
Local protons enhance photocatalytic CO2 reduction by porphyrinic zirconium-organic frameworks†
Xue
Zhao
a,
Chang-Yan
Zhu
b,
Jun-Sheng
Qin
 *ac,
Heng
Rao
ac,
Dong-Ying
Du
b,
Min
Zhang
*b,
Ping
She
ac,
Li
Li
a and
Zhong-Min
Su
*a
*ac,
Heng
Rao
ac,
Dong-Ying
Du
b,
Min
Zhang
*b,
Ping
She
ac,
Li
Li
a and
Zhong-Min
Su
*a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: qin@jlu.edu.cn; suzhongmin@jlu.edu.cn
bDepartment of Chemistry, Northeast Normal University, Changchun 130024, P. R. China. E-mail: mzhang@nenu.edu.cn
cInternational Center of Future Science, Jilin University, Changchun 130012, P. R. China
First published on 18th April 2024
Abstract
The immobilization of molecular catalysts based on porphyrin fragments within metal–organic frameworks (MOFs) offers a promising approach for achieving sustainable and stable photocatalytic activity. In this study, we presented the synthesis of a phenolic hydroxy-modified iron-porphyrinic zirconium-based MOF, Zr6O4(OH)4(FeTCBPP-OH)3, named MOF-OH (FeTCBPP-OH = iron 5,10,15,20-tetrakis[4-(4′-carboxyphenyl)-2,6-dihydroxylphenyl]porphyrin), through post-synthetic modification of a precursor MOF called MOF-OCH3 (Zr6O4(OH)4(FeTCBPP-OCH3)3, FeTCBPP-OCH3 = iron 5,10,15,20-tetrakis[4-(4′-carboxyphenyl)-2,6-dimethoxyphenyl]porphyrin). Initially, we attempted the direct assembly of Zr4+ centers and FeTCBPP-OH ligands; however, this approach was unsuccessful in obtaining MOF-OH. This perhaps resulted from the high number of hydroxyl groups on the polyphenolic porphyrinic fragments, which exhibited a stronger binding affinity towards zirconium centers. Consequently, we achieved MOF-OH by selectively modifying the partial methoxy positions of the FeTCBPP-OCH3 fragments in MOF-OCH3 through demethylation. To evaluate the photocatalytic performance of MOF-OH, we conducted CO2 reduction experiments without any additional photosensitizer. Remarkably, after 72 hours, the yield of CO reached a high value of 26.8 mmol g−1. Notably, the CO production of MOF-OH was significantly higher than that of MOF-OCH3, possibly due to the presence of phenolic hydroxyl substituents, which led to higher local proton concentrations. Furthermore, MOF-OH exhibited excellent stability, as demonstrated by the consistent CO production observed during four consecutive runs of CO2 reduction. To gain insights into the photocatalytic CO2 reduction process, we conducted a comprehensive series of characterizations and density functional theory calculations, which provided a deeper understanding of the mechanism involved.
Introduction
The conversion of CO2 into high-energy C1 products has the potential to reduce atmospheric CO2 levels while enabling sustainable strategies for obtaining valuable chemicals such as CO, HCOOH, CH3OH, and CH4.1–3 One promising pathway for this conversion is the photochemical reduction of CO2, which utilizes light instead of electricity. Sunlight, an abundant and free energy source, makes this approach particularly attractive.4,5 Researchers have dedicated considerable efforts to exploring photocatalysts capable of capturing and reducing CO2. In the past few decades, various homogeneous catalysts, including transition metal complexes,6 ionic liquids,7 and organocatalysts,8 have been employed for CO2 reduction. However, challenges related to catalyst recovery and product separation need to be overcome. Heterogeneous catalysts, such as TiO2-based systems9 and metal-doped zeolites,10 have been developed, but their practical applications were limited by the high cost of noble metal catalysts. CO2 can be reduced to CO through a two-electron and two-proton transfer reaction.11–13 However, CO2 is a highly stable molecule as it represents the most oxidized state of carbon, requiring considerable energy input for its transformation.14 Comparatively, photocatalytic systems based on non-precious metal photosensitizers and catalysts hold promise for large-scale applications. Therefore, it is particularly attractive to explore efficient and highly selective photocatalytic systems for CO2 reduction.15,16Homogeneous catalysts, such as iron tetraphenylporphyrin and its derivatives, have been used for CO2 photoreduction and have demonstrated high catalytic activities and selectivities.17,18 This can be attributed to the favourable binding of metal active sites with CO2 and the homogeneity of the photocatalytic systems.19 Recent studies have shown that hydroxyl functionalization of the macrocyclic benzene ring greatly enhances the selectivity and redox catalytic activity for CO2 reduction to CO.20–22 The hydroxyl substituents in the second coordination sphere near the catalytic centres can increase the local proton concentrations, thereby accelerate catalysis. However, homogeneous catalysts often face limitations in terms of their solubility in organic media and interfacial electron/mass transfer. In recent years, metal–organic frameworks (MOFs) have gained increasing attention as photocatalysts due to their high porosity, chemical stability, and synthetic tunability.23–29 Especially, zirconium-based MOFs can be custom-designed for specific applications under harsh conditions,30 making them potential platforms for the construction of site-isolated light-absorbing entities and single-site CO2 reduction catalysts.23,28 In this context, the immobilization of hydroxyl-modified porphyrinic fragments into Zr-MOF skeletons appears to be a judicious choice for the construction of potential photocatalysts for CO2 reduction.31,32
While the search for reusable and stable catalysts for visible light-promoted CO2 reduction continues, there is a growing gap between design and preparation that represents a critical limitation. In this study, we presented a couple of structural models in which molecular catalysts are successfully incorporated into Zr-MOFs for photocatalytic CO2 reduction. One such model is an iron porphyrin-based MOF with phenolic hydroxyl modification, called MOF-OH (Zr6O4(OH)4(FeTCBPP-OH)3, FeTCBPP-OH = iron 5,10,15,20-tetrakis[4-(4′-carboxyphenyl)-2,6-dihydroxylphenyl]porphyrin). MOF-OH can be obtained through the post-synthetic modification of MOF-OCH3 (Zr6O4(OH)4(FeTCBPP-OCH3)3, FeTCBPP-OCH3 = iron 5,10,15,20-tetrakis[4-(4′-carboxyphenyl)-2,6-dimethoxyphenyl]porphyrin). Direct synthesis of MOF-OH through the assembly of precursors was not possible due to the higher binding affinity of hydroxyl groups on the polyphenolic porphyrinic fragments towards zirconium centers.33,34 The resulting 3D microporous MOF-OH, with numerous local proton sources, was employed as a heterogeneous photocatalyst, demonstrating effective integration of CO2 capture and reduction under visible-light irradiation. For comparison, photocatalytic CO2 reduction of MOF-OCH3 was also explored to elucidate the role of hydroxyl groups attached to the skeletons between MOF-OH and MOF-OCH3. Furthermore, a comprehensive series of experimental characterizations and density functional theory (DFT) calculations were conducted to propose a mechanism for CO2 photoreduction.
