
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A synthetic strategy towards single crystals of Zr6 cluster and phosphonate-based metal–organic frameworks†
Haomiao
Xie
 a,
Kent O.
Kirlikovali
a,
Zhijie
Chen
a,
Karam B.
Idrees
a,
Timur
Islamoglu
a and
Omar K.
Farha
*ab
a,
Kent O.
Kirlikovali
a,
Zhijie
Chen
a,
Karam B.
Idrees
a,
Timur
Islamoglu
a and
Omar K.
Farha
*ab
aDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208, USA. E-mail: o-farha@northwestern.edu
bDepartment of Chemical and Biological Engineering, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA
First published on 31st January 2024
Abstract
Although metal–organic frameworks (MOFs) composed of Zr-based nodes and phosphonate-based linkers show promise in terms of their excellent stability, synthetic strategies to access them are underdeveloped due to the less reversible coordination between Zr-based nodes and phosphonate groups. In this work, we developed a generalizable strategy to access Zr–phosphonate MOFs in two steps via post-synthesis modification which allows for retaining the topology of parent Zr MOFs. Solvent-assisted ligand exchange (SALE) replaces carboxylate linkers with phosphinate linkers, and post-synthetic oxidation affords phosphonate-based linkers. We confirmed the structures through single-crystal X-ray diffraction (SCXRD) studies and fully characterized each MOF through a variety of other techniques. Water and ammonia sorption isotherms reveal that incorporation of phosphonate groups yields a more hydrophilic pore environment with increased Brønsted acidity relative to those of the carboxylate-based analogues. Overall, this strategy provides a new avenue to tune the physical and chemical properties of Zr-MOFs and further enriches the reticular chemistry of this class of materials.
Introduction
Metal–organic frameworks (MOFs), which are based on the highly ordered assembly of organic linkers and metal ion/cluster nodes, have attracted great attention in the fields of chemistry and materials science because of their potential application in gas storage, molecular separation, catalysis, chemical sensing, and drug delivery.1–7 Rational design of the organic linkers and metal nodes according to the principles of reticular chemistry enables precise, nano-scale control of the porous structures, and the physical and chemical properties of the resulting MOF can be rationally tuned based on the properties of the individual organic and metal components.8–10 To date, the majority of MOFs in the literature are based on carboxylate-based linkers and high-valent metal ions, such as Zr(IV), Al(III), and Cr(III), which leverage the reversible binding interactions between carboxylate groups and metal ions/clusters to generate crystalline materials, as well as the strong metal–oxygen coordination bonds that afford robust structures.11–13 In principle, phosphonate-based linkers should generate even stronger coordination bonds than carboxylate-based linkers to metal nodes composed of high-valent metal ions, suggesting great promise of phosphonate-based MOFs in applications such as catalysis, gas storage and separation, and ion conductors.14 However, this class of MOFs is relatively unexplored since strong phosphonate–metal coordination bonds limit the reversible binding interactions that are required for the formation of crystalline materials (i.e., these irreversible reactions result in the rapid formation of amorphous materials with no long-range order); as a result, few phosphonate–metal secondary building units (SBUs) have been reported (Scheme 1).15–18 Thus, the development of a general strategy to synthesize phosphonate-based MOFs would offer access to a new frontier in MOF synthesis.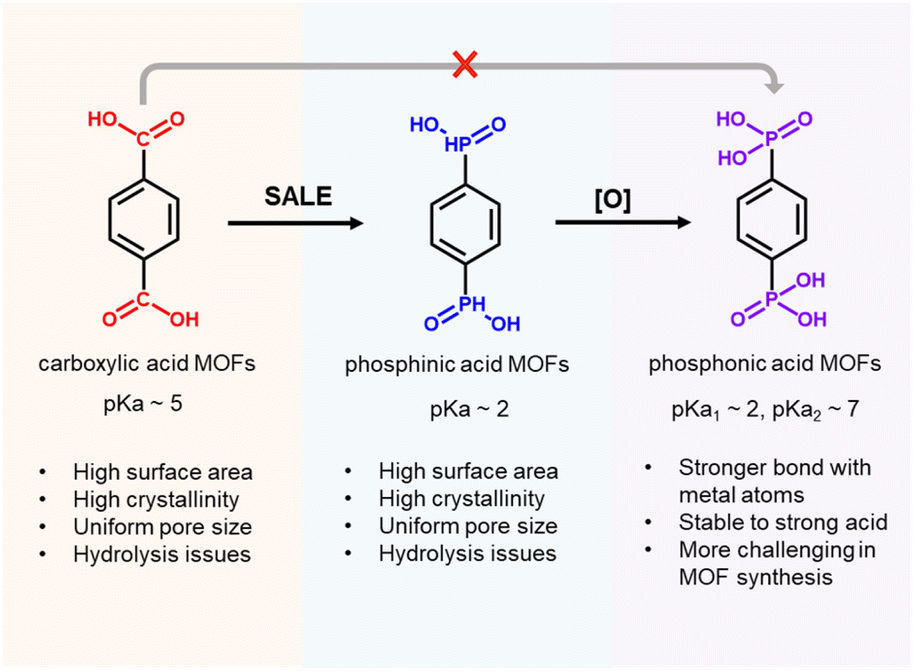 | ||
| Scheme 1 Advantages of Zr–phosphinate/phosphonate MOFs. | ||
