
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDispersing uncharged cellulose nanocrystals through a precipitation surface modification route using oligosaccharides†
Megan G.
Roberts‡
 a,
Elina
Niinivaara‡
ab,
Timo
Pääkkönen
c,
Cameron W.
King
a,
Eero
Kontturi
b and
Emily D.
Cranston
*ade
a,
Elina
Niinivaara‡
ab,
Timo
Pääkkönen
c,
Cameron W.
King
a,
Eero
Kontturi
b and
Emily D.
Cranston
*ade
aDepartment of Wood Science, University of British Columbia, 2424 Main Mall, Vancouver, BC V6T 1Z4, Canada. E-mail: emily.cranston@ubc.ca
bDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, P.O. Box 16300, FI-0076 Aalto, Espoo, Finland
cNordic Bioproducts Group Oy, Tietotie 1, 02150 Espoo, Finland
dDepartment of Chemical and Biological Engineering, University of British Columbia, 2424 Main Mall, Vancouver, BC V6T 1Z4, Canada
eUBC Bioproducts Institute, 2385 East Mall, Vancouver BC, V6T 1Z4, Canada
First published on 30th January 2024
Abstract
The trend to replace petrochemical materials with sustainable alternatives has increased interest in plant-based particles like cellulose nanocrystals (CNCs). A remarkably simple and effective method for producing uncharged CNCs involves solid-state hydrolysis using hydrochloric acid gas (HCl(g)). While this chemistry results in HCl(g)-CNCs produced at very high yields (>97%), they cannot be easily dispersed as individual nanoparticles. Here, the potential of using oligosaccharide surface modifiers as dispersing agents for HCl(g)-CNCs to yield isolated and colloidally stable CNCs is investigated. Importantly, the cello-oligosaccharide surface modifiers used were externally-produced and had very low charge. By increasing the amount of oligosaccharide added relative to HCl(g)-CNCs, it was possible to proportionally increase the degree to which the CNC surface was modified. This surface modification resulted in ubiquitous improvements to the dispersibility of HCl(g)-CNCs. We also applied this surface modification to uncharged CNCs produced using aqueous hydrochloric acid (i.e., HCl(aq)-CNCs) and observed marked improvements to their colloidal stability in aqueous media that did not trend with increasing charge but rather with oligosaccharide content. Overall, this study indicates the applicability of an easily scalable modification route that opens the door for expanded CNC functionality and tailoring colloidal stability of these versatile materials.
Introduction
Cellulose nanocrystals (CNCs) are a sustainable platform at the forefront of current research initiatives towards green polymer materials. With a typical length of 50 to 350 nm and width of 5 to 20 nm,1 these nanoscale particles have been used to improve the performance of cement,2 cosmetic products,3 emulsion-based coatings, adhesives, and latexes4,5 as well as water remediation filters.6 They have also shown promise as the basis for drug delivery and medical imaging agents in addition to scaffolds for tissue regeneration.7,8The most common method for producing uniform CNCs from cellulose involves partial hydrolysis using 64 wt% H2SO4. This method chemically modifies the cellulose surface, generating partially sulphated (and thus charged) nanoparticles through esterification (S-CNCs). Suspensions of charged S-CNCs form stable and translucent aqueous dispersions at low concentrations, carrying a blueish hue that passes to semi-opaque with increasing concentration.
An alternative to producing CNCs using H2SO4(aq) is hydrolysis with aqueous HCl.9,10 This method has the advantage of generating CNCs with unmodified surface chemistry (i.e., “native cellulose” nanoparticles). The resulting nanoparticles are comparable to S-CNCs in both crystallinity and dimension; however, because they are uncharged, these CNCs are colloidally unstable and aggregate and sediment quickly when dispersed in aqueous media.11
Recently, Kontturi and coworkers described a remarkably simple, yet efficient method for producing high quality CNCs from Whatman 1 filter paper using hydrochloric acid gas.12 In this case, surface-bound ambient water serves as the solvent for hydrolysis, the hydrochloric acid used is recyclable, and HCl(g)-CNC yields approach quantitative. This reaction has even been scaled up from its origins using benchtop desiccators to now involve large, dedicated reactors which pressurize hydrochloric acid to reduce the necessary reaction times from 24 to 1.5 hours.13 Nevertheless, the hydrolyzed material generated using this technique does not contain isolated nanoparticles. In fact, individual HCl(g)-CNCs produced in this way cannot be isolated without excessive mechanical energy input or post-hydrolysis surface modification. The initial reports from Kontturi et al. describe the necessity of formic acid as a dispersant combined with prolonged bath sonication (i.e., tens of hours) to liberate individual nanoparticles from the hydrolyzed cellulose,12 while other attempts with alternative dispersion methods have failed to give appreciable yields of discrete nanoparticles.13,14
In 2019 though, a report described using 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) radical-mediated oxidation in combination with hydrochloric acid gas hydrolysis to produce carboxylated HCl(g)-CNCs with a yield >80%.15 These CNCs “unhinged” in water to form stable aqueous dispersions. While far from ideal in terms of cost, sustainability, and ease-of-purification, the use of TEMPO-mediated oxidation is the current “gold standard” to enable the aqueous dispersion of CNCs produced without charge. This follows from the fact that TEMPO-mediated oxidation is a common pre-treatment in the production of cellulose nanofibrils (CNFs) where the introduced surface charge facilitates defibrillation of nano-width fibres from a macroscopic cellulose matrix.16,17 Another report from 2023 indicated that a phosphoric acid/monosodium phosphate dihydrate/urea pre-treatment of the cellulose fibers before the HCl(g) hydrolysis yielded equally liberated CNCs but with exceptionally high surface charge (ca. 2000 mmol kg−1).18
In 2021, our group published a proof-of-concept paper demonstrating that the surface of S-CNCs could be modified in a controlled manner in situ during the acid hydrolysis process by using externally produced cellulose phosphate oligosaccharides.19 This was achieved by leveraging the selective solubility of oligosaccharides, which are soluble at a low pH (i.e., during the cellulose hydrolysis) but become insoluble and precipitate onto CNC surfaces when the pH increases during quenching of the reaction with water. It was even possible to tune the CNC surface coverage by varying the degree of polymerization (DP) of the oligosaccharides. This modification resulted in CNCs with altered surface functionality, surface charge content, water binding capacity, and suspension viscosity because of the deposited oligosaccharides, while apparent particle size, zeta potential, crystallinity, and colloidal behavior remained unchanged.
Here, we investigate the potential of using oligosaccharide surface modifiers as dispersing agents for HCl(g)-CNCs in efforts to yield isolated and colloidally stable CNCs in lieu of TEMPO-mediated oxidation. Importantly, these cello-oligosaccharide surface modifiers were externally-produced and had very low sulphate and phosphate group degrees of substitution. Because it is not possible to use our in situ method to precipitate oligosaccharides on HCl(g)-CNCs that are made entirely in the gas phase, a post-hydrolysis method in liquid was developed. We hypothesize that the deposited oligosaccharides introduce surface charge, and potentially steric stabilization, to uncharged CNCs. By increasing the amount of oligosaccharide added relative to HCl(g)-CNCs, it was possible to proportionally increase the degree to which the CNC surface was modified. This surface modification resulted in ubiquitous improvements to the isolation and dispersibility of HCl(g)-CNCs produced using hydrochloric acid gas as well as the colloidal stability of uncharged CNCs produced using aqueous hydrochloric acid (i.e., HCl(aq)-CNCs).
Materials and methods
Materials
Whatman glass microfiber filter papers (GF/B and GF/D) were purchased from GE Healthcare Life Sciences Canada. All of the following chemicals/consumables were purchased from Sigma-Aldrich (Canada): Avicel PH-101 microcrystalline cellulose (MCC), cellulose membrane dialysis tubing (14 kDa Mw cut-off), 85 wt% H3PO4, 96 wt% H2SO4, NaCl, 30% H2O2, DMSO, methanol and phenyl isocyanate (>98%) as well as TraceCERT® standards for ICP-OES, 1000 mg L−1 P in H2O and 1000 mg mL−1 S in H2O. Compressed HCl gas (99.8% purity) was purchased from Linde (former AGA, Sweden), NaOH was purchased from AKA Chemicals (Finland), and poly(allylamine hydrochloride) (PAH, Mw 120 000–200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was purchased from PolySciences (PA, USA) and used as received. All water used was MilliQ grade (Barnstead™ GenPure™ Pro, Thermo Fisher Scientific, MA, USA) with a resistivity of 18.2 MΩ cm (referred to henceforth as ‘purified water’).
000 g mol−1) was purchased from PolySciences (PA, USA) and used as received. All water used was MilliQ grade (Barnstead™ GenPure™ Pro, Thermo Fisher Scientific, MA, USA) with a resistivity of 18.2 MΩ cm (referred to henceforth as ‘purified water’).