Results and discussion
Syntheses and characterization
The solvothermal reaction of FeTCBPP-OCH3, ZrCl4, and benzoic acid in N,N-diethylformamide (DEF) at 120 °C for 48 h yielded dark purple cubic crystals with the framework formula of [Zr6O4(OH)4(FeTCBPP-OCH3)3] (noted as MOF-OCH3, Fig. 1). Furthermore, the direct assembly of the Zr4+ centres and the FeTCBPP-OH fragments under similar conditions was unsuccessful. Fortunately, MOF-OH ([Zr6O4(OH)4(FeTCBPP-OH)3]) was achieved by post-synthetic modification of MOF-OCH3 with demethylation reaction. There are two types of cages constructed by Zr6O4(OH)4 clusters (Zr6, Fig. S1a, ESI†) and FeTCBPP-OCH3 fragments (Fig. S1b, ESI†) in MOF-OCH3: (i) large cubic cages (Fig. S1c, ESI†), in which each vertex is occupied by a typical 12-connected Zr6 cluster, and each face is a 4-connected FeTCBPP-OCH3, and (ii) small cages (Fig. S1d, ESI†), which consist of two Zr6 clusters on the axial sites and four FeTCBPP-OCH3 on the longitude positions. Thus, the expected (4,12)-connected ftw topology is formed when the Zr6 units are considered as 12-c nodes and the organic linkers as 4-c nodes. Furthermore, the space of an individual net is large enough to result in a two-fold interpenetration in MOF-OCH3 (Fig. S1e and f, ESI†). MOF-OH is conformationally identical to MOF-OCH3, except that part of the methoxy groups of the FeTCBPP-OCH3 fragments was demethylated in MOF-OH. After demethylation, the microenvironment was changed, resulting in a couple of structural models with the introduction of iron tetraphenylporphyrin fragments into Zr-MOFs for a potential study on photocatalytic CO2 reduction.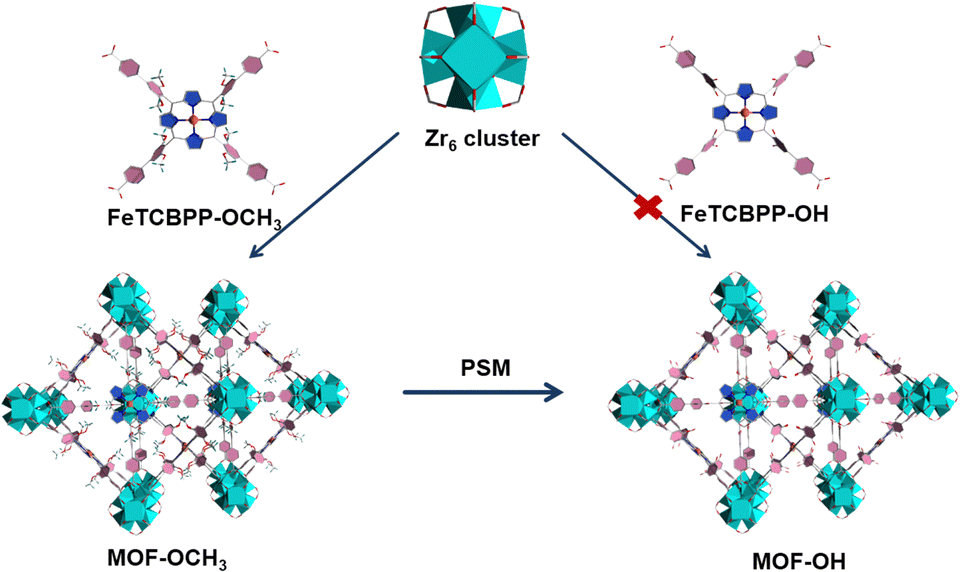 | ||
| Fig. 1 Schematic diagram for the preparation of MOF-OCH3 and MOF-OH. Colour codes: ZrO8 polyhedron, cyan polyhedra; C, gray; O, red; N, blue; Fe, orange; H, olive or omitted for clarity. | ||
After the analysis of mass spectra and 1H NMR spectra of MOF-OH and MOF-OCH3 (Fig. S2 and S3, ESI†), it was observed that FeTCBPP-OCH3 was partially converted to FeTCBPP-OH in MOF-OH. According to quantitative calculations from 1H NMR results, approximately 62.5% of the methoxy groups in MOF-OCH3 were converted to hydroxyl groups. The powder X-ray diffraction (PXRD) patterns of MOF-OCH3 and MOF-OH matched well with the simulated ones, which confirmed the purities of these two MOFs (Fig. 2a). The porosities of these MOFs were confirmed by N2 adsorption at 77 K (Fig. 2b). MOF-OCH3 and MOF-OH exhibited type I isotherms, due to the micropore nature of the interpenetrated structures. The Brunauer–Emmett–Teller (BET) surface areas were 1613 and 493 m2 g−1 for MOF-OCH3 and MOF-OH, respectively. The pore size and BET surface area of MOF-OH were reduced after the post-synthetic modification (Fig. S4, ESI†), which could be attributed to capillary-force-driven pore collapsing during desorption.35,36 This result can be assigned to the preparation conditions of MOF-OH, which was synthesized from MOF-OCH3 through BBr3 cleavage ether reaction and post-treatment with water. Thermal-gravimetric analysis (TGA) confirmed that both MOF-OH and MOF-OCH3 exhibited excellent thermal stabilities, and no obvious weight loss was observed until 400 °C (Fig. S5, ESI†).
