO−, CH3, COOH*, CO2 and C–H species respectively (Fig. 3b).52 The presence of COOH* species at 1660 cm−1 confirmed that the electroreduction was preceded through a CO pathway.
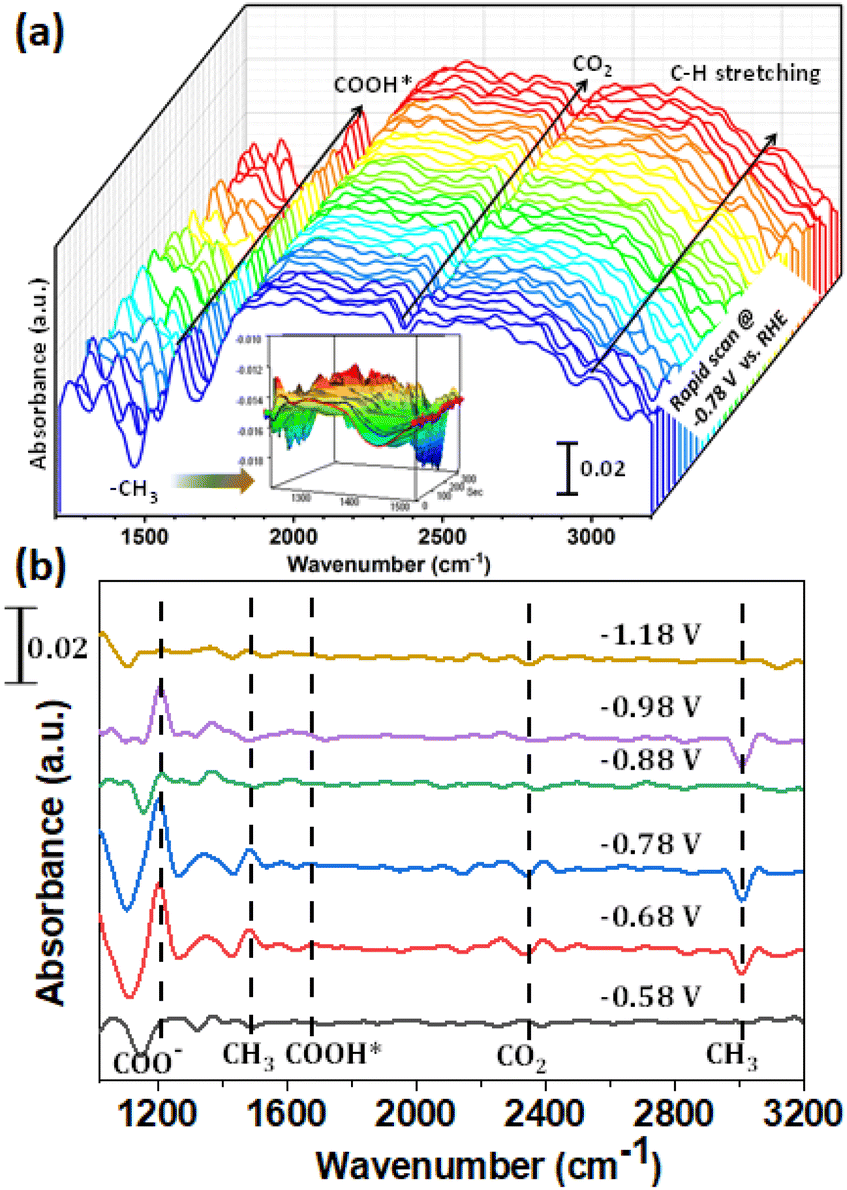 | ||
| Fig. 3 (a) In situ FTIR rapid scan measurement during the CO2RR at −0.78 V vs. RHE for 5 min (inset shows a magnified 3D plot at 1450 cm−1 showing the CH3 stretching). (b) Repeated measurement at different potentials during the in situ FTIR study. | ||
Additionally, in situ Raman was performed to further get an insight of the CO2RR pathway to investigate the vibrational characteristics and functional groups present in the triphenylamine-based covalent organic nanosheet (CON) catalyst during CO2 reduction (Fig. 4 and S19†). The obtained Raman spectra revealed distinct peaks at 219, 409, 640, 888, 1988, 2073, and 2563 cm−1. The peak at 219 cm−1 is assigned to lattice vibrations of the catalyst material. The peak at 409 cm−1 is associated with the stretching vibrations of the CO2 molecule attached to the imine bond.53 The peak at 640 cm−1 may correspond to a specific aromatic vibrational mode of the triphenylamine-based COF. The peak at 888 cm−1 is attributed to characteristic vibrational modes of COOH* intermediates.54–56 The peaks at 1988, 2073, and 2563 cm−1 are likely associated with stretching modes of the imine bond, free CO and COOH* intermediate, respectively.57 These assignments provide valuable insights into the vibrational properties of the catalyst and its potential role in CO2 reduction reactions.
