
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cell dynamics and metabolism of the foreign body response: characterizing host-biomaterial interactions for next-generation medical implant biocompatibility
Neal I.
Callaghan†
 a,
Christian N.
Rempe†
b,
Zachary S. C. S.
Froom
c,
Kyle T.
Medd
c and
Locke
Davenport Huyer
*bcde
a,
Christian N.
Rempe†
b,
Zachary S. C. S.
Froom
c,
Kyle T.
Medd
c and
Locke
Davenport Huyer
*bcde
aFaculty of Medicine, Dalhousie University, Halifax, NS B3H 4R2, Canada
bDepartment of Microbiology & Immunology, Faculty of Medicine, Dalhousie University, Halifax, NS B3H 4R2, Canada. E-mail: l.davenporthuyer@dal.ca
cSchool of Biomedical Engineering, Faculties of Medicine and Engineering, Dalhousie University, Halifax, NS B3H 4R2, Canada
dDepartment of Biomaterials & Applied Oral Sciences, Faculty of Dentistry, Dalhousie University, Halifax, NS B3H 4R2, Canada
eNova Scotia Health, Halifax, NS, Canada
First published on 27th July 2024
Abstract
Implantable medical devices (IMDs) collectively represent a critical mainstay in modern medicine. Used in many chronic diseases and in acute surgical interventions, IMDs are often associated with improvements in disease progression, quality of life, and mortality rates. Despite the positive impacts of IMD implementation, excessive fibrosis driven by the foreign body response (FBR) is frequently associated with the development of complications and failure. These complications in turn result in surgical revisions and removals, which represent a significant burden to healthcare costs and surgical wait-times in countries with elevated IMD usage rates. IMD complications are exacerbated by limitations to treatment options and limited availability of biocompatible materials. Novel treatment development is equally hampered by the complexity of the FBR, wherein complex cellular behaviors defy canonical immunological classification systems. In this review, current understandings of cellular dynamics and kinetics within the FBR are summarized, with a specific focus on the relationship between immunometabolic regulation and pathological fibrotic processes across various cell behaviours in the FBR. This review also explores promising emerging in vitro and in vivo techniques of FBR characterization, and highlights biomaterial properties associated with alterations in FBR outcomes. Finally, this review explores current and future approaches to biocompatible material development, highlighting immune-metabolic control as a therapeutic approach to mitigating the FBR.
1. The foreign body response to implanted materials: burden and clinical implications
Implantable medical devices (IMDs) have broad utility in the medical field, with applications spanning sutures, structural meshes, soft tissue fillers, orthopedic and craniofacial prosthetics, cerebral shunts, vascular stents, valvular prostheses, cardiac and neural stimulators, biosensors, contraceptive devices, and long-term drug eluting devices. For example, the single most common surgical procedure is the repair of primary or incisional ventral hernia, which is a prototypical example of soft tissue implant; current USA caseloads are approximately 611![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per year and with a total cost of $9.7 billion USD yearly.1 When combined with inguinal and femoral hernias, the incidence of medically significant hernia cases remain undertreated, with combined incidence of approximately 13 million cases per year and representing a major source of morbidity, mortality, complications, and recurrence, as well as a significant component of healthcare spending.2,3 With an aging population in the Western hemisphere and increased medical spending, along with continuous surgical innovation and the release of new IMDs, rates of surgical implantation are projected to increase for the foreseeable future.
000 per year and with a total cost of $9.7 billion USD yearly.1 When combined with inguinal and femoral hernias, the incidence of medically significant hernia cases remain undertreated, with combined incidence of approximately 13 million cases per year and representing a major source of morbidity, mortality, complications, and recurrence, as well as a significant component of healthcare spending.2,3 With an aging population in the Western hemisphere and increased medical spending, along with continuous surgical innovation and the release of new IMDs, rates of surgical implantation are projected to increase for the foreseeable future.
Generally, IMDs positively impact recipient quality of life (e.g., hernia repair via a polypropylene surgical mesh), but also carry complication risks.4 Specifically, IMDs are generally subject to some degree of chronic inflammation associated with the foreign body response (FBR) to implanted materials. The FBR is an expected inflammatory reaction, mediated by the immune system, that arises following implantation of a biomaterial within a host organism,5 and that is characterized by the persistence of immune cells and the development of a fibrotic layer encapsulating and isolating the implant.6,7 Based on patient, implant, and environmental factors, the FBR can manifest on a spectrum of severity. The FBR in a successful medical device application may present with no discernable impact on device function or patient quality of life. Conversely, a deleterious FBR can completely impair device function, severely incapacitate patients with pain or reduced quality of life, leads to systemic sequelae, or even threatens patient life altogether.4 This pathology is characterized by a prolonged period of chronic inflammation, and the deposition of fibrotic matrix around the implant to isolate the area from prolonged tissue damage. This damage results from inflammatory molecules secreted by the immune system; generally, the more prolonged the inflammation resulting from implantation, the thicker the eventual fibrosis will be.5 However, the immune system is also a powerful driver of tissue growth and healing, and in the future will likely be directly manipulated and harnessed in regenerative approaches.8
Despite the clinical ubiquity of IMDs and their respective complications, the mechanisms underlying the FBR are poorly understood. It is accepted that primary drivers of the FBR include chronic inflammation and the pathological recruitment of a variety of myeloid cells9 (Fig. 1). These cells drive excess fibrosis and limit tissue integration of the implant.9 Typically, pathological inflammation and fibrosis manifests clinically as capsular contracture, resulting in pain and in extreme cases alteration of an implant's structure; symptoms may require surgical revision or explantation. Additionally, pathological fibrosis can interfere with implant function. For example, fibrotic depositions can alter drug elution rates in insulin delivery devices,10 or implanted electrode function.11–13 Though clear clinical complications are associated with pathological FBR fibrosis, the complement of involved cells, their activators and regulators, and their correlation to the final disposition of the FBR remain undercapitalized. More rare adverse outcomes can include the development of autoimmune diseases (e.g., breast implant illness) and even malignancy; these are generally thought to be result from prolonged local or systemic immunostimulation, but their study is hampered by little direct clinical study or model development.14
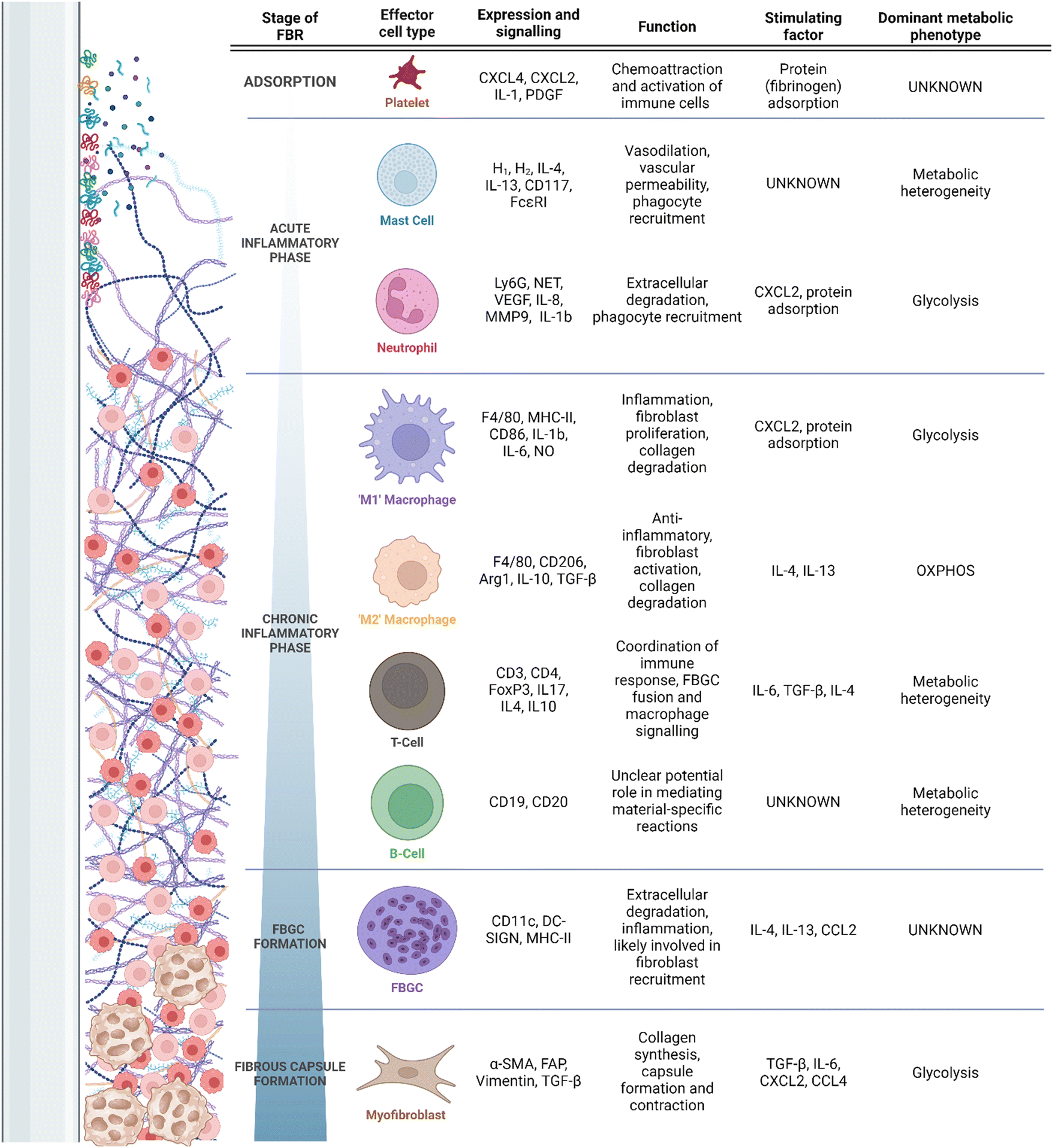 | ||
| Fig. 1 Timeline of the FBR, recruitment of associated cells, and their secretory/metabolic phenotypes. | ||
In this review, we discuss the mechanisms of the FBR, with focus on the dynamics and metabolism of participating cells in the peri-implant environment and in environments out of direct contact with systemic circulation. We review recent literature that elucidates the provenance of these cells, and the checkpoints that mediate inflammatory resolution and implant integration, or alternatively lead to chronic inflammation. Finally, we discuss novel scientific advancements in the techniques that query patient explants and in vivo and in vitro models of the FBR, as well as metabolically-based solutions to the FBR and associated pathology.
2. Physiology of the foreign body response
The most harmonious explanation for the physiological utility of the FBR is twofold: the rapid repair of the wounded tissue including protection from the external environment, and the neutralization and sequestration of both active and latent pathogens introduced with the foreign body. The placement of an IMD generates a wound environment, including significant tissue damage, and thus drives signaling of canonical wound repair processes. In models of incisional skin wound healing (arguably the most studied of tissue wound healing processes), the first week of healing is characterized by a hemostatic plug that allows for inflammatory cell infiltration and sterilization, but imparts minimal tensile strength to the wound closure. This strength greatly increases in the second week, and progresses over the course of further weeks to months to a plateau of ∼80% of normal dermis.15,16 At a fundamental level, the FBR can be considered in the context of the wound healing paradigm, and the mechanistic drive for wound closure.Early phases of wound healing utilize inflammatory pathways to eliminate damaged tissue, and address pathogens in the microenvironment. Pathogen exposure is a minimizable but unavoidable consequence of material implantation, and attributable to both endogenous (patient-colonizing) and exogenous sources (airborne particles in the operating room, films or particles from IMDs, surgical tools, and operators).17 Considering abdominal wall hernia repair meshes as an example, surgical site infection occurrence rates range enormously depending on wound and mesh size, mesh characteristics, surgical technique, environment, prophylaxis, dressing, and active wound management, as well as patient demographic factors.17–25 The development of a fibrous barrier allows for spatial limitation of pathogen growth, as well as the protection of surrounding tissue from the cytotoxic anti-pathogen response from infiltrating immune cells during the FBR.
When considering the utility of implantable devices in surgical techniques, the FBR is critical for implanted materials to achieve stability in tissue and carry out their functional roles. The utility of controlled fibrosis increasing tissue hardness and stiffness is especially crucial in the face of recent medical devices. In the context of hernias, mesh repairs generally outperform non-mesh repairs in rate of primary recurrence,26–28 generally attributable to the increased fibrosis at and around the surgical mesh; this supraphysiological stiffness of the mesh and hypertrophic scar tissue together allow for sufficient integration and tensile strength to ensure patency. Similarly, in silicone breast reconstruction, fibrosis is required for successful tissue integration and implant stability to minimize risk of displacement.4,29,30 Without the FBR, implantable medical devices of all designations would be at risk for dislodgement and loss of function or even active pathology.
However, careful attention to detail is warranted in considering tissue and IMD mechanics. For example, the adaptation of polymer meshes originally designed for use in abdominal wall hernias for gynecological application in pelvic floor dysfunction, specifically in the adoption of transvaginal meshes, has been met with significant rates of complications.31 These generally result from both differential elasticity under deformation between native tissue and the implanted material. As implantable meshes plastically deform, their interaction with softer surrounding native tissues often leads to erosion, which is often complicated by chronic pain, infection, dyspareunia, and autoexplantation or extrusion leading to perforation of surrounding organs, prolapse recurrence, or excess fibrosis.31 These complications occur much more frequently in the gynecological setting than the original abdominal wall setting, leading to widespread cancellation of medical device approvals by regulatory agencies, and the ongoing tempering of their recommendations by professional medical societies.32–34 This differential outcome between the use of similar devices for different indications underscores the importance of application specific considerations (i.e. microenvironmental signaling and mechanics) in designing surgical solutions. To understand the factors underlying these critical mechanics of the host-wound interface, a thorough understanding of relevant biology is warranted.
