
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidatively-induced C(sp3)–C(sp3) bond formation at a tucked-in iron(III) complex†
Joseph A.
Zurakowski
 ab,
Connor S.
Durfy
a,
Noah B.
Stocek
c,
Giovanni
Fanchini
ac and
Marcus W.
Drover
*a
ab,
Connor S.
Durfy
a,
Noah B.
Stocek
c,
Giovanni
Fanchini
ac and
Marcus W.
Drover
*a
aDepartment of Chemistry, Western University, 1151 Richmond Street, London, ON N8K 3G6, Canada. E-mail: marcus.drover@uwo.ca
bDepartment of Chemistry and Biochemistry, University of Windsor, 401 Sunset Avenue, Windsor, ON N9B 3P4, Canada
cDepartment of Physics and Astronomy, Western University, 1151 Richmond Street, London, ON N6A 3K7, Canada
First published on 14th June 2024
Abstract
Carbon–carbon (C–C) bond formation is a cornerstone of synthetic chemistry, relying on routes such as transition-metal mediated cross-coupling for the introduction of new carbon-based functionality. For {[M]n+–C} (M = metal) structural units, studies that offer well-defined relationships between metal oxidation state, hydrocarbon strain, and {[M]n+–C} bond thermochemistry are thus informative, providing a means to reliably access new product classes. Here, we show that one-electron oxidation of the iron tucked-in complex [(η6-C5Me4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)Fe(dnppe)] (dnppe = 1,2-bis(di-n-propylphosphino)ethane) results in C(sp3)–C(sp3) bond formation giving unique {Fe2} dimers. Freeze-quenched CW X-band EPR spectroscopy allowed for spectroscopic identification of the reactive [(η6-C5Me4CH2)Fe(dnppe)]+ intermediate. Density functional theory (DFT) calculations reveal a primarily Fe-centered radical and a weak {[Fe]–C} bond (BDE[Fe]–C = 24.5 kcal mol−1, c.f. BDEC–C(ethane) = 90 kcal mol−1). For comparison, a structurally analogous Fe(III) methyl complex was prepared, [Cp*Fe(dnppe)(CH3)]+ (Cp* = C5Me5−), where C(sp3)–C(sp3) coupling was not observed, consistent with a larger calculated BDE[Fe]–C value of 47.8 kcal mol−1. These data are analogized to the simple hydrocarbons ethane and cyclopropane, where a strain-induced BDEC–C decrease of 33 kcal mol−1 is witnessed on cyclization.
CH2)Fe(dnppe)] (dnppe = 1,2-bis(di-n-propylphosphino)ethane) results in C(sp3)–C(sp3) bond formation giving unique {Fe2} dimers. Freeze-quenched CW X-band EPR spectroscopy allowed for spectroscopic identification of the reactive [(η6-C5Me4CH2)Fe(dnppe)]+ intermediate. Density functional theory (DFT) calculations reveal a primarily Fe-centered radical and a weak {[Fe]–C} bond (BDE[Fe]–C = 24.5 kcal mol−1, c.f. BDEC–C(ethane) = 90 kcal mol−1). For comparison, a structurally analogous Fe(III) methyl complex was prepared, [Cp*Fe(dnppe)(CH3)]+ (Cp* = C5Me5−), where C(sp3)–C(sp3) coupling was not observed, consistent with a larger calculated BDE[Fe]–C value of 47.8 kcal mol−1. These data are analogized to the simple hydrocarbons ethane and cyclopropane, where a strain-induced BDEC–C decrease of 33 kcal mol−1 is witnessed on cyclization.
Introduction
Carbon–carbon (C–C) bond forming reactions are a cornerstone of synthetic chemistry. Most pharmaceutically active compounds contain a C–C bond, along with countless materials relevant to industry and everyday life. It thus comes as no surprise that transition metal mediated C–C bond forming reactions have been the focus of two Nobel prizes.1,2 Using transition metals, one can form C–C bonds through cross-coupling – a sequence that proceeds via stepwise oxidative addition, transmetalation, and reductive elimination.3 In recent years, there has been a surge of reports that have driven such transformations using electro-4–7 or photochemistry;8–11 many of these studies have focused on cheaper and more abundant base transition elements as feedstocks.12Reductive elimination is the main step associated with C–C bond formation.13 When reductive elimination from {[M]n+–C} is unfavourable and prohibits C–C bond generation, oxidation of the metal-center can be used to induce so called oxidatively-induced reductive elimination (ORE; Fig. 1A).14–16 In this way, oxidation increases the tendency to undergo elimination (e.g., homolytic bond cleavage), thus returning the metal to a stable d-electron count.17,18
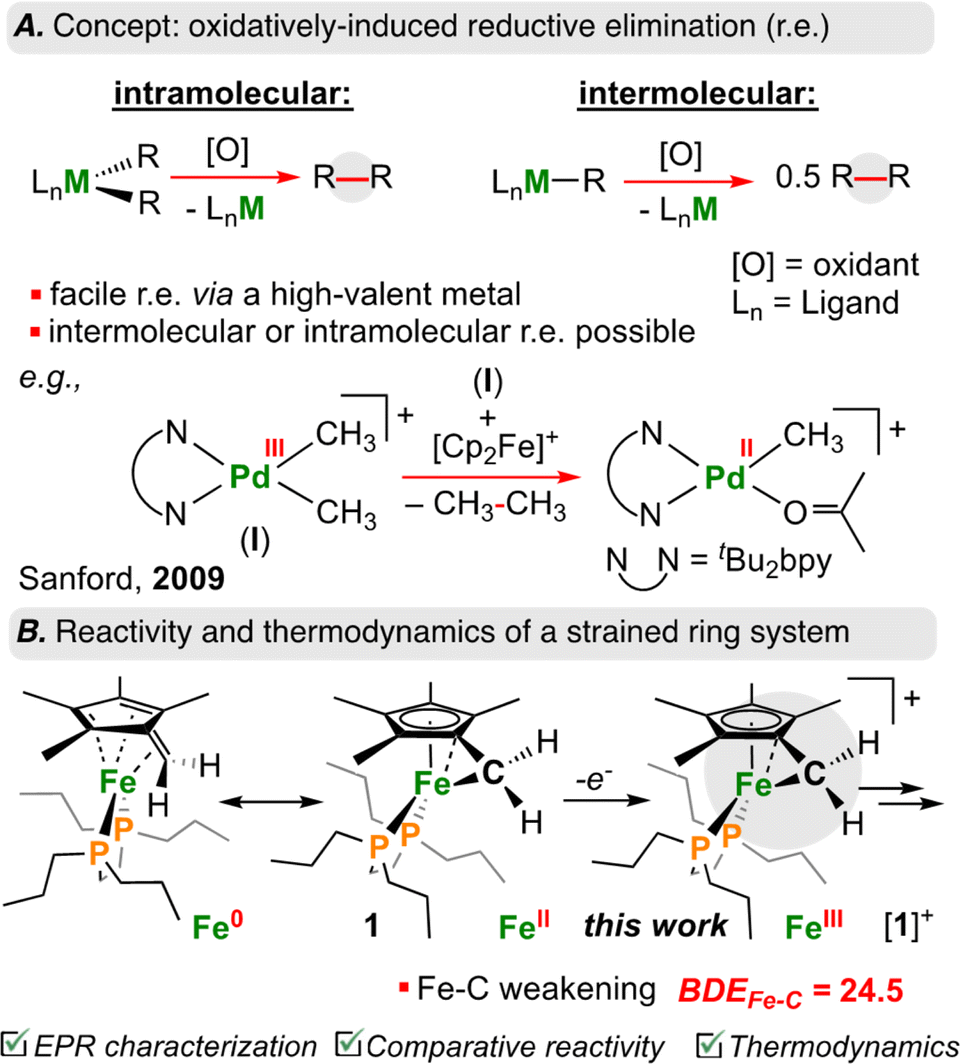 | ||
| Fig. 1 (A) Overview of intra/intermolecular oxidatively induced reductive elimination with transition metals. (B) Decrease in BDEC–C in cyclopropane upon oxidation, and this work. | ||
Sanford and co-workers have shown that one-electron oxidation of [(tBu2bpy)PdIII(CH3)2]+ (tBu2bpy = 4,4′-di-tert-butyl-2,2′-bipyridine) results in C(sp3)–C(sp3) reductive elimination of ethane (CH3CH3) from an intermediate {[PdIV]-(CH3)3} species (Fig. 1A).19 Similarly, Baik, Chang, and co-workers showed that oxidation of an Ir(III) complex to Ir(IV) led to a 19 kcal mol−1 reduction in the barrier associated with reductive elimination in a C–H arylation reaction.20 These examples illustrate the power of ORE in C–C bond formation, providing opportunities for otherwise unfavourable bond construction.