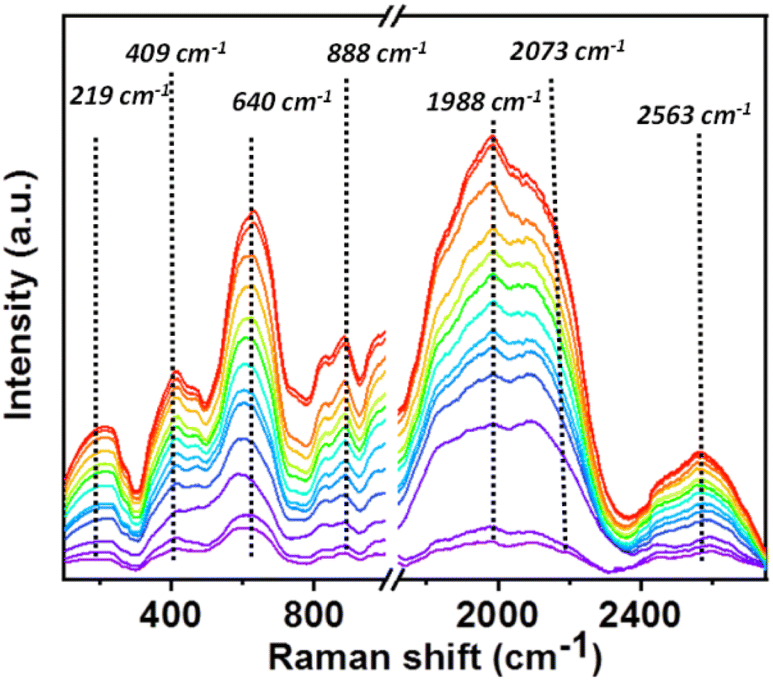 | ||
| Fig. 4 In situ Raman study for the electrochemical CO2RR. | ||
Moreover, density functional theory (DFT) calculations were performed to unveil the mechanistic aspects behind the electrocatalytic CO2 reduction to CH3OH catalyzed by the TPA–TPA COF. With the help of earlier literature reports32,37,47 and, different operando studies, we have performed density functional theory (DFT) calculations which guided us to establish a possible catalytic pathway based on relative Gibbs free energy change plot (Fig. 5 and S20–S22†). For DFT calculations, we have considered the smallest possible repetitive unit having one triphenylamine unit of each type connected through the imine bond and we have represented it as TPA–TPA, where the two TPAs correspond to the TPA–amine fragment and TPA–aldehyde fragment, respectively. There is a slight difference in electron density between the two types of TPA units; the TPA–amine fragment is electron rich whereas the TPA–aldehyde fragment is electron deficient. DFT calculations revealed that the electrocatalytic process will be initiated with CO2-binding accompanied by a proton-coupled electron transfer to generate *COOH (2), which is the rate-determining step (ΔG = +0.86 eV). We have also identified the most suitable active center for COOH-binding through DFT calculations, which revealed that the imine nitrogen will bind COOH in the *COOH (2) intermediate (Fig. S20†). In the next step, the intermediate *COOH (2) will undergo further proton-coupled reduction leading to the formation of the intermediate *CO (3) after water elimination, which is an exergonic process (ΔG = −0.21 eV). In the following steps of the catalytic process, the intermediate *CO (3) will be converted to *CHO (4) (ΔG = −0.99 eV) to *OCH2 (5) (ΔG = +0.03 eV) to *OCH3 (6) (ΔG = −0.42 eV) after successive proton-coupled electron transfer reactions (Fig. S21 and S22†). Finally, proton-coupled reduction of the intermediate *OCH3(6) will result in the rapid removal of the produced CH3OH to regenerate the initial catalyst TPA–TPA (*) (1) (ΔG = −1.90 eV). In addition, DFT calculations further revealed that the CH3OH production step from intermediate *OCH3(6) is highly feasible (ΔG = −1.90 eV) compared to the less feasible CH4 production step (ΔG = −0.24 eV), which explains the non-production of CH4 (Fig. 5).