Oligosaccharide preparation
Cellulose phosphate oligosaccharides were prepared through a H3PO4(aq) hydrolysis of MCC as previously described.20 Briefly, 48 g of MCC (Avicel PH-101) was wetted with 35 g of purified water and added to 900 mL of 85 wt% H3PO4. After 18 h of hydrolysis at room temperature, the mixture was filtered using a Büchner funnel with a GF/B grade glass fibre filter paper to remove any insoluble cellulose fragments. This removal is important because otherwise the solids gel and make the substrate inaccessible. After four weeks of hydrolysis, the mixture was again filtered using a Büchner funnel but with a GF/D grade glass fibre filter paper. The acid dissolved oligosaccharides were precipitated by adding 900 mL of purified water to the mixture and the precipitate was collected via filtration through a GF/B grade glass fibre filter paper. The precipitated oligosaccharides were then redispersed into a small volume of purified water, placed in cellulose membrane dialysis tubing and dialysed against purified water to remove any excess acid. To determine molecular weight data for this sample, a cellulose carbanilation reaction was performed to produce cellulose tricarbanilate (tricarbanilated anhydroglucose unit (AGU) molecular weight = 519 g mol−1) using conditions optimized for a full phenyl isocyanate substitution with minimal cellulose degradation20 making DMSO-soluble oligosaccharide tricarbanilates (GPC (DMSO/LiBr): Mw,GPC = 3200 g mol−1, Đ = 1.5). For full experimental details refer to the experimental section on molecular weight distribution and gel permeation chromatography (GPC). The degree of substitution (DS) of phosphorus for these oligosaccharides was determined to be 0.0028 ± 0.0001 using elemental analysis.HCl gas hydrolysis of cellulose
Hydrogen chloride gas hydrolysis to prepare cellulose at its leveling-off degree of polymerization (LODP; HCl(g)-CNCs) was carried out as reported elsewhere.13 Briefly, 10 pieces of Whatman ® qualitative filter paper, Grade 1 filter paper were placed into a 1 L pressure resistant glass media bottle (Duran pressure plus (−1 to 1.5 bar), Sigma Aldrich) reactor equipped with an inlet and outlet valve. A vacuum was created inside the reactor and the filter paper was degassed 5 times with HCl gas. Once the degassing was finished, 1 bar of HCl gas was added, and the reactor was removed from the gas line. The gas line was thoroughly flushed using both compressed air and nitrogen, and excess HCl gas was neutralized by bubbling it directly into an alkaline solution (50% NaOH). After 20 h of hydrolysis, the reactor was placed back on the gas line, the HCl gas was evacuated from the reactor and the reactor was flushed with compressed air. The hydrolysed Whatman filter paper was immediately submerged into an excess of purified water and rinsed twice for 15 minutes to remove any residual acid. The samples were then dried in a fumehood overnight, and pulverized using a Wiley Mill (Model M02, Arthur H. Thomas Company, PA, USA) with a 30 size mesh screen.Aqueous HCl hydrolysis of cellulose
HCl(aq)-CNCs were prepared using a slightly modified method to that reported by Abitbol et al.21 Briefly, 10 g of MCC was mixed with 100 mL of 8 wt% HCl at 100 °C, and hydrolysis was allowed to proceed for 15 min. The hydrolysis reaction was then stopped by abruptly cooling the mixture by immersing it into an ice bath. The hydrolysis mixture was then filtered through a GF/B grade glass fibre filter paper to remove the acid. The solids were washed with an excess of purified water until the pH of the filtrate was above pH 5. The washed solids were then blended in 200 mL purified water for 30 min to liberate the CNCs from any residual, unhydrolyzed MCC. To separate the isolated HCl(aq)-CNCs from the unhydrolyzed MCC, the blended mixture was centrifuged for 5 min at 600 × g and the supernatant containing CNCs was collected. The solid fraction was resuspended in 100–150 mL purified water and centrifuged again, after which the supernatant was once again collected. This process was repeated until the supernatant remained clear, indicating CNCs were no longer being separated from the solids fraction.Oligosaccharide precipitation onto CNCs
Both HCl(g)-CNCs and HCl(aq)-CNCs were modified with cellulose phosphate oligosaccharides in varying mass ratios: 1:0.2, 1:0.5 and 1:1 CNCs to oligosaccharides. Fig. 1 shows a schematic representation of this post-hydrolysis modification of HCl-CNCs. Due to the lack of surface charge on CNCs hydrolysed using HCl in either the gas or aqueous state, a significant amount of sonication energy was first introduced to the CNC dispersions to improve CNC surface accessibility for the oligosaccharides. To begin, 0.18 wt% CNC dispersions (i.e., 90 mg of CNCs in 50 g of purified water) were sonicated for a total of 15 min (using a repeated cycle with 1 min sonication, 1 min rest) at 60% amplitude using a microtip sonicator (Sonifier 450 Probe Sonicator, Branson Ultrasonics, CT, USA) for a total energy input of 60 kJ g−1 mL−1 cellulose. Next, either 18 mg (1 CNC:0.2 oligosaccharide mass ratio), 45 mg (1 CNC:0.5 oligosaccharide ratio), or 90 mg (1 CNC:1 oligosaccharide ratio) of oligosaccharides was dissolved in 5 mL of 64 wt% H2SO4 by bath sonicating (Symphony Ultrasonic Cleaner, VWR, PA, USA) for 4 min at 23 °C. The oligosaccharide solution was then slowly added to the CNC dispersion which was under continuous stirring. The drastic change in pH upon dilution of the oligosaccharide solution resulted in the precipitation of the oligosaccharides onto the available CNC surfaces. The oligosaccharide modified CNC dispersions were then probe sonicated at 60% amplitude for an additional 2 min (using a repeated cycle with 1 min sonication, 1 min rest), after which the dispersions were placed inside cellulose membrane dialysis tubing, and dialysed against purified water to remove any residual H2SO4(aq). Dialysis was stopped when the conductivity of the dialysis water remained constant. No visual sedimentation of insoluble oligosaccharides or unhydrolyzed cellulose was visible in the dialysis tubing at the end of the purification.