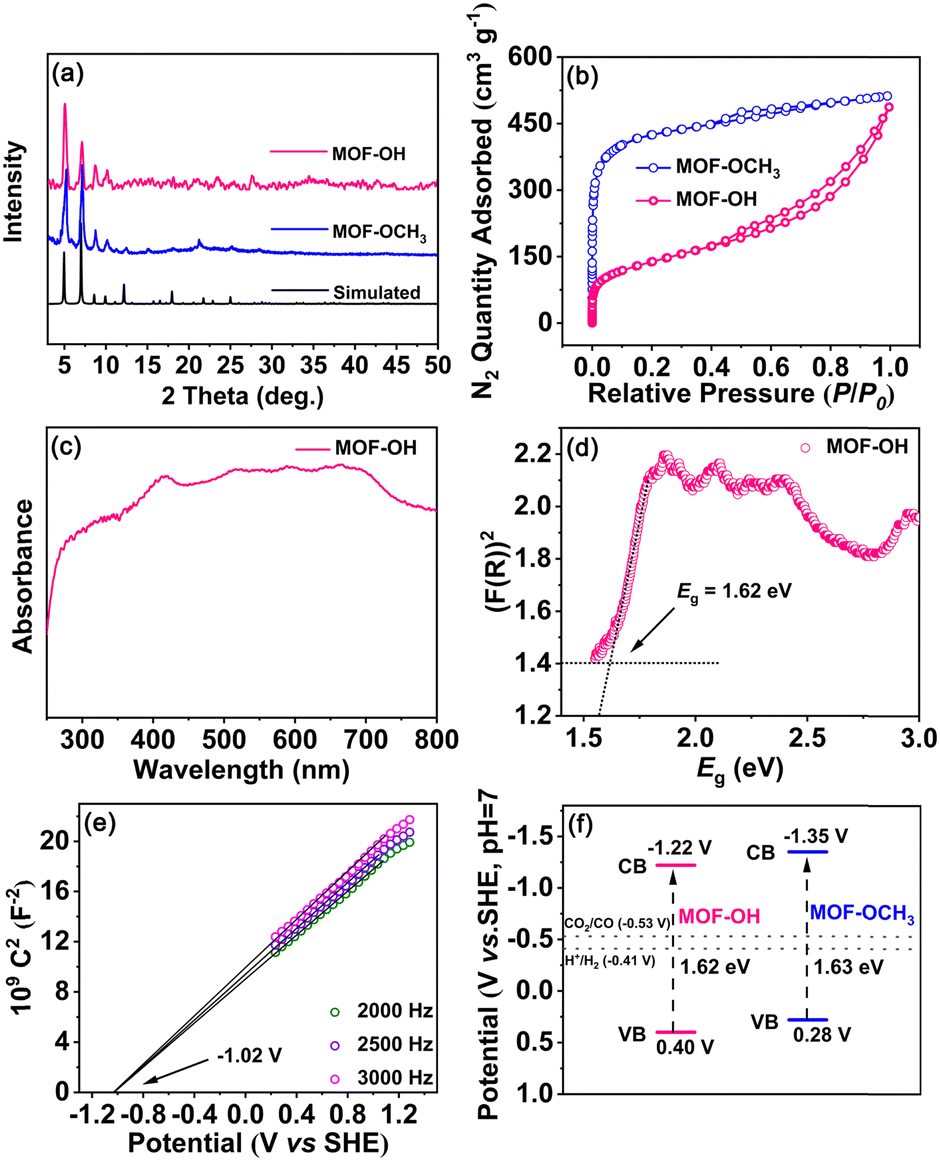 | ||
| Fig. 2 (a) PXRD patterns and (b) N2 isotherms at 77 K of MOF-OH and MOF-OCH3, respectively. (c) UV-vis spectrum and (d) diffuse reflectance spectrum via K–M transformation of MOF-OH, (e) Mott–Schottky plots of the as-prepared MOF-OH electrode, and (f) calculated redox potentials of MOF-OH and MOF-OCH3, respectively. | ||
Thermodynamic feasibility for CO2 reduction
In the UV-vis spectra of MOF-OH and MOF-OCH3 (Fig. 2c and Fig. S6, ESI†), a broad and strong absorption was observed in the range from 250 to 800 nm, which inherits the feature of the porphyrin fragments. These results perhaps allowed for promoting the electrons of MOF-OH and MOF-OCH3 to an excited state upon visible-light irradiation.37 The band gap (Eg) was tested by fitting the Tauc function to the K–M absorbance above the Urbach tail.38 Accordingly, the Eg values of MOF-OH and MOF-OCH3 can be estimated to ∼1.62 and ∼1.63 eV (Fig. 2d and Fig. S7, ESI†), indicating the semiconductivities of these two materials. In addition, Mott–Schottky measurements of MOF-OH and MOF-OCH3 were conducted at frequencies of 2000, 2500, and 3000 Hz (Fig. 2e and Fig. S8, ESI†), respectively. Furthermore, the conduction-band (CB) position was estimated by measuring the flatband potential (Efb) via Mott–Schottky plots. The positive slope of the plots indicates the n-type semiconductors of MOF-OH and MOF-OCH3,39 and Efb were evaluated to be approx. −1.02 and −1.15 V vs. SHE, respectively. Since Efb of n-type semiconductors was considered to be ∼0.2 V more negative than the CB minima,40 the CB of MOF-OH and MOF-OCH3 were estimated to be approx. −1.22 and −1.35 V vs. SHE (Fig. 2f), which is more negative than the redox potentials of CO/CO2 (−0.53 V vs. SHE, pH 7).41 Thus, MOF-OH and MOF-OCH3 possess strong thermodynamic driving forces to realize CO2 reduction.Catalytic performance in overall CO2 photoreduction
In light of the good semiconductor characteristics and excellent stabilities of Zr-MOFs, MOF-OH and MOF-OCH3 were utilized as photocatalysts for CO2 reduction. Photocatalytic reduction of CO2 over MOF-OH was conducted with 0.025 M BIH and 3 vol% 2,2,2-trifluoroethanol (TFE) in a 5 mL acetonitrile (MeCN) as solvent under an LED lamp irradiation (λ = 420 nm). Remarkably, MOF-OH exhibited significant photocatalytic CO2 reduction activity. The yield of CO increased with time and was up to 26.8 mmol g−1 at 72 h (Fig. 3a), which was much higher than those of the homogeneous molecular systems.42 In addition, only a minor amount of H2 was observed by gas chromatography. No other products could be detected in either the gas or liquid phases, suggesting that MOF-OH possessed a high CO selectivity of 93% toward the CO2 conversion. As a comparison, the performance of MOF-OCH3 was also carried out under the same conditions. The photocatalytic activity of MOF-OH was much higher than that of MOF-OCH3, which suggested the critical role of the phenol substituents on the phenyl rings in orienting the photocatalysis towards CO2 reduction. Furthermore, no CO was detected without MOF-OH (Fig. 3b), demonstrating the crucial role in the reaction. In the absence of BIH, no CO was produced, proving that BIH was essential as a sacrificial electron donor for CO2 reduction.43 A decrease in CO production was observed without TFE, suggesting that proper weak Brønsted acids significantly improved the catalytic process.44,45 Moreover, no CO was produced when the parallel reaction was carried out in the dark, revealing a truly photocatalytic behaviour. To confirm the origin of CO, Ar as a reactant was added to the reaction system instead of CO2 under a similar condition. However, no CO was detected.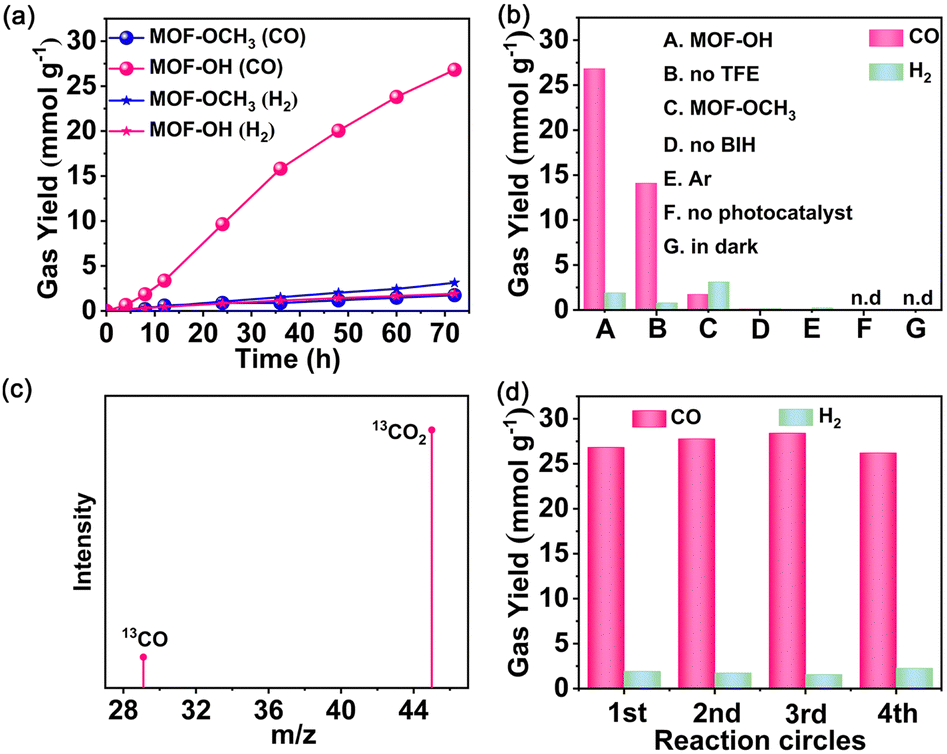 | ||