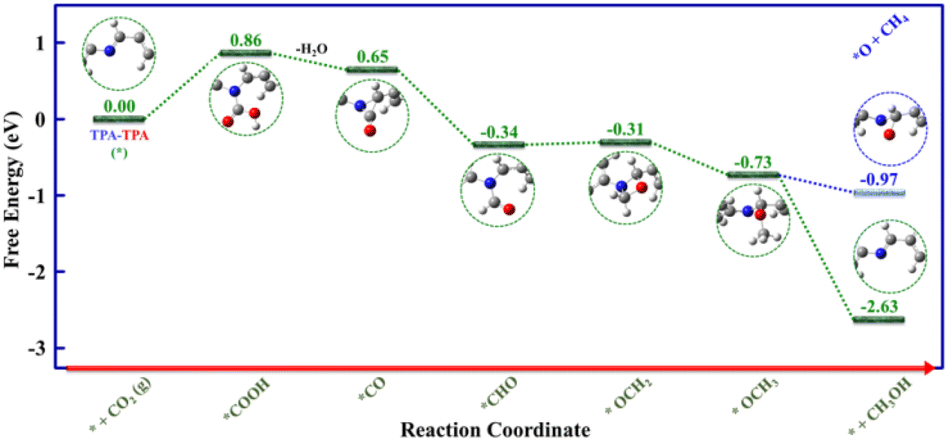 | ||
| Fig. 5 Relative Gibbs free energy diagram for electrocatalytic CO2 to CH3OH reduction on TPA–TPA COF. | ||
Conclusion
In conclusion, our research identified a remarkable CO2 reduction electrocatalyst, the triphenylamine-based redox-active CONs, for efficient and sustainable liquid fuel production under mild conditions. The pivotal catalytic role of the N-centre within the imine functionality has been elegantly demonstrated through an intricate interplay of several operando experimental analysis and sophisticated theoretical calculations. This unequivocal evidence solidifies its position as the catalytically active site, underscoring the scientific rigor underpinning our findings. Comparative studies established the catalyst's superiority in methanol formation with an impressive 51.6% faradaic efficiency and a minimal 210 mV overpotential, all achieved without metallic components. This advancement paves the way for energy-efficient electrocatalysis, offering a new pathway for sustainable fuel synthesis. The TPA–TPA-COF catalyst showcases exceptional performance, operational efficiency, and electronic modulation, holding great promise for a greener energy future.Author contributions
SB and TKM designed the concept of this work. SB, AD, and VRB performed major experiments. SB and TKM analyzed the experimental data and wrote the manuscript. FAR performed the computational study. AD and RJ performed the in situ measurement. AG performed the AFM study. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TKM gratefully acknowledges the SERB, Department of Science and Technology (DST), Govt. of India, Project No. – SPR/2021/000592, for financial support. TKM is also grateful to RAK-CAM (from UAE), SSL, ICMS, and JNCASR for financial support. SAMat Research Facility, Sheikh Saqr Senior Fellowship (TKM) is also gratefully acknowledged. S. B. acknowledges JNCASR for the support and DST for the Inspire Faculty Fellowship and funding (DST/INSPIRE/04/2022/002447). AD, FAR, and RJ are thankful to the CSIR, Government of India for the fellowship. VRB and AG are thankful to JNCASR for fellowship. The support and the resources provided by “PARAM Yukti Facility” under the National Supercomputing Mission, Government of India, at the Jawaharlal Nehru Centre for Advanced Scientific Research are gratefully acknowledged.References
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. Stephens, K. Chan and C. Hahn, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- J. Du, S. Li, S. Liu, Y. Xin, B. Chen, H. Liu and B. Han, Chem. Sci., 2020, 11, 5098–5104 RSC.