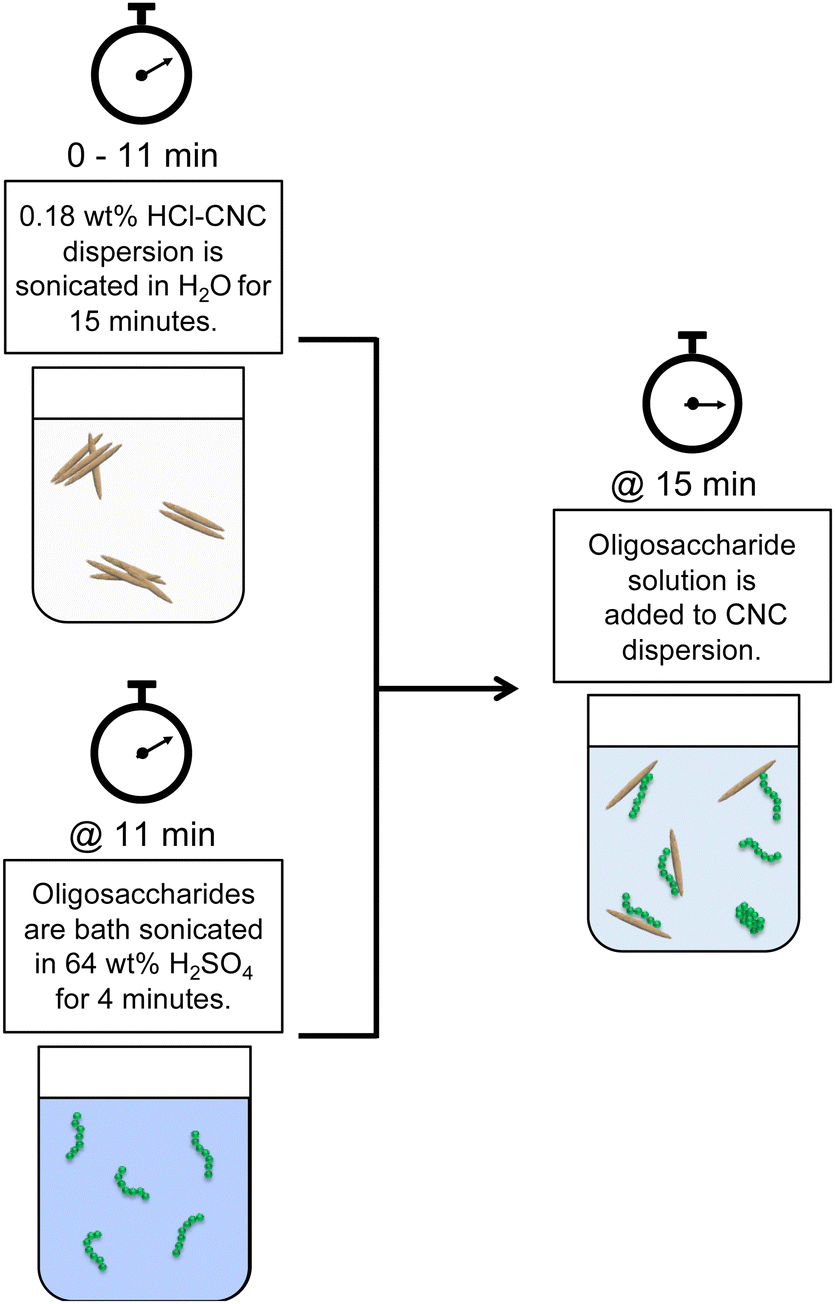 | ||
| Fig. 1 Schematic representation of the post-hydrolysis CNC surface modification through oligosaccharide precipitation, where the externally produced oligosaccharides are dissolved in 64 wt% H2SO4(aq) in a separate beaker and then added to a dispersion of HCl(g)-CNCs which have been extensively probe sonicated. | ||
CNC control samples
To have a comparable control sample to the oligosaccharide modified CNCs, both the HCl(g)-CNCs and the HCl(aq)-CNCs were exposed to the same H2SO4 concentration as present during the precipitation modification. As such, 5 mL of 64 wt% H2SO4 was slowly added to sonicated 0.18 wt% CNC dispersions (i.e., 90 mg of CNCs in 50 g of dispersion) whereby the final concentration of H2SO4 was ca. 8.6 wt%. The CNC dispersions were then probe sonicated at 60% amplitude for an additional 2 min (using a repeated cycle with 1 min sonication, 1 min rest). The dispersions were then placed into cellulose membrane dialysis tubing and dialysed against purified water to remove any residual H2SO4. Dialysis was stopped when the conductivity of the dialysis water remained constant.Dynamic light scattering (DLS)
DLS was used to measure changes in apparent particle size in dispersion upon oligosaccharide precipitation. DLS measurements were carried out on 0.025 wt% CNC suspensions in disposable polystyrene cuvettes using a Zetasizer Nano-ZS (Malvern Panalytical, Malvern, U.K.). The measured diffusion coefficient was converted to apparent hydrodynamic particle size using the Stokes–Einstein equation with the assumption that the CNCs are spherical. Since CNCs are rod-shaped particles, it is important to note that DLS data only provides relative values in this context.22Atomic force microscopy (AFM)
AFM was used to visualize the degree of dispersion of CNCs before and after the oligosaccharide surface modification. Sample substrates were prepared by first cutting ca. 1 cm2 pieces of silica wafer (Pure Wafer, CA, USA), which were then cleaned using an acidic piranha solution (three parts 96 wt% H2SO4 to one part 30% H2O2). The substrates were immersed in the piranha solution for 15 min, rinsed thoroughly with purified water and dried with N2 gas. To prevent the aggregation of CNCs during film drying, a cationic adlayer was first spin coated onto the clean substrates using a 0.1 wt% PAH at 3000 rpm for 30 s with an acceleration of 2300 s−1 (SPS 150 spin coater, SPS-Europe, Gelderland, Netherlands), and rinsed with purified water.23–25 Finally, CNC films were spin coated using the same parameters from a 0.025 wt% CNC dispersion. The films were imaged using a Jupiter XR AFM (Asylum Research – Oxford Instruments, Santa Barbara, CA, USA) in air in tapping mode using AC160TS-R3 probes (resonance frequency: 300 kHz; spring constant: 26 N m−1; tip material: Si; Asylum Research – Oxford Instruments).Molecular weight distribution and gel permeation chromatography (GPC) of oligosaccharides and CNCs
In order to solubilize both the CNCs and oligosaccharides in DMSO (i.e., the GPC solvent), a cellulose carbanilation reaction was performed to produce cellulose tricarbanilate (tricarbanilated AGU molecular weight = 519 g mol−1). Carbanilation was carried out as described elsewhere, using conditions optimized for a full phenyl isocyanate substitution with minimal cellulose degradation.20 In short, any residual moisture was first removed from freeze-dried (Labconco, MI, USA) CNC samples by placing them into the oven overnight at 80 °C. A mixture of 10 mL of DMSO and 1 mL of phenyl isocyanate was then added to each sample (25 mg) and mixed vigorously by hand. The reaction vials were then placed into an oil bath at 70 °C and the carbanilation reaction was continued for 40 h, with each vial being shaken by hand every 5–10 h. To terminate the reaction, 2 mL of methanol was added to the reaction mixture and shaken. Lastly, the reaction vials were left open in the fumehood to allow for the evaporation of any excess methanol. The carbanilated samples were filtered prior to GPC measurements using a 0.45 μm pore size syringe filter (Chromatographic Specialties Inc., ON, Canada). The concentration of carbanilated samples was 2.5 g of cellulose per liter of DMSO. GPC measurements were performed on an Agilent 1100 GPC (Agilent Technologies, California) equipped with an autoloader, with a DMSO and 5% LiBr elution solvent flowing at a rate of 0.5 mL min−1. Samples were analyzed using a Wyatt Optilab T-Rex refractive index detector (Wyatt Technology, CA, USA) with a wavelength of 785 nm at 35 °C. Weight average molecular weights (Mw) of the samples were processed using Wyatt ASTRA 6.0 software (Wyatt Technology, CA, USA) calibrated with polystyrene sulfonate standards of Mw = 0.891, 2, 4.29, 10, 29.5, and 140 kDa. Weight-average degree of polymerization (DPw) values were based on the assumption of a complete substitution of the AGU, and calculated by dividing the Mw of each sample by the molecular weight of a tricarbanilated AGU (519 g mol−1).Quartz crystal microbalance with dissipation monitoring (QCM-D)
QCM-D was used to probe the effect of surface oligosaccharides on the water adsorption capacity of the CNCs, as described elsewhere.19,26 To begin, silicon dioxide coated quartz crystal sensors (Biolin Scientific, Gothenburg, Sweden) were cleaned of dust using dry N2 followed by 15 min inside a UV Ozone Cleaner ProCleaner (BioForce Nanosciences, QC, Canada). After cleaning, the sensors were spin coated using 1 wt% CNC suspensions at 3000 rpm for 60 s with an acceleration of 2300 s−1. The CNC films were then annealed overnight in an oven at 80 °C.Due to the heterogeneous nature of CNC thin films, the spin coated sensors required overnight stabilization in the QCM-D measurement chamber under a constant flow of purified water (0.1 μL min−1). Solvent exchange measurements were then carried out by passing purified water through the chamber for 20 min, followed by deuterium oxide for 20 min and a second water rinse for an additional 20 min all at a flow rate of at 0.1 mL min−1. The temperature was kept constant at 25 °C throughout the measurement. Changes in the resonance frequency (Δf) and dissipation of resonance (ΔD) of the sensor resulting from changes in the mass of the sensor were collected simultaneously throughout the duration of the measurement.