Among potential phosphonate-based MOFs, strong coordination bonds between high-valent Zr4+ centers and −PO32− groups should endow Zr–phosphonate-based MOFs with excellent stability.16,19 So far, only a few examples of Zr–phosphonate MOFs have been reported in the literature, presumably due to strong, irreversible coordination bonds between Zr(IV)–phosphonate resulting in the rapid precipitation of amorphous products. For instance, “unconventional” Zr–phosphonate MOFs (UMOFs) were among the first reported for this class of MOFs, but they are mainly compact 1-D or 2-D structures with poor crystallinities and relatively low porosities.20–22 In comparison to these lower dimensional Zr–phosphonate MOF structures, Zr–phosphonate MOFs with 3-D framework structures are relatively rare in the literature. In one example, Wang and coauthors developed strategies to synthesize 3 highly crystalline Zr–phosphonate MOFs, denoted as SZ-1, SZ-2, and SZ-3, by using linkers with a tetrahedral geometry to prevent the formation of 2-D layers, as well as harsh ionothermal conditions with HF as the modulator. Notably, SZ-1, -2, and -3 contained three previously unreported Zr–phosphonate SBUs with chemical formulae of Zr5P8, Zr3P4 and Zr7P8,23 which contrast the Zr6-based nodes typically found in Zr–carboxylate MOFs. Despite the structural diversity of Zr–phosphonate SBUs, Zr–phosphonate MOFs composed of Zr6-based nodes have yet to be reported. In this context, we considered that a methodology that provides access to Zr–phosphonate MOFs with conventional Zr6 nodes would catalyze studies on these materials since the well-studied reticular chemistry of Zr–carboxylate MOFs could then be applied to Zr–phosphonate MOFs. In this work, we developed a generalizable strategy to synthesize Zr–phosphonate MOFs with Zr6 clusters as SBUs, which should offer the MOF community straightforward access to a diverse range of previously unknown Zr–phosphonate MOFs.
Results and discussion
The synthetic strategy is summarized in Scheme 2. To begin our investigation, we first synthesized the parent Zr–carboxylate MOF with fcu topology, DUT-52, in both single crystal and powder forms according to literature methods24 with naphthalene-2,6-dicarboxylic acid (H2NDC). Next, we performed SALE by incubating DUT-52 in a DMF solution containing excess diphosphinic acid linker, H2NDPi (2.6 eq. based on the carboxylic linker), and 0.8% TFA as a modulator for 2 hours at 100 °C. The powder X-ray diffraction (PXRD) patterns of bulk, polycrystalline samples illustrate a shift for the diffraction peaks towards lower angles, which indicates an expansion of the unit cell parameters. Following digestion in NaOD, analysis of the 1H NMR spectrum confirms complete exchange from NDC to NDPi (Fig. S10†). We then employed single crystal X-ray diffraction (SCXRD) to confirm the structure of the product from this reaction, NU-406. Analysis of the diffraction data collected at 100 K reveals that while the space group (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and topology of NU-406 remain the same as those of DUT-52, the unit cell parameter of NU-406 slightly increases to a = 24.6280(3) Å (Table S1,†a = 23.910(3) Å for DUT-52),24 which corroborates the shifts in the diffraction peaks observed in the PXRD patterns for the bulk samples (Fig. 1(a)). Notably, the phosphinate linker in NU-406 adopts a different geometry relative to the linear coordination geometry observed for the carboxylate linker in DUT-52. Specifically, the phosphinate ligand coordinates to the Zr6-based node with a tilted angle due to the tetrahedral geometry of the sp3 phosphorus atom in the phosphinate group in NU-406 (Fig. 1(c)), which results in a positional disorder that correlates to the mirror planes at the D2h symmetry sites.
m) and topology of NU-406 remain the same as those of DUT-52, the unit cell parameter of NU-406 slightly increases to a = 24.6280(3) Å (Table S1,†a = 23.910(3) Å for DUT-52),24 which corroborates the shifts in the diffraction peaks observed in the PXRD patterns for the bulk samples (Fig. 1(a)). Notably, the phosphinate linker in NU-406 adopts a different geometry relative to the linear coordination geometry observed for the carboxylate linker in DUT-52. Specifically, the phosphinate ligand coordinates to the Zr6-based node with a tilted angle due to the tetrahedral geometry of the sp3 phosphorus atom in the phosphinate group in NU-406 (Fig. 1(c)), which results in a positional disorder that correlates to the mirror planes at the D2h symmetry sites.