- R.-B. Lin and B. Chen, Joule, 2018, 2, 1030–1032 CrossRef.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- Z. Lin, C. Han, G. E. O'Connell and X. Lu, Angew. Chem., Int. Ed., 2023, e202301435 CAS.
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55, 134–144 CrossRef CAS PubMed.
- S. Karmakar, S. Barman, F. A. Rahimi, D. Rambabu, S. Nath and T. K. Maji, Nat. Commun., 2023, 14, 4508 CrossRef CAS PubMed.
- S. Karmakar, S. Barman, F. A. Rahimi, S. Biswas, S. Nath and T. K. Maji, Energy Environ. Sci., 2023, 16, 2187–2198 RSC.
- S. Karmakar, S. Barman, F. A. Rahimi and T. K. Maji, Energy Environ. Sci., 2021, 14, 2429–2440 RSC.
- W. Ni, Y. Guan, H. Chen, Y. Zhang, S. Wang and S. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202303233 CrossRef CAS PubMed.
- B. Pan, X. Zhu, Y. Wu, T. Liu, X. Bi, K. Feng, N. Han, J. Zhong, J. Lu, Y. Li and Y. Li, Adv. Sci., 2020, 7, 2001002 CrossRef CAS PubMed.
- L. Li, Y. Huang and Y. Li, EnergyChem, 2020, 2, 100024 CrossRef.
- T. Ma, Q. Fan, X. Li, J. Qiu, T. Wu and Z. Sun, J. CO2 Util., 2019, 30, 168–182 CrossRef CAS.
- J. Wu, T. Sharifi, Y. Gao, T. Zhang and P. M. Ajayan, Adv. Mater., 2019, 31, 1804257 CrossRef PubMed.
- R. Paul, L. Zhu, H. Chen, J. Qu and L. Dai, Adv. Mater., 2019, 31, 1806403 CrossRef PubMed.
- S. Barman, A. Singh, F. A. Rahimi and T. K. Maji, J. Am. Chem. Soc., 2021, 143, 16284–16292 CrossRef CAS PubMed.
- H.-J. Zhu, M. Lu, Y.-R. Wang, S.-J. Yao, M. Zhang, Y.-H. Kan, J. Liu, Y. Chen, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2020, 11, 497 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- S. Huang, K. Chen and T.-T. Li, Coord. Chem. Rev., 2022, 464, 214563 CrossRef CAS.
- S. Biswas, A. Dey, F. A. Rahimi, S. Barman and T. K. Maji, ACS Catal., 2023, 13, 5926–5937 CrossRef CAS.
- A. Dey, F. A. Rahimi, S. Barman, A. Hazra and T. K. Maji, J. Mater. Chem. A, 2023, 11, 13615–13622 RSC.
- A. Singh, S. Barman, F. A. Rahimi, A. Dey, R. Jena, R. Kumar, N. Mathew, D. Bhattacharyya and T. K. Maji, Energy Environ. Sci., 2024, 17, 2315–2325 RSC.
- B. Han, X. Ding, B. Yu, H. Wu, W. Zhou, W. Liu, C. Wei, B. Chen, D. Qi and H. Wang, J. Am. Chem. Soc., 2021, 143, 7104–7113 CrossRef CAS PubMed.
- Q. Wu, R.-K. Xie, M.-J. Mao, G.-L. Chai, J.-D. Yi, S.-S. Zhao, Y.-B. Huang and R. Cao, ACS Energy Lett., 2020, 5, 1005–1012 CrossRef CAS.
- Y.-R. Wang, Q. Huang, C.-T. He, Y. Chen, J. Liu, F.-C. Shen and Y.-Q. Lan, Nat. Commun., 2018, 9, 4466 CrossRef PubMed.
- D. Rodríguez-San-Miguel, C. Montoro and F. Zamora, Chem. Soc. Rev., 2020, 49, 2291–2302 RSC.
- W. Wang, W. Zhao, H. Xu, S. Liu, W. Huang and Q. Zhao, Coord. Chem. Rev., 2021, 429, 213616 CrossRef CAS.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- J. Albo, M. Alvarez-Guerra, P. Castaño and A. Irabien, Green Chem., 2015, 17, 2304–2324 RSC.