2.1. Timeline of the foreign body response
Placement of an IMD interacts with a wound healing response, wherein the device is placed in an environment of acute tissue damage. After the initial abrasion is created, and the implant is introduced into the body, an adsorption period begins,5 during which host proteins, extracellular matrix (ECM) and cell debris adsorb and desorb fluidly at the surface of the implant.7 Due to the tissue damage associated with implantation, adsorption occurs effectively immediately (i.e., reaching adsorbed homeostasis in under 30 min) to an implant surface.35 The damaged tissues and denatured proteins surrounding the implant serve as damage-associated molecular patterns (DAMPs), which engage the cells of the immune system through pattern recognition receptors (PRRs) to respond to tissue damage.36 Common DAMPs include ECM proteins such as fibrinogen, fibronectins and heparan sulfate, all of which initiate immune signaling through Toll-like receptors (TLRs) 437–39 as well as intracellular components (DNA, RNA, histones, etc.)40 and adsorbed immune proteins such as complement elements and antibodies.7 Proteins adsorbed at the surface of the implant promote the recruitment of platelets, induction of complement cascading, and immune cell chemotaxis through cytokine signalling.36 Receptor binding to TLRs underlies early inflammation through induction of inflammatory mediators such as IL-6 and interferons, and appropriately each TLR recognizes a finite number of ligands. Differential conformation and adsorption profiles of DAMPs may be an important factor in the inflammatory profiles associated with different materials.41 In the FBR, adsorbed DAMPs (namely fibronectin and fibrinogen) have implicated both TLR2 and TLR4 as activating PRRs6,42 with downstream activation of NF-κB and pro-inflammatory mediators. However, the breadth of DAMP signalling engaged during the FBR likely involves other endogenous ligand-binding TLRs (e.g., TLR3, TLR7, and TLR9) and C-type lectin receptors.36 Ultimately, more research is required to identify the entirety of PRRs engaged during the FBR.As soon as biochemical absorption has occurred, cell reaction local to the foreign body induces resident immune cell activation and recruitment of more distal or systemically circulating immune cells. These events constitute the acute inflammatory phase of the FBR, lasting for hours to days5 and characterized by nonspecific cell activity optimized for rapid neutralization of invading organisms and systemic immune escalation and recruitment (Table 1). Neutrophils are the ‘first responders’ of the FBR and secrete pro-inflammatory cytokines.5,7,36 Neutrophil extracellular trap (NET) activity, reactive oxygen species (ROS) production and chemoattractant production are also induced by neutrophils.5,7,36 To date, the balance of evidence suggests key roles of these functions43–45 in propagating deleterious FBRs, although differences may be minimal or only manifest later in the course of healing and fibrosis.43–46 Additionally, mast cells have long been recognized as critical to the early FBR, as well as to the fibrotic process; early mast cell degranulation is ostensibly responsible for neutrophil and monocyte extravasation and chemotaxis to the peri-implant environment in the context of the FBR.47,48 The mast cell response, although somewhat lacking in recent study, has been linked specifically to near-instantaneous fibrinogen binding and activation on the surface of implanted biomaterials,49 with modulation of this pathway robustly affecting the early response with a negligible to moderate effect on fibrotic outcome.38,47,48 Despite long-standing knowledge, mechanistic study of mast cell contributions to the FBR have been hampered by challenges associated with in vitro study and in modulating the mast cell response experimentally in vivo.50
| Cytokine | Cellular origin | Target cell | Function | Source(s) |
|---|---|---|---|---|
| TGF-β | Macrophages, fibroblasts | Fibroblasts, macrophages | Promotes activation of fibroblasts. Promotes ‘anti-inflammatory’ profile in macrophages. | 5, 51 and 52 |
| TNFα | ‘M1-like’ macrophages, neutrophils | Macrophages | Promotes differentiation and proliferation of myofibroblasts. Promotes inflammation. | 53 and 54 |
| PDGF | Macrophages, platelets, fibroblasts | Macrophages, fibroblasts | Macrophage and fibroblast recruitment. Promotes myofibroblast differentiation and angiogenesis. | 7, 11, 55 and 56 |
| VEGF | ‘M1-like’ macrophages, FBGCs | Endothelial cells | Promotes formation of neo-vasculature. Associated with elevated fibrosis. | 9, 11 and 56 |
| IL-1β | ‘M1-like’ macrophages, neutrophils | Macrophages | Induces alterations to MMP secretion profiles. Promotes inflammation and expression of pro-fibrotic mediators. | 57 and 58 |
| IL-4 | Mast cells, T-cells | Macrophages | Promotes alternative ‘anti-inflammatory’ activation of macrophages. Believed to play a role in cell fusion. | 7, 59 and 60 |
| IL-6 | Macrophages | Macrophages, fibroblasts | Early pro-inflammatory immune activation. Promotes activation of fibroblasts and stimulates paracrine TGF-β signalling. Primes macrophages for anti-inflammatory signaling later in wound healing. | 61–63 |
| IL-10 | ‘M2-like’ macrophages | Macrophages, myofibroblasts | Reduces collagen and α-SMA expression in activated fibroblasts. Promotes anti-inflammatory macrophage phenotype. | 5 and 64–68 |
| IL-13 | Mast cells, T-cells | Macrophages | Promotes alternative ‘anti-inflammatory’ activation of macrophages. Believed to play a role in cell fusion. | 7, 59 and 60 |
| IL-17 | Th17 cells | Neutrophils, macrophages, monocytes | Undifferentiated role in promoting chronic fibrosis. Associated with increased monocyte, macrophage, and neutrophil presence. | 69 |
The release of inflammatory mediators during the acute inflammatory phase contributes to the recruitment of macrophages, critically involved in the FBR;5,7,36 the continued recruitment of these cells and persistence beyond that of mast cells and neutrophils, and forms the chronic phase as specific to the field of biomaterials and the FBR.5,70 Macrophages, in turn, respond to DAMPs associated with the implant surface through secretion of inflammatory molecules.5,7,36,71 During normal wound healing processes, inflammatory macrophages are short-lived. This is in contrast with macrophages in the FBR, which remain inflammatory due to the persistence of the implant and denatured proteins within the host. In the absence of foreign material, this chronic phase gives way to a resolution phase; continued exposure to foreign material instead induces granulation in the FBGC formation phase, which will be discussed below.
To protect the body from the harmful products being released by immune cells that direct the chronic inflammatory response at the implant interface, macrophages secrete pro-fibrotic cytokines, and recruit fibroblasts which deposit a collagenous layer of tissue around the implant.5,7,36,71 This collagenous layer is referred to as the fibrotic capsule. The fibrotic capsule plays a critical role in determining the negative outcomes of the FBR, including discomfort, pain, capsular contracture and implant failure.4 This process of fibrotic encapsulation and ongoing immune reaction at the implant-interface constitute the final phase of the FBR, the fibrous capsule phase.5,7,36,71 As the capsule continues to form, a variety of pro-inflammatory macrophage- and FBGC-secreted cytokines, in particular vascular endothelial growth factor (VEGF), mediate angiogenesis and recruitment/activation of myofibroblastic processes.9 Finally, although the formation of a foreign body granuloma does not require adaptive immunity,72 adaptive immune cells (i.e., lymphocytes) are present in large quantity in the local region.43,73,74 There is currently little evidence to support direct antigenicity of synthetic polymeric biomaterials, but FBRs have featured T- and B-cells interacting either directly with biomaterials or in response to macrophage secretions, and then acting as intermediaries and amplifiers to cytokines and chemokines by other cells.69,73,75–77
2.2. Macrophage phenotypes associated with the foreign body response
There is an increasingly pervasive skepticism towards this classification system however, particularly in the context of complex environments like the FBR, as focused investigation reveals that macrophage phenotypes have indicated a greater number of exceptions to the M1/M2 rule than not.78,79,83 In the tissue-implant microenvironment, macrophages show elements of both M1 and M2, due to their involvement in inflammation and remodelling;83 this may help to explain the generally-accepted observation that a “runaway” M2 response is considered to be a contributor to the pro-fibrotic component of the FBR. Macrophages may first require inflammatory activation to develop a robust M2-like response, which may contribute to this phenotype in the chronic inflammatory environment of fibrosis.84 Oversimplification of macrophage subsets is further complicated by heterogeneity in phenotypes in the fibrotic tissue microenvironments; macrophages of both characteristic phenotypes that may be arranged in a spatially distinct fashion, contributing to the macroscopic phenotype of the environment.85
Further nuance can be attributed to tissue-specific macrophage metabolic preferences in inflammation,86 findings of systemic immunity or its interactors (e.g. gut microbiome) influencing the response to implanted materials,87 and findings that various macrophages functions (e.g., secretion of individual cytokines) seem dependent on different metabolic pathways and again differential per activation profile.88 Efforts to establish an FBR-specific macrophage phenotype are progressing, although clear models of function remain challenged by discrepancies between different experimental models and techniques.89 In the FBR, macrophages produce pro-inflammatory cytokines90 such as tumor necrosis factor (TNF), interleukin-6 (IL-6), and interleukin-1β (IL-1β) as well as pro-reparative cytokines such as transforming growth factor β (TGFβ), leading to speculation that macrophages in this environment might represent a novel phenotype, ill-defined by the M1/M2 system,78,79,83 but characterized by an environment rich in IL-4, IL-17, and IL-34.69,91
In contrast, ‘reparative’ (i.e., M2) late stage wound healing macrophages are putatively characterized by intact TCA cycles (OXPHOS metabolism), associated with reduced ROS production.100 These cells can utilize glycolysis to generate succinate as fuel for the TCA cycle, but have demonstrated the ability to maintain TCA cycle usage during glycolytic inhibition, likely through increased reliance on fatty acid oxidation (FAO).101 These changes are once again functional. Reparative macrophages are not as reliant on rapid energy turnover and the availability of synthetic intermediates, and as such do not require upregulated glycolysis and PPP to the same degree as inflammatory cells. FAO, which is notably increased in ‘M2’ macrophages, is typically associated with longer cellular lifespans, which would be useful in orchestrating prolonged periods of tissue remodeling.
Additionally, reparative signals and oxidative phosphorylation are linked. Various studies have pointed to the reduction in OXPHOS following IL-10 suppression, pointing to IL-10 as having a role in metabolic orchestration. Induction of these metabolic pathways are believed to contribute to the synthesis of wound-healing intermediates such as collagen.102 IL-10 supports successful wound healing, and has been documented as anti-fibrotic by acting on myofibroblasts to reduce collagen gene expression and lower α-SMA production.64 Conversely, other studies have indicated reductions in IL-10 signaling following OXPHOS interruption, suggesting that there is reciprocity to the regulation of IL-10 on OXPHOS.103 Mechanistically, these pathways are still unclear. Further, the precise metabolic distinctions between M2 subgroups remain ambiguous. For example, the M2b subgroup of macrophages are implicated in “M2-mediated inflammation”, and as such it is unlikely that these cells rely on anti-inflammatory OXPHOS and FAO pathways to the same extent as M2a macrophages, which are more strongly implicated in tissue remodeling. A 2023 publication demonstrated the capacity for strong HIF1α-associated glycolytic reprogramming of dually IL-4- and IL-13-stimulated macrophages. Though these cells showed glycolytic upregulation and succinate accumulation characteristic of M1 metabolic programming, they also maintained certain metabolic and phenotypic features characteristic of the M2 lineage, namely arginase 1 (Arg1) activity.104 This study is useful in illustrating the potential incongruence between classification by metabolic behavior and canonical phenotyping.
Critically, within the context of the FBR, the phenotypic characteristics of macrophages are ill-defined, and reliable information regarding the metabolic behavior of these cells is even more evasive. Thus, any inferences with regards to the metabolic behavior of macrophages during the FBR are speculative and require further demonstration. However, the engagement of key cellular pathways during the FBR may shed light on the metabolic behavior of macrophages during this reaction. For example, NLRP3 inhibition has been observed to reduce implant-associated fibrosis following nerve injury in mice.90 The links between NLRP3-mediated inflammation, IL-1β expression, and glycolysis would implicate glycolysis in the chronic inflammation associated with implants. The role of glycolysis in the FBR is also supported by the high-flux HIF-1α-mediated glycolytic metabolism observed in early wound-healing macrophages, though the persistence of HIF-1α signaling in chronic FBR-involved tissues remains to be demonstrated.37,94
Though the induction of TGF-β signaling by implant-associated macrophages is universally believed to play a key role in the pathology of the FBR,5 the cellular populations responsible for excessive TGF-β signaling in the FBR remain unclear.105 This is potentially once again attributable to the cells in the FBR displaying phenotypic plasticity, with ‘M1’-like cells and ‘M2’-like cells being equally implicated in TGF-β signaling. Interestingly, despite being largely attributed to ‘M2’-like cells (which are primarily oxidative), HIF-1α upregulation has been shown to induce TGF-β production by human macrophages in hypoxic conditions,51 and specifically has a documented supporting role in TGF-β mediated transcription of fibrotic genes by alveolar macrophages during bleomycin-induced lung fibrosis in mice.52 This could imply a relationship between hypoxia, glycolysis and fibrosis, though metabolic differences between the macrophage phenotypes studied would require confirmation in the FBR context. Adding further complexity, IL-10 has been observed to reduce TGF-β production by alveolar macrophages, and ultimately reduce bleomycin-induced lung fibrosis, despite the commonly held belief that both of these cytokines stem from the same cellular origins.65 Ultimately, further analyses are required to determine the links between metabolism and excessive TGF-β signaling.
Finally, classification of macrophages in the FBR is further impaired by their formation of foreign body giant cells (FBGCs). FBGCs are large, terminally differentiated, multinucleated cells with poor phagocytic, and enhanced lysosomal abilities, suggesting a role in debris clearance and extracellular degradation; they are the hallmark of the FBR, and persist at the tissue-implant interface for the remainder of the implant life cycle.5,7,36,71 Multiple studies have demonstrated functional roles for IL-4 and IL-13 in macrophage fusion and FBGC formation through the upregulation of mannose receptors,7,59,60 but the exact mechanisms and conditions through which macrophage fusion occurs in the FBR are still relatively unclear. The functional role of FBGCs, however, primarily involves extracellular degradation and phagocytosis, two processes which are strongly linked to glycolysis through HIF1α, though this link requires further study.