Of {[M]n+–C} organometallics, sandwich complexes ([Cp/Cp*]2M; Cp = C5H5−; Cp* = C5Me5−) represent a well-studied area owing to robust syntheses and product stability.21,22 Lesser studied, however, are related “tucked-in” complexes, where a H+/H˙/H− from the Cp*–CH3 ligand has been removed, encouraging high reactivity.23 This is especially true for first row late transition elements. As a salient example, it was reported that the ferrocenyl carbocation, [((η6-C5Me4)CH2)Fe(Cp*)]+ is reactive at temperatures >−82 °C.24–26
Recently, we reported the first example of an Fe diphosphine tucked-in complex, [((η6-C5Me4)CH2)Fe(dnppe)] (dnppe = 1,2-bis(di-n-propylphosphino)ethane) (1) (Fig. 1B) and its reactivity with electrophiles including CO2, HBCy2 (Cy = cyclohexyl), Br–AuI-PPh3, benzaldehyde, and B(C6F5)3.27 Herein, we examine the redox behaviour of 1 using electrochemical, photochemical, and chemical means (Fig. 2A). Oxidation of the Fe-center in 1 led to rapid C(sp3)–C(sp3) formation between CH2 groups of the η6-C5Me4CH2 ligand. Model studies and theoretical calculations were used to rationalize iron–carbon {[Fe]n+–C} bond strength (i.e., bond dissociation energy (BDE)) as a function of both iron oxidation state and ring-strain. These observations are analogized to cyclic organic compounds (e.g., cyclopropane) that also bear weak C–C bonds due to strain – a characteristic that has been leveraged for oxidatively-induced functionalization.28,29
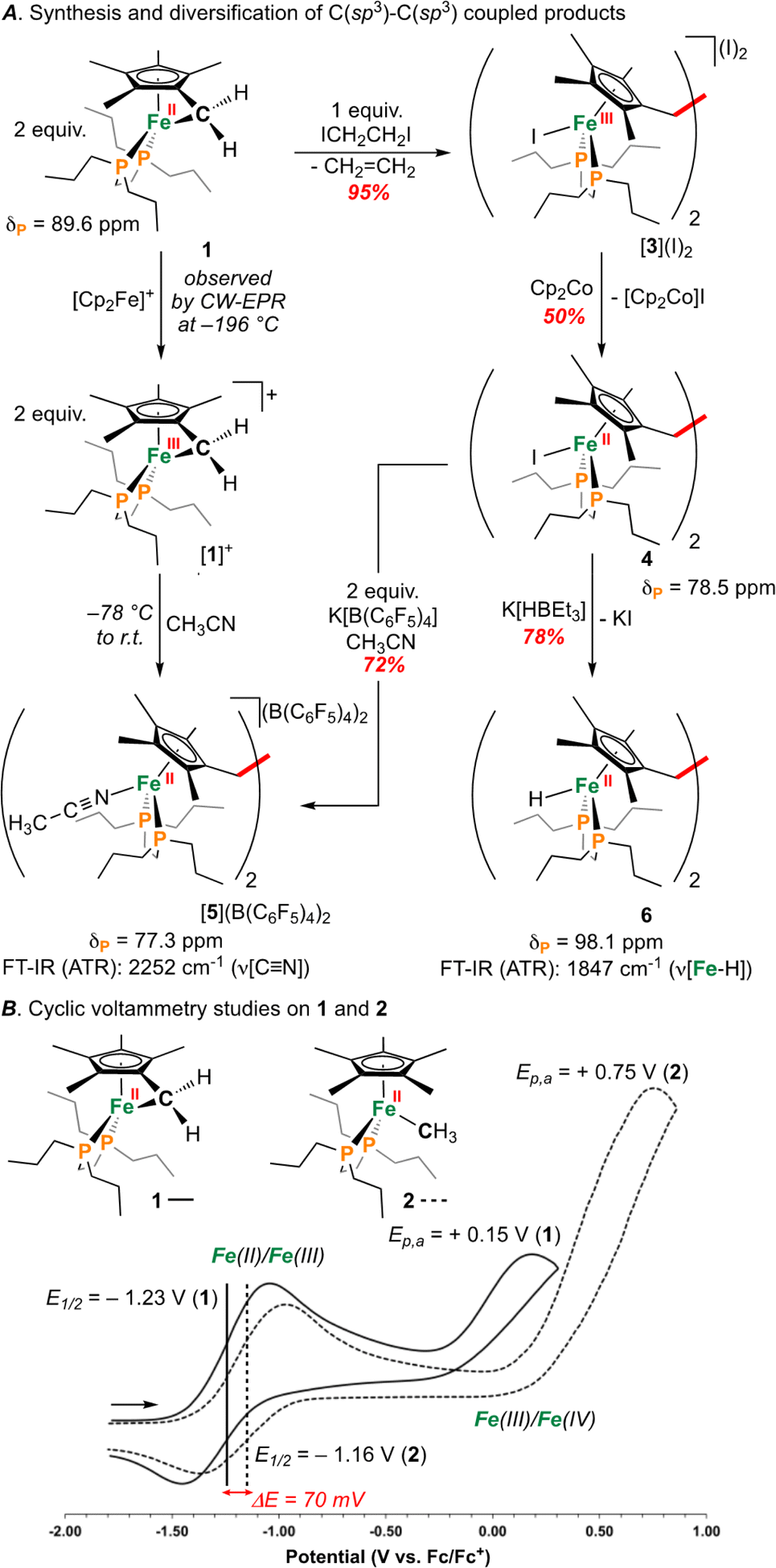 | ||
| Fig. 2 (A) Synthesis of [1]+ and C(sp3)–C(sp3) coupled products. (B) Cyclic voltammograms for 1 and 2. | ||
Results and discussion
An investigation into the oxidative chemistry of 1 was commenced by means of cyclic voltammetry (CV). Scanning from −1.90 V to +0.50 V in THF showed a quasi-reversible feature at E1/2 = −1.23 V (iredP/ioxP = 0.89 at 200 mV s−1; Fig. S43†)30 and an irreversible feature at Ep,a = +0.15 V vs. Fc/Fc+ (Fc = ferrocene) (Fig. 2B); these are assigned as Fe(II/III) and Fe(III/IV) events, respectively.31 Varying the scan rate from 50 to 500 mV s−1 did not change voltammogram shape (Fig. S44†). For other known Fe(II) half-sandwich complexes, [Cp*FeII(dppe)(X)] (dppe = 1,2-bis(diphenylphosphino)ethane; X = H, CH3, F, Cl, Br, I), the Fe(II/III) couple was found to be −0.54 > E1/2 > −0.88 V, indicating the E1/2 value for 1 to be comparably cathodically shifted.32,33 To probe the effect of hydrocarbon strain on electrochemical oxidation, an analogous {[FeII]–CH3} complex [Cp*FeII(dnppe)(CH3)] (2) (Fig. 2B) was examined.34 Compound 2 showed a reversible feature at E1/2 = −1.16 V with iredP/ioxP near 1.00 at 200 mV s−1 (Fig. S45†), and an irreversible feature at Ep,a = +0.35 V vs. Fc/Fc+ in THF, indicating that the tucked-in η6-C5Me4CH2 ligand contributes to a 70 mV cathodic shift in the Fe(II/III) couple. For both 1 and 2, the Fe(III/IV) couple was irreversible – a reported outcome for [Cp*Fe(diphosphine)(CH3)]+-type complexes.32
As a result of our observations from the CV data, complex 1 was next exposed to 1 equiv. of [Cp2Fe]B(C6F5)4 at 25 °C (Fig. 2A) in CH3CN. Monitoring this reaction by 31P{1H} NMR spectroscopy showed the formation of diamagnetic products in the range of ca. δP = 78 ppm, consistent with complexes of the type [Cp*Fe(diphosphine)(NCCH3)]+.33 This hypothesis was further supported by FT-IR spectroscopy, providing a band at ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) = 2252 cm−1 (c.f., ν(CN) = 2234 cm−1) for [Cp*Fe(dnppe)(NCCH3)]+, prepared independently (vide infra; [7]+) (Fig. S28†).