- E. Boutin, M. Wang, J. C. Lin, M. Mesnage, D. Mendoza, B. Lassalle-Kaiser, C. Hahn, T. F. Jaramillo and M. Robert, Angew. Chem., Int. Ed., 2019, 58, 16172–16176 CrossRef CAS PubMed.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- P. Li, J. Bi, J. Liu, Q. Zhu, C. Chen, X. Sun, J. Zhang and B. Han, Nat. Commun., 2022, 13, 1965 CrossRef CAS PubMed.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Nat. Commun., 2019, 10, 677 CrossRef CAS PubMed.
- X. Ren, J. Zhao, X. Li, J. Shao, B. Pan, A. Salamé, E. Boutin, T. Groizard, S. Wang, J. Ding, X. Zhang, W.-Y. Huang, W.-J. Zeng, C. Liu, Y. Li, S.-F. Hung, Y. Huang, M. Robert and B. Liu, Nat. Commun., 2023, 14, 3401 CrossRef CAS PubMed.
- Y. Peng, Y. Huang, Y. Zhu, B. Chen, L. Wang, Z. Lai, Z. Zhang, M. Zhao, C. Tan and N. Yang, J. Am. Chem. Soc., 2017, 139, 8698–8704 CrossRef CAS PubMed.
- A. F. El-Mahdy, C.-H. Kuo, A. Alshehri, C. Young, Y. Yamauchi, J. Kim and S.-W. Kuo, J. Mater. Chem. A, 2018, 6, 19532–19541 RSC.
- A. Singh, D. Samanta and T. K. Maji, ChemElectroChem, 2019, 6, 3756–3763 CrossRef CAS.
- S. Roy, A. Bandyopadhyay, M. Das, P. P. Ray, S. K. Pati and T. K. Maji, J. Mater. Chem. A, 2018, 6, 5587–5591 RSC.
- C. Dai, L. Zhong, X. Gong, L. Zeng, C. Xue, S. Li and B. Liu, Green Chem., 2019, 21, 6606–6610 RSC.
- Y. Peng, Y. Huang, Y. Zhu, B. Chen, L. Wang, Z. Lai, Z. Zhang, M. Zhao, C. Tan and N. Yang, J. Am. Chem. Soc., 2017, 139, 8698–8704 CrossRef CAS PubMed.
- A. F. El-Mahdy, C.-H. Kuo, A. Alshehri, C. Young, Y. Yamauchi, J. Kim and S.-W. Kuo, J. Mater. Chem. A, 2018, 6, 19532–19541 RSC.
- A. Singh, D. Samanta and T. K. Maji, ChemElectroChem, 2019, 6, 3756–3763 CrossRef CAS.
- L. Ji, L. Li, X. Ji, Y. Zhang, S. Mou, T. Wu, Q. Liu, B. Li, X. Zhu and Y. Luo, Angew. Chem., Int. Ed., 2020, 132, 768–772 CrossRef.
- Q. H. Low, N. W. X. Loo, F. Calle-Vallejo and B. S. Yeo, Angew. Chem., Int. Ed., 2019, 58, 2256–2260 CrossRef CAS PubMed.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean and R. E. Winans, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- N. J. Firet and W. A. Smith, ACS Catal., 2017, 7, 606–612 CrossRef CAS.
- X. Ye, C. Yang, X. Pan, J. Ma, Y. Zhang, Y. Ren, X. Liu, L. Li and Y. Huang, J. Am. Chem. Soc., 2020, 142, 19001–19005 CrossRef CAS PubMed.
- C. Yang, R. Mu, G. Wang, J. Song, H. Tian, Z.-J. Zhao and J. Gong, Chem. Sci., 2019, 10, 3161–3167 RSC.
- M. Fang, L. Xu, H. Zhang, Y. Zhu and W.-Y. Wong, J. Am. Chem. Soc., 2022, 144, 15143–15154 CrossRef CAS PubMed.
- R. Daiyan, W. H. Saputera, H. Masood, J. Leverett, X. Lu and R. Amal, Adv. Energy Mater., 2020, 10, 1902106 CrossRef CAS.
- D. Bohra, I. Ledezma-Yanez, G. Li, W. de Jong, E. A. Pidko and W. A. Smith, Angew. Chem., Int. Ed., 2019, 131, 1359–1363 CrossRef.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li and Y. Wang, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00737a |
| This journal is © The Royal Society of Chemistry 2024 |