2.3. Foreign body giant cells
The hallmark histologic feature of the FBR is the persistent presence of both macrophages and the multinucleated giant cell (MNGC) or FBGC, the latter of which are optimal for the phagocytosis and breakdown of large particles (e.g., 45 μm or even larger106) that eclipse the capacity of macrophages. There are multiple types of MNGC in the human body, which can be differentiated both by mechanism of formation as well as function or profiling. The best-characterized MNGs are arguably osteoclasts (mediating physiological bone remodeling in pathogen-free local environments) and Langhans giant cells (mediating granuloma formation in the presence of persistent and recalcitrant microbes such as M. tuberculosis and M. leprae as well as Bacillus Calmette–Guérin (BCG) vaccine).107 FBGCs are poorly-characterized in contrast to these other cell types, but have been positively identified due to different cytoplasmic structure and function as distinct from both osteoclast and infectious granulomatous MNGCs.72 FBGCs have emerged as key mediators of the acute and chronic FBR with wide-ranging sensing and activity,108 including demonstrated roles in extracellular degradation via protease excretion, large particle phagocytosis, chemokine and cytokine release for immunomodulation, and antigen presentation.6,7,107,109–112Despite longstanding awareness of the existence of FBGCs, little is known about both their formation and their resolution. Consensus from both in vivo and in vitro study, including in vitro fusion protocols and microscopic and molecular evidence, strongly supports the fusion of macrophages as the major factor in MNGC formation.106,111–115 However, a recent study suggested that pattern recognition receptor-induced polyploidy and frustrated mitosis were the major drivers of MNGC formation in a model of BCG granulomatosis;116 these mechanisms would merit further investigation in a model of sterile granuloma such as the FBR. In the case of material implantation specifically, foreign body MNGCs are induced by IL-4 and IL-13106,111,115 secreted ostensibly from a non-T-cell source117 and aspects of this activity have been recapitulated in vitro,42,118 although the precise mechanism at play and co-stimulators in vivo are unclear.108 It is equally noteworthy that while mast cells are well-documented producers of IL-4 and IL-13,119 macrophage fusion has been documented following implantation in mast-cell deficient mice, further complicating the understanding of fusion signals resulting in FBGC formation.120
Biomechanical signaling has also been implicated via various integrins7,112,114,121 and TRPV4-sensed stiffness122 in combination with soluble cytokines,123 which represent potential control points for improving implant biocompatibility. Additionally, TLR-mediated detection of adsorbed self proteins (i.e., DAMPs)7,42,124,125 is correlated with GC formation or function. Again in the context of an infectious granuloma, complement opsonization (e.g., C3) is a potent trigger for MNGC function;106 FBGCs may retain this activity, which could even be elicited in the context of non-opsonization sterile complement adsorption to protein-naïve biomaterial surfaces as part of the protein corona during the tissue damage associated with implantation, and so alter the course of the FBR and the material-specific safety profile.7,112,126 T-cell contributions to FBR development and MNGC formation have been noted in both infectious (e.g., tuberculosis and parasite) and autoimmune pathology.72 T-cell antigen response may be observed in certain instances of foreign body introduction, such as chronic beryllium disease, but other reactions such as silicosis have not yet demonstrated antigen-directed T-cell response beyond nonspecific pro- and anti-inflammatory helper T-cell and regulatory T-cell involvement.72
Many MNGCs display aspects of both M1 and M2 between function and metabolic phenotype.127 One mechanism might be macrophage plasticity, in which classically-activated (“M1”) macrophages become resistant to TLR signaling and limit their pro-inflammatory activity while maintaining anti-inflammatory IL-10 release.112 This complements both the observation that FBGCs develop after high proportions of M1 cells in the early FBR,42 and the hypothesis that pro-inflammatory macrophages may take on anti-inflammatory properties and fuse upon stimulation with IL-4, Il-13 or vitamin E or tocopherol in vivo.115 Here, the TLR2 pathway frustration may be synergistic or even priming to cytokine-mediated transdifferentiation and cell fusion.128 Fibrinogen dependency of FBGC occurrence has been noted,38 which correlates to mast cell degranulation during introduction of the material.48 Material properties have long been known to influence FBGC activity, density per unit area, and ploidy.129 Given the context dependence on the varied functionalities of FBGCs (including cytokine secretion, phagocytosis, and enzyme secretion), it is likely difficult to directly correlate FBGC frequency in situ to the ultimate outcome of an implant across conditions. Instead, the trajectory of cell function likely remains the gold standard by which to assess the relevance of FBGC presence to FBR course.
2.4. Clinical models of macrophage and giant cell function and dysfunction: the placenta and infectious vs. non-infectious granulation
Complexities of correlating histological occurrence to tissue function notwithstanding, macrophage and GC presence have long been correlated to chronic inflammation and disease outcomes based on their persistence and density. Given their often deleterious roles in pathological FBR occurrences, it is understandable that there is considerable effort to better understand the factors that mechanistically underly GC occurrence and resolution to allow for design of materials that minimize these factors of the FBR and thus improve clinical outcomes.125,130,131 Therefore, additional insight in GC formation, persistence, and resolution may be found in the physiological and pathological models of the placenta and sarcoidosis, respectively.The placenta is a high-throughput fetal-maternal interface for the exchange of dissolved O2, nutrients, and waste, forming from the embryonic trophoblast. It primarily shields the mother from the fetus to prevent development of anti-fetus immunity, although it also restricts certain antibody classes and cell-mediated immunity. This shielding often breaks down in the presence of previous RhD antigen alloimmunization, but generally is of exceptionally high quality. Of note, the placenta features a high concentration of GCs.132 As in the FBR, M1 and M2 distinctions break down in the placenta. There appears to be a time course of dominant signatures, but there remain both M1 and M2 markers throughout the course of normal pregnancy, and both molecular signatures and function of both M1 and all M2 subtypes persist in the healthy placenta across all stages of pregnancy.132,133 M1 markers are more strongly expressed in the first trimester and aid in implantation before M2 markers demonstrate a receptivity to trophoblast invasion, vascularization, and placental development.133 Later in pregnancy, high M1 activity is correlated with preeclampsia,132,134 drawing similarities to sustained inflammation and systemic sequelae in sterile FBR-related illness.
In response to infectious insults, granulation can often be effective at indefinitely sequestering an agent, or even eliminating it completely and resulting with a sterile granuloma. In contrast, these reactions as well as sterile autoimmune reactions such as Crohn disease, sarcoidosis, vasculitides, or various hypersensitivities manifesting in granulation can progress to highly deleterious impact even based solely on mass effect and resulting tissue dysfunction,72 separate from any systemic effect of chronic immune activation. In many cases, the provoking antigen or insult is never found. Further understanding of the induction, propagation, and growth of granulation processes may be highly valuable to clinical resolution of these sterile diseases as well as the FBR.
2.5. Macrophage direction of myofibroblast activity
Myofibroblasts are most responsible for maladaptive fibrotic deposition during FBR. Though direct TGFβ signaling from macrophages is known to promote myofibroblast induction, macrophages participate in promoting fibrosis through many pathways.135,136 Early wound macrophages release IL-6, which induces paracrine TGFβ signaling in fibroblasts, and promotes differentiation into myofibroblasts.61 IL-6 can also induce myofibroblasts directly, by promoting α-SMA via a JAK1-ERK signaling pathway.62 PDGF, another well-documented macrophage-associated inducer of fibrosis, promotes myofibroblast activation seemingly through promoting paracrine TGF-β signaling.55 Finally, macrophages can seemingly induce myofibroblast activity through TGF-β independent mechanisms. Specifically, RELMα expression induces α-SMA expression in fibroblasts through Notch1 signaling.137 TNF has also demonstrated pro-fibrotic roles in several pathologies, through the induction of collagen synthesis, proliferation and activation of myofibroblasts.53,54 IL-1β further drives fibrotic outcomes through indirect promotion of IL-6, TGF-β and PDGF,57 although the mechanism is less obvious and warrants further study.58 FGF-2, TGFβ, TNFα, PDGF as secreted by macrophages and FBGCs have been associated with myofibroblastic activity and wound/implant fibrosis and fibrotic disease pathology.11,37,126,138 Heightened local VEGF levels have been associated with both increased9 and decreased139,140 fibrosis. Local ischemia can induce VEGF secretion, which can both support increasing capsular formation, or stabilize healthy healing interstitium; VEGF control can therefore potentiate both pro-fibrotic and pro-regenerative FBRs based on other physical, chemical, and temporal cues.56The interaction between macrophages and fibroblasts extends beyond the promotion of fibrosis through myofibroblast induction. Macrophages act as potent sources of matrix metalloproteinases (MMPs), which can impact fibrosis directly via ECM degradation, or indirectly through cellular signaling and regulation of inflammation.141 The roles of MMPs in fibrotic control are extensive and highly context dependent. For example, MMP-9 expression or activity has variably shown pro-fibrotic, neutral, or anti-fibrotic roles among different models of lung fibrosis.141 The roles of MMPs in FBR-specific fibrosis are not entirely understood. Current understandings are summarized in Table 2. Additionally, macrophages can also limit fibrosis through IL-10 signaling, which reduces collagen production in activated myofibroblasts.5,64–68 Arg1 metabolic flux in macrophages has also been associated with reduced fibrosis. Though the mechanism remains unclear, a possible explanation could involve ‘substrate stealing’, wherein the highly metabolically active oxidative macrophages reduce available arginine for collagen production by fibroblasts.45,102
| MMP | Classification | Macrophage phenotype | Co-associated genes/products | Temporal trend in expression | Niche/implant location | Species | Material | Technique | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 2 | Gelatinase | (Gross analysis of explanted collagen disk) | IL-1α, TNFα, TGFβ | Progressive increase | Subcutaneous | Mouse | Bovine collagen | PCR gel | 2 |
| CD163+/CD206+ | Col-I | 0.6–6 years post-implantation | Abdominal wall | Human | Polypropylene | Immunofluorescence microscopy | 3 | ||
| 8 | Collagenase | (Gross analysis of explanted collagen disk) | IL-1β, IL-10, CXCL1, CXCL2 | Acute; decline after 1 week | Subcutaneous | Mouse | Bovine collagen | PCR gel | 2 |
| 9 | Gelatinase | (Gross analysis of explanted collagen disk) | IL-1α, TNFα, TGFβ | Appearance after 2 weeks | Subcutaneous | Mouse | Bovine collagen | PCR gel | 2 |
| Proliferative macrophage (“MD2”) | Proliferation-associated products | [Not characterized] | Subcutaneous | Rat | Silk | scRNAseq | 4 | ||
| Adherent macrophage and FBGCs | Adhesion-associated products | Significant increase in 3 days | In vitro | Human | PET, and modified PET | Antibody array and quantitative ELISA | 142 | ||
| (Gross analysis of explanted capsule breast implant tissue) | Rac2 | Increasing with increased Baker score | Breast | Human | Silicone | Bulk RNAseq | 143 | ||
| FBGCs | Macrophage fusion, ECM degradation | [Not characterized] | Subcutaneous | Mouse | Mixed cellulose ester disks and polyvinyl alcohol sponges | Immunohistochemistry antibody array | 144 | ||
| 12 | Stromelysin | Giant cell (“C7”) | ECM degradation, macrophage fusion, complement receptors, glycolysis | Progressive increase; resolution between 2–4 weeks in resorbable silk but not sponge | Peritoneal | Mouse | Silk and sponge (putatively cellulose) | scRNAseq | 5 |
| Giant cell -like (“MD1”) | ECM degradation | Intermediate | Subcutaneous | Rat | Silk | scRNAseq and histology | 4 | ||
| Giant cell-like (“MD3”) | Macrophage fusion, oxidative function | Progressive increase to 2 weeks | Subcutaneous | Rat | Silk | scRNAseq and histology | 4 | ||
| (Gros analysis of explanted capsule breast implant tissue) | Il-8, Tnsf11 | [Not characterized] | Breast | Human | Silicone | qPCR | 30 | ||
| 13 | Collagenase | (Gross analysis of explanted collagen disk) | IL-1α, TNFα, TGFβ | Increasing after 2–3 weeks | Subcutaneous | Mouse | Bovine collagen | PCR gel | 2 |
| 14 | Membrane-type | Epithelioid (“C6”) | Pro-inflammatory (IL1b), ECM, chemokines | Progressive increase; resolution between 2–4 weeks in resorbable silk but not sponge | Peritoneal | Mouse | Silk and sponge (putatively cellulose) | scRNAseq | 5 |
| (Gross analysis of explanted collagen disk) | IL-1α, TNFα, TGFβ | Increasing 2–3 weeks post implant | Subcutaneous | Mouse | Bovine collagen | PCR gel | 2 | ||
| (Gross analysis of explanted CC breast implant tissue) | Rac2 | Increasing with increased Baker | Breast | Human | Silicone | Bulk RNAseq | 143 | ||
| 19 | Not classified | Epithelioid (“C6”) | Pro-inflammatory (IL1b), ECM, chemokines | Progressive increase; resolution between 2–4 weeks in resorbable silk but not sponge | Peritoneal | Mouse | Silk and sponge (putatively cellulose) | scRNAseq | 5 |
| 16 (MT3) | Membrane-type | Giant cell-like (“MD3”) | Macrophage fusion, oxidative function | Progressive increase to 2 weeks | Subcutaneous | Rat | Silk | scRNAseq and histology | 4 |
3. Advances in profiling and mitigating the foreign body response
Growing clinical evidence continues to establish causation and is demonstrating the significant burden of pathology attributable to the foreign body response. With increased attention, rapid advancements in both clinical and translational science are outlining new ways to interrogate and mitigate the FBR. Below, we outline exciting new approaches to profiling clinical disease, new in vitro and in vivo models of the FBR, and therapeutic strategies in development to avoid or treat FBR incidence.3.1. Investigating the foreign body response: models and techniques
As a complex phenomenon involving many lineages of cells and a highly context-dependent timeline, most mechanistic investigations of the FBR have benefitted from the use of animal models, informed by available clinical explants. These animal models and associated analyses have recently been expertly summarized in the context of hernia meshes, a well-studied and clinically significant example of the FBR,145 with efforts to better model metabolic and biomechanical considerations often prompting the use of larger animal models.146 With growing concern about the clinical adaptation of hernia mesh to pelvic organ prolapse, similar animal models of vaginal mesh application have been developed,147,148 as has a model of neural implant FBR,90 and models of silicone-implant associated capsular contracture.29,149 Concerning the development of biocompatible materials to ameliorate the FBR, studies are often complicated by differences between and even within standardized animal lines by individual variability and by batch effects of implantation runs, both in terms of animal and implant batches.76,150 The use of well-designed and statistically-informed experimental and surgical procedures can mitigate many of these complications, such as in the combinatorial screening of multiple materials in mice, which was successfully translated to primate usage.151 From both model system and patient explants, single-cell techniques for functional profiling continue to be developed and standardized for replication and translation;152 findings from multi-omics studies may be transferrable across species, experiments, and lines with the appropriate standardization and controls.153From both in vivo models and with clinical explants, improved characterization techniques are helping to elucidate spatial relationships in the FBR. Cell profiling in situ with histology can allow for more precise profiling of cell phenotype and interactions in the local region,43,74 while spatial transcriptomics of granulomas allow for cell interaction profiling.154 Intravital microscopy, by allowing repeated individual measures over time, allows for highly mechanistic studies to also inform factors of individualized response.9 Individual genetics, medical history, and these intersecting and non-linear contributors to an individual's immune response are also more understood than ever, and inquiry and design with these concepts in mind will help to account for and harness individual variability in immunity.155
While in vivo models of the FBR provide valuable translational direction and holistic biometrics, they often fail to provide detailed mechanistic insight or quantitative metrics for optimization of therapeutic design. Immunology has benefitted significantly from recent advances in reproducible in vitro experimentation, and investigation of the FBR specifically is likely to benefit similarly from the level of mechanistic resolution that in vitro approaches provide. For example, new models of FBGC fusion in vitro42,156 will allow for understanding of the factors leading to FBGC formation, as well as elucidate their functionality and amenability to productive control to then be trialled in vivo. Approaches to screening materials are well-established in vitro, although immunoregulatory applications are still very novel; macrophage M1/M2 differentiation was successfully profiled on combinatorial copolymer libraries in a recent study,157 an approach that will prove instructive for further efforts. Similarly, statistically informed approaches reach a level of throughput that in vivo approaches cannot practically accommodate. Studies have revealed multifunctionality of individual cell phenotypes, and complex bidirectional relationships between macrophages and other stromal cells. Use of high-factor multivariate optimization encompassing genes, soluble excreted protein expression (e.g. VEGF), and microscopy-derived morphometrics (e.g. vascular tube formation and cell diameter) allowed for multiparametric optimization of macrophage-directed interstitial cell function in vitro.158 These techniques can be used not only to model the peri-implant environment for material and therapeutic development, but also to derive quantitative multifactorial cause and effect mechanisms of immune function in situ.