The amount of bound surface water (ΓH2O) was calculated according to the Sauerbrey equation:
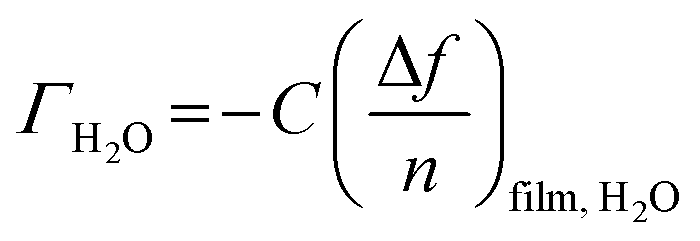 | (1) |
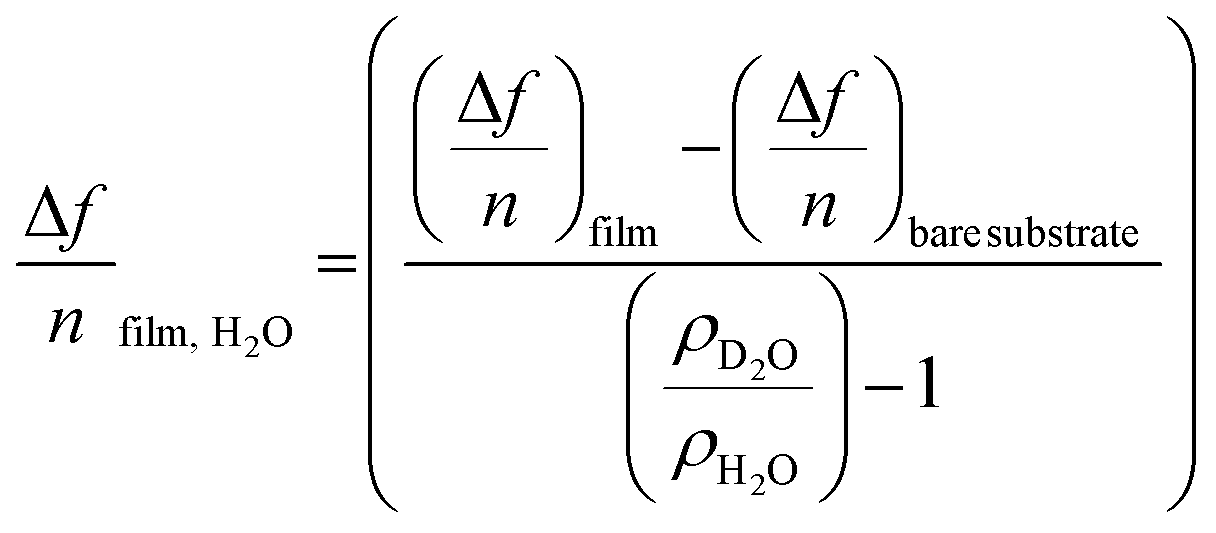 | (2) |
substrate is the measured Δf of a pristine SiO2 coated QCM-D sensor (−54 Hz) due to a H2O/D2O solvent exchange, ρD2O the density of D2O (1.104 g cm−3 at 25 °C) and ρH2O the density of water (0.977 g cm−3 at 25 °C).
Elemental analysis
Elemental analysis for phosphorus and sulphur was performed on the oligosaccharide and CNC samples at the Pacific Centre for Isotopic and Geochemical Research in the Department of Earth, Ocean and Atmospheric Sciences at The University of British Columbia with a Varian 725ES inductively coupled plasma-optical emission spectrometry (ICP-OES) system. Measurements were done by placing ca. 50 mg of freeze-dried sample into a Teflon digestion tube with a few drops of purified water, 1 mL of HF(aq), and 6 mL of a 60–70 wt% HNO3(aq). The samples were then digested using a CEM MARS 6TM Microwave Digestion System, after which they were diluted in purified water to a volume of 25 mL for ICP-OES detection of phosphorus and sulphur. At the same time, a series of calibration standards of phosphorus and sulphur were prepared (from 0 to 100 ppm) and analyzed such that ICP-OES signal could be converted to element concentration for our CNC samples.Results and discussion
HCl(g)-CNC surface modification through oligosaccharide precipitation
Contrary to our original proof-of-concept work in which S-CNCs were modified in situ during their hydrolysis,19 the modification method implemented here is a post-hydrolysis method (Fig. 1 and 2a). Oligosaccharides were dissolved in concentrated H2SO4 and the solution was slowly introduced to an aqueous dispersion of HCl(g)-CNCs. Upon contact with the aqueous phase, the drastic change in pH of the oligosaccharide solution resulted in the instantaneous precipitation of the oligosaccharides onto the available CNC surfaces. A post-hydrolysis surface modification was necessary in the case of HCl(g)-CNCs as the gas hydrolysis is carried out on dry cellulosic substrate, and as such does not facilitate an in situ modification (Fig. 2b).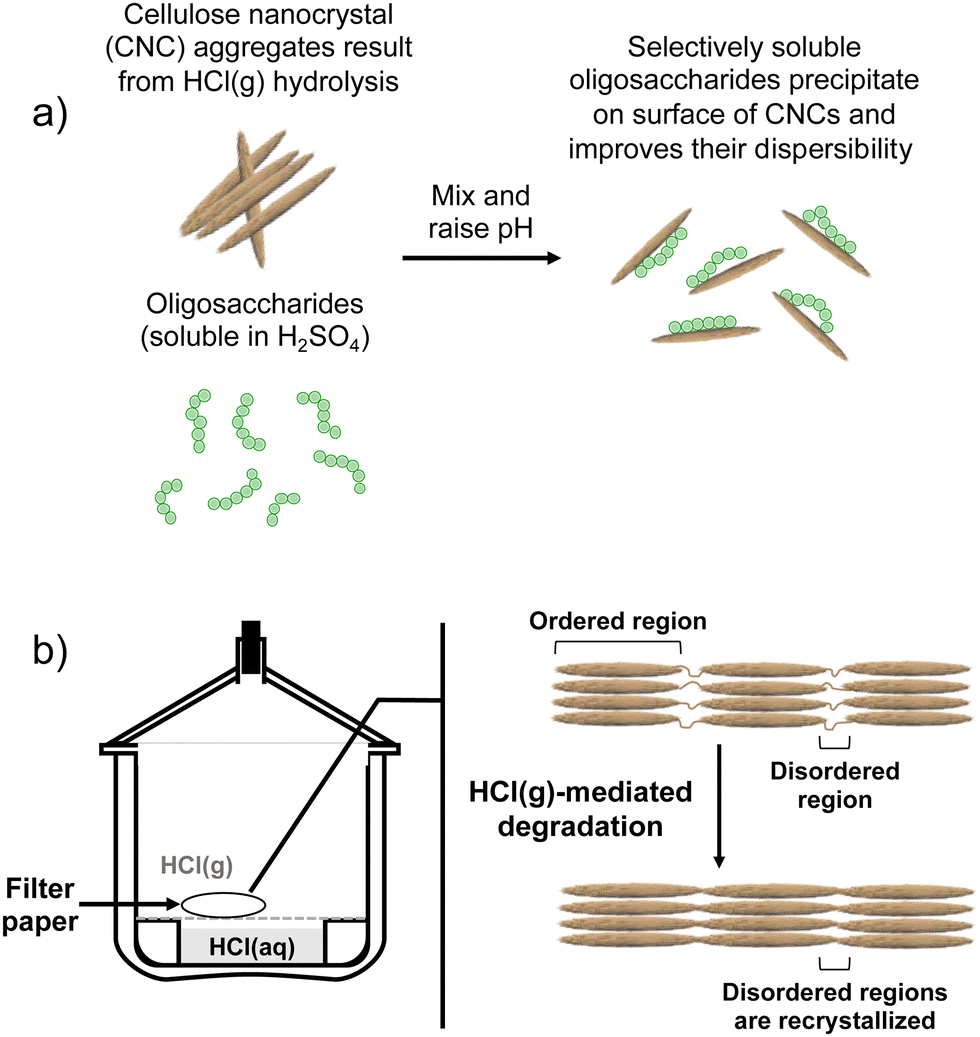 | ||
| Fig. 2 The process of (a) precipitation of water-insoluble oligosaccharides onto the surface of CNCs as a result of raising the pH and (b) HCl(g)-mediated hydrolysis of cellulose to produce HCl(g)-CNCs after which intense mechanical treatment is required to disperse CNCs well enough for the modification represented in (a). We note that since there was no observable sedimentation of insoluble oligosaccharides or unhydrolyzed cellulose visible in the dialysis tubing once dialysis was complete, that any oligosaccharides (DPw,GPC = 6, Mw = 1000 g mol−1) that did not precipitate to the surface of CNCs were likely removed during dialysis (MWCO = 14000 g mol−1). | ||
The effectiveness of the deposited cellulose phosphate oligosaccharides as dispersants for HCl(g)-CNCs was investigated using three different CNC to oligosaccharide mass ratios (i.e., 1:0.2, 1:0.5 and 1:1). Fig. 3 shows the molecular weight distributions of the control (HCl(g)-CNCs + short H2SO4 treatment) as well as the three CNC samples modified with increasing amounts of oligosaccharides. As expected, modification of the HCl(g)-CNCs resulted in an additional shoulder peak in the low molecular weight region of the chromatograph, the intensity of which increased with increasing oligosaccharide fraction (Fig. 3). This shoulder can be seen to correspond directly to the retention time of the oligosaccharide surface modifiers (black dotted trace) and is not present in the control HCl(g)-CNC sample. Based on work published by our group in 2021, we selected to use oligosaccharides of DPw = 6 for these experiments since they provided more CNC surface coverage than other oligosaccharides tested with both larger (DPw = 10) and smaller (DPw = 5) values for DPw.19
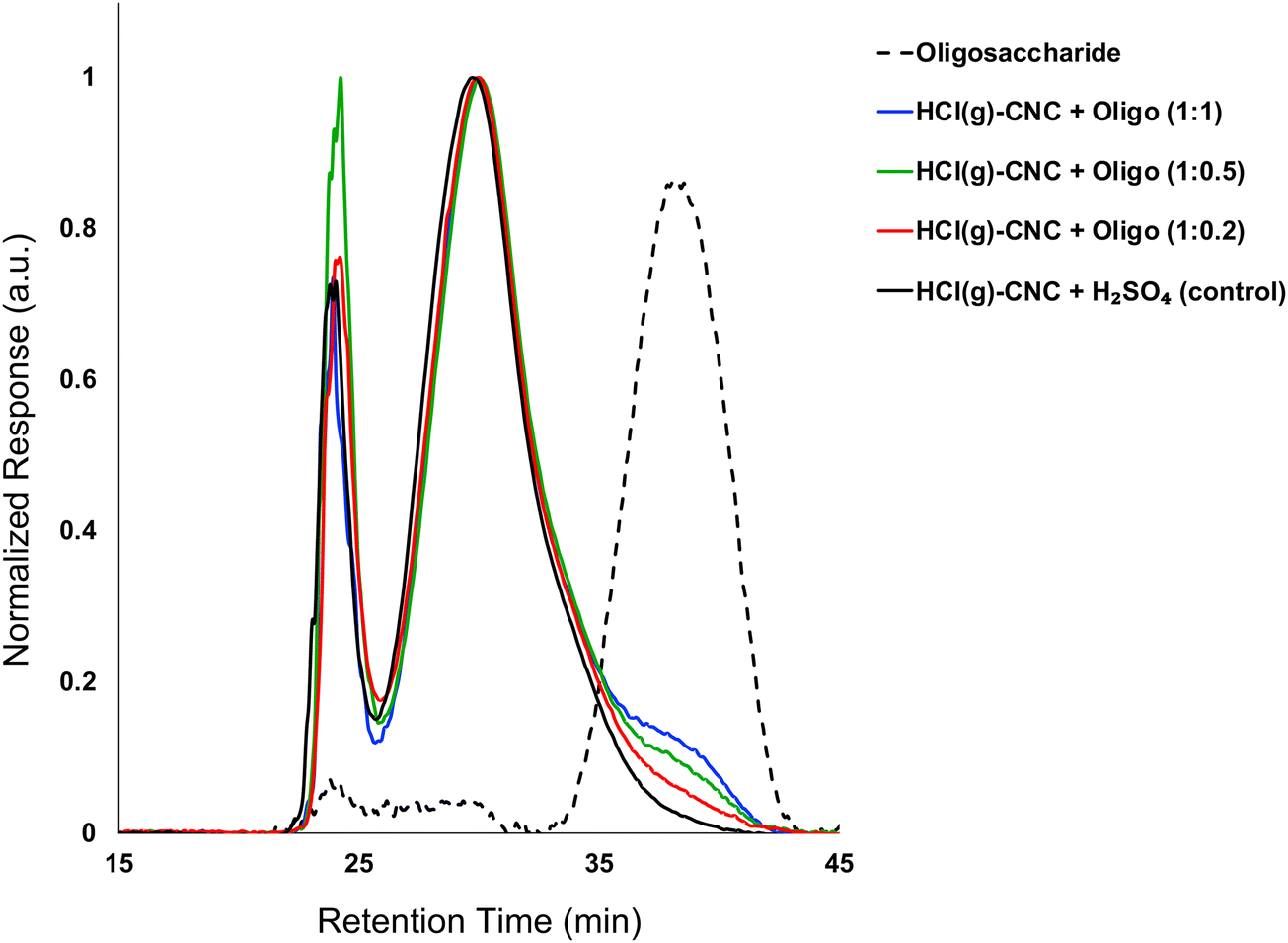 | ||
| Fig. 3 Molecular weight distribution from GPC (DMSO/LiBr) of HCl(g)-CNCs after exposure to H2SO4(aq) and after oligosaccharide surface modification. Also shown, is the molecular weight distribution of the oligosaccharide surface modifiers. | ||