| Fig. 3 Visible-light-driven CO2 reduction performance of MOF-OH and MOF-OCH3: (a) time-dependent CO/H2 yields over MOF-OH (pink) and MOF-OCH3 (blue), (b) the yields of CO/H2 under different conditions, (c) mass spectrum of produced 13CO via isotope 13CO2 reduction under visible light, and (d) the yields of CO/H2 over MOF-OH of four runs. | ||
In addition, isotope labelling experiments were performed by using 13CO2 under the same conditions (Fig. 3c). A clear signal at an m/z ratio of 29 was observed that corresponding to 13CO, which verified the source of CO. No obvious change in CO production occurred during the four runs, demonstrated the stability of MOF-OH (Fig. 3d). The PXRD patterns further manifested that the skeleton of MOF-OH was retained after CO2 reduction (Fig. S9, ESI†).
Mechanism study
The charge separation efficiency is considered to be of paramount importance for determining the photocatalytic activity.46 The photoelectrochemical properties were performed with the combination of transient photocurrent measurements and electrochemical impedance spectroscopy (EIS). Both MOF-OCH3 and MOF-OH exhibited obvious photocurrent signals and demonstrated excellent reproducibility of the response intensity throughout the on–off cycles (Fig. S10a, ESI†). However, the transient photocurrent intensity of MOF-OH is ∼3 times that of MOF-OCH3. In addition, the EIS spectra revealed that MOF-OH exhibited a smaller semicircle radius compared to MOF-OCH3 (Fig. S10b, ESI†). These results indicated the presence of a higher separation and transfer efficiency of charge carriers in MOF-OH. In addition, photoluminescence (PL) spectra and time-resolved PL spectra were analysed to investigate the charge migration dynamics. The fluorescence intensity of MOF-OH was lower than that of MOF-OCH3 (Fig. S11a, ESI†), indicating that the charge recombination was suppressed in MOF-OH. The average lifetime of MOF-OH (2.32 ns) was shorter compared to MOF-OCH3 (4.46 ns) (Fig. S11b, ESI†), suggesting that the efficient electron–hole pairs separation in MOF-OH and therefore more favourable for photocatalytic CO2 reduction.47,48To further investigate the rapid charge separation of photocatalysts, we recorded the femtosecond time-resolved transient absorption (fs-TA) spectra that is a powerful tool for tracking carrier dynamics in nanosystems in real time. For the measurements, a pump–probe scheme with visible-light pumping and white-light continuous probing was used. The fs-TA spectra of MOF-OH and MOF-OCH3 were recorded in the wavelength range of 450–750 nm under the excitation with a 400 nm laser pulse (Fig. 4a and b). For MOF-OH, pulsed laser excitation resulted in a bleaching signal in 500–530 nm, which was a ground-state bleaching. The absorption signal in the excited state appeared at 530–750 nm. By probing the absorption intensity at λ = 700 nm after band edge excitation, the kinetic curves for MOF-OH and MOF-OCH3 were shown in Fig. 4c and d. Most importantly, the average lifetime (τave) of MOF-OH (94.0 ± 8.1 ps) was shorter than that of MOF-OCH3 (225.6 ± 15.5 ps). The fast component may be attributed to fast electron transfer from the conduction-band minimum of the MOF to the trap state, suggesting that the accelerated TA decay kinetics suppressed electron–hole binding. This result was also consistent with the transient photocurrent measurements and EIS results.