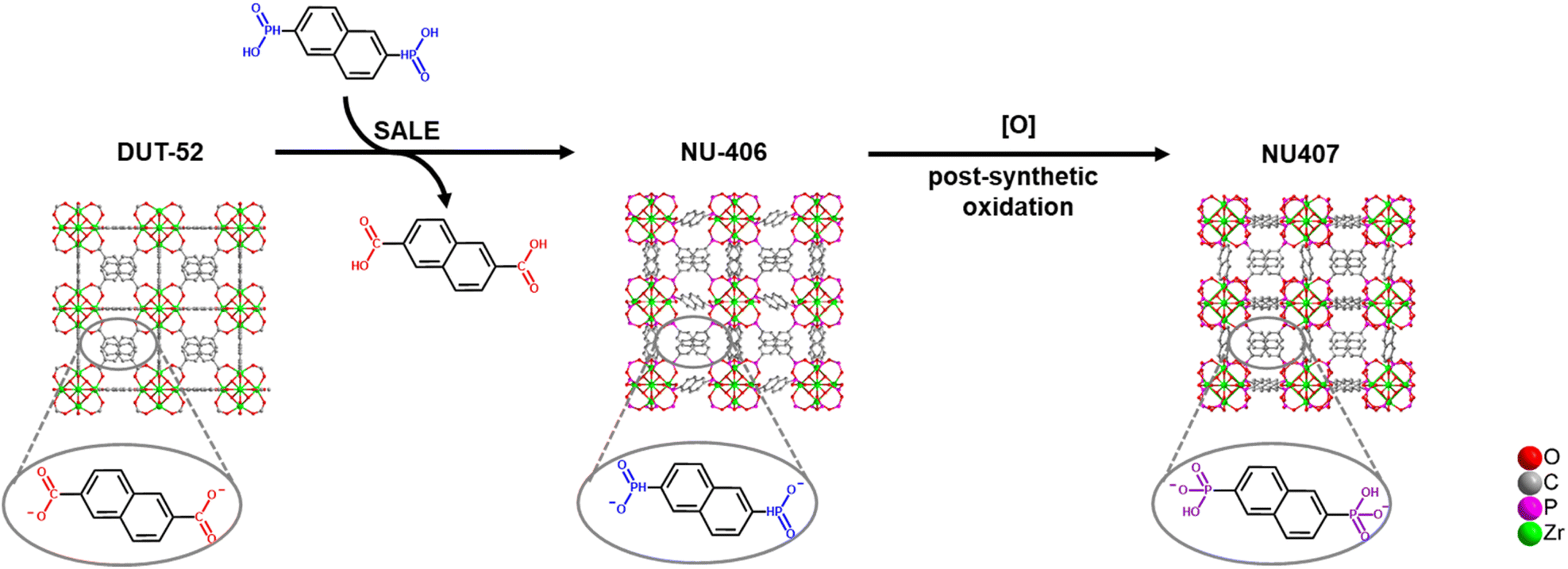 | ||
| Scheme 2 Strategy for Zr–phosphinate/phosphonate MOF synthesis. | ||
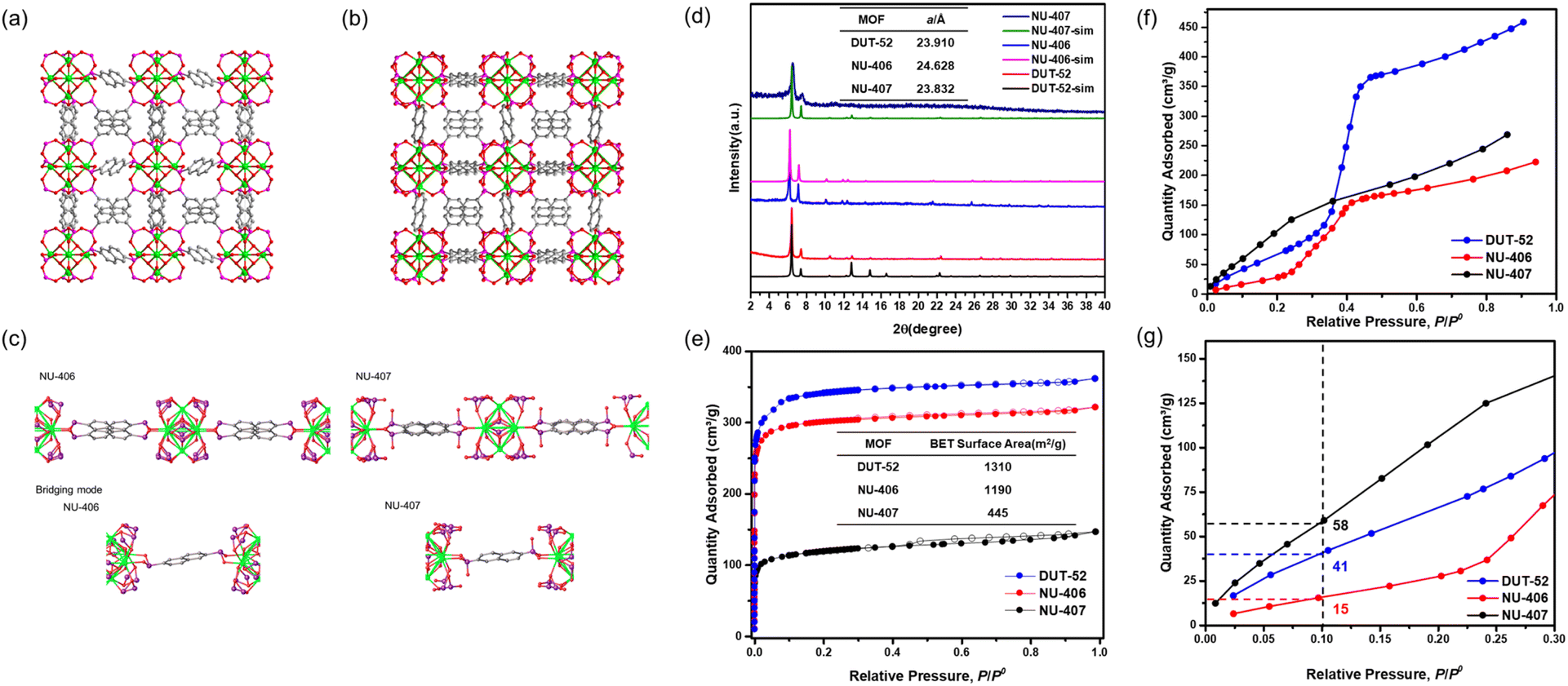 | ||
| Fig. 1 Single-crystal structures of (a) NU-406 and (b) NU-407, (c) bridging modes of ligands in NU-406 and NU-407, and (d) powder XRD pattern of DUT-52, NU-406 and NU-406 (color code for atoms: zirconium (green), phosphor (purple), carbon (grey), and oxygen (red)). (e) N2 sorption at 77 K of DUT-52, NU-406 and NU-407. (f) Water sorption at 298 K of DUT-52, NU-406 and NU-407 and (g) the zoomed-in view of the low relative pressure region. | ||
To access the phosphonate analogue, NU-407, we performed one-step oxidation of the phosphinate linkers by immersing NU-406 in an acetonitrile solution with 112 equivalents of the oxidizing agent tBuOOH, as well as HCl (4.4 eq.), at 80 °C for 72 hours. During the reaction, the color of the crystalline powder changed from white to pale orange. The main peaks in the PXRD pattern of NU-407 remain similar to those of NU-406 but shift to higher 2θ angles (Fig. 1(d)), which implies preservation of the fcu topology of NU-406 but with a slightly shrunken unit cell parameter. We performed 31P NMR spectroscopic analysis of samples digested in NaOD to monitor the conversion of the phosphinate linkers to the phosphonate form, as the chemical shift for the phosphonate group (δ = 10.95 ppm) is upfield from that of the phosphinate group (δ = 18.32 ppm) and provides a diagnostic handle (Fig. S13†).