As immunometabolism is a rapidly-growing field, so too are approaches by which to profile it. Traditional extracellular flux approaches such as those offered by Agilent Seahorse systems have successfully been used to profile specific pathway control in immune populations.159,160 However, flow cytometry-based approaches to profile single-cell metabolism not only offer greater resolution of rare populations and limited samples in vitro or in vivo, but allow for correlation to cell phenotype by coupling to simultaneous traditional flow cytometric analyses, such as in the SCENITH method of profiling central carbon metabolism161 and the QUAS-R method of profiling glutamine uptake.162 Finally, metabolomic profiling allows for targeted pathway flux analysis,163,164 which can be adapted for use in FBR characterization.
3.2. Material properties influencing the foreign body response
A degree of fibrosis is an expected outcome of wound healing, and thus will be present following implantation with any biomaterial. In fact, a physiological level of fibrosis is desired for mechanical stabilization and tissue integration of most implants.165 Nevertheless, aberrant FBR outcomes are associated with costly implant revisions, highlighting the importance of understanding the dynamics of the biomaterial-host interactions. The extent to which material properties can alter fibrous encapsulation during the FBR has been outlined in previous reviews.11,36,112,166 The mechanical properties of implanted materials are considered paramount to stable integration with host tissues. Mismatch between the stiffness of host tissue and an implant result in mechanical disruptions and worsen fibrosis.12,167 Additionally, implant topography and scale can significantly alter the FBR. Jagged edges and macroscopic texturing that promote continuous tissue disruptions are associated with severe FBR outcomes.11 Conversely, micro- and nano-texturing can impact adhesion and orientation of proteins, the patterning of cells, and ultimately mitigate adverse FBR outcomes.168,169 A material's degradative profile is also believed to impact the FBR. Where material degradation is possible, implant fragmentation is conducive to both phagocytosis and discontinuous capsule formation, contributing to lower rates of capsular contracture and implant failure.Material properties which influence the rate of protein adsorption at the implant interface are thought to be equally important in determining the outcome of the FBR. Some of the commonly identified characteristics that alter adsorption rates (and thus potentially affect the FBR) include surface roughness, surface charge and hydrophobia/hydrophilia.5 Hydrophobicity/wettability will partially determine the degree of protein adsorption to its surface; hydrophilic surfaces resist protein adsorption relative to hydrophobic surfaces and are accordingly associated with less severe fibrotic reactions.170,171 The surface charge of the material also in part determines the degree of protein adsorption to an implant, where increased charge increases protein adsorption.172 Fibroblast traction based on both material stiffness and protein adsorption can influence macrophage migration and thus potentially chemotactic gradient formation, potentially creating positive feedback loops;173 implants or factors that influence ECM deposition could therefore have outsized effects. Hence, implants of different materials might result in different levels of inflammation, and consequently the fibrosis following implantation. Understanding the metabolic differentiation in macrophages for different materials could elucidate some of the metabolic processes involved in a successful FBR outcome. However, the generation of particulates and degradation products from the material, or metabolites released from cells responding to the material, may result in local toxicity leading to unintended effects. Careful and systematic characterization of material disposition and mechanism of action of therapy will be required to ensure safety and efficacy of novel therapeutic materials.
Additionally, a better understanding of effector material properties will be critical in identifying potential targets for novel therapies, in addition to improving our understanding of the interplay between metabolism, signalling and effector function. Among the most commonly used medical implant materials are: poly(lactic-co-glycolic) acid (PLGA), polypropylene (PP), polyethylene (PE), polycaprolactone (PCL), polyethylene glycol diacrylate (PEGDA) and polydimethylsiloxane (PDMS). PP is preferentially used for permanent support-lending applications such as structural meshes and sutures.174 PE implants are used in facial reconstruction as well as in joint replacement surgeries.175 PCL implants are often used in cranioplasty,176 and PDMS is commonly used for breast implants.4 PLGA has a wide range of popular applications including tissue engineering scaffolding and micro- and nanoparticle delivery systems, is bio-degradable, and as such is considered to be highly biocompatible.177 With a variety of materials available, each likely with inherent physicochemical properties, there is much to be learned about macrophages in the response to different IMD materials.
3.3. Biomaterial chemistry-based strategies to mitigate FBR pathology: physical, chemical, and immune-signaling
Significant effort has been expended in profiling various physicochemical approaches to minimizing immunoreactivity and fibrosis to implanted materials. Alterations to physical properties outlined above, including wettability, stiffness, charge, (nano)topography and porosity have demonstrated clear trends in mitigating the FBR through altered rates of protein adsorption (Fig. 2).166 Additionally, patterning of the surface that mimics features of the surrounding tissue has been shown to lead to a reduction of fibrosis through biomimicry, though this is not always possible or beneficial.178 Changes to surface topography, such as micro-texturing, have demonstrated success in reducing implant-induced fibrosis in vivo, with relevant translation into the clinical setting for breast reconstructive surgeries. However, alarming links between micro-textured implants and the development of breast-implant-associated anaplastic large cell lymphoma (BIA-ALCL) have resulted in a market recall.179,180 Similarly, polyurethane foam coatings for silicone breast implants have been associated with reductions in capsular contracture rates in cases where the coatings remain intact. Here, the porosity and texturing of the foam is thought to disrupt spatially continuous capsule formation and effectively dissipates mechanical tension,181 albeit with risks of pathology such as ALCL as discussed above. However, concerns with regards to material degradation and toxicity have resulted in caution toward such implementations; the benefit of similar form designs with other materials remain undercapitalized. | ||
| Fig. 2 Biomaterial design strategies to mitigate the immune-driven foreign body response. | ||
Implant surface coatings have demonstrated the highest degree of success in reducing implant fibrosis. Zwitterionic materials/coatings and polyethylene glycol (PEG) surface conjugation have exhibited antifouling properties through decreasing adsorption of host proteins (i.e. ECM and DAMPs) to the implant surface.35,126,182 This leads to a reduction in immune cell recruitment, macrophage recognition and activation, and proinflammatory cytokines.151,183,184 Non-synthetic protein (e.g., gelatin and fibronectin),39,185,186 polysaccharide (hyaluronic acid, alginate, pectin, and heparin),151,186–191 and cytokine192 polymer coatings have been used to reduce cellular activation by improving biocompatibility between the implant surface and tissue microenvironment. Similarly, use of a xeno/allo-derived adipose ECM extract showed improved biocompatibility and CD4 T-cell/M2 macrophage activation relative to gross fat when implanted in soft tissue.193 While immunoevasion or biomimicry methods are useful in decreasing the acute FBR, they do not fully prevent host protein, ECM, and DAMP adsorption to the material, and downstream chronic immune cell activation. Therefore, strategies to target the FBR over long-term implantation are required; immunomodulation has become the major strategy to improve implant material outcomes in chronic settings.194
Research into immunomodulation to combat the FBR has been focused primarily on anti-fibrotic drug release/elution from material surfaces. In contrast to immunoevading materials, which either evade or integrate the inflammatory response, immunomodulating materials/systems actively supress the inflammatory response through drug delivery. The simplest of these systems employ anti-fibrotic drug harbouring coatings which passively elute drug from the material surface.195,196 Most of these models focus on the release of dexamethasone as a drug of choice due to its potent anti-inflammatory properties197–199 and utility in the case of long-lasting implants.200 Dexamethasone serves as an excellent model of coating chemistries and hydrophobic drug release,13,150,197–200 and is highly effective at minimizing FBR. However, there are two key weaknesses in the translation of passive dexamethasone-eluting materials: (1) non-discriminating local immunosuppression is contraindicated for real-world surgeries due to the inevitability of occasional surgical site infections,201 (2) dexamethasone impairs pro-regenerative responses.90 Apart from dexamethasone, many other anti-fibrotic drugs such as methotrexate,202 pirfenidone,203 triamcinolone,204–206 and tranilast207 have shown efficacy in reducing implant induced fibrosis. Additionally, some non-traditional anti-fibrotic molecules such as kynurenic acid,208 colony-stimulating factor 1-inhibitor GW2580,209 NLRP3 inhibitor MCC950,90 rapamycin210 and sirolimus,211 cytokines (e.g., IL-4 eluting materials),212 and immunotherapies (e.g., parasite antigens)213 have all been shown to reduce implant induced fibrosis over various models.
Finally, justified concern of pathological fibrosis and surgical site infection has spurred the continued development of biologically derived implantable materials. These are most visible in applications for structural support, such as in surgical sutures and biological meshes for use in various hernia repairs (e.g., primary or incisional abdominal hernias, hiatal hernias, and inguinal hernias), as more specialized functional implants do not have biological options. Suture choice is heavily dependent on the tissue in question, as well as surgeon comfort or preference. Silk and gut sutures remain in active use for specialized indications; with the former being effectively permanent. Silk sutures offer advantages in handling, but are rarely strictly biologic in modern times as they often carry synthetic coatings. Overall, silk sutures offer little advantage in terms of both fibrosis (they remain highly immunogenic) and infection risk (as their braided design increases pathogen growth capacity).214–216 In terms of meshes for surgical plane repair, absorbable meshes, both biological and synthetic, have existed for decades. Biological meshes vary in terms of animal and tissue source, chemical crosslinking, and additives, and are designed with the intention to facilitate host tissue ingrowth before resorption renders them structurally noncontributory. These meshes are also often used in contaminated procedures in an effort to reduce the risk of surgical site infection, however infection rates, recurrence, and chronic pain suggest that biological meshes have no benefit over synthetics.217–219 Absorbable synthetic meshes, although contentious in terms of their risk of hernia reoccurrrence,220 are generally growing in acceptance,146,221–223 although judicious implant selection is still required when balancing patient demographics, anatomical considerations, and infection risks. In light of the high cost and uncertain benefit of biological materials in such a niche, it is likely that newer generations of chemically-defined synthetic products, both permanent and absorbable, will continue to grow in utility.
3.4. Immunometabolism as a promising therapeutic target to control the FBR
Recent advancements in the field of immunometabolism highlight the potential of multiple small molecule metabolites to regulate the FBR and associated material-induced fibrosis.224 Small molecules such as TCA metabolites have all shown promise in regulating the FBR.163,225–229 In particular, the metabolite itaconate (IA) has emerged as a potent regulator of macrophage phenotype.230–232 Itaconate has diverse direct and indirect immunomodulatory roles, including inhibition of both glycolytic and TCA pathways, multipronged inhibition of the NLRP3 inflammasome, and inhibition of myofibroblast activity, together allowing IA and its isomers to participate in highly specific contextual immunoregulation.230,231,233–235 IA can target classically activated macrophages due to its potent anti-inflammatory effects, and can equally target alternatively activated macrophages by blocking monocyte differentiation into M(IL-4).226,236–238 In addition to TCA metabolites, glycolytic metabolites and analogues also demonstrate promising immunoregulatory activity. For example, incorporation of the glycolytic inhibitors 2-deoxyglucose and aminooxyacetic acid in subcutaneous polylactide (PLA) implants induced an anti-inflammatory phenotype.239 PLGA has equally shown the ability to intrinsically exert metabolic control over local cells.240 As mentioned above, when working with such chiral metabolites, validation of the stereochemistry of the formulation in question is crucial, and requires further standardization and control in studies.241Despite its promise in precise and context-specific control of the immune response, immunometabolism presents several inherent challenges before it can be effectively leveraged in clinical practice. As demonstrated above, metabolites require relatively high concentrations before reaching therapeutic concentrations relative to many other bioactive molecules. This therefore poses a difficult engineering challenge to deliver doses safely to the target region. Chemical stability of bioactive components is also a consideration for all designs. Finally, these therapeutics need to be chemically and logistically compatible with the IMD in question. A potential solution to all of these individual challenges lies in the design of smart biomaterials. For example, metabolites such as IA require relatively high dose delivery to effectively modulate the immune system (5–10 mmol L−1).242,243 Passive drug-eluting surfaces fail to sustain effective high concentration drug delivery over long-term inflammation.244,245 As such, with limited loading capacity, passive-drug eluting systems could be tailored to either deliver short-term high dose, or long-term low dose drug release. In the case of small molecule delivery, such as IA, a different approach to drug delivery needs to be taken to allow for high dose release in the long-term. IA, as well as other TCA metabolites, can be synthesized into the backbone of polyester polymetric biomaterials.244 As the material is degraded in the host environment, the eluted metabolites regulate the fibrotic microenvironment. This method of drug delivery has two advantages over passive diffusion. Firstly, it greatly increases the loading capacity of the material.245,246 Secondly, polymeric degradation can be tunable for temporal specific increased drug delivery.246–248 Polymeric degradation kinetics rely on a variety of intrinsic polymer characteristics. Co-polymer ratio, molecular weights, polydispersity, material viscosity, transition temperatures, polymer endcaps, and hierarchical structuring (e.g., branching) are all useful tools in modulating degradation profiles and drug delivery.249 This may provide a novel biomimetic avenue to reduce FBR-associated complications in future work.
4. Conclusions and future perspectives
Ultimately, the current understandings of FBR-associated fibrosis summarized in this paper indicate complex cellular heterogeneity and dynamic behaviours. The macrophage-fibroblast signalling axis is key to the development of capsular contracture and implant failures, but significant gaps remain in our understanding of the underlying mechanism and therefore potential targets. Critically, FBGC formation is a notable hallmark progression of the FBR but these cells share features of both M1 and M2 macrophages and defy canonical classification. Here, immunometabolism offers a promising new approach to cellular phenotyping in the implant microenvironment. Where the conventional immunological paradigm fails to capture the totality of cellular dynamics in the FBR, evidence supporting links between metabolic behavior and pathological fibrotic signalling is abundant, underscoring the need for a comprehensive immunophenotyping of the tissue-implant microenvironment. Availability of effective treatment options to mitigate pathological fibrosis in the FBR is equally limited. The functionality of minor levels of fibrosis in improving tissue-implant integration, as well as the putative importance of early inflammation in promoting angiogenesis and tissue sterilization should caution the usage of broad-spectrum anti-inflammatory drugs such as dexamethasone. Additionally, currently available treatments lack tuneable release systems with sufficient reservoirs needed to promote extended release of anti-inflammatory drugs during the chronic stages of inflammation associated with pathological fibrosis in the FBR. The development of novel treatments is required to allow the temporal control of pharmacological interventions required to treat pathological inflammation and fibrosis in the FBR. Critically, FBR study is challenged by the lack of in vitro models available to recapitulate the complexity of the tissue-implant microenvironment, to overcome both the logistical and inherent immunological challenges in translating animal studies. Efforts are required to improve and develop better models of the FBR, with improved clinical translatability. Finally, major outstanding questions remain as to the clinical risk factors associated with undesirable FBR outcomes. Robust prospective analyses describing early patient phenotypes correlated to downstream clinical outcomes would help identify risk factors associated with FBR complications.Ultimately, this review has served to summarize current understandings of the FBR to implanted biomaterials, as well as identify critical gaps requiring future investigation and development. This review has benefitted from significant recent attention to the mechanisms underlying immunometabolic regulation, as well as efforts in translating this knowledge to practical effect.