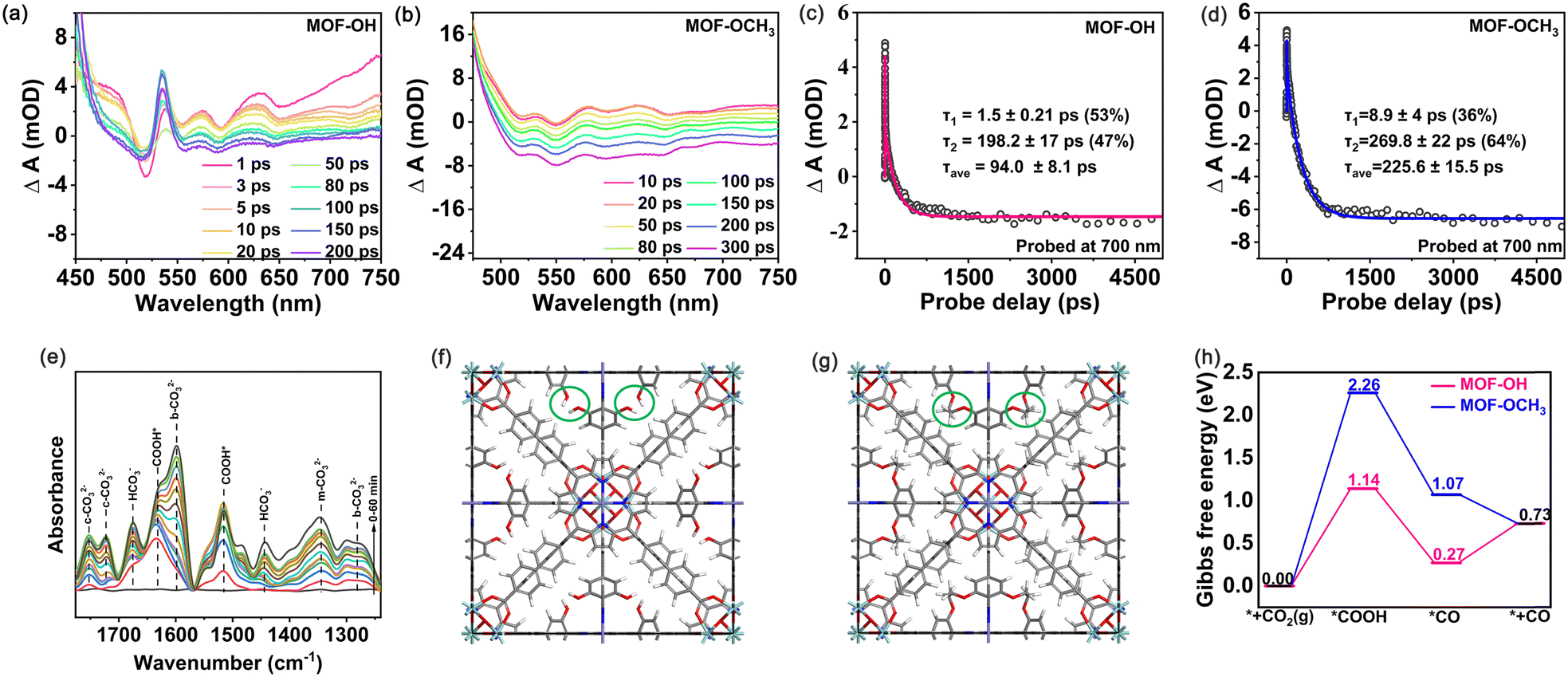 | ||
| Fig. 4 (a) and (b) fs-TA data of MOF-OH and MOF-OCH3 excited at 400 nm showing the temporal changes at different time constants. (c) and (d) Representative TA kinetics of MOF-OH and MOF-OCH3 taken at the probing wavelength of 700 nm. (e) In situ ATR-FTIR measurement using MOF-OH as a catalyst in the presence of CO2 and H2O vapor under visible light irradiation (Xe lamp, λ > 420 nm). Optimized structural models of (f) MOF-OH and (g) MOF-OCH3. The green circles indicated the hydroxy and methoxy. (h) Gibbs free energy diagrams for CO2 reduction on MOF-OH and MOF-OCH3, respectively. | ||
To investigate the conversion pathway and mechanism of CO2 in MOF-OH, the intermediates on the surface of the photocatalysts were analysed by using in situ attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR). As shown in Fig. 4e, the carbonate species, i.e. monodentate carbonate (m-CO32−, at 1345 cm−1), bidentate carbonate (b-CO32−, at 1598 and 1282 cm−1),49 and bicarbonate (c-CO32−, at 1752 and 1721 cm−1) were identified. Additionally, the peaks at 1676 and 1444 cm−1 can be assigned to asymmetric stretching of *HCO3− and symmetric stretching of *HCO3−, respectively.50 The peaks at 1631, 1515 cm−1 emerged, which were attributed to the formation of COOH*,49,51 a critical intermediate during the photoreduction of CO2 to CO.
Furthermore, DFT calculations were carried out to investigate the mechanism of CO2 reduction to CO over MOF-OH and MOF-OCH3 (computational details can be found in the ESI†). The fully optimized geometric models of MOF-OH and MOF-OCH3 were shown in Fig. 4f and g, where their difference of building groups was marked in green circles. The central Fe atom of the porphyrin ring was well-accepted to be the active site for CO2 reduction, which was consistent with previous reports.52,53 The second coordination spheres of polyhydroxy groups offer a high local concentration of protons, which enhanced the photocatalytic efficiencies in terms of both rate constant and catalytic selectivity. To further understand the better catalytic performance of MOF-OH than that of MOF-OCH3, the Gibbs free energy diagrams for CO2 reduction to CO were systematically considered and explored (Fig. 4h). The CO2 reduction starts with forming *C–Fe bonding at the active site and the subsequent proton-coupled electron transfer process to produce *COOH intermediate. The computed ΔG values for this elementary reaction on MOF-OH and MOF-OCH3 were 1.14 and 2.26 eV, respectively. Subsequently, the *COOH intermediate was further hydrogenated to generate *CO intermediate. This process is exothermic in the Gibbs free energy diagram. The formed *CO intermediate on MOF-OH can easily release the CO gas by overcoming a small barrier by 0.46 eV. These results confirmed the better catalytic activity of MOF-OH, which further explained the much higher CO yield of MOF-OH than that of MOF-OCH3.
On the basis of the above investigations, we proposed the mechanism of overall CO2 photoreduction over MOF-OH (Scheme 1). CO2 was first complexed with the triply reduced Fe0 species (forming FeIICO2), which after protonation, produced a FeIICO intermediate, which is stabilized by internal hydrogen bonding.54,55 The release of CO requires the breaking of the C–O bond and therefore an additional electron. In addition, it was noted that the electron-rich Fe0 species can react with H+ to generate FeII and the unrequired byproduct H2, although it remained a minor pathway in optimized conditions. On the other hand, a high concentration of Fe0 active species in solution that would ensue from highly efficient electron transfers from the sensitizer can also support H2 evolution.