Fig. 3 shows that the CNC core did not change as a result of oligosaccharide precipitation, i.e., the primary peak at 30 min in the chromatogram of each modified sample resembled that of the control. There is another significant peak at ca. 24 min that represents a higher molecular weight polymer presence. We hypothesized in our previous work that this may be the result of a cellulose carbanilation aggregation artefact.19 Additional discussion relevant to this artifact is included in the ESI.†
It is important to note that there are no low molecular weight sugars (or oligosaccharides) represented in the molecular weight distribution of the control samples (Fig. 3, black solid trace). To obtain S-CNCs lacking in naturally occurring surface oligosaccharides, the acid hydrolysis conditions must be sufficiently harsh to degrade the disordered regions of the cellulose microfibril to water soluble sugars (i.e., oligosaccharides with DP < 6).20 If this is not accomplished, water insoluble sugars (i.e., DP > 6) that are a by-product of the hydrolysis, will precipitate onto the surface of the CNCs as a result of the sudden increase in pH when the reaction is quenched with excess water. Kontturi et al. hypothesized that in the case of the HCl gas hydrolysis of cellulose, the disordered cellulose regions in fact become arranged and thus crystallize rather than degrade to individual sugars (Fig. 2). This is suggested by the unprecedented yield of the hydrolysis (97.4%) along with the increase in degree of crystallinity of the material after hydrolysis.12 The lack of low molecular weight sugars (or oligosaccharides) in the molecular weight distribution of the control samples provides further support for this hypothesis.
The degree of CNC surface modification was further investigated by elemental analysis to quantify both sulphur and phosphorus contents (Table 1). Excluding the pristine HCl(g)-CNCs, all the samples had similar sulphur content with an average of 1980 ± 70 ppm S. While this is unsurprising as each of the samples (inclusive of the HCl(g)-CNCs + H2SO4 control) was exposed to the same concentration of H2SO4(aq) during the mixing/quenching step (ca. 9 wt%), it does confirm the reproducibility of the method since there is only ca. 3.5% deviation in sulphur content between samples. It is important to note, however, that these values are still low relative to S-CNCs produced using conventional H2SO4(aq) hydrolysis where the sulphur content is commonly five times higher, around 10000 ppm.19 It is also useful to note that the total surface charge (Table 1) does not trend with increasing oligosaccharide content meaning that the useful properties imparted to these HCl(g)-CNCs are not the result of charge added during the post-hydrolysis surface modification.
| Sulphur content (ppm) | Phosphorus content (ppm) | % Yield of precipitation | Total surface charge calculation (mmol kg−1 cellulose) | |
|---|---|---|---|---|
| Oligosaccharide | 25.1 ± 0.4 | 573 ± 5 | 19.3 ± 0.2 | |
| HCl(g)-CNCs | 8.3 ± 0.4 | 6 ± 2 | 0.45 ± 0.08 | |
| HCl(g)-CNCs + H2SO4(control) | 2030 ± 20 | 33 ± 7 | 65 ± 1 | |
| HCl(g)-CNCs + Oligo (1:0.2) |
1810 ± 10 | 39 ± 9 | 32 | 57 ± 1 |
| HCl(g)-CNCs + Oligo (1:0.5) |
2090 ± 20 | 99 ± 6 | 46 | 69 ± 1 |
| HCl(g)-CNCs + Oligo (1:1) |
1980 ± 20 | 170 ± 10 | 56 | 67 ± 1 |
Similar to the oligosaccharide GPC peak, the phosphorus content of the surface modified samples increased with increasing oligosaccharide content (Table 1). The phosphorus content of the HCl(g)-CNCs + Oligo (1:0.2) showed only a slight increase as compared to the control sample (33 ppm vs. 39 ppm, respectively), which is also in good agreement with the GPC results. Importantly, exposure of the HCl(g)-CNCs to dilute H2SO4(aq) and subsequent ultrasonication does not result in any further degradation of the cellulose (i.e., Mw remains constant) and as such, any oligosaccharides present on the CNC surfaces are the direct result of the precipitation surface modification (ESI,† Fig. S1). It should also be noted that exposure to dilute H2SO4(aq) (and the associated decrease in suspension pH) did not result in further degradation of either the CNC core nor the oligosaccharides as no shift in their corresponding peaks is visible in the molecular weight distributions (Fig. 3 and ESI,† Fig. S1).