N) = 2252 cm−1 (c.f., ν(CN) = 2234 cm−1) for [Cp*Fe(dnppe)(NCCH3)]+, prepared independently (vide infra; [7]+) (Fig. S28†).
Supposing the main product from oxidation of 1 to be the C(sp3)–C(sp3) coupled {μ-H2CCH2−} dimer, [{η5-C5Me4–(μ-CH2)}FeII(dnppe)(NCCH3)]22+ ([5]2+), the redox behaviour of 1 was probed in the presence of 4 mL of THF with 100 μL CH3CN added.35 Consistent with an electrochemical–chemical (EC) reaction (onwards reactivity), scanning oxidatively from −2.00 to +1.00 V showed the previously discussed quasi-reversible couple at E1/2 = −1.23 V, with a decreased value of iredP/ioxP = 0.62 (c.f., 0.89 in THF at 200 mV s−1). In addition to the observed signature for 1, an additional wave at E1/2 = −0.16 V was assigned to the Fe(II/III) couple of [5]2+, confirmed through independent preparation (vide infra; Fig. S51†).
Outlining an alternative preparation of [5]2+, we envisaged treatment of 1 with a halide-based oxidant to initiate C(sp3)–C(sp3) coupling, followed by halide abstraction in CH3CN (Fig. 2A). Diiodoethane (ICH2CH2I), previously shown to oxidize {Fe(diphosphine)x} complexes,36 was selected as the candidate oxidant, supplying iodide groups that could later be removed. In accord, exposure of 1 to 1 equiv. of ICH2CH2I showed an immediate colour change from orange to dark red (Fig. 2A). Analysis by 31P{1H} NMR spectroscopy, however, showed an absence of signals, suggestive of paramagnetism. Growth of crystals from a THF solution layered with pentane at −35 °C overnight provided material appropriate for an Evans method measurement, giving μeff = 2.66 μB (298 K, CDCl3), falling within the expected range of a molecule containing two unpaired electrons. Further analysis by single-crystal X-ray diffraction (scXRD) revealed a connectivity map that supported a dimeric structure of [{η5-C5Me4–(μ-CH2)}FeIII(dnppe)(I)]2(I)2 ([3](I)2) (Fig. S54†). This molecule features a bridging {μ-H2CCH2−} unit that links two {(C5Me4–(CH2))FeIII(dnppe)(I)} moieties. Consistent with the μeff value obtained from Evans method and the two exogeneous iodide counteranions observed by scXRD, both Fe-centers are formally Fe(III). High-resolution electrospray ionization mass-spectrometry (HRMS-ESI) additionally provides a signal for [3]+ at m/z = 1158.299 (calcd. m/z = 1158.293).
Formation of [3](I)2 is a four-electron process involving two successive oxidations of 1. Considering the Fe(II) resonance contributor for 1 (Fig. 1B), oxidation leads to homolytic bond cleavage, forming an S = 1 Fe(II) center37 and a {C5Me4–(·CH2)} radical (vide infra). Recombination of two {C5Me4–(·CH2)} radicals results in C(sp3)–C(sp3) bond formation. Iodide coordination and subsequent oxidation to Fe(III) (by excess ICH2CH2I) thus provides the observed product [3](I)2 (Fig. 2A). To provide the Fe(II) complex [{η5-C5Me4–(μ-CH2)}FeII(dnppe)(I)]2 (4), 0.5 equiv. of ICH2CH2I was added to 1 (to preclude oxidation from Fe(II) to Fe(III)). However, this reaction resulted in mixtures of products, with at least two 31P{1H} resonances appearing in the range of 4 (δP = ca. +80 ppm), unreacted 1 (δP = +89.6 ppm), and free dnppe (δP = −28.3 ppm) (Fig. S30†).
As an alternative route towards 4, reduction of [3](I)2 was explored using cobaltocene ([Cp2Co]). Addition of 2 equiv. of [Cp2Co] to a THF solution of [3](I)2 produced an insoluble yellow precipitate and a purple supernatant over 3 h. Extraction of the purple product into toluene and analysis by NMR spectroscopy revealed a diamagnetic product. By 31P{1H} NMR spectroscopy, one signal at δP = +78.5 ppm was observed, standard of [Cp*FeII(dnppe)(halide)]-type compounds (e.g., for halide = Cl, δP = +79.4 ppm).34 Analysis of the 1H NMR spectrum also revealed a characteristic signal for the bridging {μ-H2CCH2−} moiety (δH = 2.57 ppm) as well as two pairs of signals for the unsymmetric methyl 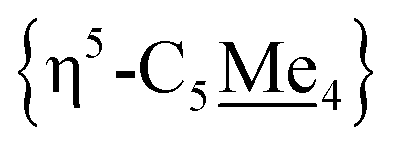 groups (δH = 1.89 and 1.82 ppm). Crystals of 4 were grown from slow evaporation of a benzene solution overnight; analysis by scXRD confirmed its structural identity as [{η5-C5Me4–(μ-CH2)}FeII(dnppe)(I)]2 (4) (Fig. 3). Offering context, a Cambridge Structural Database (CSD) search provides fewer than 5 examples of structurally-authenticated Cp*-based compounds featuring a bridging {–H2CCH2−} unit.24,38–43 Using pre-formed ligand constructs, Wang and co-workers reported a set of polymeric Ir complexes bridged by {C5Me4–(CH2)n–C5Me4} (n = 2, 3, 4);40 while Brintzinger and co-workers synthesized monometallic complexes of the type [(C5Me4–(CH2)2–C5Me4)MCl2] (M = Ti, Zr).41 Another example has been accessed by Cp*{H˙} removal – Severin and co-workers utilized [OAl(C6F5)3]˙− in the presence of Al(C6F5)3 to perform H-atom abstraction of
groups (δH = 1.89 and 1.82 ppm). Crystals of 4 were grown from slow evaporation of a benzene solution overnight; analysis by scXRD confirmed its structural identity as [{η5-C5Me4–(μ-CH2)}FeII(dnppe)(I)]2 (4) (Fig. 3). Offering context, a Cambridge Structural Database (CSD) search provides fewer than 5 examples of structurally-authenticated Cp*-based compounds featuring a bridging {–H2CCH2−} unit.24,38–43 Using pre-formed ligand constructs, Wang and co-workers reported a set of polymeric Ir complexes bridged by {C5Me4–(CH2)n–C5Me4} (n = 2, 3, 4);40 while Brintzinger and co-workers synthesized monometallic complexes of the type [(C5Me4–(CH2)2–C5Me4)MCl2] (M = Ti, Zr).41 Another example has been accessed by Cp*{H˙} removal – Severin and co-workers utilized [OAl(C6F5)3]˙− in the presence of Al(C6F5)3 to perform H-atom abstraction of 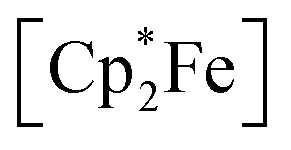 , generating a [(η5-C5Me4)(·CH2)]Fe(Cp*)] radical,38 which dimerized to give [{η5-C5Me4–(μ-CH2)}FeII(Cp*)]2. This example, however, does not require Fe oxidation nor does it inform on the relationship between iron-ring-strain and C–C coupling.