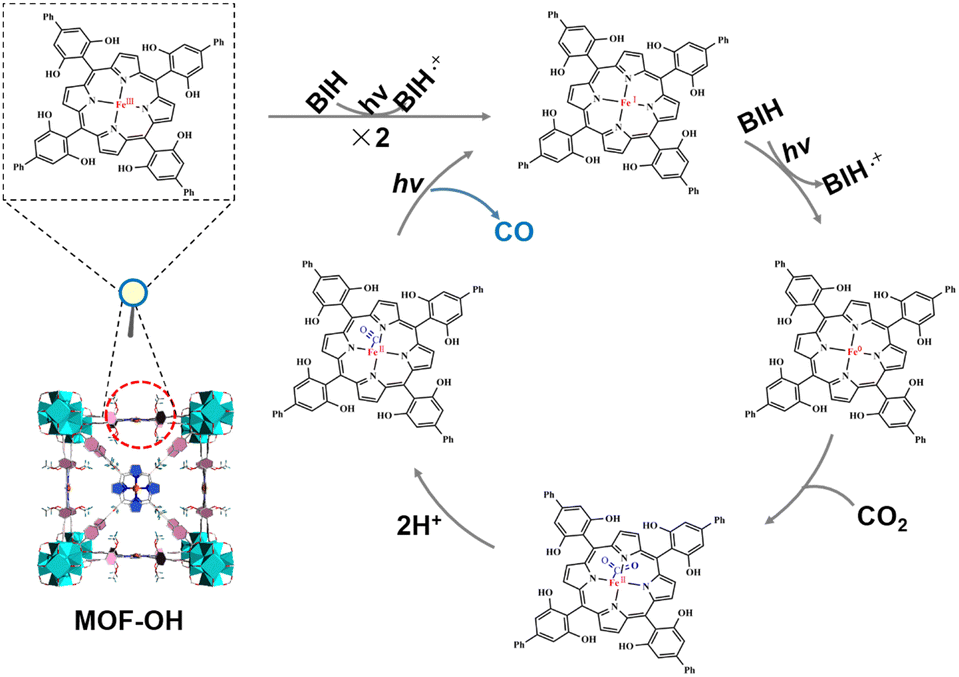 | ||
| Scheme 1 Proposed mechanism for the overall CO2 photoreduction of MOF-OH. | ||
Conclusions
In conclusion, we achieved the production of MOF-OH through a demethylation-driven post-synthetic modification process. This involved modifying the methoxy groups of the FeTCBPP-OCH3 fragments in MOF-OCH3. The yield of CO reached an impressive 26.8 mmol g−1 after 72 hours using MOF-OH as the photocatalyst for CO2 reduction, even without any photosensitizer. This yield was significantly higher than that achieved with MOF-OCH3. Comprehensive studies on the kinetic mechanism revealed that the exceptional photocatalytic performance of MOF-OH can be primarily attributed to the iron porphyrin ligand with phenolic hydroxyl groups. These groups successfully immobilized the catalytic centre within the MOFs, facilitating ultrafast electron migration and efficient charge separation. Furthermore, the introduction of phenolic hydroxyl groups resulted in a high concentration of local protons, greatly enhancing the photocatalytic efficiencies. Additionally, DFT calculations demonstrated that MOF-OH can easily release CO gas by overcoming a small potential barrier in the formed *CO intermediate, compared to MOF-OCH3. This work not only provides a solid foundation for the accurate design of MOF photocatalysts at the molecular level but also presents successful MOF models that can be used to investigate the relationship between structures and photocatalytic performances. These findings strongly emphasize the need for further studies to explore the multifunctional coordination sphere of MOF photocatalysts. Moreover, ongoing investigations in our laboratory aim to delve into the design and performance of the pore microenvironment.Author contributions
Xue Zhao: validation, formal analysis, investigation, data curation, writing – original draft. Chang-Yan Zhu: data curation, writing – original draft. Jun-Sheng Qin: conceptualization, funding acquisition, project administration. Heng Rao: conceptualization, funding acquisition. Dong-Ying Du: conceptualization, validation, writing – review & editing. Min Zhang: data curation, conceptualization. Ping She: conceptualization, funding acquisition. Li Li: conceptualization. Zhong-Min Su: project administration.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported in part by the National Natural Science Foundation of China (Grant No. 21901084, 21905106, 22301099 and 22279041), the Natural Science Foundation of Jilin Province (No. SKL202302017), the National Key Research and Development Program of China (No. 2022YFC2105800) and the 111 Project (B17020).References
- T. Kong, Y. Jiang and Y. Xiong, Photocatalytic CO2 conversion: What can we learn from conventional COx hydrogenation?, Chem. Soc. Rev., 2020, 49, 6579–6591 RSC
.
- T. Nakajima, Y. Tamaki, K. Ueno, E. Kato, T. Nishikawa, K. Ohkubo, Y. Yamazaki, T. Morimoto and O. Ishitani, Photocatalytic Reduction of Low Concentration of CO2, J. Am. Chem. Soc., 2016, 138, 13818–13821 CrossRef CAS PubMed
- J. S. Qin, S. Yuan, L. Zhang, B. Li, D. Y. Du, N. Huang, W. Guan, H. F. Drake, J. Pang, Y. Q. Lan, A. Alsalme and H. C. Zhou, Creating Well-Defined Hexabenzocoronene in Zirconium Metal-Organic Framework by Postsynthetic Annulation, J. Am. Chem. Soc., 2019, 141, 2054–2060 CrossRef CAS PubMed
- J. Tian, R. Han, Q. Guo, Z. Zhao and N. Sha, Direct Conversion of CO2 into Hydrocarbon Solar Fuels by a Synergistic Photothermal Catalysis, Catalysts, 2022, 12, 612 CrossRef CAS
- K. Li, X. An, K. H. Park, M. Khraisheh and J. Tang, A critical review of CO2 photoconversion: Catalysts and reactors, Catal. Today, 2014, 224, 3–12 CrossRef CAS
- H. Yuan, B. Cheng, J. Lei, L. Jiang and Z. Han, Promoting photocatalytic CO2 reduction with a molecular copper purpurin chromophore, Nat. Commun., 2021, 12, 1835 CrossRef CAS PubMed
- F. D. Bobbink and P. J. Dyson, Synthesis of carbonates and related compounds incorporating CO2 using ionic liquid-type catalysts: State-of-the-art and beyond, J. Catal., 2016, 343, 52–61 CrossRef CAS
- S.-H. Wang, F. Khurshid, P.-Z. Chen, Y.-R. Lai, C.-W. Cai, P.-W. Chung, M. Hayashi, R.-J. Jeng, S.-P. Rwei and L. Wang, Solution-Processable Naphthalene Diimide-Based Conjugated Polymers as Organocatalysts for Photocatalytic CO2 Reaction with Extremely Stable Catalytic Activity for Over 330 Hours, Chem. Mater., 2022, 34, 4955–4963 CrossRef CAS
- S. Kreft, D. Wei, H. Junge and M. Beller, Recent advances on TiO2-based photocatalytic CO2 reduction, EnergyChem, 2020, 2, 100044 CrossRef
- Ş. Neaţu, J. Maciá-Agulló and H. Garcia, Solar Light Photocatalytic CO2 Reduction: General Considerations and Selected Bench-Mark Photocatalysts, Int. J. Mol. Sci., 2014, 15, 5246–5262 CrossRef PubMed
- A. Muthuperiyanayagam, A. G. Nabi, Q. Zhao, R. Aman Ur and D. D. Tommaso, Adsorption, activation, and conversion of carbon dioxide on small copper-tin nanoclusters, Phys. Chem. Chem. Phys., 2023, 25, 13429–13441 RSC
- A. Vidal-López, S. Posada-Pérez, M. Solà and A. Poater, Au Single Metal Atom for Carbon Dioxide Reduction Reaction, Chemistry, 2023, 5, 1395–1406 CrossRef