Author contributions
NIC conceived the manuscript. NIC, CR, ZF, KM wrote the manuscript. NIC, CR, ZF, KM and LDH edited the manuscript and approved the final version.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research is supported by the Dalhousie Medical Research Foundation Faculty of Dentistry Early Career Research Award, Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant (RGPIN-2022-03666), and New Frontiers in Research Fund – Exploration Fund (NFRFE-2022-00313). NC was supported by an award from the Webster Family Fund for Research in Immunology and Genetics. ZSCSF was supported by an NSERC Canada Graduate Scholarship – Masters, Nova Scotia Graduate Scholarship, and Killam Predoctoral Scholarship (Masters).References
- K. A. Schlosser, S. M. Renshaw, R. M. Tamer, S. A. Strassels and B. K. Poulose, Hernia, 2022, 27, 415–421 CrossRef PubMed.
- Q. Ma, W. Jing, X. Liu, J. Liu, M. Liu and J. Chen, Int. J. Surg., 2023, 109(3), 333–342 CrossRef PubMed.
- M. P. Simons, M. Smietanski, H. J. Bonjer, R. Bittner, M. Miserez, T. J. Aufenacker, R. J. Fitzgibbons, P. K. Chowbey, H. M. Tran, R. Sani, F. Berrevoet, J. Bingener, T. Bisgaard, K. Bury, G. Campanelli, D. C. Chen, J. Conze, D. Cuccurullo, A. C. de Beaux, H. H. Eker, R. H. Fortelny, J. F. Gillion, B. J. van den Heuvel, W. W. Hope, L. N. Jorgensen, U. Klinge, F. Köckerling, J. F. Kukleta, I. Konate, A. L. Liem, D. Lomanto, M. J. A. Loos, M. Lopez-Cano, M. C. Misra, A. Montgomery, S. Morales-Conde, F. E. Muysoms, H. Niebuhr, P. Nordin, M. Pawlak, G. H. van Ramshorst, W. M. J. Reinpold, D. L. Sanders, N. Schouten, S. Smedberg, R. K. J. Simmermacher, S. Tumtavitikul, N. van Veenendaal, D. Weyhe, A. R. Wijsmuller and T. H. Group, Hernia, 2018, 22, 1–165 Search PubMed.
- H. Headon, A. Kasem and K. Mokbel, Arch. Plast. Surg., 2015, 42, 532–543 CrossRef PubMed.
- R. Klopfleisch and F. Jung, J. Biomed. Mater. Res., Part A, 2017, 105, 927–940 CrossRef CAS PubMed.
- O. Veiseh and A. J. Vegas, Adv. Drug Delivery Rev., 2019, 144, 148–161 CrossRef CAS PubMed.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS PubMed.
- J. Elisseeff, S. F. Badylak and J. D. Boeke, N. Engl. J. Med., 2021, 385, 2451–2462 CrossRef CAS PubMed.
- E. Dondossola, B. M. Holzapfel, S. Alexander, S. Filippini, D. W. Hutmacher and P. Friedl, Nat. Biomed. Eng., 2017, 1, 0007 CrossRef CAS PubMed.
- S. Gentile, F. Strollo, T. Della Corte, G. Marino and G. Guarino, Diabetes Res. Clin. Pract., 2018, 138, 284–287 CrossRef CAS PubMed.
- A. Carnicer-Lombarte, S. T. Chen, G. G. Malliaras and D. G. Barone, Front. Bioeng. Biotechnol., 2021, 9, 1–22 Search PubMed.
- M. Gori, G. Vadalà, S. M. Giannitelli, V. Denaro and G. Di Pino, Front. Bioeng. Biotechnol., 2021, 9, 659033 CrossRef PubMed.
- N. De la Oliva, X. Navarro and J. del Valle, Anat. Rec., 2018, 301, 1722–1733 CrossRef CAS PubMed.
- J. W. C. Tervaert, Y. Shoenfeld, C. Cruciani, C. Scarpa and F. Bassetto, Autoimmun. Rev., 2023, 2, 103448 Search PubMed.
- J. E. Ireton, J. G. Unger and R. J. Rohrich, Plast. Reconstr. Surg. Glob. Open, 2013, 1, 1–10 Search PubMed.
- C. L. Baum and C. J. Arpey, Dermatol. Surg., 2005, 31, 674–686 CrossRef CAS PubMed.
- R. B. Wilson and Y. Farooque, J. Gastrointest. Surg., 2022, 26, 950–964 CrossRef PubMed.
- A. L. Vorst, World J. Gastrointest. Surg., 2015, 7, 293 CrossRef PubMed.
- M. J. Tolino, D. E. Tripoloni, R. Ratto and M. I. García, Hernia, 2009, 13, 631–637 CrossRef CAS PubMed.
- D. J. Tubre, A. D. Schroeder, J. Estes, J. Eisenga and R. J. Fitzgibbons, Hernia, 2018, 22, 1003–1013 CrossRef CAS PubMed.
- D. Wouters, G. Cavallaro, K. K. Jensen, B. East, B. Jíšová, L. N. Jorgensen, M. López-Cano, V. Rodrigues-Gonçalves, C. Stabilini and F. Berrevoet, Front. Surg., 2022, 9, 847279 CrossRef CAS PubMed.
- A. Ríos, J. M. Rodríguez, V. Munitiz, P. Alcaraz, D. Pérez Flores and P. Parrilla, Hernia, 2001, 5, 148–152 CrossRef PubMed.
- F. Ali, G. Sandblom, A. Wikner and G. Wallin, Hernia, 2022, 26, 635–646 CrossRef CAS PubMed.
- M. Cevasco and K. M. F. Itani, Surg. Infect., 2012, 13, 209–215 CrossRef PubMed.
- K. Baylón, P. Rodríguez-Camarillo, A. Elías-Zúñiga, J. A. Díaz-Elizondo, R. Gilkerson and K. Lozano, Membranes, 2017, 7, 1–23 CrossRef PubMed.
- K. Lockhart, D. Dunn, S. Teo, J. Y. Ng, M. Dhillon, E. Teo and M. L. van Driel, Cochrane Database Syst. Rev., 2018, 2018, CD011517 Search PubMed.
- S. M. Smith, A. A. Khoja, J. H. W. Jacobsen, J. G. Kovoor, D. R. Tivey, W. J. Babidge, H. S. Chandraratna, D. R. Fletcher, C. Hensman, A. Karatassas, K. W. Loi, K. M. F. McKertich, J. M. A. Yin and G. J. Maddern, Aust. N. Z. J. Surg., 2022, 92, 2492–2499 CrossRef PubMed.
- M. T. Nguyen, R. L. Berger, S. C. Hicks, J. A. Davila, L. T. Li, L. S. Kao and M. K. Liang, JAMA Surg., 2014, 149, 415–421 CrossRef PubMed.
- F. T. Foroushani, K. Dzobo, N. P. Khumalo, V. Z. Mora, R. de Mezerville and A. Bayat, Biomater. Res., 2022, 26, 1–27 Search PubMed.
- I. Brigaud, C. Garabédian, N. Bricout, L. Pieuchot, A. Ponche, R. Deltombe, R. Delille, M. Atlan, M. Bigerelle and K. Anselme, Plast. Reconstr. Surg., 2020, 145, 542e–551e CrossRef CAS PubMed.
- A. Seifalian, Z. Basma, A. Digesu and V. Khullar, Biomedicines, 2023, 11, 741 CrossRef CAS PubMed.
- Independent Medicines and Medical Devices Safety Review, First Do No Harm: The report of the Independent Medicines and Medical Devices Safety Review, 2020.
- A. Padoa, A. Braga, T. Fligelman, S. Athanasiou, C. Phillips, S. Salvatore and M. Serati, Urogynecology, 2023, 29, 703–716 CrossRef PubMed.
- B. Welk, K. V. Carlson, R. J. Baverstock, S. S. Steele, G. G. Bailly and D. R. Hickling, Can. Urol. Assoc. J., 2017, 11, 105 CrossRef PubMed.
- M. D. Swartzlander, C. A. Barnes, A. K. Blakney, J. L. Kaar, T. R. Kyriakides and S. J. Bryant, Biomaterials, 2015, 41, 26–36 CrossRef CAS PubMed.
- E. Mariani, G. Lisignoli, R. M. Borzì and L. Pulsatelli, Int. J. Mol. Sci., 2019, 20(3), 636 CrossRef CAS PubMed.
- N. Noskovicova, B. Hinz and P. Pakshir, Cells, 2021, 10(7), 1794 CrossRef CAS PubMed.
- A. Balabiyev, N. P. Podolnikova, J. A. Kilbourne, D. P. Baluch, D. Lowry, A. Zare, R. Ros, M. J. Flick and T. P. Ugarova, Biomaterials, 2021, 277, 121087 CrossRef CAS PubMed.
- B. G. Keselowsky, A. W. Bridges, K. L. Burns, C. C. Tate, J. E. Babensee, M. C. LaPlaca and A. J. García, Biomaterials, 2007, 28, 3626–3631 CrossRef CAS PubMed.
- J. S. Roh and D. H. Sohn, Immune Network, 2018, 18, e27 CrossRef PubMed.
- Q. Wei, T. Becherer, S. Angioletti-Uberti, J. Dzubiella, C. Wischke, A. T. Neffe, A. Lendlein, M. Ballauff and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 8004–8031 CrossRef CAS PubMed.
- A. Kaushal, Y. Zhang, L. L. Ballantyne and L. E. Fitzpatrick, Front. Bioeng. Biotechnol., 2022, 10, 1–19 Search PubMed.
- U. Klinge, A. Dievernich and J. Stegmaier, Front. Med., 2022, 9, 1–15 Search PubMed.
- J. l Crossley, S. Ostashevskaya-Gohstand, S. Comazzetto, J. S. Hook, L. Guo, N. Vishlaghi, C. Juan, L. Xu, A. R. Horswill, G. Hoxhaj, J. G. Moreland, R. J. Tower and B. Levi, JCI Insight, 2023, 8(20), e169208 CrossRef PubMed.
- L. Moretti, J. Stalfort, T. H. Barker and D. Abebayehu, J. Biol. Chem., 2022, 298, 101530 CrossRef CAS PubMed.
- D. M. Simpson and R. Ross, J. Clin. Invest., 1972, 51, 2009–2023 CrossRef CAS PubMed.
- P. T. Thevenot, D. W. Baker, H. Weng, M. W. Sun and L. Tang, Biomaterials, 2011, 32, 8394–8403 CrossRef CAS PubMed.
- L. Tang, T. A. Jennings and J. W. Eaton, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8841–8846 CrossRef CAS PubMed.
- L. Tang and J. W. Eaton, Mol. Med., 1999, 5, 351–358 CAS.
- E. W. Ozpinar, A. L. Frey, G. Cruse and D. O. Freytes, Tissue Eng., Part B, 2021, 27, 590–603 CrossRef CAS PubMed.
- J. C. White, Z. L. Jiang, M. P. Diamond and G. M. Saed, Fertil. Steril., 2011, 96, 758–763.e3 CrossRef CAS PubMed.
- M. Ueno, T. Maeno, M. Nomura, K. Aoyagi-Ikeda, H. Matsui, K. Hara, T. Tanaka, T. Iso, T. Suga and M. Kurabayashi, Am. J. Physiol., 2011, 300, L740–L752 CAS.
- A. L. Theiss, J. G. Simmons, C. Jobin and P. K. Lund, J. Biol. Chem., 2005, 280, 36099–36109 CrossRef CAS PubMed.
- L. S. Verjee, J. S. N. Verhoekx, J. K. K. Chan, T. Krausgruber, V. Nicolaidou, D. Izadi, D. Davidson, M. Feldmann, K. S. Midwood and J. Nanchahal, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(10), E928–E937 CrossRef CAS PubMed.
- N. Takamura, L. Renaud, W. A. da Silveira and C. Feghali-Bostwick, Front. Immunol., 2021, 12 Search PubMed.
- W. K. Ward, J. Diabetes Sci. Technol., 2008, 2, 768–777 CrossRef PubMed.
- M. Kolb, P. J. Margetts, D. C. Anthony, F. Pitossi and J. Gauldie, J. Clin. Invest., 2001, 107, 1529–1536 CrossRef CAS PubMed.
- M. M. Mia, M. Boersema and R. A. Bank, PLoS One, 2014, 9, e91559 CrossRef PubMed.
- K. M. DeFife, C. R. Jenney, A. K. McNally, E. Colton and J. M. Anderson, J. Immunol., 1997, 158, 3385–3390 CrossRef CAS.
- A. K. McNally and J. M. Anderson, Am. J. Pathol., 1995, 147, 1487–1499 CAS.
- Y. Li, J. Zhao, Y. Yin, K. Li, C. Zhang and Y. Zheng, Int. J. Biol. Sci., 2022, 18, 5405–5414 CrossRef CAS PubMed.
- R. M. Gallucci, E. G. Lee and J. J. Tomasek, J. Invest. Dermatol., 2006, 126, 561–568 CrossRef CAS PubMed.
- F. Ma, Y. Li, L. Jia, Y. Han, J. Cheng, H. Li, Y. Qi and J. Du, PLoS One, 2012, 7, e35144 CrossRef CAS PubMed.
- J.-H. Shi, H. Guan, S. Shi, W.-X. Cai, X.-Z. Bai, X.-L. Hu, X.-B. Fang, J.-Q. Liu, K. Tao, X.-X. Zhu, C.-W. Tang and D.-H. Hu, Arch. Dermatol. Res., 2013, 305, 341–352 CrossRef CAS PubMed.
- K. Nakagome, M. Dohi, K. Okunishi, R. Tanaka, J. Miyazaki and J. Yamamoto, Thorax, 2006, 61, 886–894 CrossRef CAS PubMed.
- A. S. Caldwell, V. V. Rao, A. C. Golden, D. J. Bell, J. C. Grim and K. S. Anseth, Bioeng. Transl. Med., 2021, 6, 1–13 Search PubMed.
- T. Truong and K. S. Jones, J. Biomed. Mater. Res., Part A, 2018, 106, 2424–2432 CrossRef CAS PubMed.
- D. T. Luttikhuizen, M. J. van Amerongen, P. C. de Feijter, A. H. Petersen, M. C. Harmsen and M. J. A. van Luyn, Biomaterials, 2006, 27, 5763–5770 CrossRef CAS PubMed.
- L. Chung, D. R. Maestas, A. Lebid, A. Mageau, G. D. Rosson, X. Wu, M. T. Wolf, A. J. Tam, I. Vanderzee, X. Wang, J. I. Andorko, H. Zhang, R. Narain, K. Sadtler, H. Fan, D. Čiháková, C. J. Le Saux, F. Housseau, D. M. Pardoll and J. H. Elisseeff, Sci. Transl. Med., 2020, 12, 539 Search PubMed.
- J. M. Anderson, J. Mater. Sci.: Mater. Med., 2015, 26, 121 CrossRef PubMed.
- L. B. Moore and T. R. Kyriakides, Adv. Exp. Med. Biol., 2015, 865, 109–122 CrossRef CAS PubMed.
- A. J. Pagán and L. Ramakrishnan, Annu. Rev. Immunol., 2018, 36, 639–665 CrossRef PubMed.
- T. R. Kyriakides, H. J. Kim, C. Zheng, L. Harkins, W. Tao and E. Deschenes, Biomed. Mater., 2022, 17(2), 022007 CrossRef CAS PubMed.