, generating a [(η5-C5Me4)(·CH2)]Fe(Cp*)] radical,38 which dimerized to give [{η5-C5Me4–(μ-CH2)}FeII(Cp*)]2. This example, however, does not require Fe oxidation nor does it inform on the relationship between iron-ring-strain and C–C coupling.
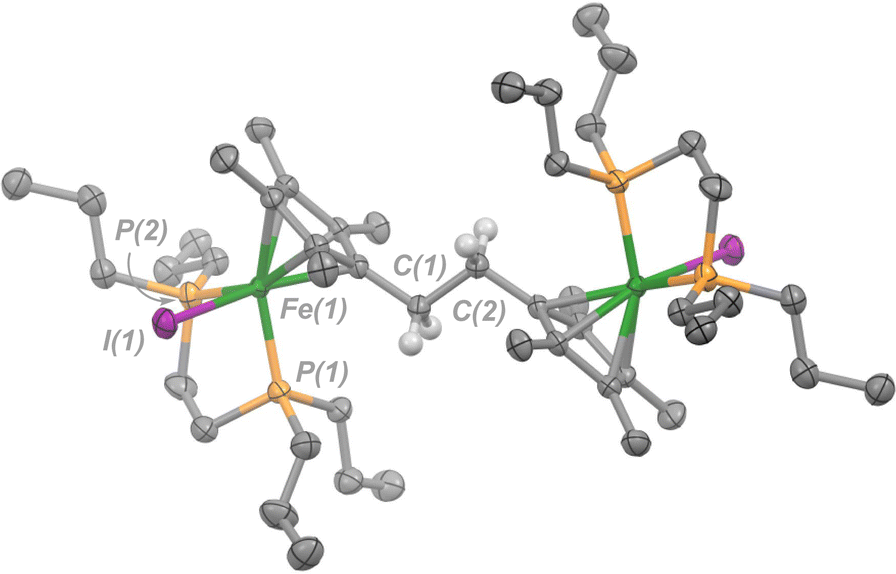 | ||
| Fig. 3 Molecular structure of 4 with ellipsoids drawn at 50% probability. Hydrogen atoms omitted except for those on C(1) and C(2). Solvent molecule (benzene) has been omitted for clarity. | ||
Compound 4 is a viable candidate for halide abstraction in CH3CN. Reaction of a CH3CN solution of 4 with 2 equiv. K[B(C6F5)4] (Fig. 2A) produced an immediate colour change from purple to orange. After workup, FT-IR (ATR) spectroscopic analysis showed a stretch at ν(NC) = 2252 cm−1, corroborating formation of [{η5-C5Me4-(μ-CH2)}FeII(dnppe)(NCCH3)]2[B(C6F5)4]2 ([5](B(C6F5)4)2). The 1H NMR spectrum of [5]2+ also showed the expected inequivalent 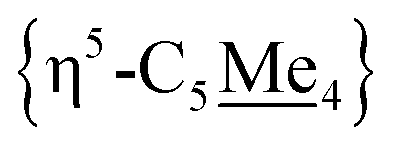 signals (δH = 1.63 and 1.48 ppm), a {μ-CH2} signal (δH = 2.22 ppm), and a peak for [Fe]-NCC
signals (δH = 1.63 and 1.48 ppm), a {μ-CH2} signal (δH = 2.22 ppm), and a peak for [Fe]-NCC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3 (δH = 2.41 ppm; c.f., δH = 1.91 ppm for [Cp*Fe(dnppe)(NCCH3)]+ ([7]+)). Comparison of the FT-IR spectra for the reaction between 1 and [Cp2Fe]B(C6F5)4, [5]2+, [7]+, and neat CH3CN, confirmed that [5]2+ was formed from oxidation of 1 (Fig. 4) with an observed stretch at ν(NC) = 2252 cm−1. Notably, the combination band for neat CH3CN (ν = 2300 cm−1) also disappears upon CH3CN coordination.44,45 In addition to our observations from IR spectroscopy, a THF solution of 4 exposed to 100 μL of CH3CN and analyzed by CV displayed a predominant wave at E1/2 = −0.16 V, akin to the voltammogram acquired from the addition of 100 μL CH3CN to 1, noted above (Fig. S51†).
3 (δH = 2.41 ppm; c.f., δH = 1.91 ppm for [Cp*Fe(dnppe)(NCCH3)]+ ([7]+)). Comparison of the FT-IR spectra for the reaction between 1 and [Cp2Fe]B(C6F5)4, [5]2+, [7]+, and neat CH3CN, confirmed that [5]2+ was formed from oxidation of 1 (Fig. 4) with an observed stretch at ν(NC) = 2252 cm−1. Notably, the combination band for neat CH3CN (ν = 2300 cm−1) also disappears upon CH3CN coordination.44,45 In addition to our observations from IR spectroscopy, a THF solution of 4 exposed to 100 μL of CH3CN and analyzed by CV displayed a predominant wave at E1/2 = −0.16 V, akin to the voltammogram acquired from the addition of 100 μL CH3CN to 1, noted above (Fig. S51†).
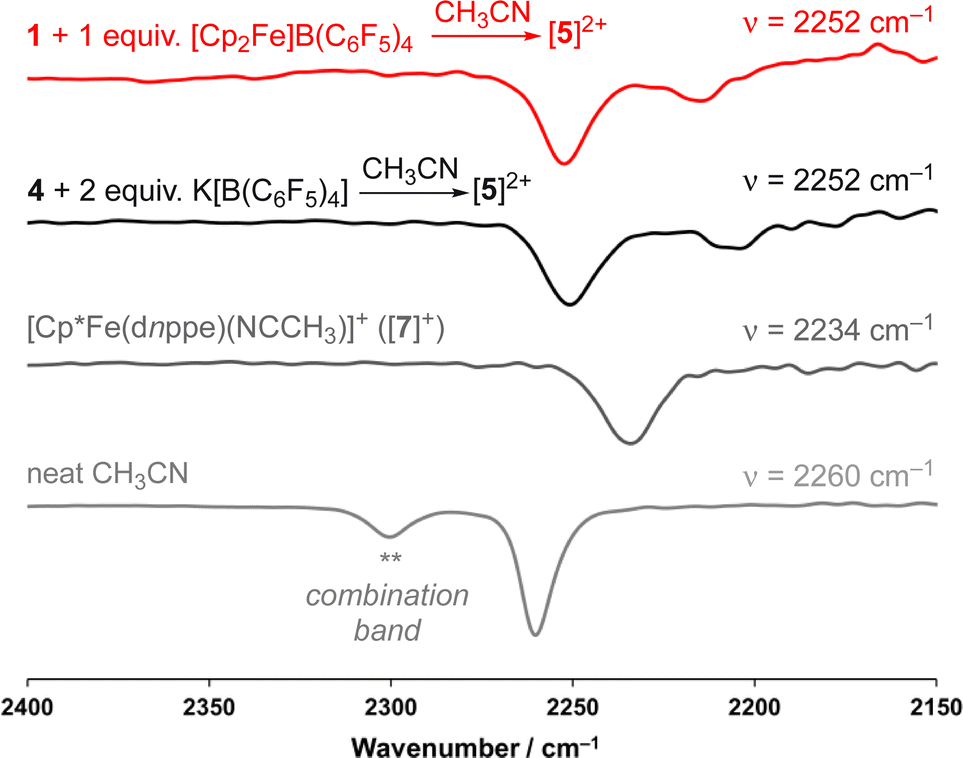 | ||
| Fig. 4 Enhanced view of the region between 2400–2150 cm−1 for the FT-IR (ATR) spectra for the reaction between 1 and 1 equiv. of [Cp2Fe]B(C6F5)4 (red), [5]2+ (black), [7]+ (dark grey), and neat CH3CN (light grey). | ||
Compound 4 is also a valuable synthon for clean access to dimeric iron hydrides. For example, exposure of a THF solution of 4 to 2 equiv. of K[HBEt3] led to clean formation of the bis-hydride complex [{η5-C5Me4–(μ-CH2)}FeII(dnppe)(H)]2 (6) (Fig. 2A).46 By 1H NMR spectroscopy, the bridging {μ-H2CCH2−} group was located at δH = 2.62 and the inequivalent 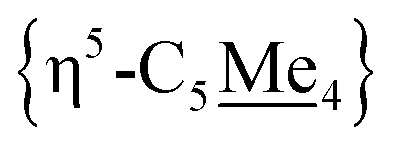 signals were observed at δH = 2.05 and 1.96 ppm. Successful [Fe]–H formation was further established by locating the [Fe]– signal as a triplet at δH = −17.81 ppm (2JH–P = 70.2 Hz) (c.f., δH = −17.90 ppm (2JH–P = 70.2 Hz) for the monomeric [Cp*Fe(dnppe)(H)]).34 FT-IR spectroscopy also revealed a characteristic ν(Fe–H) stretch at 1847 cm−1.