To gain deeper insight into how oxidation to the phosphonate form affects the structure of the Zr6-based nodes, we characterized the structure of NU-407 using SCXRD analysis (Fig. 1(a)). Both the space group (Fmm) and topology of NU-407 are the same as those of the parent MOF, NU-406, but with a slightly decreased unit cell parameter (from a = 24.46280(3) Å for NU-406 to a = 23.832(2) Å for NU-407). Similar to the phosphinate linker in NU-406, the sp3 phosphorus atom on the phosphonate ligand adopts a tetrahedral geometry but contains a “free” hydroxyl group instead of a hydrogen atom (Fig. 1(c)). Next, we evaluated the Brunauer–Emmett–Teller (BET) surface areas of DUT-52, NU-406, and NU-407 using N2 sorption isotherms at 77 K to confirm that these MOFs maintain the intrinsic porosity of the parent DUT-52 following the SALE and subsequent oxidation steps (Fig. 1(d)). The isotherms reveal BET surface areas of 1310, 1190, and 445 cm2 g−1 with a N2 uptake of 361, 321, and 144 cm3 g−1 at P/P0 = 0.99 for DUT-52, NU-406, and NU-407, respectively (Table S6).† Table S6† also contains the pore volumes of the three MOFs calculated from the N2 sorption isotherm at P/P0 = 0.99, which are comparable to the theoretical pore volumes calculated from the single crystal data. For NU-407, both the slight contraction of the unit cell and the increase in the molecular weight of the phosphonate linker likely contribute to observed decrease in porosity relative to NU-406.