- A. Dievernich, P. Achenbach, L. Davies and U. Klinge, Hernia, 2022, 26, 309–323 CrossRef CAS PubMed.
- J. C. Doloff, O. Veiseh, A. J. Vegas, H. H. Tam, S. Farah, M. Ma, J. Li, A. Bader, A. Chiu, A. Sadraei, S. Aresta-Dasilva, M. Griffin, S. Jhunjhunwala, M. Webber, S. Siebert, K. Tang, M. Chen, E. Langan, N. Dholokia, R. Thakrar, M. Qi, J. Oberholzer, D. L. Greiner, R. Langer and D. G. Anderson, Nat. Mater., 2017, 16, 671–680 CrossRef CAS PubMed.
- B. Yang, N. Rutkowski and J. Elisseeff, Biomater. Sci., 2023, 11, 7730–7747 RSC.
- E. M. Moore, D. R. Maestas, C. C. Cherry, J. A. Garcia, H. Y. Comeau, L. Davenport Huyer, S. H. Kelly, A. N. Peña, R. L. Blosser, G. D. Rosson and J. H. Elisseeff, Sci. Adv., 2021, 7, 1–13 Search PubMed.
- J. M. Daley, S. K. Brancato, A. A. Thomay, J. S. Reichner and J. E. Albina, J. Leukocyte Biol., 2009, 87, 59–67 CrossRef PubMed.
- F. O. Martinez and S. Gordon, F1000Prime Rep., 2014, 6, 1–13 Search PubMed.
- M. E. Ogle, C. E. Segar, S. Sridhar and E. A. Botchwey, Exp. Biol. Med., 2016, 241, 1084–1097 CrossRef CAS PubMed.
- M. Orecchioni, Y. Ghosheh, A. B. Pramod and K. Ley, Front. Immunol., 2019, 10, 1–14 CrossRef PubMed.
- Y. Shintani, T. Ito, L. Fields, M. Shiraishi, Y. Ichihara, N. Sato, M. Podaru, S. Kainuma, H. Tanaka and K. Suzuki, Sci. Rep., 2017, 7, 1–14 CrossRef CAS PubMed.
- Y. Liu and T. Segura, Front. Bioeng. Biotechnol., 2020, 8, 1–14 CrossRef PubMed.
- E. M. O’Brien and K. L. Spiller, J. Leukocyte Biol., 2022, 111, 989–1000 CrossRef PubMed.
- F. Heymann, K. T. Von Trotha, C. Preisinger, P. Lynen-Jansen, A. A. Roeth, M. Geiger, L. J. Geisler, A. K. Frank, J. Conze, T. Luedde, C. Trautwein, M. Binnebösel, U. P. Neumann and F. Tacke, JCI Insight, 2019, 4(2), e123862 CrossRef PubMed.
- S. K. Wculek, I. Heras-Murillo, A. Mastrangelo, D. Mañanes, M. Galán, V. Miguel, A. Curtabbi, C. Barbas, N. S. Chandel, J. A. Enríquez, S. Lamas and D. Sancho, Immunity, 2023, 56, 516–530.e9 CrossRef CAS PubMed.
- S.-L. Chen, D. J. Lundy, S.-C. Ruan, H.-C. Chen, Y.-K. Chao, Y.-Y. Cheng, R. P. Prajnamitra, C.-C. Liao, C.-Y. Lin, J. J. Lai and P. C. H. Hsieh, Biomaterials, 2022, 289, 121807 CrossRef CAS PubMed.
- R. Thibaut, L. Orliaguet, T. Ejlalmanesh, N. Venteclef and F. Alzaid, Front. Immunol., 2022, 13, 918747 CrossRef CAS PubMed.
- K. E. Martin and A. J. García, Acta Biomater., 2021, 133, 4–16 CrossRef CAS PubMed.
- D. G. Barone, A. Carnicer-Lombarte, P. Tourlomousis, R. S. Hamilton, M. Prater, A. L. Rutz, I. B. Dimov, G. G. Malliaras, S. P. Lacour, A. A. B. Robertson, K. Franze, J. W. Fawcett and C. E. Bryant, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, 1–10 CrossRef PubMed.
- S. D. Sommerfeld, C. Cherry, R. M. Schwab, L. Chung, D. R. Maestas, P. Laffont, J. E. Stein, A. Tam, S. Ganguly, F. Housseau, J. M. Taube, D. M. Pardoll, P. Cahan and J. H. Elisseeff, Sci. Immunol., 2019, 4(40), eaax4783 CrossRef CAS PubMed.
- T. Rőszer, Mediators Inflammation, 2015, 2015, 816460 CrossRef PubMed.
- A. Viola, F. Munari, R. Sánchez-Rodríguez, T. Scolaro and A. Castegna, Front. Immunol., 2019, 10, 1–16 CrossRef PubMed.
- S. Willenborg, D. E. Sanin, A. Jais, X. Ding, T. Ulas, J. Nüchel, M. Popović, T. MacVicar, T. Langer, J. L. Schultze, A. Gerbaulet, A. Roers, E. J. Pearce, J. C. Brüning, A. Trifunovic and S. A. Eming, Cell Metab., 2021, 33, 2398–2414.e9 CrossRef CAS PubMed.
- J. R. Erlich, E. E. To, R. Luong, F. Liong, S. Liong, O. Oseghale, M. A. Miles, S. Bozinovski, R. D. Brooks, R. Vlahos, S. Chan, J. J. O’Leary, D. A. Brooks and S. Selemidis, Antioxidants, 2022, 11, 1488 CrossRef CAS PubMed.
- J. Huang, J. Xia, L.-L. Huang and Y.-C. Li, Mol. Med. Rep., 2019, 20, 3424–3432 CAS.
- W. Chou, E. Rampanelli, X. Li and J. P.-Y. Ting, Cell. Mol. Immunol., 2022, 19, 337–351 CrossRef CAS PubMed.
- R. van der Burgh and M. Boes, Trends Endocrinol. Metab., 2015, 26, 263–271 CrossRef CAS PubMed.
- A. Olona, S. Leishman and P. K. Anand, Trends Immunol., 2022, 43, 978–989 CrossRef CAS PubMed.
- E. L. Mills, B. Kelly, A. Logan, A. S. H. Costa, M. Varma, C. E. Bryant, P. Tourlomousis, J. H. M. Däbritz, E. Gottlieb, I. Latorre, S. C. Corr, G. McManus, D. Ryan, H. T. Jacobs, M. Szibor, R. J. Xavier, T. Braun, C. Frezza, M. P. Murphy and L. A. O’Neill, Cell, 2016, 167, 457–470.e13 CrossRef CAS PubMed.
- F. Wang, S. Zhang, I. Vuckovic, R. Jeon, A. Lerman, C. D. Folmes, P. P. Dzeja and J. Herrmann, Cell Metab., 2018, 28, 463–475.e4 CrossRef CAS PubMed.
- G. Caputa, L. J. Flachsmann and A. M. Cameron, Immunol. Cell Biol., 2019, 97, 268–278 CrossRef CAS PubMed.
- Y. Zhu, X. Zhang, S. Xie, W. Bao, J. Chen, Q. Wu, X. Lai, L. Liu, S. Xiong and Y. Peng, Immunology, 2022, 167, 576–589 CrossRef CAS PubMed.
- B. Dang, Q. Gao, L. Zhang, J. Zhang, H. Cai, Y. Zhu, Q. Zhong, J. Liu, Y. Niu, K. Mao, N. Xiao, W.-H. Liu, S. Lin, J. Huang, S. C.-C. Huang, P.-C. Ho and S.-C. Cheng, Cell Rep., 2023, 42, 112471 CrossRef CAS PubMed.
- R. Klopfleisch, Acta Biomater., 2016, 43, 3–13 CrossRef CAS PubMed.
- R. Milde, J. Ritter, G. A. Tennent, A. Loesch, F. O. Martinez, S. Gordon, M. B. Pepys, A. Verschoor and L. Helming, Cell Rep., 2015, 13, 1937–1948 CrossRef CAS PubMed.
- K. Ahmadzadeh, M. Vanoppen, C. D. Rose, P. Matthys and C. H. Wouters, Front. Cell Dev. Biol., 2022, 10, 1–22 Search PubMed.
- F. Eslami-Kaliji, N. Hedayat Nia, J. R. T. Lakey, A. M. Smink and M. Mohammadi, Polymers, 2023, 15, 1313 CrossRef CAS PubMed.
- J. Koopsen, S. M. Van Putten, H. Van Veen, D. I. Picavet, T. J. De Vries, R. A. Bank and V. Everts, J. Struct. Biol., 2016, 195, 31–40 CrossRef PubMed.
- R. J. Miron and D. D. Bosshardt, Tissue Eng., Part B, 2018, 24, 53–65 CrossRef PubMed.
- P. J. Brooks, M. Glogauer and C. A. McCulloch, Am. J. Pathol., 2019, 189, 1145–1158 CrossRef CAS PubMed.
- Z. Sheikh, P. J. Brooks, O. Barzilay, N. Fine and M. Glogauer, Materials, 2015, 8, 5671–5701 CrossRef CAS PubMed.
- O. Dufrançais, R. Mascarau, R. Poincloux, I. Maridonneau-Parini, B. Raynaud-Messina and C. Vérollet, Cell. Mol. Life Sci., 2021, 78, 6087–6104 CrossRef PubMed.
- J. Takito and M. Nakamura, Int. J. Mol. Sci., 2020, 21, 6629 CrossRef CAS PubMed.
- A. K. McNally and J. M. Anderson, Cell Fusion in Health and Disease, 2011, vol. 5, pp. 97–111 Search PubMed.
- L. Herrtwich, I. Nanda, K. Evangelou, T. Nikolova, V. Horn, D. Sagar, J. Erny, J. Stefanowski, L. Rogell, C. Klein, K. Gharun, M. Follo, M. Seidl, B. Kremer, N. Münke, J. Senges, M. Fliegauf, T. Aschman, D. Pfeifer, S. Sarrazin, M. H. Sieweke, D. Wagner, C. Dierks, T. Haaf, T. Ness, M. M. Zaiss, R. E. Voll, S. D. Deshmukh, M. Prinz, T. Goldmann, C. Hölscher, A. E. Hauser, A. J. Lopez-Contreras, D. Grün, V. Gorgoulis, A. Diefenbach, P. Henneke and A. Triantafyllopoulou, Cell, 2016, 167, 1264–1280.e18 CrossRef CAS PubMed.
- A. Rodriguez, S. R. MacEwan, H. Meyerson, J. T. Kirk and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2009, 90A, 106–113 CrossRef CAS PubMed.
- A. K. McNally and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2015, 103, 1380–1390 CrossRef PubMed.
- J. J. A. McLeod, B. Baker and J. J. Ryan, Cytokine, 2015, 75, 57–61 CrossRef CAS PubMed.
- J. Yang, B. Jao, A. K. McNally and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2014, 102, 2017–2023 CrossRef PubMed.
- A. K. McNally and J. M. Anderson, Exp. Mol. Pathol., 2011, 91, 673–681 CrossRef CAS PubMed.
- R. Goswami, R. K. Arya, S. Sharma, B. Dutta, D. R. Stamov, X. Zhu and S. O. Rahaman, Sci. Signaling, 2021, 14(707), eabd4077 CrossRef CAS PubMed.
- J. M. Anderson, K. Defife, A. Mcnally, T. Collier and C. Jenney, J. Mater. Sci.: Mater. Med., 1999, 10, 579–588 CrossRef CAS PubMed.
- S. N. Christo, K. R. Diener, J. Manavis, M. A. Grimbaldeston, A. Bachhuka, K. Vasilev and J. D. Hayball, Sci. Rep., 2016, 6, 1–14 CrossRef PubMed.
- L. A. McKiel, K. A. Woodhouse and L. E. Fitzpatrick, MRS Commun., 2020, 10, 55–68 CrossRef CAS.
- G. Zhou and T. Groth, Macromol. Biosci., 2018, 18, 1–15 CrossRef PubMed.
- Y.-S. Kim, S. Shin, E. J. Choi, S. W. Moon, C. K. Jung, Y.-J. Chung and S. H. Lee, J. Invest. Dermatol., 2022, 2, 1–16 CrossRef.
- L. A. McKiel and L. E. Fitzpatrick, ACS Biomater. Sci. Eng., 2018, 4, 3792–3801 CrossRef CAS PubMed.
- Q. H. Zhao, J. M. Anderson, A. Hiltner, G. A. Lodoen and C. R. Payet, J. Biomed. Mater. Res., 1992, 26, 1019–1038 CrossRef CAS PubMed.
- S. Chen, J. A. Jones, Y. Xu, H.-Y. Low, J. M. Anderson and K. W. Leong, Biomaterials, 2010, 31, 3479–3491 CrossRef CAS PubMed.
- W. G. Brodbeck, J. Patel, G. Voskerician, E. Christenson, M. S. Shive, Y. Nakayama, T. Matsuda, N. P. Ziats and J. M. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10287–10292 CrossRef CAS PubMed.
- S. Mezouar, M. Katsogiannou, A. Ben Amara, F. Bretelle and J.-L. Mege, Placenta, 2021, 103, 94–103 CrossRef CAS PubMed.
- L. Reyes and T. G. Golos, Front. Immunol., 2018, 9, 1–8 CrossRef PubMed.
- Y. Ma, Y. Ye, J. Zhang, C. C. Ruan and P. J. Gao, Medicine, 2019, 98, 1–6 Search PubMed.
- M. B. Buechler, W. Fu and S. J. Turley, Immunity, 2021, 54, 903–915 CrossRef CAS PubMed.
- S. Van Linthout, K. Miteva and C. Tschöpe, Cardiovasc. Res., 2014, 102, 258–269 CrossRef CAS PubMed.
- T. A. Wynn and L. Barron, Semin. Liver Dis., 2010, 30, 245–257 CrossRef CAS PubMed.
- J. C. Bonner, Cytokine Growth Factor Rev., 2004, 15, 255–273 CrossRef CAS PubMed.
- D. H. Kim, Y. S. Song, S. Y. Song and C. H. Kim, Arch. Aesthetic Plast. Surg., 2016, 22, 129 CrossRef.
- U. Klueh, D. I. Dorsky and D. L. Kreutzer, Biomaterials, 2005, 26, 1155–1163 CrossRef CAS PubMed.
- M. Giannandrea and W. C. Parks, Dis. Models Mech., 2014, 7, 193–203 CrossRef CAS PubMed.
- J. A. Jones, A. K. McNally, D. T. Chang, L. A. Qin, H. Meyerson, E. Colton, I. L. K. Kwon, T. Matsuda and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2008, 84A, 158–166 CrossRef CAS PubMed.
- J. Padmanabhan, K. Chen, D. Sivaraj, D. Henn, B. A. Kuehlmann, H. C. Kussie, E. T. Zhao, A. Kahn, C. A. Bonham, T. Dohi, T. C. Beck, A. A. Trotsyuk, Z. A. Stern-Buchbinder, P. A. Than, H. S. Hosseini, J. A. Barrera, N. J. Magbual, M. C. Leeolou, K. S. Fischer, S. S. Tigchelaar, J. Q. Lin, D. P. Perrault, M. R. Borrelli, S. H. Kwon, Z. N. Maan, J. C. Y. Dunn, R. Nazerali, M. Januszyk, L. Prantl and G. C. Gurtner, Nat. Biomed. Eng., 2023, 7, 1419–1436 CrossRef CAS PubMed.