- W. Deng, P. Zhang, B. Seger and J. Gong, Unraveling the rate-limiting step of two-electron transfer electrochemical reduction of carbon dioxide, Nat. Commun., 2022, 13, 803 CrossRef CAS PubMed
- J. Kim and E. E. Kwon, Photoconversion of carbon dioxide into fuels using semiconductors, J. CO2 Util., 2019, 33, 72–82 CrossRef CAS
- J. W. Wang, F. Ma, T. Jin, P. He, Z. M. Luo, S. Kupfer, M. Karnahl, F. Zhao, Z. Xu, T. Jin, T. Lian, Y. L. Huang, L. Jiang, L. Z. Fu, G. Ouyang and X. Y. Yi, Homoleptic Al(III) Photosensitizers for Durable CO2 Photoreduction, J. Am. Chem. Soc., 2023, 145, 676–688 CrossRef CAS PubMed
- H. Takeda, K. Ohashi, A. Sekine and O. Ishitani, Photocatalytic CO2 Reduction Using Cu(I) Photosensitizers with a Fe(II) Catalyst, J. Am. Chem. Soc., 2016, 138, 4354–4357 CrossRef CAS PubMed
- H. Rao, C. H. Lim, J. Bonin, G. M. Miyake and M. Robert, Visible-Light-Driven Conversion of CO2 to CH4 with an Organic Sensitizer and an Iron Porphyrin Catalyst, J. Am. Chem. Soc., 2018, 140, 17830–17834 CrossRef CAS PubMed
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Visible-light-driven methane formation from CO2 with a molecular iron catalyst, Nature, 2017, 548, 74–77 CrossRef CAS PubMed
- R. Wan, D. G. Ha, J. H. Dou, W. S. Lee, T. Chen, J. J. Oppenheim, J. Li, W. A. Tisdale and M. Dinca, Dipole-mediated exciton management strategy enabled by reticular chemistry, Chem. Sci., 2022, 13, 10792–10797 RSC
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst, Science, 2012, 338, 90–94 CrossRef CAS PubMed
- C. Costentin, G. Passard, M. Robert and J. M. Saveant, Pendant acid-base groups in molecular catalysts: H-bond promoters or proton relays? Mechanisms of the conversion of CO2 to CO by electrogenerated iron(0)porphyrins bearing prepositioned phenol functionalities, J. Am. Chem. Soc., 2014, 136, 11821–11829 CrossRef CAS PubMed
- C. Costentin, G. Passard, M. Robert and J. M. Saveant, Ultraefficient homogeneous catalyst for the CO2-to-CO electrochemical conversion, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14990–14994 CrossRef CAS PubMed
- Z. B. Fang, T. T. Liu, J. Liu, S. Jin, X. P. Wu, X. Q. Gong, K. Wang, Q. Yin, T. F. Liu, R. Cao and H. C. Zhou, Boosting Interfacial Charge-Transfer Kinetics for Efficient Overall CO2 Photoreduction via Rational Design of Coordination Spheres on Metal-Organic Frameworks, J. Am. Chem. Soc., 2020, 142, 12515–12523 CrossRef CAS PubMed
- X.-H. Liang, A.-X. Yu, X.-J. Bo, D.-Y. Du and Z.-M. Su, Metal/covalent-organic frameworks-based electrochemical sensors for the detection of ascorbic acid, dopamine and uric acid, Coord. Chem. Rev., 2023, 497, 215427 CrossRef CAS
- G. Y. Qiao, D. Guan, S. Yuan, H. Rao, X. Chen, J. A. Wang, J. S. Qin, J. J. Xu and J. Yu, Perovskite Quantum Dots Encapsulated in a Mesoporous Metal-Organic Framework as Synergistic Photocathode Materials, J. Am. Chem. Soc., 2021, 143, 14253–14260 CrossRef CAS PubMed
- S. Kampouri, M. Zhang, T. Chen, J. J. Oppenheim, A. C. Brown, M. T. Payne, J. L. Andrews, J. Sun and M. Dinca, Pyrogallate-Based Metal-Organic Framework with a Two-Dimensional Secondary Building Unit, Angew. Chem., Int. Ed., 2022, 61, e202213960 CrossRef CAS PubMed
- A. Iliescu, J. L. Andrews, J. J. Oppenheim and M. Dinca, A Solid Zn-Ion Conductor from an All-Zinc Metal-Organic Framework Replete with Mobile Zn2+ Cations, J. Am. Chem. Soc., 2023, 145, 25962–25965 CrossRef CAS PubMed
- H. Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S. H. Yu and H. L. Jiang, Visible-Light Photoreduction of CO2 in a Metal-Organic Framework: Boosting Electron-Hole Separation via Electron Trap States, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed
- L. Feng, J. Pang, P. She, J. L. Li, J. S. Qin, D. Y. Du and H. C. Zhou, Metal-Organic Frameworks Based on Group 3 and 4 Metals, Adv. Mater., 2020, 32, e2004414 CrossRef PubMed
- G. Y. Qiao, S. Yuan, J. Pang, H. Rao, C. T. Lollar, D. Dang, J. S. Qin, H. C. Zhou and J. Yu, Functionalization of Zirconium-Based Metal-Organic Layers with Tailored Pore Environments for Heterogeneous Catalysis, Angew. Chem., Int. Ed., 2020, 59, 18224–18228 CrossRef CAS PubMed
- M. E. Kosal, J.-H. Chou, S. R. Wilson and K. S. Suslick, A functional zeolite analogue assembled from metalloporphyrins, Nat. Mater., 2002, 1, 118–121 CrossRef CAS PubMed
- D. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. Wei and H. C. Zhou, Zirconium-metalloporphyrin PCN-222: mesoporous metal-organic frameworks with ultrahigh stability as biomimetic catalysts, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed
- J. Aguilera-Sigalat, A. Fox-Charles and D. Bradshaw, Direct photo-hydroxylation of the Zr-based framework UiO-66, Chem. Commun., 2014, 50, 15453–15456 RSC
- F. M. Al-Sogair, B. P. Operschall, A. Sigel, H. Sigel, J. Schnabl and R. K. Sigel, Probing the metal-ion-binding strength of the hydroxyl group, Chem. Rev., 2011, 111, 4964–5003 CrossRef CAS PubMed