To probe how converting carboxylate linkers to phosphinate and then to phosphonate linkers affects the internal surface properties of these MOFs, we collected water sorption isotherms of DUT-52, NU-406, and NU-407 at 298 K (Fig. 1(f) and (g)). These isotherms reveal that the condensation point shifts to lower relative pressures at each step, starting at P/P0 = 0.38 for DUT-52 and then shifting to P/P0 = 0.32 for NU-406 and P/P0 = 0.012 for NU-407. This shift indicates a significant increase in hydrophilicity for NU-407 following the post-synthetic oxidation of the phosphinate-based linkers, which can likely be attributed to the capability of the phosphonate linkers in NU-407 to engage in enhanced hydrogen bonding interactions between hydroxyl groups on phosphonate linkers and water molecules in the MOF cavities.
In addition to increasing hydrophilicity of the internal surface, we anticipated that the presence of the hydroxyl group on the phosphonate-based linker in NU-407 would result in increased Brønsted acidity. To confirm this, we collected NH3 sorption isotherms at 298 K for DUT-52, NU-406, and NU-407 (Fig. S16†). The NH3 uptake of NU-407 at extremely low pressure (1.0 mmol g−1 at P = 0.1 mbar) is significantly higher than that of both DUT-52 and NU-406 (0.16 and 0.30 mmol g−1, respectively), which suggests that the phosphonate linker in NU-407 exhibits a higher affinity for NH3 than its carboxylate and phosphinate analogues. Moreover, three consecutive adsorption–desorption cycles with activation under vacuum at room temperature between cycles reveal that NH3 uptake at P = 0.1 mbar for NU-407 drops from 1.0 mmol g−1 for the first cycle to 0.3 mmol g−1 for the second cycle, and then it remains unchanged between the second and the third cycle (Fig. S17†). This decrease in adsorption capacity between the first and second cycles suggests chemisorption of NH3 to generate NH4+ ions at the phosphonate linkers. This is consistent with X-ray photoelectron spectroscopy (XPS) measurements that confirm the formation of NH4+ ions following NH3 adsorption (Fig. S21†). In contrast, there are no significant changes observed for NH3 uptake at P < 0.5 mbar during consecutive adsorption–desorption cycles for DUT-52, suggesting minimal chemisorption that is consistent with the lack of Brønsted acid groups on the carboxylate-based linker. Finally, NH3 uptake at P = 0.1 mbar for NU-406 also drops slightly from 0.3 mmol g−1 during the first adsorption cycle to 0.1 mmol g−1 for the second adsorption cycle, which may be due to the presence of a small number of phosphonate groups that are generated during the sample processing.
To explore the isoreticular chemistry of the Zr6-based phosphonite/phosphonate MOFs, we performed similar SALE reactions with two other fcu Zr–carboxylate MOFs, UiO-66 and UiO-67. Heating DMF solutions of each MOF with ∼9 equivalents of benzene-1,4-diphosphinic acid (H2BDPi) or 4,4′-biphenyldisphosphinic acid (H2BPDPi) at 80 °C affords NU-408 and NU-410, respectively. Notably, the SALE reaction for NU-408 requires 24 hours to achieve full conversion compared to only 2 hours for NU-406 and NU-410, which might be caused by the smaller pores limiting the diffusion of the exchanging linkers in the case of NU-408. 1H NMR spectroscopic analysis of digested samples of NU-408 and NU-410 confirms complete exchange of the carboxylate linkers to phosphinate linkers (Fig. S11 and 12†). Similar to the case of NU-406, the low-angle peaks in the PXRD patterns for NU-408 and NU-410 shift to lower 2θ angles (Fig. 2(c) and 3(c)), suggesting that the phosphinate-based MOFs have larger unit cells relative to their carboxylate analogues. SCXRD analysis confirms the structure of NU-410 (Fig. 3(a)), which exhibits the same space group (Fmm) and topology of UiO-67 but with a slightly larger unit cell parameter, a = 27.5232(5) Å (a = 26.8809(3) Å for UiO-67). Similar to NU-406, the phosphinate ligands coordinate to the Zr6-based nodes with a tilt angle due to the tetrahedral geometry of sp3 phosphorus atoms on the phosphinate groups (Fig. S18†). Due to the extremely slow exchange rate for the single-crystal UiO-66 sample compared to the powder sample with nano-scale particle size (Fig. S19†), we were unable to obtain the single-crystal structure of NU-408.