- S. MacLauchlan, E. A. Skokos, N. Meznarich, D. H. Zhu, S. Raoof, J. M. Shipley, R. M. Senior, P. Bornstein and T. R. Kyriakides, J. Leukocyte Biol., 2009, 85, 617–626 CrossRef CAS PubMed.
- T. Whitehead-Clarke, R. Karanjia, J. Banks, V. Beynon, S. Parker, D. Sanders, V. Mudera, A. Windsor and A. Kureshi, Hernia, 2022, 26, 297–307 CrossRef CAS PubMed.
- G. Pascual, M. Rodríguez, B. Pérez-Köhler, S. Benito-Martínez, B. Calvo, F. García-Moreno and J. M. Bellón, Hernia, 2020, 24, 1159–1173 CrossRef CAS PubMed.
- R. Liang, A. Fisk, G. King, L. Meyn, X. Xiao and P. Moalli, Acta Biomater., 2022, 143, 310–319 CrossRef CAS PubMed.
- A. M. Artsen, M. Rytel, R. Liang, G. E. King, L. Meyn, S. D. Abramowitch and P. A. Moalli, Acta Biomater., 2019, 96, 203–210 CrossRef CAS PubMed.
- E. E. Anderson, E. Perilli, C. J. Carati and K. J. Reynolds, J. Biomed. Mater. Res., Part B, 2017, 105, 1447–1452 CrossRef CAS PubMed.
- M. Kastellorizios, F. Papadimitrakopoulos and D. J. Burgess, J. Controlled Release, 2015, 202, 101–107 CrossRef CAS PubMed.
- A. J. Vegas, O. Veiseh, J. C. Doloff, M. Ma, H. H. Tam, K. Bratlie, J. Li, A. R. Bader, E. Langan, K. Olejnik, P. Fenton, J. W. Kang, J. Hollister-Locke, M. A. Bochenek, A. Chiu, S. Siebert, K. Tang, S. Jhunjhunwala, S. Aresta-Dasilva, N. Dholakia, R. Thakrar, T. Vietti, M. Chen, J. Cohen, K. Siniakowicz, M. Qi, J. McGarrigle, S. Lyle, D. M. Harlan, D. L. Greiner, J. Oberholzer, G. C. Weir, R. Langer and D. G. Anderson, Nat. Biotechnol., 2016, 34, 345–352 CrossRef CAS PubMed.
- K. Sadtler and J. H. Elisseeff, Biomater. Sci., 2019, 7, 4472–4481 RSC.
- C. Cherry, J. I. Andorko, K. Krishnan, J. C. Mejías, H. H. Nguyen, K. B. Stivers, E. F. Gray-Gaillard, A. Ruta, J. Han, N. Hamada, M. Hamada, I. Sturmlechner, S. Trewartha, J. H. Michel, L. Davenport Huyer, M. T. Wolf, A. J. Tam, A. N. Peña, S. Keerthivasan, C. J. Le Saux, E. J. Fertig, D. J. Baker, F. Housseau, J. M. van Deursen, D. M. Pardoll and J. H. Elisseeff, GeroScience, 2023, 45, 2559–2587 CrossRef CAS PubMed.
- T. Krausgruber, A. Redl, D. Barreca, K. Doberer, D. Romanovskaia, L. Dobnikar, M. Guarini, L. Unterluggauer, L. Kleissl, D. Atzmüller, C. Mayerhofer, A. Kopf, S. Saluzzo, C. X. Lim, P. Rexie, T. Weichhart, C. Bock and G. Stary, Immunity, 2023, 56, 289–306.e7 CrossRef CAS PubMed.
- E. M. Moore, D. R. Maestas, H. Y. Comeau and J. H. Elisseeff, Tissue Eng., Part B, 2021, 27, 39–47 CrossRef CAS PubMed.
- L. S. Saleh and S. J. Bryant, Drug Discovery Today Dis. Model., 2017, 24, 13–21 CrossRef PubMed.
- H. M. Rostam, L. E. Fisher, A. L. Hook, L. Burroughs, J. C. Luckett, G. P. Figueredo, C. Mbadugha, A. C. K. Teo, A. Latif, L. Kämmerling, M. Day, K. Lawler, D. Barrett, S. Elsheikh, M. Ilyas, D. A. Winkler, M. R. Alexander and A. M. Ghaemmaghami, Matter, 2020, 2, 1564–1581 CrossRef.
- K. P. Robb, J. Audet, R. Gandhi and S. Viswanathan, Front. Immunol., 2022, 13, 1–22 Search PubMed.
- K. E. de Goede, K. J. Harber, F. S. Gorki, S. G. S. Verberk, L. A. Groh, E. D. Keuning, E. A. Struys, M. van Weeghel, A. Haschemi, M. P. J. de Winther, X. A. M. H. van Dierendonck and J. Van den Bossche, Biochim. Biophys. Acta, Mol. Basis Dis., 2022, 1868, 166427 CrossRef CAS PubMed.
- S. Koo, B. Szczesny, X. Wan, N. Putluri and N. J. Garg, Front. Immunol., 2018, 9, 202 CrossRef PubMed.
- R. J. Argüello, A. J. Combes, R. Char, J.-P. Gigan, A. I. Baaziz, E. Bousiquot, V. Camosseto, B. Samad, J. Tsui, P. Yan, S. Boissonneau, D. Figarella-Branger, E. Gatti, E. Tabouret, M. F. Krummel and P. Pierre, Cell Metab., 2020, 32, 1063–1075.e7 CrossRef PubMed.
- L. R. Pelgrom, G. M. Davis, S. O’Shaughnessy, E. J. M. Wezenberg, S. I. Van Kasteren, D. K. Finlay and L. V. Sinclair, Cell Rep., 2023, 42, 112828 CrossRef CAS PubMed.
- R. J. W. Arts, B. Novakovic, R. ter Horst, A. Carvalho, S. Bekkering, E. Lachmandas, F. Rodrigues, R. Silvestre, S. C. Cheng, S. Y. Wang, E. Habibi, L. G. Gonçalves, I. Mesquita, C. Cunha, A. van Laarhoven, F. L. van de Veerdonk, D. L. Williams, J. W. M. van der Meer, C. Logie, L. A. O’Neill, C. A. Dinarello, N. P. Riksen, R. van Crevel, C. Clish, R. A. Notebaart, L. A. B. Joosten, H. G. Stunnenberg, R. J. Xavier and M. G. Netea, Cell Metab., 2016, 24, 807–819 CrossRef CAS PubMed.
- S. Bekkering, R. J. W. Arts, B. Novakovic, I. Kourtzelis, C. D. C. C. van der Heijden, Y. Li, C. D. Popa, R. ter Horst, J. van Tuijl, R. T. Netea-Maier, F. L. van de Veerdonk, T. Chavakis, L. A. B. Joosten, J. W. M. van der Meer, H. Stunnenberg, N. P. Riksen and M. G. Netea, Cell, 2018, 172, 135–146.e9 CrossRef CAS PubMed.
- A. Gavlin, A. S. Kierans, J. Chen, C. Song, P. Guniganti and F. S. Mazzariol, Radiographics, 2020, 40, 432–453 CrossRef PubMed.
- S. Capuani, G. Malgir, C. Y. X. Chua and A. Grattoni, Bioeng. Transl. Med., 2022, 1–22 Search PubMed.
- E. N. Zhang, J. Clément, A. Alameri, A. Ng, T. E. Kennedy and D. Juncker, Adv. Mater. Technol., 2021, 6(3), 2000909 CrossRef CAS.
- N. O. Monteiro, M. R. Casanova, J. F. Fangueiro, R. L. Reis and N. M. Neves, Biomed. Mater., 2023, 18, 035008 CrossRef PubMed.
- F. Robotti, S. Bottan, F. Fraschetti, A. Mallone, G. Pellegrini, N. Lindenblatt, C. Starck, V. Falk, D. Poulikakos and A. Ferrari, Sci. Rep., 2018, 8, 10887 CrossRef PubMed.
- E. A. Vogler, Biomaterials, 2012, 33, 1201–1237 CrossRef CAS PubMed.
- L.-C. Xu and C. A. Siedlecki, Biomaterials, 2007, 28, 3273–3283 CrossRef CAS PubMed.
- R. M. Shelton, A. C. Rasmussen and J. E. Davies, Biomaterials, 1988, 9, 24–29 CrossRef CAS PubMed.
- P. Pakshir, M. Alizadehgiashi, B. Wong, N. M. Coelho, X. Chen, Z. Gong, V. B. Shenoy, C. A. McCulloch and B. Hinz, Nat. Commun., 2019, 10, 1850 CrossRef PubMed.
- C. Tanasescu, A. Moisin, A. Mihetiu, D. Serban, A. Costache and D. Bratu, Exp. Ther. Med., 2021, 22, 1–6 Search PubMed.
- J. L. Frodel and S. Lee, Arch. Otolaryngol., Head Neck Surg., 1998, 124, 1219–1223 CrossRef CAS PubMed.
- H. Park, J. W. Choi and W. S. Jeong, J. Craniofac. Surg., 2022, 33, 1394–1399 CrossRef PubMed.
- M. J. R. Virlan, D. Miricescu, A. Totan, M. Greabu, C. Tanase, C. M. Sabliov, C. Caruntu and B. Calenic, J. Chem., 2015 DOI:10.1155/2015/525832.
- T. Weigel, T. Schmitz, T. Pfister, S. Gaetzner, M. Jannasch, R. Al-Hijailan, S. Schürlein, S. Suliman, K. Mustafa and J. Hansmann, Sci. Rep., 2018, 8, 14545 CrossRef PubMed.
- F. Santanelli di Pompeo, M. Sorotos, M. W. Clemens, G. Firmani, E. Athanasopoulos, K. Arctander, B. Berenguer, K. Bozikov, A. Cardoso, Å. E. Nord, C. Filip, A. Georgeskou Romania, C. Heitman, O. Kaarela, M. Kolenda, M. Hamdi, L. Lantieri, D. Lumenta, N. Mercer, E. Ruegg, F. Santanelli di Pompeo, Z. Stanec, R. Van Der Hulst and J. J. Vranckx, Aesthetic Surg. J., 2021, 41, 1014–1025 CrossRef PubMed.
- E. B. Lynch, R. C. DeCoster, K. S. Vyas, B. D. Rinker, M. Yang, H. C. Vasconez and M. W. Clemens, Ann. Breast Surg., 2021, 5, 30 CrossRef PubMed.
- N. Castel, T. Soon-Sutton, P. Deptula, A. Flaherty and F. D. Parsa, Arch. Plast. Surg., 2015, 42, 186–193 CrossRef PubMed.
- N. G. Welch, D. A. Winkler and H. Thissen, Adv. Drug Delivery Rev., 2020, 167, 109–120 CrossRef CAS PubMed.
- D. Dong, C. Tsao, H. C. Hung, F. Yao, C. Tang, L. Niu, J. Ma, J. MacArthur, A. Sinclair, K. Wu, P. Jain, M. R. Hansen, D. Ly, S. G. H. Tang, T. M. Luu, P. Jain and S. Jiang, Sci. Adv., 2021, 7, 1–10 Search PubMed.
- L. Zhang, Z. Cao, T. Bai, L. Carr, J.-R. Ella-Menye, C. Irvin, B. D. Ratner and S. Jiang, Nat. Biotechnol., 2013, 31, 553–556 CrossRef CAS PubMed.
- D. Faulón Marruecos, L. S. Saleh, H. H. Kim, S. J. Bryant, D. K. Schwartz and J. L. Kaar, ACS Appl. Bio Mater., 2019, 4698–4702 CrossRef PubMed.
- K. Burugapalli, S. Wijesuriya, N. Wang and W. Song, J. Biomed. Mater. Res., Part A, 2018, 106, 1072–1081 CrossRef CAS PubMed.
- S. M. Ruppert, T. R. Hawn, A. Arrigoni, T. N. Wight and P. L. Bollyky, Immunol. Res., 2014, 58, 186–192 CrossRef CAS PubMed.
- H. AlKhoury, A. Hautmann, F. Erdmann, G. Zhou, S. Stojanović, S. Najman and T. Groth, J. Biomed. Mater. Res., Part A, 2020, 108, 1099–1111 CrossRef CAS PubMed.
- S. Hu, F. D. Martinez-Garcia, B. N. Moeun, J. K. Burgess, M. C. Harmsen, C. Hoesli and P. de Vos, Mater. Sci. Eng., C, 2021, 123, 112009 CrossRef CAS PubMed.
- S. Mukherjee, B. Kim, L. Y. Cheng, M. D. Doerfert, J. Li, A. Hernandez, L. Liang, M. I. Jarvis, P. D. Rios, S. Ghani, I. Joshi, D. Isa, T. Ray, T. Terlier, C. Fell, P. Song, R. N. Miranda, J. Oberholzer, D. Y. Zhang and O. Veiseh, Nat. Biomed. Eng., 2023, 7, 867–886 CrossRef CAS PubMed.
- D. A. Alagpulinsa, J. J. L. Cao, R. K. Driscoll, R. F. Sîrbulescu, M. F. E. Penson, M. Sremac, E. N. Engquist, T. A. Brauns, J. F. Markmann, D. A. Melton and M. C. Poznansky, Am. J. Transplant., 2019, 19, 1930–1940 CrossRef CAS PubMed.
- R. P. Tan, N. Hallahan, E. Kosobrodova, P. L. Michael, F. Wei, M. Santos, Y. T. Lam, A. H. P. Chan, Y. Xiao, M. M. M. Bilek, P. Thorn and S. G. Wise, ACS Appl. Mater. Interfaces, 2020, 12, 56908–56923 CrossRef CAS PubMed.
- A. E. Anderson, I. Wu, A. J. Parrillo, M. T. Wolf, D. R. Maestas, I. Graham, A. J. Tam, R. M. Payne, J. Aston, C. M. Cooney, P. Byrne, D. S. Cooney and J. H. Elisseeff, npj Regener. Med., 2022, 7, 6 CrossRef CAS PubMed.
- J. S. Lewis, K. Roy and B. G. Keselowsky, MRS Bull., 2014, 39, 25–34 CrossRef PubMed.
- L. W. Norton, H. E. Koschwanez, N. A. Wisniewski, B. Klitzman and W. M. Reichert, J. Biomed. Mater. Res., Part A, 2007, 81A, 858–869 CrossRef CAS PubMed.
- Y. Zhong and R. V. Bellamkonda, Brain Res., 2007, 1148, 15–27 CrossRef CAS PubMed.
- K. Battiston, I. Parrag, M. Statham, D. Louka, H. Fischer, G. Mackey, A. Daley, F. Gu, E. Baldwin, B. Yang, B. Muirhead, E. A. Hicks, H. Sheardown, L. Kalachev, C. Crean, J. Edelman, J. P. Santerre and W. Naimark, Nat. Commun., 2021, 12, 1–17 CrossRef PubMed.