Based on the theoretical maximum sulphur and phosphorus contents of the modified HCl(g)-CNCs (i.e., assuming 100% precipitation of the oligosaccharides added to the CNCs), an approximation was made for the yield of precipitation on the CNCs (Table 1). The results show that the yield of precipitation may be improved by increasing the fraction of oligosaccharides introduced to the aqueous CNC mixture. Also of note is that both the intensity of the oligosaccharide peak in the GPC chromatograph and the phosphorus content as measured by elemental analysis increase linearly with increasing oligosaccharide fraction (ESI,† Fig. S2a and b, respectively). Even at a 1:1 mass ratio of CNCs to oligosaccharides, there is no apparent decrease in slope, indicating the limit of this surface modification route has yet to be reached. We consider these observations may be further illustrated using fundamentals of physical chemistry as they pertain to solid–liquid separation.27,28 We know that while the oligosaccharides used for this study are soluble in strong acids, as the pH increases (in the absence of CNCs), they crystallize and precipitate. Precipitation and crystallization are analogous to the extent that their processes consist of two major events: nucleation and growth. Both nucleation as well as growth are driven by supersaturation and, depending on the conditions, either process can dominate. Given the results from our GPC and elemental analysis experiments, we hypothesize that our proximity to the supersaturation point of the oligosaccharide solution is increased by increasing the concentration of oligosaccharide. Therefore, since surface-precipitation yields (Table 1) increase with increasing oligosaccharide fraction, we consider that, under these conditions, crystal growth from the CNC “seeds” is favoured over oligosaccharide nucleation. We also expect that at the point supersaturation changes in favour of crystal-nucleation over seed-growth, we would note a change in slope for the plots in Fig. S2a and b (ESI†) as the surface coverage approaches a theoretical maximum. This change would indicate a limit to the potential of this surface modification route.
While X-ray diffraction data supports the idea that a co-crystallization mechanism predominates for this surface modification method,19 it is possible and thus important to note that the oligosaccharides could adopt a twofold screw conformation similar to the cellulose crystal without actually adding to the crystal. This would be analogous to how xylan adsorbs to the surface of cellulose.29 In a 2017 report from Falcoz-Vigne et al., only the first monolayer of xylan adapted the twofold screw conformation while further layers adopted their bulk state threefold screws instead.29 A more recent study used solid-state 13C NMR to elucidate molecular structures present in softwood and showed that xylan adopted this twofold screw when in close proximity to cellulose microfibrils indicating that hemicellulose strongly binds to the hydrophilic face of cellulose.30 In the context of this broader research, we have recently begun experiments to characterize the conformation of natural and externally-produced oligosaccharides precipitated following different pathways onto CNCs.
Effect of oligosaccharide surface modification on dispersibility of HCl(g)-CNCs
The complete hydrolysis of cellulose to its LODP using HCl gas has been demonstrated to be highly effective from both a process efficiency and product yield perspective.12,13,31 However, the resulting material does not lend itself to being utilized as a nanoscaled cellulose as the hydrolysis itself is void of mixing and mass transfer processes and does not yield “liberated” individual CNCs (Fig. 2a and ESI,† Fig. S3a and b). Primarily, this is due to the inherent lack of surface charge of the cellulose which when present, greatly facilitates the unhinging and separation of cellulose fibrils and crystals. While surface modification methods that introduce charge to nanocelluloses are available (i.e., esterification, oxidation, and polyelectrolyte adsorption), these methods often require the use of harsh chemicals and extensive purification protocols.32,33 The addition of oligosaccharides to HCl(g)-CNCs through a precipitation route circumvents these issues and presents a simple yet efficient way to facilitate their dispersion in aqueous media.
Fig. 4 shows that the precipitation of oligosaccharides to the surface of HCl(g)-CNCs improves their dispersibility in water. In fact, as the mass fraction of oligosaccharides increases, so does the degree of dispersion of the HCl(g)-CNCs. As can be seen, exposure of the HCl(g)-CNCs to ca. 9 wt% H2SO4 only (i.e., no oligosaccharides) and subsequent ultrasonication resulted in the liberation of CNC bundles from the bulk material shown in Fig. S3a (ESI†) (Fig. 4a). Nevertheless, further isolation of individual nanocrystals did not occur and the CNCs remained tethered to one another. Interestingly, modification of the HCl(g)-CNCs with the lowest mass fraction of oligosaccharides (i.e., HCl(g)-CNCs + Oligo (1:0.2)) did not result in a noticeable increase in their dispersibility (Fig. 4b). There is a notable threshold overcome, however, once the oligosaccharide mass fraction surpasses 0.5 to 1 with Fig. 4c and d showing an increase in the liberation of individual CNCs from the bundled aggregates that trends with increasing oligosaccharide fraction. These results are somewhat surprising considering the overall charge content does not increase significantly with increasing surface oligosaccharide content. Even at the highest oligosaccharide concentration (1:1, Fig. 4d), where we observe the most individual, CNC-like particles in the image, the combined concentration of charge groups (sulphur and phosphorus) for this sample was only 2150 ± 30 ppm (i.e., 67 mmol kg−1 CNC). This is not a very different value from the control HCl(g)-CNC that were exposed to just H2SO4(aq) (charge content of 65 mmol kg−1 CNC). Therefore, we consider the effect of the surface oligosaccharides introduced to the HCl(g)-CNCs through the surface modification to be two-fold. First, the added charge groups impart repulsion between aggregated CNCs which weakens the cellulose–cellulose interactions and facilitates the breaking apart of CNC aggregates. However, given there is no significant increase in charge content between the control HCl(g)-CNC + H2SO4 and the HCl(g)-CNC + Oligo (1:1) imaged in Fig. 4a and d, respectively, this improved dispersibility cannot be the result of added electrostatic repulsion on its own. We thus infer that, when present at high enough concentrations, the oligosaccharides sterically disrupt the aggregation resulting in individual CNCs being liberated from the bulk hydrolyzed cellulose. We also infer that the increase in electrostatic repulsion due to the exposure of the HCl(g)-CNCs to H2SO4 for 15 minutes is not sufficient to liberate individual CNCs and the surface modification with the neutralized oligosaccharides is necessary to produce nanoparticles with some degree of dispersibility.
 | ||
| Fig. 4 AFM images showing the improved degree of dispersion of HCl(g)-CNCs (a) after exposure to ca. 9 wt% H2SO4 and with (b)–(d) increasing surface oligosaccharide content after surface modification. | ||
With the post-hydrolysis oligosaccharide modification introduced here, we were able to greatly improve the dispersibility of HCl(g)-CNCs with 15-fold lower surface charge contents than the previously reported, gold-standard TEMPO-mediated oxidation method, without the necessity for complex, oxidative chemistries.15 This work by Pääkkönen et al. showed HCl(g)-CNCs from bacterial cellulose to be successfully dispersed in aqueous media after introducing carboxyl charge groups to the CNC surface via TEMPO-mediated oxidation. The authors reported a surface charge content of 1000 mmol COOH/kg CNC and showed that most of the hydrolyzed material could be isolated as individual nanoparticles.
DLS analysis showed that the apparent hydrodynamic size decreased with increasing oligosaccharide content, with the smallest particle size detected at a 1:1 CNC to oligosaccharide ratio (251 ± 6 nm) (as shown in Fig. 5). These results suggest that the surface modification was indeed successful in enabling the isolation of HCl(g)-CNCs in water, but that increasing the oligosaccharide concentration resulted in even further CNC dispersibility. This can also be seen in the photographs of Fig. 5 where the control sample shows CNC sedimentation from the aqueous phase (and as such no colloidal stability of the CNCs) regardless of the exposure to H2SO4(aq), whereas the oligosaccharide-modified CNCs are clearly suspended in solution. Interestingly, the characteristic blue hue of the CNCs dispersions increases with increasing oligosaccharide content from which we can infer an increasing concentration of suspended (and smaller) particles. Lastly, it is also important to note the lack of a visible precipitate at the bottom of the vials which would indicate a large fraction of oligosaccharide precipitate, very large CNC aggregates, or a combination of the two.
 | ||
| Fig. 5 Photographs demonstrating the colloidal stability of 0.1 wt% dispersions of HCl(g)-CNCs after exposure to H2SO4(aq) and after modification through oligosaccharide precipitation with varying amounts of oligosaccharides after 26 days. The blue hue of the dispersions indicates dispersed CNCs in the aqueous phase. Also shown is the DLS apparent hydrodynamic size of the HCl(g)-CNCs suggesting that more oligosaccharides lead to better dispersed CNCs. | ||
Extending surface modification through oligosaccharide precipitation to HCl(aq)-CNCs
In the experiments described above, we showed oligosaccharide precipitation to be an effective surface modification technique for facilitating the dispersion of HCl(g)-CNCs produced using HCl(g) hydrolysis. In an effort to illustrate the broader applicability of this surface modification technique, we turned to HCl(aq)-CNCs isolated using the more established aqueous hydrochloric acid hydrolysis. Oligosaccharides were added to dispersions of HCl(aq)-CNCs post-hydrolysis at the three different cellulose-to-oligosaccharide mass ratios (i.e., 1:0.2, 1:0.5 and 1:1) as earlier.