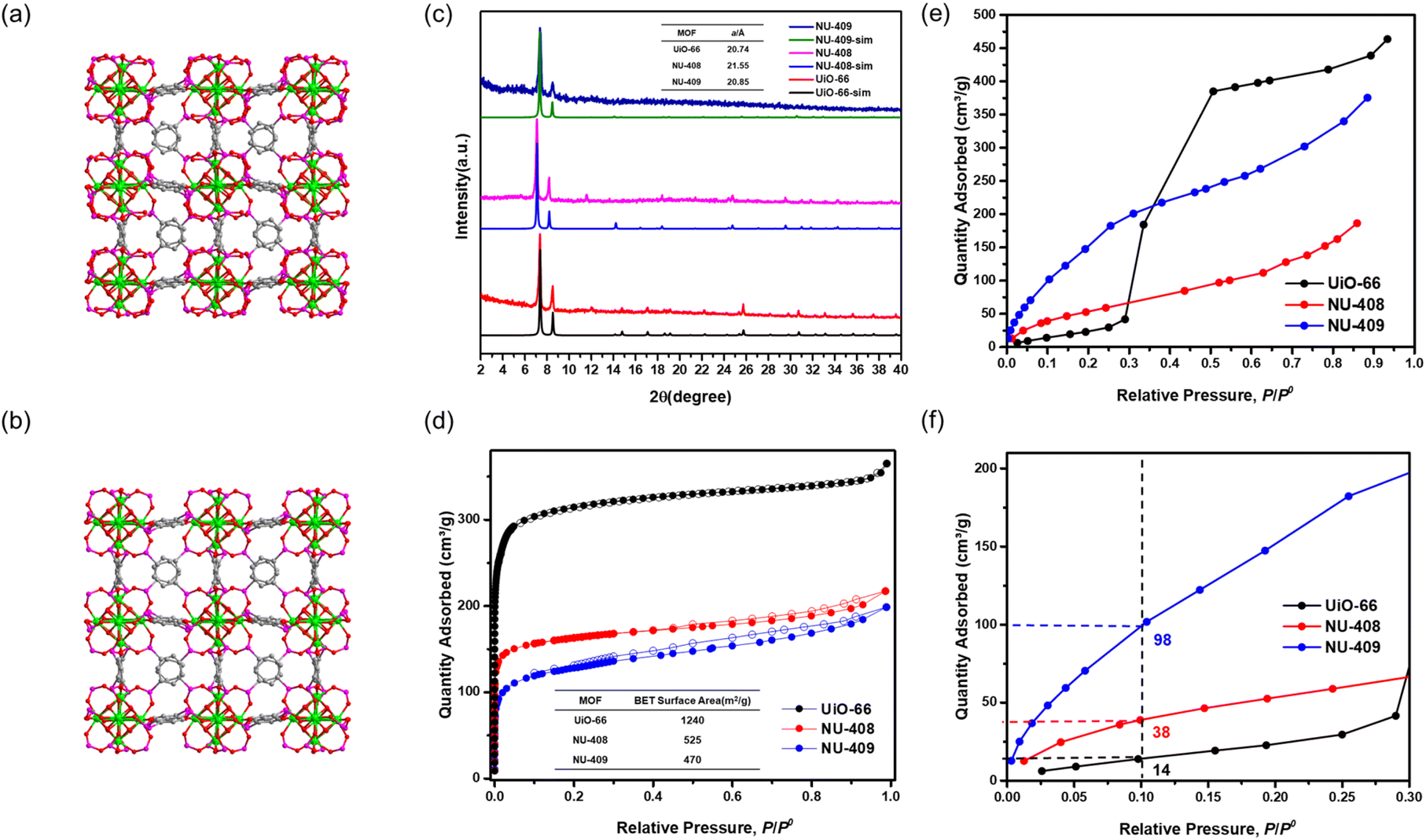 | ||
| Fig. 2 Simulated structure of (a) NU-408 and (b) NU-409 (color code for atoms: zirconium (green), phosphor (purple), carbon (grey), and oxygen (red)). (c) Powder XRD pattern of UiO-66, NU-408 and NU-409 and their unit cell parameters. (d) N2 sorption at 77 K of UiO-66, NU-408 and NU-409. (e) Water sorption at 298 K of UiO-66, NU-408 and NU-409 and (f) the zoomed-in of the 0–0.3 P/P0 range. | ||
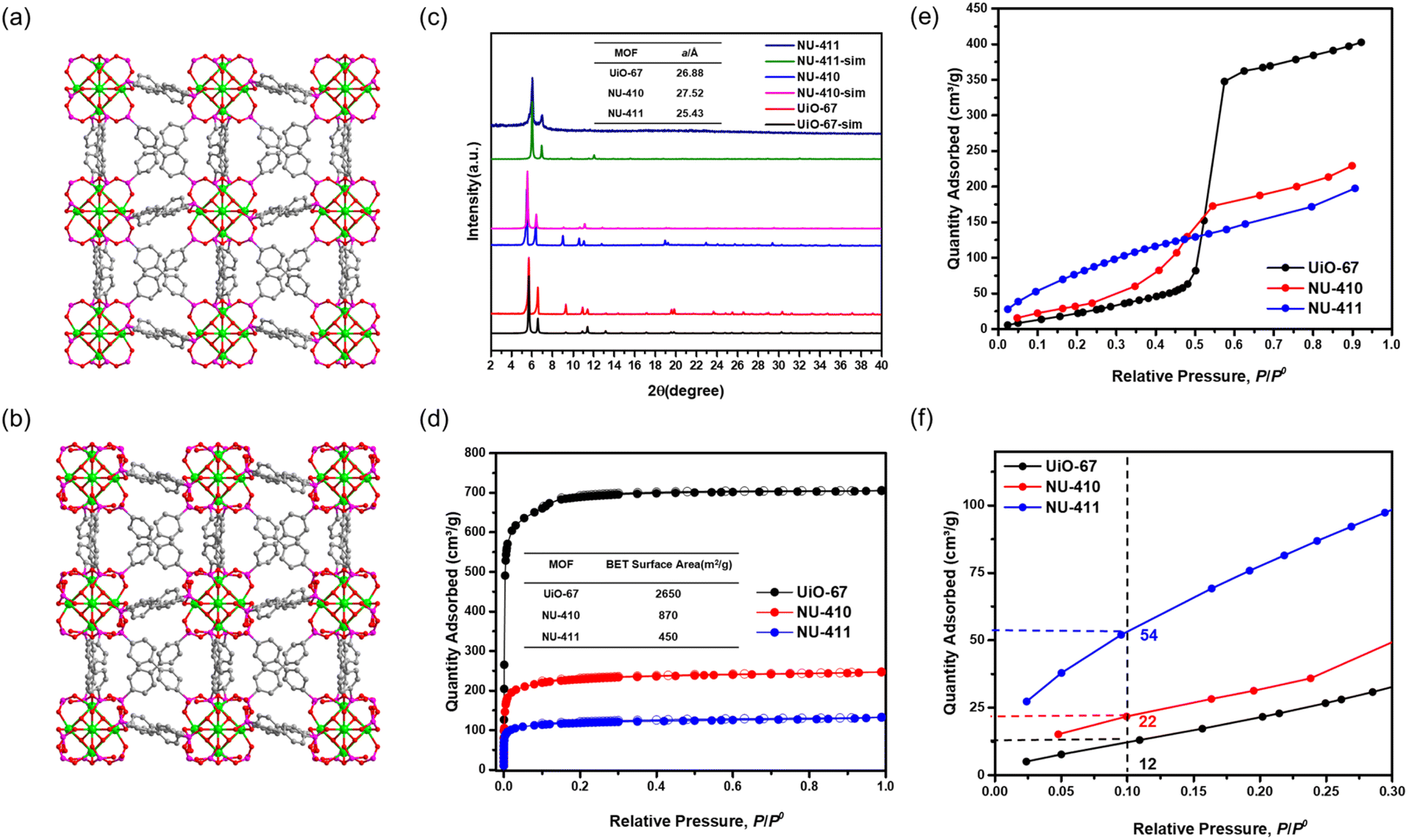 | ||
| Fig. 3 Single-crystal structure of (a) NU-410 and simulated structure of (b) NU-411 (color code for atoms: zirconium (green), phosphor (purple), carbon (grey), and oxygen (red)). (c) Powder XRD pattern of UiO-67, NU-410 and NU-411 and their unit cell parameters. (d) N2 sorption at 77 K of UiO-67, NU-410 and NU-411. (e) Water sorption at 298 K of UiO-67, NU-410 and NU-411 and (f) the zoomed-in of the 0–0.3 P/P0 range. | ||
We collected N2 sorption isotherms of both phosphinate-based MOFs, and the BET surface areas of NU-408 (Fig. 2(d)) and NU-410 (Fig. 3(d)) are 525 m2 g−1 and 870 m2 g−1, respectively. The decrease in the BET area of NU-410 might be due to the structural deformation of the framework during activation and the higher molecular weights of the linkers. Next, we performed post-synthetic oxidation of NU-408 and NU-410 to NU-409 and NU-411, respectively, and confirmed successful oxidation by 31P NMR spectroscopy (Fig. S14 and S15†). The low-angle peaks in the PXRD patterns for NU-409 and NU-411 shift to higher angles compared to NU-408 and NU-410 (Fig. 2(c) and 3(c)), respectively, which indicates a relative decrease in the unit cell and is consistent with observations for NU-407 (Fig. 1(d)). The BET area of NU-409 decreases slightly to 470 m2 g−1 due to the increase in the molecular weight of the linker and the decrease in the unit cell volume. On the other hand, the BET area for NU-411 significantly decreases to 450 m2 g−1, which might be caused by the partial loss of the cavity after the oxidation steps. Water sorption isotherms also suggest that NU-409 (Fig. 2(e) and (f)) and NU-411 (Fig. 3(e) and (f)) are more hydrophilic in the range of 0.001 to 0.30 P/P0 than UiO-66/NU-408 and UiO-67/NU-410, which is consistent with the NU-407 case and suggests the formation of the non-coordinating –OH group on the phosphonate linker.