signals were observed at δH = 2.05 and 1.96 ppm. Successful [Fe]–H formation was further established by locating the [Fe]– signal as a triplet at δH = −17.81 ppm (2JH–P = 70.2 Hz) (c.f., δH = −17.90 ppm (2JH–P = 70.2 Hz) for the monomeric [Cp*Fe(dnppe)(H)]).34 FT-IR spectroscopy also revealed a characteristic ν(Fe–H) stretch at 1847 cm−1.
Having characterized the terminal C(sp3)–C(sp3) coupled product, we next sought to characterize the reactive oxidized precursor, [1]+. Oxidation of 1 was thus carried out in 2-MeTHF using [Cp2Fe]+ at −78 °C. Following 5 min of reaction time, freeze-quenched continuous wave (CW) X-band EPR spectroscopy provided a spectrum with rhombic symmetry (g = [2.0004, 2.0526, 2.2134]) (Fig. 5A) – this represents the first example of a spectroscopically characterized Fe(III) tucked-in complex by EPR. Compound [1]+ can likewise be photochemically generated in a 2-MeTHF glass using 1 and a 405 nm laser (350 mW mm−2) following 10 min of irradiation (Fig. 5B). To provide a comparative basis, the {[FeIII]–CH3} complex [2]+ was also prepared by reaction of 2 with 1 equiv. of [Cp2Fe]+ in 2-MeTHF at −78 °C. By CW X-band EPR spectroscopy, a rhombic signal was also observed (g = [1.9827, 2.0450, 2.3968]) (Fig. 5A), again consistent with an S = 1/2 Fe(III) radical species ([2]+).47 The gmax shift between compounds [1]+ and [2]+ is attributed to a difference in geometry about Fe. For [1]+, g-values are all greater than 2.0 with a spread of 0.05 to 0.16 – characteristic of the unpaired electron predominately residing in a dz2 orbital.48 For [2]+, a larger value of gmax is consistent with an unpaired electron residing in a dx2−y2 orbital.49 Calculated spin density plots for [1]+ and [2]+ additionally corroborate that the unpaired electron predominately resides on Fe, with populations of 0.917e− for [1]+ and 0.987e− for [2]+ (Fig. 5C). The marked geometry change noted above (between [1]+ and [2]+) is best visualized from an overlay plot of their DFT-optimized structures (with coincident Fe and P atoms) (Fig. 6A). This data also showcases an elongated Fe–C bond in [1]+ (d(Fe–C) = 2.24 Å) as compared to [2]+ (d(Fe–C) = 2.01 Å), indicating weaker Fe–C overlap in [1]+. The ring-strain inherent to [1]+ is additionally described by an Fe–C–C triangle with angles of 38.4°, 60.5°, and 81.1° (Fig. 6A).
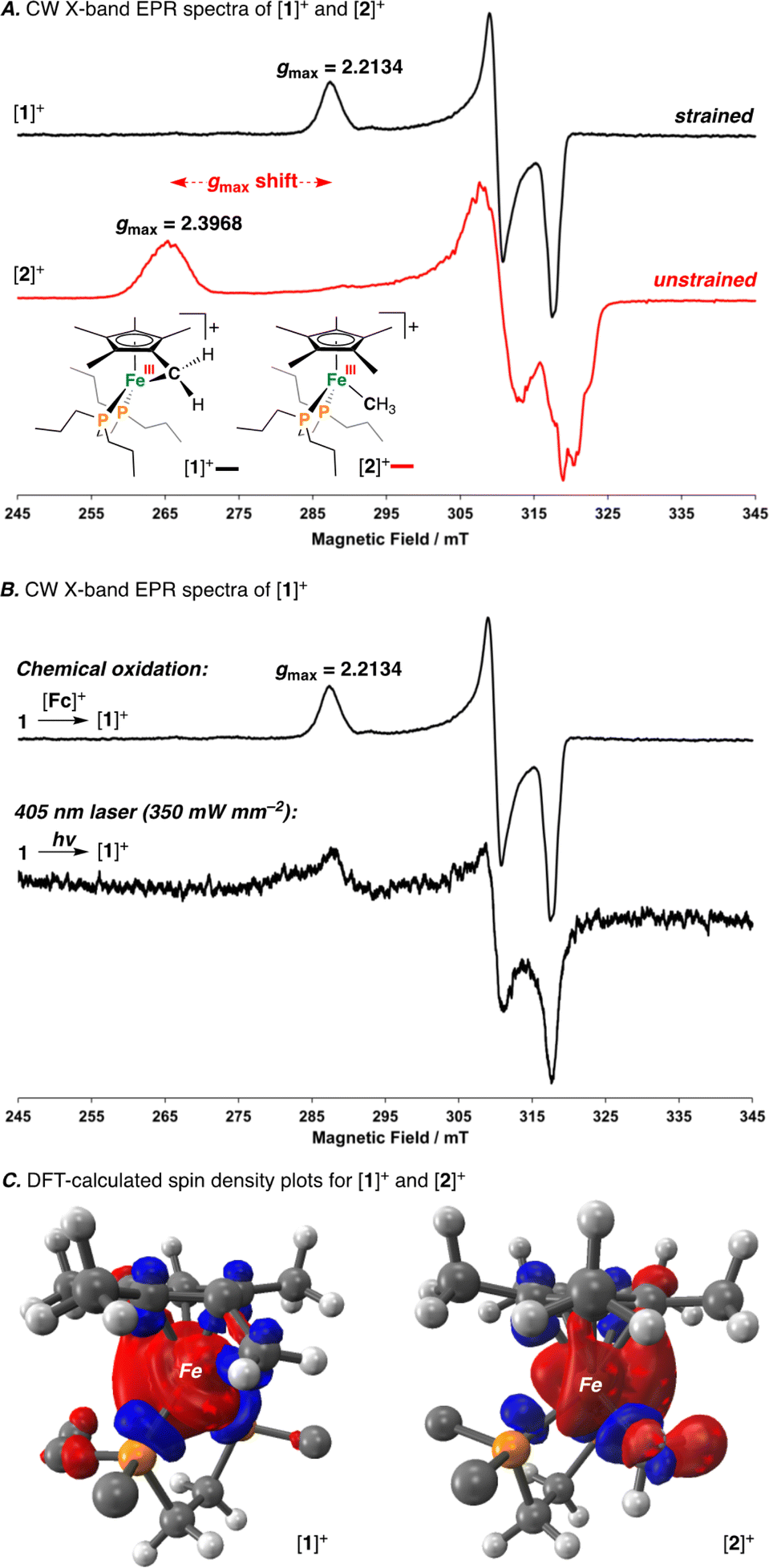 | ||
| Fig. 5 A) Freeze-quenched CW X-band EPR spectra (8.885 GHz) recorded in 2-MeTHF glass at 77 K for [1]+ (black) and [2]+ (red). (B) Freeze-quenched CW X-band EPR spectra for [1]+ (top) and a photooxidized sample of 1 under 405 nm (350 mW mm−2) irradiation (bottom). (C) DFT-calculated spin-density plots for [1]+ and [2]+ (all C and H atoms from the dnppe ligand, except those C atoms directly attached to P, have been omitted for clarity). Fc = Cp2Fe. | ||