Fig. 6 shows the overlayed molecular weight distributions of HCl(aq)-CNCs after exposure to H2SO4 (control) or oligosaccharide solutions. Here again, there is a clear difference between the control and the oligosaccharide-modified samples. The control shows a broad, high molecular weight peak with a maximum at ca. 30 minutes retention time but no peak in the oligosaccharide molecular weight range at ca. 40 minutes retention time. The dotted-line molecular weight distribution for the oligosaccharide modifier is included to help guide the eye to its elution time. In contrast, the surface modified samples have broad, high molecular weight peaks matching the control but with additional shoulder peaks in the oligosaccharide range. Again, the profile of the CNC core was not affected by the oligosaccharide precipitation or the associated decrease in suspension pH. The relative intensity of the oligosaccharide shoulder peaks increased with cellulose-to-oligosaccharide mass ratio, in exactly the same way as the modification of HCl(g)-CNCs described above (Fig. 3).
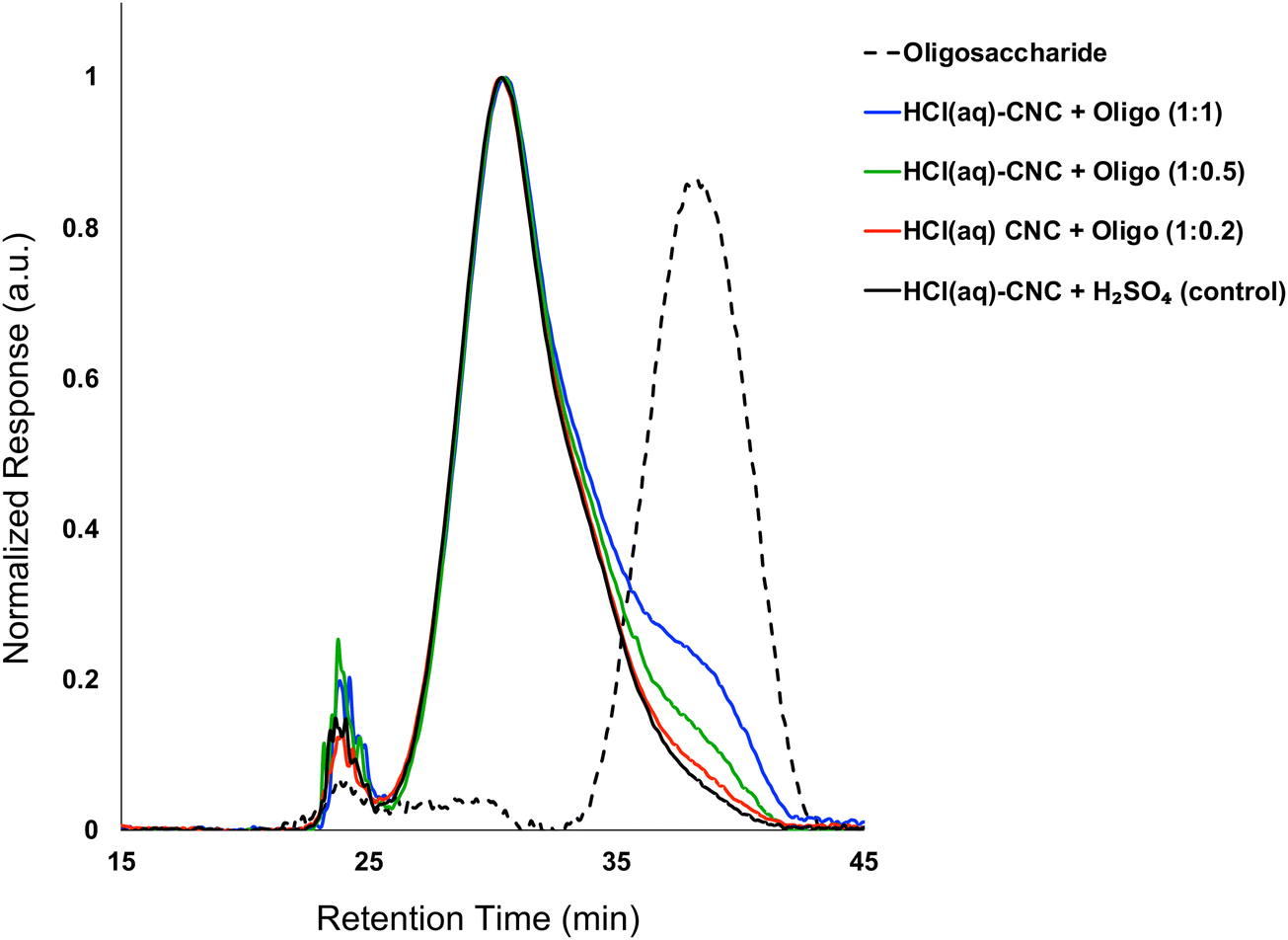 | ||
| Fig. 6 Molecular weight distribution of HCl(aq)-CNCs from GPC (DMSO/LiBr) after exposure to H2SO4(aq) and after oligosaccharide surface modification. Also shown is the molecular weight distribution of the oligosaccharide surface modifiers. | ||
Elemental analysis (Table S1, ESI†) also supported that a higher cellulose-to-oligosaccharide mass ratio led to more surface deposited oligosaccharides on CNCs. This makes sense, as higher concentration relates to higher supersaturation and thus higher driving forces for CNC crystal growth. Just like with HCl(g)-CNC modification, degree of surface modification for HCl(aq)-CNCs increased linearly with increasing concentration of oligosaccharide relative to CNC in the modification mixture. This trend is observed for both the oligosaccharide peak-intensity determined using GPC and phosphorus content determined using ICP-OES (Fig. S4a and b, respectively ESI†).
We used a quartz crystal microbalance with dissipation monitoring (QCM-D) to examine the water binding capacity of the oligosaccharide modified HCl(aq)-CNCs. Microgravimetry revealed that the content of bound surface water of HCl(aq)-CNCs decreased with increasing oligosaccharide content (ESI,† Fig. S5), suggesting changes to the HCl(aq)-CNC surfaces upon oligosaccharide deposition. If a composite film of HCl(aq)-CNCs and oligosaccharide aggregates had formed instead of homogenous films of oligosaccharide-coated HCl(aq)-CNCs, one may expect the available surface area for water adsorption to increase with increasing oligosaccharide content. However, because we observe the opposite trend and microgravimetry detects a decrease in bound surface water with increasing oligosaccharide content, we know that the oligosaccharides are in fact associated with the CNCs and as such impede their ability to interact with water in line with past results.19 This result is supported by earlier work demonstrating the effects of surface oligosaccharides on the processability of CNCs. Bouchard et al. noted that CNC suspension viscosity significantly decreased as a function of increasing oligosaccharide content.20 They attributed this effect to a decrease in the capacity of CNCs to bind water to their surfaces when oligosaccharides already occupy their potential binding sites, and thereby decreasing their apparent size in suspension. Similar phenomena have been reported for other colloidal particles such as alumina34–36 and other oxide nanoparticle suspensions.37 Similarly, we showed that both dispersion viscosity and water binding capacity could be decreased through the intentional precipitation of externally-produced oligosaccharides onto the surface of S-CNCs.19
While pristine HCl(aq)-CNCs can be relatively well-dispersed in aqueous media, their colloidal stability is notoriously poor.11 As such, we note less dramatic apparent differences to the microscopic dispersity of oligosaccharide-modified and unmodified HCl(aq)-CNCs (Fig. 7) as compared with the HCl(g)-CNCs (Fig. 4). Nonetheless, as was the case with HCl(g)-CNCs, the AFM images of Fig. 7a(i) and b(i) do show that the presence of oligosaccharides aids in the isolation of individual CNCs. As can be seen, prior to modification, the HCl(aq)-CNCs are – to a large extent – tethered to one another whereas after modification a substantially larger fraction of individual CNCs can be identified in the image.
 | ||
| Fig. 7 AFM images of (a) HCl(aq)-CNCs after exposure to H2SO4(aq) (control) and (c) after a 1:1 CNC-to-oligosaccharide surface modification. Also shown are photographs to demonstrate the colloidal stability of 0.1 wt% (b) HCl(aq)-CNCs after exposure to H2SO4(aq) and (d) HCl(aq)-CNCs + Oligo (1:1) after 24 h without salt and with 5 mM NaCl. | ||
We also observe a significant improvement in colloidal stability of the HCl(aq)-CNCs as a result of oligosaccharide precipitation. When no salt is added, dispersions of the modified and unmodified HCl(aq)-CNCs are indistinguishable. However, in the presence of 5 mM NaCl there is a clear difference in colloidal behaviour between the samples (Fig. 7a(ii) and d(ii)). Before 24 h, the unmodified HCl(aq)-CNCs had precipitated out of suspension whereas their oligosaccharide-modified counterparts remained suspended.