- J. E. Mondloch, M. J. Katz, N. Planas, D. Semrouni, L. Gagliardi, J. T. Hupp and O. K. Farha, Are Zr6-based MOFs water stable? Linker hydrolysis vs. capillary-force-driven channel collapse, Chem. Commun., 2014, 50, 8944–8946 RSC
- T. Y. Luo, S. Park, T. H. Chen, Prerna, R. Patel, X. Li, J. Ilja Siepmann, S. Caratzoulas, Z. Xia and M. Tsapatsis, Simultaneously Enhanced Hydrophilicity and Stability of a Metal-Organic Framework via Post-Synthetic Modification for Water Vapor Sorption/Desorption, Angew. Chem., Int. Ed., 2022, 61, e202209034 CrossRef CAS PubMed
- J. Jin, Porphyrin-based metal–organic framework catalysts for photoreduction of CO2: understanding the effect of node connectivity and linker metalation on activity, New J. Chem., 2020, 44, 15362–15368 RSC
- N. Sharma, K. Prabakar, S. Ilango, S. Dash and A. K. Tyagi, Optical band-gap and associated Urbach energy tails in defected AlN thin films grown by ion beam sputter deposition: Effect of assisted ion energy, Adv. Mater. Proccess., 2021, 2, 342–346 CrossRef
- Y. Gao, J. Duan, X. Zhai, F. Guan, X. Wang, J. Zhang and B. Hou, Photocatalytic Degradation and Antibacterial Properties of Fe3+-Doped Alkalized Carbon Nitride, Nanomaterials, 2020, 10, 1751 CrossRef CAS PubMed
- L. Shi, P. Li, W. Zhou, T. Wang, K. Chang, H. Zhang, T. Kako, G. Liu and J. Ye, n-type boron phosphide as a highly stable, metal-free, visible-light-active photocatalyst for hydrogen evolution, Nano Energy, 2016, 28, 158–163 CrossRef CAS
- Y. Cao, Q. Chen, C. Shen and L. He, Polyoxometalate-Based Catalysts for CO2 Conversion, Molecules, 2019, 24, 2069 CrossRef CAS PubMed
- J. Bonin, M. Robert and M. Routier, Selective and Efficient Photocatalytic CO2 Reduction to CO Using Visible Light and an Iron-Based Homogeneous Catalyst, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS PubMed
- S. Choi, W. J. Jung, K. Park, S. Y. Kim, J. O. Baeg, C. H. Kim, H. J. Son, C. Pac and S. O. Kang, Rapid Exciton Migration and Amplified Funneling Effects of Multi-Porphyrin Arrays in a Re(I)/Porphyrinic MOF Hybrid for Photocatalytic CO2 Reduction, ACS Appl. Mater. Interfaces, 2021, 13, 2710–2722 CrossRef CAS PubMed
- Y. Kuramochi and A. Satake, Photocatalytic CO2 Reductions Catalyzed by meso-(1,10-Phenanthrolin-2-yl)-Porphyrins Having a Rhenium(I) Tricarbonyl Complex, Chem. – Eur. J., 2020, 26, 16365–16373 CrossRef CAS PubMed
- M. Lupa-Myszkowska, M. Oszajca and D. Matoga, From non-conductive MOF to proton-conducting metal-HOFs: a new class of reversible transformations induced by solvent-free mechanochemistry, Chem. Sci., 2023, 14, 14176–14181 RSC
- J. Gu, Q. Huang, Y. Yuan, K.-H. Ye, Z. Wang and W. Mai, In situ growth of a TiO2 layer on a flexible Ti substrate targeting the interface recombination issue of BiVO4 photoanodes for efficient solar water splitting, J. Mater. Chem. A, 2017, 5, 20195–20201 RSC
- X. Gao, B. Guo, C. Guo, Q. Meng, J. Liang and J. Liu, Zirconium-Based Metal-Organic Framework for Efficient Photocatalytic Reduction of CO2 to CO: The Influence of Doped Metal Ions, ACS Appl. Mater. Interfaces, 2020, 12, 24059–24065 CrossRef CAS PubMed
- R. Hariri and S. Dehghanpour, Effective visible-light CO2 photoreduction over (metallo)porphyrin-based metal-organic frameworks to achieve useful hydrocarbons, Appl. Organomet. Chem., 2021, 35, e6422 CrossRef CAS
- J. Zhou, J. Li, L. Kan, L. Zhang, Q. Huang, Y. Yan, Y. Chen, J. Liu, S. L. Li and Y. Q. Lan, Linking oxidative and reductive clusters to prepare crystalline porous catalysts for photocatalytic CO2 reduction with H2O, Nat. Commun., 2022, 13, 4681 CrossRef CAS PubMed
- L. Ran, Z. Li, B. Ran, J. Cao, Y. Zhao, T. Shao, Y. Song, M. K. H. Leung, L. Sun and J. Hou, Engineering Single-Atom Active Sites on Covalent Organic Frameworks for Boosting CO2 Photoreduction, J. Am. Chem. Soc., 2022, 144, 17097–17109 CrossRef CAS PubMed
- Y. He, Y. Zhao, X. Wang, Z. Liu, Y. Yu and L. Li, Multiple Heteroatom-Hydrogen Bonds Bridging Electron Transport in Covalent Organic Framework-Based Supramolecular System for Photoreduction of CO2, Angew. Chem., Int. Ed., 2023, 62, e202307160 CrossRef PubMed
- L. Lin, C. Hou, X. Zhang, Y. Wang, Y. Chen and T. He, Highly efficient visible-light driven photocatalytic reduction of CO2 over g-C3N4 nanosheets/tetra(4-carboxyphenyl)porphyrin iron(III) chloride heterogeneous catalysts, Appl. Catal., B, 2018, 221, 312–319 CrossRef CAS
- J. Y. Chen, M. Li and R. Z. Liao, Mechanistic Insights into Photochemical CO2 Reduction to CH4 by a Molecular Iron-Porphyrin Catalyst, Inorg. Chem., 2023, 62, 9400–9417 CrossRef CAS PubMed
- J. Bonin, A. Maurin and M. Robert, Molecular catalysis of the electrochemical and photochemical reduction of CO2 with Fe and Co metal based complexes. Recent advances, Coord. Chem. Rev., 2017, 334, 184–198 CrossRef CAS
- H. Rao, J. Bonin and M. Robert, Non-sensitized selective photochemical reduction of CO2 to CO under visible light with an iron molecular catalyst, Chem. Commun., 2017, 53, 2830–2833 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, computational details and figures. See DOI: https://doi.org/10.1039/d4qm00187g |
| This journal is © the Partner Organisations 2024 |