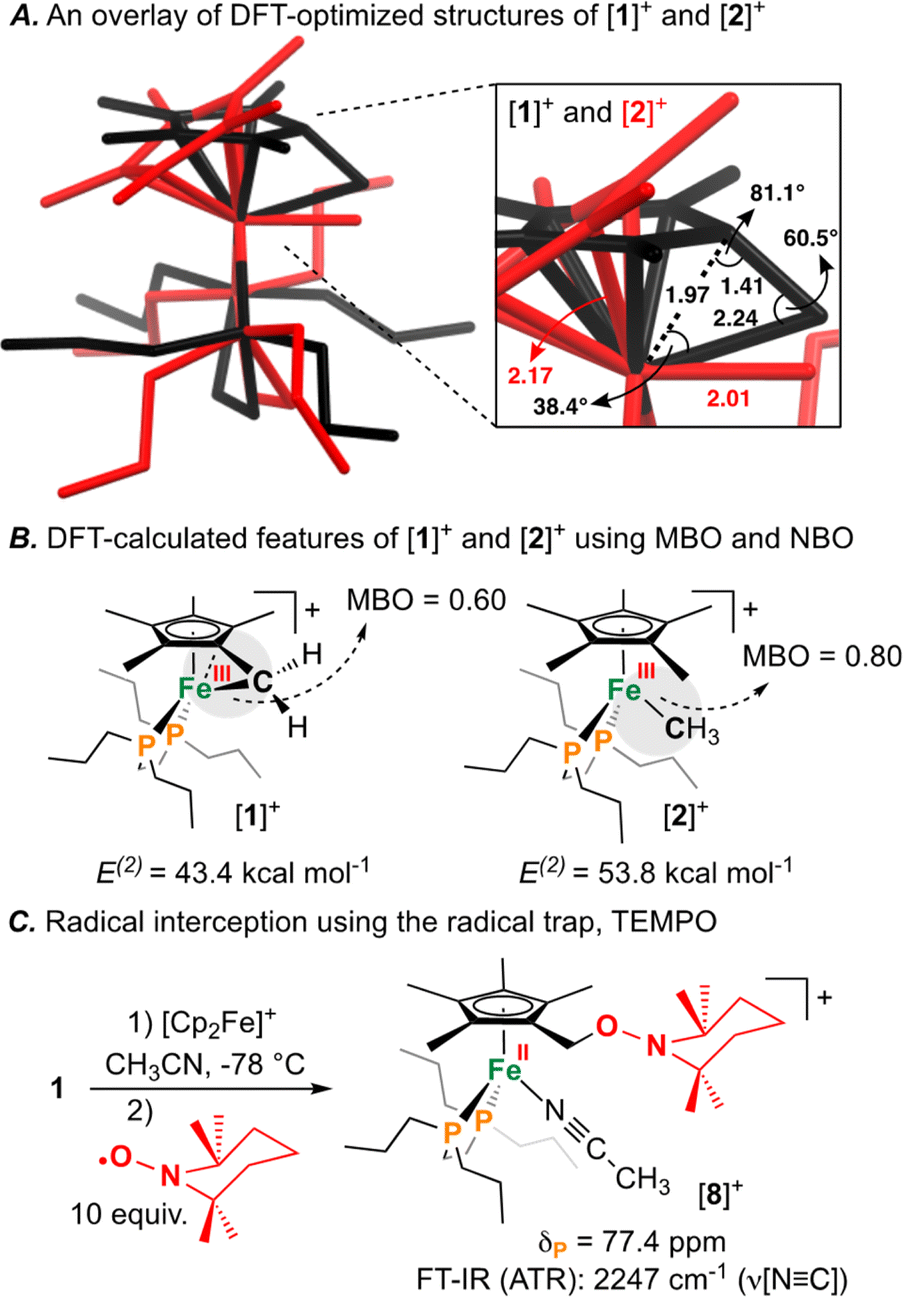 | ||
| Fig. 6 (A) Structural overlay (capped sticks) of [1]+ (black) and [2]+ (red). Select angles (°) and select bond lengths (Å) are provided. (B) Mayer bond order (MBO) and natural bond orbital (NBO) analysis providing E(2) values between [Fe]–C. (C) Reaction of [1]+ with (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) forming [8]+. | ||
Mayer bond order (MBO) and natural bond orbital (NBO) analysis provide further insight that helps to rationalize C(sp3)–C(sp3) coupling in [1]+ (Fig. 6B).50 To determine the change in bond order upon oxidation, MBOs were calculated. For 1/[1]+, the MBO decreased by 0.06 on oxidation (1: 0.66/[1]+: 0.60), while for 2/[2]+, the MBO was found to slightly increase (2: 0.73/[2]+: 0.80). NBO calculations provided second order perturbation energies (E(2)) which showed a similar trend. For the C(lp) → Fe(lv) (lp = lone pair, lv = lone valence) interaction, energies were found to be 43.4 and 53.8 kcal mol−1 for [1]+ and [2]+, respectively (Fig. 6B). This data indicates that (1) the Fe–C bond becomes weaker on going from 1 → [1]+ and (2) that the Fe–C bond of 1/[1]+ is weaker than 2/[2]+ – a consequence of ring-strain.
The radical character intrinsic to [1]+ is also evidenced by its chemical reactivity with 10 equiv. of the radical trap, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), which gives [{η5-C5Me4–(CH2–ONC9H18)}FeII(dnppe)(NCCH3)]B(C6F5)4 ([8]B(C6F5)4) (Fig. 6C). Compound [8]+ was characterized by signals in the 1H NMR spectrum corresponding to {C2–ONC9H18} at δH = 4.18 ppm and Fe-bound NCCH3 at δH = 2.40 ppm. Confirmation of an Fe-bound NCCH3 group was determined via FT-IR spectroscopy, with a characteristic ν(NC) stretch at 2247 cm−1. The 13C{1H} NMR spectrum also showed a signal at δC = 71.0 ppm, corresponding to the TEMPO-bound CH2 from the η5-C5Me4–(CH2–ONC9H18) ligand. HRMS-ESI also provided a signal corresponding to [8]+ at 649.407 (calcd; 649.407). These data corroborate a radical mechanism, whereby the generated {C5Me4–(·CH2)} group has been trapped by TEMPO.51
To assess the effect of ring-strain on {[Fe]–C} bond homolysis, we next sought information regarding the BDE[Fe]–C of [1]+. For reference, the BDEC–C associated with the simplest cyclical saturated hydrocarbon (cyclopropane; (CH2)3) is substantially lower than ethane due to ring strain (BDEC–C(cyclopropane) = 57 kcal mol−1vs. BDEC–C(ethane) = 90 kcal mol−1).52–54 Given this, and assuming CH3CH3 and RCH2CH2R (R = C5Me4−) to have similar bond strengths, C–C coupling should be favoured for {[Fe]–CH2R} when BDE[Fe]–C ≤ 45 kcal mol−1.