- J. C. Quarterman, S. M. Geary and A. K. Salem, Eur. J. Pharm. Biopharm., 2021, 159, 21–35 CrossRef CAS PubMed.
- J. D. Weaver, Y. Song, E. Y. Yang, C. Ricordi, A. Pileggi, P. Buchwald and C. L. Stabler, Tissue Eng., Part A, 2015, 21, 2250–2261 CrossRef CAS PubMed.
- P. Pakshir, F. Younesi, K.-A. Wootton, K. Battiston, G. Whitton, B. Ilagan, D. Louka, M. Statham, G. Mackey, A. Daley, I. Parrag, W. Naimark and B. Hinz, Biomaterials, 2022, 286, 121586 CrossRef CAS PubMed.
- J. A. W. Polderman, V. Farhang-Razi, S. van Dieren, P. Kranke, J. H. DeVries, M. W. Hollmann, B. Preckel and J. Hermanides, Anaesthesia, 2019, 74, 929–939 CrossRef CAS PubMed.
- L. Nabai, A. Ghahary and J. Jackson, Bioengineering, 2023, 10, 298 CrossRef CAS PubMed.
- A. Fayzullin, S. Churbanov, N. Ignatieva, O. Zakharkina, M. Tokarev, D. Mudryak, Y. Khristidis, M. Balyasin, A. Kurkov, E. N. Golubeva, N. A. Aksenova, T. Dyuzheva, P. Timashev, A. Guller and A. Shekhter, Biomedicines, 2021, 9, 853 CrossRef CAS PubMed.
- B. S. Jeon, B. H. Shin, B. K. Huh, B. H. Kim, S.-N. Kim, H. B. Ji, S. H. Lee, S. I. Kang, J. H. Shim, S. M. Kang, J. C. Lee, K. S. Lee, C. Y. Heo and Y. Bin Choy, J. Ind. Eng. Chem., 2018, 63, 168–180 CrossRef CAS.
- S.-Y. Nam, H. B. Ji, B. H. Shin, P. N. Chien, N. Donmez, X. R. Zhang, B. K. Huh, M. J. Kim, Y. Bin Choy and C. Y. Heo, Materials, 2021, 14, 3917 CrossRef CAS PubMed.
- W. Li, B. He, W. Dai, Q. Zhang and Y. Liu, Int. Ophthalmol., 2014, 34, 465–476 CrossRef PubMed.
- S. Park, M. Park, B. H. Kim, J. E. Lee, H. J. Park, S. H. Lee, C. G. Park, M. H. Kim, R. Kim, E. H. Kim, C. Y. Heo and Y. Bin Choy, J. Controlled Release, 2015, 200, 125–137 CrossRef CAS PubMed.
- L. Nabai, A. Ghahary and J. Jackson, Pharmaceutics, 2022, 14, 1546 CrossRef CAS PubMed.
- S. Farah, J. C. Doloff, P. Müller, A. Sadraei, H. J. Han, K. Olafson, K. Vyas, H. H. Tam, J. Hollister-Lock, P. S. Kowalski, M. Griffin, A. Meng, M. McAvoy, A. C. Graham, J. McGarrigle, J. Oberholzer, G. C. Weir, D. L. Greiner, R. Langer and D. G. Anderson, Nat. Mater., 2019, 18, 892–904 CrossRef CAS PubMed.
- M. Duvvuri, K. Motz, M. Murphy, M. Feeley, D. Ding, A. Lee, J. H. Elisseeff and A. T. Hillel, Biomater. Sci., 2019, 7, 1863–1874 RSC.
- K. M. Motz, I. A. Lina, I. Samad, M. K. Murphy, M. Duvvuri, R. J. Davis, A. Gelbard, L. Chung, Y. Chan-Li, S. Collins, J. D. Powell, J. H. Elisseeff, M. R. Horton and A. T. Hillel, JCI Insight, 2023, 8(11), e158456 CrossRef PubMed.
- D. Hachim, S. T. LoPresti, C. C. Yates and B. N. Brown, Biomaterials, 2017, 112, 95–107 CrossRef CAS PubMed.
- D. R. Maestas, L. Chung, J. Han, X. Wang, S. D. Sommerfeld, S. H. Kelly, E. Moore, H. H. Nguyen, J. C. Mejías, A. N. Peña, H. Zhang, J. S. T. Hooks, A. F. Chin, J. I. Andorko, C. A. Berlinicke, K. Krishnan, Y. Choi, A. E. Anderson, R. Mahatme, C. Mejia, M. Eric, J. Woo, S. Ganguly, D. J. Zack, L. Zhao, E. J. Pearce, F. Housseau, D. M. Pardoll and J. H. Elisseeff, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(8), e2211703120 CrossRef CAS PubMed.
- M. Dragovic, M. Pejovic, J. Stepic, S. Colic, B. Dozic, S. Dragovic, M. Lazarevic, N. Nikolic, J. Milasin and B. Milicic, Clin. Oral Investig., 2020, 24, 1527–1541 CrossRef PubMed.
- F. Selvi, S. Cakarer, T. Can, S. İ. Kirli Topcu, A. Palancioglu, B. Keskin, B. Bilgic, M. Yaltirik and C. Keskin, J. Istanbul Univ. Fac. Dent., 2016, 50, 35–42 CAS.
- M. Alves de Oliveira, A. Arcanjo, F. Castro, J. C. Fernandes and G. V. Fernandes, Surgeries, 2024, 5, 350–366 CrossRef.
- F. Köckerling, N. N. Alam, S. A. Antoniou, I. R. Daniels, F. Famiglietti, R. H. Fortelny, M. M. Heiss, F. Kallinowski, I. Kyle-Leinhase, F. Mayer, M. Miserez, A. Montgomery, S. Morales-Conde, F. Muysoms, S. K. Narang, A. Petter-Puchner, W. Reinpold, H. Scheuerlein, M. Smietanski, B. Stechemesser, C. Strey, G. Woeste and N. J. Smart, Hernia, 2018, 22, 249–269 CrossRef PubMed.
- S. Trippoli, E. Caccese, G. Tulli, P. Ipponi, C. Marinai and A. Messori, Int. J. Surg., 2018, 52, 278–284 CrossRef PubMed.
- S. K. Kamarajah, S. J. Chapman, J. Glasbey, D. Morton, N. Smart, T. Pinkney and A. Bhangu, BJS Open, 2018, 2, 371–380 CrossRef CAS PubMed.
- F. Köckerling, N. N. Alam, S. K. Narang, I. R. Daniels and N. J. Smart, Front. Surg., 2015, 2, 1–5 Search PubMed.
- G. Passot, J. Margier, A. Kefleyesus, P. Rousset, P. Ortega-Deballon, Y. Renard, S. Bin and L. Villeneuve, BMJ Open, 2022, 12, e061184 CrossRef PubMed.
- U. Klinge, B. Klosterhalfen, M. Müller and V. Schumpelick, Eur. J. Surg., 1999, 165, 665–673 CrossRef CAS PubMed.
- S. Öberg, K. Andresen and J. Rosenberg, Surg. Innov., 2017, 24, 289–298 CrossRef PubMed.
- J. Henderson and S. O’Reilly, Trends Endocrinol. Metab., 2021, 32, 639–653 CrossRef CAS PubMed.
- A. Hooftman, C. G. Peace, D. G. Ryan, E. A. Day, M. Yang, A. F. McGettrick, M. Yin, E. N. Montano, L. Huo, J. E. Toller-Kawahisa, V. Zecchini, T. A. J. Ryan, A. Bolado-Carrancio, A. M. Casey, H. A. Prag, A. S. H. Costa, G. De Los Santos, M. Ishimori, D. J. Wallace, S. Venuturupalli, E. Nikitopoulou, N. Frizzell, C. Johansson, A. Von Kriegsheim, M. P. Murphy, C. Jefferies, C. Frezza and L. A. J. O’Neill, Nature, 2023, 615, 490–498 CrossRef CAS PubMed.
- C. Diskin, A. Zotta, S. E. Corcoran, V. J. Tyrrell, Z. Zaslona, V. B. O’Donnell and L. A. J. O’Neill, J. Immunol., 2021, 207, 2561–2569 CrossRef CAS PubMed.
- M. D. Kornberg, P. Bhargava, P. M. Kim, V. Putluri, A. M. Snowman, N. Putluri, P. A. Calabresi and S. H. Snyder, Science, 2018, 360, 449–453 CrossRef CAS PubMed.
- D. G. Ryan, M. P. Murphy, C. Frezza, H. A. Prag, E. T. Chouchani, L. A. O’Neill and E. L. Mills, Nat. Metab., 2019, 1, 16–33 CrossRef CAS PubMed.
- D. G. Ryan and L. A. J. O’Neill, Annu. Rev. Immunol., 2020, 38, 289–313 CrossRef CAS PubMed.
- L. A. J. O’Neill and M. N. Artyomov, Nat. Rev. Immunol., 2019, 19, 273–281 CrossRef PubMed.
- C. G. Peace and L. A. J. O’Neill, J. Clin. Invest., 2022, 132(2), e148548 CrossRef CAS PubMed.
- J. Lin, J. Ren, D. S. Gao, Y. Dai and L. Yu, Front. Chem., 2021, 9 Search PubMed.
- J. Shi and C. Cai, Front. Immunol., 2022, 13, 1–9 Search PubMed.
- W. He, A. Henne, M. Lauterbach, E. Geißmar, F. Nikolka, C. Kho, A. Heinz, C. Dostert, M. Grusdat, T. Cordes, J. Härm, O. Goldmann, A. Ewen, C. Verschueren, J. Blay-Cadanet, R. Geffers, H. Garritsen, M. Kneiling, C. K. Holm, C. M. Metallo, E. Medina, Z. Abdullah, E. Latz, D. Brenner and K. Hiller, Nat. Metab., 2022, 4, 524–533 CrossRef CAS PubMed.
- F. Chen, W. A. M. Elgaher, M. Winterhoff, K. Büssow, F. H. Waqas, E. Graner, Y. Pires-Afonso, L. Casares Perez, L. de la Vega, N. Sahini, L. Czichon, W. Zobl, T. Zillinger, M. Shehata, S. Pleschka, H. Bähre, C. Falk, A. Michelucci, S. Schuchardt, W. Blankenfeldt, A. K. H. Hirsch and F. Pessler, Nat. Metab., 2022, 4, 534–546 CrossRef CAS PubMed.
- M. Bambouskova, L. Gorvel, V. Lampropoulou, A. Sergushichev, E. Loginicheva, K. Johnson, D. Korenfeld, M. E. Mathyer, H. Kim, L.-H. Huang, D. Duncan, H. Bregman, A. Keskin, A. Santeford, R. S. Apte, R. Sehgal, B. Johnson, G. K. Amarasinghe, M. P. Soares, T. Satoh, S. Akira, T. Hai, C. de Guzman Strong, K. Auclair, T. P. Roddy, S. A. Biller, M. Jovanovic, E. Klechevsky, K. M. Stewart, G. J. Randolph and M. N. Artyomov, Nature, 2018, 556, 501–504 CrossRef CAS PubMed.
- E. L. Mills, D. G. Ryan, H. A. Prag, D. Dikovskaya, D. Menon, Z. Zaslona, M. P. Jedrychowski, A. S. H. Costa, M. Higgins, E. Hams, J. Szpyt, M. C. Runtsch, M. S. King, J. F. McGouran, R. Fischer, B. M. Kessler, A. F. McGettrick, M. M. Hughes, R. G. Carroll, L. M. Booty, E. V. Knatko, P. J. Meakin, M. L. J. Ashford, L. K. Modis, G. Brunori, D. C. Sévin, P. G. Fallon, S. T. Caldwell, E. R. S. Kunji, E. T. Chouchani, C. Frezza, A. T. Dinkova-Kostova, R. C. Hartley, M. P. Murphy and L. A. O’Neill, Nature, 2018, 556, 113–117 CrossRef CAS PubMed.
- M. C. Runtsch, S. Angiari, A. Hooftman, R. Wadhwa, Y. Zhang, Y. Zheng, J. S. Spina, M. C. Ruzek, M. A. Argiriadi, A. F. McGettrick, R. S. Mendez, A. Zotta, C. G. Peace, A. Walsh, R. Chirillo, E. Hams, P. G. Fallon, R. Jayaraman, K. Dua, A. C. Brown, R. Y. Kim, J. C. Horvat, P. M. Hansbro, C. Wang and L. A. J. O’Neill, Cell Metab., 2022, 34, 487–501.e8 CrossRef CAS PubMed.
- C. V. Maduka, A. V. Makela, A. Tundo, E. Ural, K. B. Stivers, M. M. Kuhnert, M. Alhaj, E. Hoque Apu, N. Ashammakhi, K. D. Hankenson, R. Narayan, J. H. Elisseeff and C. H. Contag, Bioact. Mater., 2024, 40, 64–73 CAS.
- R. P. Allen, A. Bolandparvaz, J. A. Ma, V. A. Manickam and J. S. Lewis, ACS Biomater. Sci. Eng., 2018, 4, 900–918 CrossRef CAS PubMed.
- C. V. Maduka, M. Alhaj, E. Ural, M. M. Kuhnert, O. M. Habeeb, A. L. Schilmiller, K. D. Hankenson, S. B. Goodman, R. Narayan and C. H. Contag, ACS Biomater. Sci. Eng., 2023, 9, 932–943 CrossRef CAS PubMed.
- X. Shi, H. Zhou, J. Wei, W. Mo, Q. Li and X. Lv, Redox Biol., 2022, 58, 102553 CrossRef CAS PubMed.
- L. Tretter, A. Patocs and C. Chinopoulos, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 1086–1101 CrossRef CAS PubMed.
- L. Davenport Huyer, S. Mandla, Y. Wang, S. B. Campbell, B. Yee, C. Euler, B. F. Lai, A. D. Bannerman, D. S. Y. Lin, M. Montgomery, K. Nemr, T. Bender, S. Epelman, R. Mahadevan and M. Radisic, Adv. Funct. Mater., 2021, 31, 2003341 CrossRef CAS PubMed.
- L. Davenport Huyer, S. Pascual-Gil, Y. Wang, S. Mandla, B. Yee and M. Radisic, Adv. Funct. Mater., 2020, 30(44), 1909331 CrossRef CAS.
- Y. Shou, S. B. Campbell, A. Lam, A. J. Lausch, J. P. Santerre, M. Radisic and L. Davenport Huyer, ACS Appl. Polym. Mater., 2021, 3, 1943–1955 CrossRef CAS.
- V. Arias, A. Höglund, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2014, 15, 391–402 CrossRef CAS PubMed.
- W. R. Lykins, D. A. Bernards, E. B. Schlesinger, K. Wisniewski and T. A. Desai, Polymer, 2022, 262, 125473 CrossRef CAS.
- A. I. Visan, G. Popescu-Pelin and G. Socol, Polymers, 2021, 13(8), 1272 CrossRef CAS PubMed.
Footnote |
| † Contributed equally to work. |
| This journal is © The Royal Society of Chemistry 2024 |