Conclusions
To sum up, we have introduced a general strategy to synthesize Zr–phosphonate-based MOFs in two steps from Zr–carboxylate-based MOFs. First, solvent-assisted linker exchange (SALE) replaces carboxylate linkers with phosphinate linkers, and post-synthetic oxidation of the phosphinate linkers generates phosphonate linkers. This strategy preserves the fcu topology of UiO-type MOFs after each step, and all MOFs retain the Zr6 clusters from the parent Zr–carboxylate MOFs. Single crystal X-ray diffraction (SCXRD) studies illustrate that in contrast to the linear coordination geometry of the carboxylate linkers, the phosphinate and phosphonate linkers coordinate to the Zr6-based nodes at an angle, which is due to the tetrahedral geometry of the sp3 phosphorus atom in these linkers. The phosphonate-based hydroxyl group remains uncoordinated, which results in a more hydrophilic pore environment and increased Brønsted acidity, as determined by water and ammonia sorption isotherms, respectively. We anticipate that this synthetic strategy will provide access to more Zr–phosphinate/phosphonate MOFs and further enriches the reticular chemistry of metal–organic frameworks.Author contributions
The research was primarily conducted by H. X. under the guidance of O. K. F. Isotherm data collection was assisted by Z. C. and K. B. I. The manuscript was primarily drafted by H. X. with significant contributions from K. O. K., Z. C., K. B. I., and I. T.Conflicts of interest
O. K. F. has a financial interest in NuMat Technologies, a startup company that is seeking to commercialize MOFs. All other authors declare no competing interests.Acknowledgements
This material is based on work supported by the Defense Advanced Research Projects Agency (DARPA) under contract HR001121C0015. This work made use of the IMSERC Crystallography facility at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), and Northwestern University. This work made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-2025633), the IIN, and Northwestern's MRSEC program (NSF DMR-1720139). O. K. F. acknowledges the support from the Army Research Office, under Award No. W911NF1910340. K. O. K. gratefully acknowledges support from the IIN Postdoctoral Fellowship and the Northwestern University International Institute for Nanotechnology.Notes and references
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- V. Pascanu, G. González Miera, A. K. Inge and B. Martín-Matute, J. Am. Chem. Soc., 2019, 141, 7223–7234 CrossRef CAS.
- H.-Y. Li, S.-N. Zhao, S.-Q. Zang and J. Li, Chem. Soc. Rev., 2020, 49, 6364–6401 RSC.
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. Mizrahi Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS.
- J. W. M. Osterrieth and D. Fairen-Jimenez, Biotechnol. J., 2021, 16, 2000005 CrossRef CAS PubMed.
- N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2018, 4, eaat9180 CrossRef CAS PubMed.
- W. Xu, B. Tu, Q. Liu, Y. Shu, C.-C. Liang, C. S. Diercks, O. M. Yaghi, Y.-B. Zhang, H. Deng and Q. Li, Nat. Rev. Mater., 2020, 5, 764–779 CrossRef CAS.
- M. Ding, X. Cai and H.-L. Jiang, Chem. Sci., 2019, 10, 10209–10230 RSC.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- X. Zhang, Z. Chen, X. Liu, S. L. Hanna, X. Wang, R. Taheri-Ledari, A. Maleki, P. Li and O. K. Farha, Chem. Soc. Rev., 2020, 49, 7406–7427 RSC.
- P. Tholen, Y. Zorlu, J. Beckmann and G. Yücesan, Eur. J. Inorg. Chem., 2020, 2020, 1542–1554 CrossRef CAS.
- G. K. H. Shimizu, R. Vaidhyanathan and J. M. Taylor, Chem. Soc. Rev., 2009, 38, 1430–1449 RSC.
- K. J. Gagnon, H. P. Perry and A. Clearfield, Chem. Rev., 2012, 112, 1034–1054 CrossRef CAS.
- K. Siemensmeyer, C. A. Peeples, P. Tholen, F.-J. Schmitt, B. Çoşut, G. Hanna and G. Yücesan, Adv. Mater., 2020, 32, 2000474 CrossRef CAS.
- T. Rhauderwiek, H. Zhao, P. Hirschle, M. Döblinger, B. Bueken, H. Reinsch, D. De Vos, S. Wuttke, U. Kolb and N. Stock, Chem. Sci., 2018, 9, 5467–5478 RSC.
- G. Yücesan, Y. Zorlu, M. Stricker and J. Beckmann, Coord. Chem. Rev., 2018, 369, 105–122 CrossRef.
- A. Clearfield, Dalton Trans., 2008, 6089–6102, 10.1039/B807676F.
- A. Clearfield and G. D. Smith, Inorg. Chem., 1969, 8, 431–436 CrossRef CAS.
- J. Huang, A. Subbiah, D. Pyle, A. Rowland, B. Smith and A. Clearfield, Chem. Mater., 2006, 18, 5213–5222 CrossRef CAS.
- T. Zheng, Z. Yang, D. Gui, Z. Liu, X. Wang, X. Dai, S. Liu, L. Zhang, Y. Gao, L. Chen, D. Sheng, Y. Wang, J. Diwu, J. Wang, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Nat. Commun., 2017, 8, 15369 CrossRef CAS PubMed.
- V. Bon, I. Senkovska, M. S. Weiss and S. Kaskel, CrystEngComm, 2013, 15, 9572–9577 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2260133–2260135. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ta07163d |
| This journal is © The Royal Society of Chemistry 2024 |