Computations show the difference in BDE[Fe]–C for [1]+ and [2]+ to be 23.3 kcal mol−1 (Fig. 7). The absolute values of BDE[Fe]–C: 24.5 and 47.8 kcal mol−1 for [1]+ and [2]+, respectively are also consistent with observation of C(sp3)–C(sp3) coupling for the former. These calculated values are benchmarked against ethane, which provides BDEC–C(calcd) = 88.4 kcal mol−1c.f., the experimental BDEC–C = 90 kcal mol−1.52 Notwithstanding differences in BDEC–C, both processes were calculated to be mildly exergonic (Fig. 7): ΔG° = −2.1 kcal mol−1 and −4.3 kcal mol−1 for C–C coupling from [1]+ or [2]+, respectively, suggesting a difference in kinetic barrier. Based on the above discussion, the C(sp3)–C(sp3) coupling associated with [1]+ can thus be ascribed to a minimum of two features: (1) a strained three-membered {–Fe–CH2–C–} ring and (2) a weakened {[FeIII]–CH2R} bond c.f., 1.32 This is contrasted with [2]+, where experimentally, CH3CH3 is not evolved during synthesis (Fig. 7B).
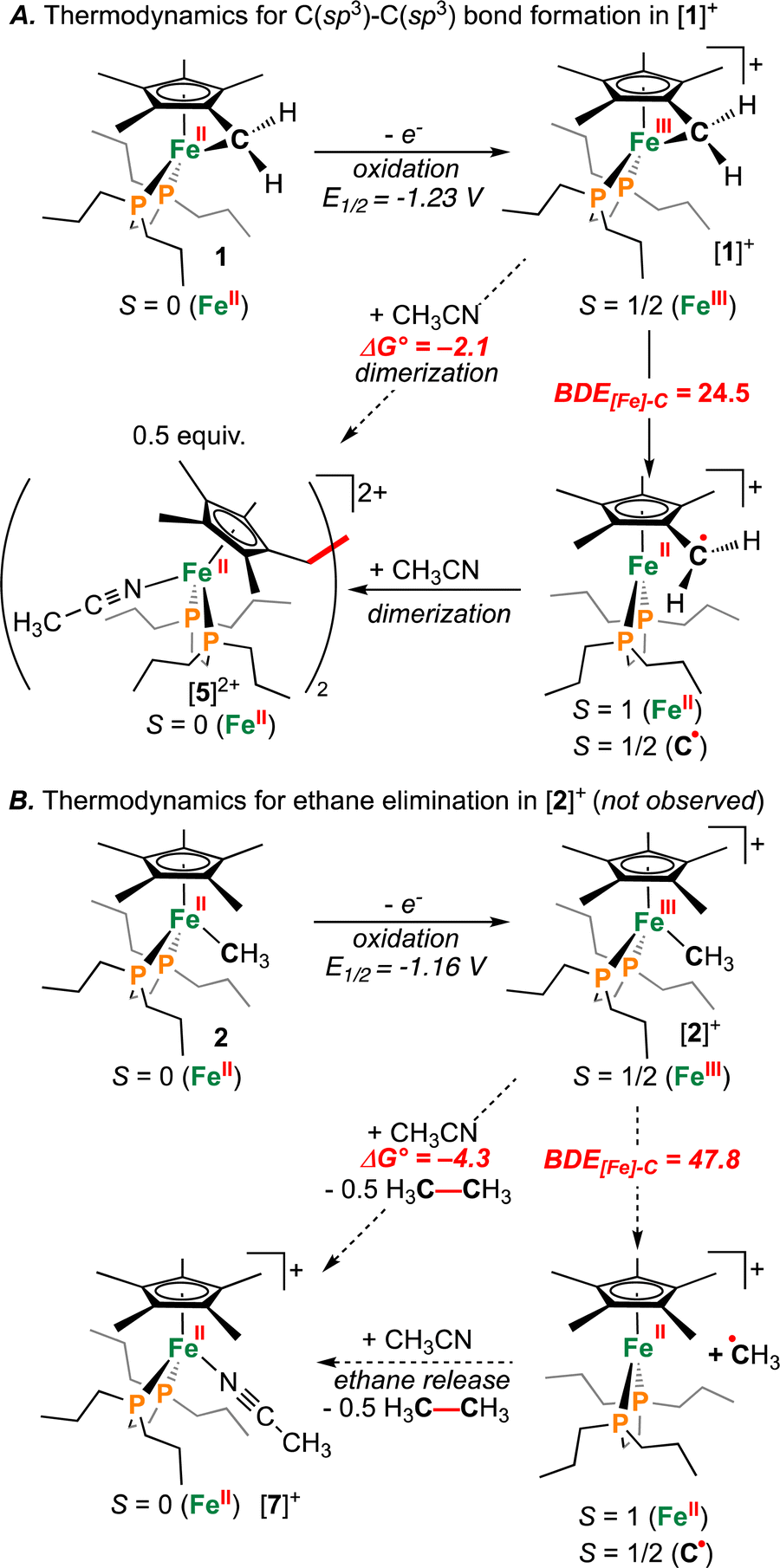 | ||
| Fig. 7 (A) Thermodynamic calculations showing energies associated with the dimerization of [1]+. (B) Analogous calculations for the elimination of ethane (CH3CH3) from [2]+. All energies are in kcal mol−1. | ||
Conclusions
Metal-mediated cross-coupling has emerged as a powerful means to construct C–C bonds. In recent years, such studies have focused predominantly on base transition elements, such as iron, cobalt, and nickel, as cheaper and more sustainable sources. Here, we have studied an oxidized iron tucked-in complex [(η6-C5Me4CH2)FeIII(dnppe)]+ ([1]+), which due to both ring-strain and iron oxidation state, promotes C(sp3)–C(sp3) bond coupling. The product {Fe2} diphosphine dimers invite reactions to give functionalized {FeIII2} or {FeII2} products having coordinated halide, hydride, or solvent equivalents. Characterization of the reactive oxidized “tucked-in” {FeIII} precursor was performed using CW X-band EPR spectroscopy, with computations revealing a weak {[Fe]III–C} bond with BDE[Fe]–C = 24.5 kcal mol−1. The observed C(sp3)–C(sp3) coupling, however, is not simply an artifact of iron oxidation state. The model, [Cp*FeIII(dnppe)(CH3)]+ [2]+, for instance, does not undergo C(sp3)–C(sp3) coupling, consistent with a higher calculated BDE[Fe]–C of 47.8 kcal mol−1. This pair of compounds and their contrasting thermochemistry/reactivity pave the way towards better understanding the intricacies of {M–C} strain and metal oxidation state on reductive elimination, offering insight into an important elementary transformation of direct relevance to C–C bond coupling.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 4 has been deposited at the CCDC under 2341812. This data can be obtained free of charge from the cambridge crystallographic data centre viahttps://www.ccdc.cam.uk/data_request/cif.Author contributions
Joseph A. Zurakowski: investigation, methodology, formal Analysis, visualization, writing – original draft, writing – review & editing. Connor S. Durfy: investigation, validation. Noah B. Stocek: investigation, methodology. Giovanni Fanchini: resources, methodology, funding acquisition. Marcus W. Drover: project administration, funding acquisition, visualization, supervision, formal analysis, conceptualization, methodology, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Western University, the Council of Ontario Universities for a John C. Polanyi award to M. W. D., the Canadian Foundation for Innovation (LOF-212442), and the Natural Sciences and Engineering Research Council of Canada (Discovery Grant, RGPIN-2020-04480 (M. W. D.), RGPIN-2020-05008 (G. F.), Discovery Launch Supplement, DGECR-2020-00183), and graduate award (CGS-D/NSERC Vanier to J. A. Z.) for funding.References
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- B. Zhang, Y. Gao, Y. Hioki, M. S. Oderinde, J. X. Qiao, K. X. Rodriguez, H.-J. Zhang, Y. Kawamata and P. S. Baran, Nature, 2022, 606, 313–318 CrossRef CAS PubMed.
- Y. Liu, P. Li, Y. Wang and Y. Qiu, Angew. Chem., Int. Ed., 2023, 62, e202306679 CrossRef CAS.
- P. Li, G. Kou, T. Feng, M. Wang and Y. Qiu, Angew. Chem., Int. Ed., 2023, 62, e202311941 CrossRef CAS.
- Y. Wang, S. Dana, H. Long, Y. Xu, Y. Li, N. Kaplaneris and L. Ackermann, Chem. Rev., 2023, 123, 11269–11335 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- M. De Abreu, P. Belmont and E. Brachet, Eur. J. Org Chem., 2020, 2020, 1327–1378 CrossRef CAS.
- L. K. G. Ackerman, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059–14063 CrossRef CAS.
- M. L. Clapson, C. S. Durfy, D. Facchinato and M. W. Drover, Cell Rep. Phys. Sci., 2023, 4, 101548 CrossRef CAS.
- X. Chen, K. M. Engle, D. Wang and J. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS.
- M. B. Watson, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2017, 139, 35–38 CrossRef CAS.
- J. Kim, K. Shin, S. Jin, D. Kim and S. Chang, J. Am. Chem. Soc., 2019, 141, 4137–4146 CrossRef CAS.
- A. Cizikovs, E. E. Basens, P. A. Zagorska, A. Kinens and L. Grigorjeva, ACS Catal., 2024, 14, 1690–1698 CrossRef CAS.
- A. Pedersen and M. Tilset, Organometallics, 1993, 12, 56–64 CrossRef CAS.
- W. Lau, J. C. Huffman and J. K. Kochi, Organometallics, 1982, 1, 155–169 CrossRef CAS.
- M. P. Lanci, M. S. Remy, W. Kaminsky, J. M. Mayer and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 15618–15620 CrossRef CAS PubMed.
- K. Shin, Y. Park, M.-H. Baik and S. Chang, Nat. Chem., 2018, 10, 218–224 CrossRef CAS PubMed.
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- P. J. Chirik, Organometallics, 2010, 29, 1500–1517 CrossRef CAS.
- P. Preethalayam, K. S. Krishnan, S. Thulasi, S. S. Chand, J. Joseph, V. Nair, F. Jaroschik and K. V. Radhakrishnan, Chem. Rev., 2017, 117, 3930–3989 CrossRef CAS.
- A. Z. Kreindlin, F. M. Dolgushin, A. I. Yanovsky, Z. A. Kerzina, P. V. Petrovskii and M. I. Rybinskaya, J. Organomet. Chem., 2000, 616, 106–111 CrossRef CAS.
- E. I. Fedin, A. L. Blumenfeld, P. V. Petrovskii, A. Z. Kreindlin, S. S. Fadeeva and M. I. Rybinskaya, J. Organomet. Chem., 1985, 292, 257–268 CrossRef CAS.
- A. Z. Kreindlin, S. S. Fadeeva and M. I. Rybinskaya, Izv. Akad. Nauk SSSR, Ser. Khim., 1984, 2, 403–406 Search PubMed.
- J. A. Zurakowski and M. W. Drover, Chem. Commun., 2023, 59, 11349–11352 RSC.
- J. P. Dinnocenzo, W. P. Todd, T. R. Simpson and I. R. Gould, J. Am. Chem. Soc., 1990, 112, 2462–2464 CrossRef CAS.
- P. Peng, X. Yan, K. Zhang, Z. Liu, L. Zeng, Y. Chen, H. Zhang and A. Lei, Nat. Commun., 2021, 12, 3075 CrossRef CAS.
- T. Krämer, M. R. Gyton, I. Bustos, M. J. G. Sinclair, S. Tan, C. J. Wedge, S. A. Macgregor and A. B. Chaplin, J. Am. Chem. Soc., 2023, 145, 14087–14100 CrossRef.
- Given the similarity of E1/2 for both 1 and 2, along with the lower reduction potentials (E1/2 ≪ −1.23 V vs. Fc/Fc+) observed for related Fe(0) species, a formal Fe(II) oxidation state seemingly better describes the observed electrochemical behaviour of 1.
- M. Tilset, I. Fjeldahl, J.-R. Hamon, P. Hamon, L. Toupet, J.-Y. Saillard, K. Costuas and A. Haynes, J. Am. Chem. Soc., 2001, 123, 9984–10000 CrossRef CAS PubMed.
- D. Patel, A. Wooles, A. D. Cornish, L. Steven, E. S. Davies, D. J. Evans, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2015, 44, 14159–14177 RSC.
- J. A. Zurakowski, K. R. Brown and M. W. Drover, Inorg. Chem., 2023, 62, 7053–7060 CrossRef CAS.
- Complex 1 is insoluble in pure acetonitrile and was found to react with CH2Cl2.
- F. S. Pick, D. B. Leznoff and M. D. Fryzuk, Dalton Trans., 2018, 47, 10925–10931 RSC.
- P. Hamon, L. Toupet, J.-R. Hamon and C. Lapinte, Organometallics, 1996, 15, 10–12 CrossRef CAS.
- Y. Liu, E. Solari, R. Scopelliti, F. Fadaei Tirani and K. Severin, Chem.–Eur. J., 2018, 24, 18809–18815 CrossRef CAS PubMed.
- M. Kessler, S. Hansen, C. Godemann, A. Spannenberg and T. Beweries, Chem.–Eur. J., 2013, 19, 6350–6357 CrossRef CAS PubMed.
- X. Tan, B. Li, S. Xu, H. Song and B. Wang, Organometallics, 2013, 32, 3253–3261 CrossRef CAS.
- F. Wochner, L. Zsolnai, G. Huttner and H. H. Brintzinger, J. Organomet. Chem., 1985, 288, 69–77 CrossRef CAS.
- D. V. Muratov, A. S. Romanov, M. Corsini, A. R. Kudinov, F. F. de Biani and W. Siebert, Chem.–Eur. J., 2017, 23, 11935–11944 CrossRef CAS PubMed.
- X. Wang, M. Sabat and R. N. Grimes, Organometallics, 1995, 14, 4668–4675 CrossRef CAS.
- Y. Kwon, C. Lee and S. Park, Chem. Phys., 2014, 445, 38–45 CrossRef CAS.
- B. Dereka, N. H. C. Lewis, J. H. Keim, S. A. Snyder and A. Tokmakoff, J. Phys. Chem. B, 2022, 126, 278–291 CrossRef CAS PubMed.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
- J. Morrow, D. Catheline, M. H. Desbois, J. M. Manriquez, J. Ruiz and D. Astruc, Organometallics, 1987, 6, 2605–2607 CrossRef CAS.
- D. Chong, W. E. Geiger, N. A. Davis, A. Weisbrich, Y. Shi, A. M. Arif and R. D. Ernst, Organometallics, 2008, 27, 430–436 CrossRef CAS.
- F. Paul and C. Lapinte, Coord. Chem. Rev., 1998, 178–180, 431–509 CrossRef CAS.
- A. J. Bridgeman, G. Cavigliasso, L. R. Ireland and J. Rothery, J. Chem. Soc., Dalton Trans., 2001, 2095–2108 RSC.
- Of note, complexes 1 and 2 exhibited no reactivity with TEMPO.
- B. Ruscic, J. Phys. Chem. A, 2015, 119, 7810–7837 CrossRef CAS.
- F. H. Seubold, J. Phys. Chem., 1954, 22, 945–946 CrossRef CAS.
- X. Lei, X. Cao, J. Wang and X. Li, Combust. Flame, 2022, 237, 111881 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, computational details. CCDC 2341812. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03292f |
| This journal is © The Royal Society of Chemistry 2024 |