Interestingly, S-CNCs modified with oligosaccharides in situ did not demonstrate a similar resistance to increased ionic strength.19 We speculated that precipitation of the oligosaccharides in situ resulted in their co-crystallizing with the CNCs as evidenced by a lack of cellulose II (i.e., precipitated oligosaccharides) detected by X-ray diffraction. The resistance of the surface modified HCl(aq)-CNCs to increased ionic strength at such low surface charge contents leads us to believe that the mechanism of precipitation post-hydrolysis might differ from that of the in situ case. It is possible that only one end of the oligosaccharide is deposited (or crystallized) on the CNC leaving the remaining oligosaccharide “tail” extending out from the CNC surface, as opposed to a flat co-crystallized deposition onto the surface, which could explain the improved colloidal stability in the presence of salt (i.e., potential steric stabilization).
Conclusions
This work presented the first successful surface modification of uncharged CNCs using a post-hydrolysis oligosaccharide deposition route. Using this technique, it was possible to proportionally increase the degree to which CNC surfaces were modified by increasing the amount of oligosaccharide added relative to uncharged CNCs (produced by both gas-phase and aqueous HCl hydrolyses). The ratio 1:0.2 of HCl(g)-CNCs to oligosaccharide presented with the least significant changes while the 1:0.5 and 1:1 samples showed proportional improvements to the liberation and dispersibility of the resulting nanoparticles. When this modification technique was extended to the HCl(aq)-CNCs their colloidal stability (resistance to ionic strength) was improved in addition to their dispersibility.
Interestingly, the yield of the precipitation modification improved with increasing oligosaccharide content for both HCl(g)-CNCs and HCl(aq)-CNCs. This result indicates an increase to the efficiency of the modification with increasing oligosaccharide concentration and makes sense, as higher concentration relates to higher supersaturation and thus higher driving forces for crystallization and specifically, crystal growth. We expect that there will be a cross-over point at a high enough oligosaccharide concentration where the formation of multiple nucleation sites would be favoured over continued crystal growth (that is considering CNCs are seen by precipitating oligosaccharides as already existing nucleation sites). Going forward it will be important for us to further explore the limits of this crystallization mechanism.
The presence of oligosaccharides on the CNC surfaces induced temporary colloidal stability of the HCl(aq)-CNCs in the presence of salt. This phenomena was not noted in our proof-of-concept work where modification was carried out in situ on sulfated CNCs. It is possible that the crystal structure of oligosaccharides deposited to the CNC surface depends on whether the modification was done in situ or post-hydrolysis. Lastly, to examine the practicality of this surface-modification technique, we are working to synthesize new oligosaccharide-based block copolymer surface modifiers that may be deposited to CNC surfaces following the methods presented here. Altogether, our hope is to present a comprehensive dataset illustrating the potential of oligosaccharide precipitation as a CNC surface modification method and ultimately, lead the development of new generations of CNC-based materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. N. acknowledges the Academy of Finland (grant no. 321801) for funding. E. K. acknowledges the support from FinnCERES Flagship Materials Bioeconomy Ecosystem. E. D. C. is grateful for financial support and recognition through the Canada Research Chairs programme, the University of British Columbia's President's Excellence Chair initiative, NSERC Discovery Grant (RGPIN-2017-05252), NSERC E.W.R. Steacie Memorial Fellowship and Canadian Foundation for Innovation (John R. Evans Leaders Fund, project number 38623). The authors acknowledge, Profs. Scott Renneckar and Feng Jiang for equipment usage. Maureen Soon and Marghaleray Amini in the Department of Earth, Ocean and Atmospheric Sciences at The University of British Columbia are thanked for aide in developing the ICP-OES experimental protocol. We appreciate Dr Li-Yang Liu, Qi Hua and Dr Oliver Musl for their guidance related to GPC.References
- L. Chen, Q. Wang, K. Hirth, C. Baez, U. P. Agarwal and J. Y. Zhu, Cellulose, 2015, 22, 1753–1762 CrossRef CAS.
- O. A. Hisseine, W. Wilson, L. Sorelli, B. Tolnai and A. Tagnit-Hamou, Constr. Build. Mater., 2019, 206, 84–96 CrossRef CAS.
- M. C. Li, Q. Wu, R. J. Moon, M. A. Hubbe and M. J. Bortner, Adv. Mater., 2021, 33, 2006052 CrossRef CAS PubMed.
- I. Kalashnikova, H. Bizot, B. Cathala and I. Capron, Biomacromolecules, 2012, 13, 267–275 CrossRef CAS PubMed.
- S. A. Kedzior, V. A. Gabriel, M. A. Dubé and E. D. Cranston, Adv. Mater., 2021, 33, 2002404 CrossRef CAS PubMed.
- P. R. Sharma, S. K. Sharma, T. Lindström and B. S. Hsiao, Adv. Sustainable Syst., 2020, 4, 1900114 CrossRef CAS.
- R. M. A. Domingues, M. E. Gomes and R. L. Reis, Biomacromolecules, 2014, 15, 2327–2346 CrossRef CAS PubMed.
- X. R. Ong, A. X. Chen, N. Li, Y. Y. Yang and H. K. Luo, Small Sci., 2023, 3, 2200076 CrossRef CAS.
- J. Araki, M. Wada and S. Kuga, Langmuir, 2001, 17, 21–27 CrossRef CAS.
- T. Abitbol, A. Palermo, J. M. Moran-Mirabal and E. D. Cranston, Biomacromolecules, 2013, 14, 3278–3284 CrossRef CAS PubMed.
- O. M. Vanderfleet and E. D. Cranston, Nat. Rev. Mater., 2021, 6, 124–144 CrossRef CAS.
- E. Kontturi, A. Meriluoto, P. A. Penttilä, N. Baccile, J.-M. Malho, A. Potthast, T. Rosenau, J. Ruokolainen, R. Serimaa, J. Laine and H. Sixta, Angew. Chem., 2016, 128, 14671–14674 CrossRef.
- T. Pääkkönen, P. Spiliopoulos, A. Knuts, K. Nieminen, L. S. Johansson, E. Enqvist and E. Kontturi, React Chem. Eng., 2018, 3, 312–318 RSC.
- W. Fang, S. Arola, J. M. Malho, E. Kontturi, M. B. Linder and P. Laaksonen, Biomacromolecules, 2016, 17, 1458–1465 CrossRef CAS PubMed.
- T. Pääkkönen, P. Spiliopoulos, Nonappa, K. S. Kontturi, P. Penttilä, M. Viljanen, K. Svedström and E. Kontturi, ACS Sustainable Chem. Eng., 2019, 7, 14384–14388 CrossRef.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- M. Kröger, O. Badara, T. Pääkkönen, I. Schlapp-Hackl, S. Hietala and E. Kontturi, Biomacromolecules, 2023, 24, 1318–1328 CrossRef PubMed.
- E. Niinivaara, O. M. Vanderfleet, E. Kontturi and E. D. Cranston, Biomacromolecules, 2021, 22, 3284–3296 CrossRef CAS PubMed.
- J. Bouchard, M. Méthot, C. Fraschini and S. Beck, Cellulose, 2016, 23, 3555–3567 CrossRef CAS.
- T. Abitbol, A. Palermo, J. M. Moran-mirabal and E. D. Cranston, Biomacromolecules, 2013, 14, 3278–3284 CrossRef CAS PubMed.
- S. Bhattacharjee, J. Control Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- M. S. Reid, M. Villalobos and E. D. Cranston, Langmuir, 2017, 33, 1583–1598 CrossRef CAS PubMed.
- G. Delepierre, O. M. Vanderfleet, E. Niinivaara, B. Zakani, C. Weder and E. D. Cranston, Langmuir, 2021, 37, 8393–8409 CrossRef CAS PubMed.
- E. J. Foster, R. J. Moon, J. Bras, S. Camarero-espinosa, K. J. Chan, M. J. D. Clift, E. D. Cranston, S. J. Eichhorn, D. M. Fox, W. Y. Hamad, L. Heux, B. Jean, M. Korey, W. Nieh, K. J. Ong, M. S. Reid, S. Renneckar, R. Roberts and A. Shatkin, Chem. Soc. Rev., 2018, 47, 2609–2679 RSC.
- J. D. Kittle, X. Du, F. Jiang, C. Qian, T. Heinze, M. Roman and A. R. Esker, Biomacromolecules, 2011, 12, 2881–2887 CrossRef CAS PubMed.
- A. S. Myerson and R. Ginde, Crystals, Crystal Growth and Nucleation, in Handbook of Industrial Crystallisation, ed. A. S. Myerson, Butterworth-Heinemann, London, 2nd edn, 2002, pp. 33–65 Search PubMed.
- J. J. De Yoreo and P. G. Vekilov, Rev. Mineral. Geochem., 2003, 54, 57–93 CrossRef CAS.
- L. Falcoz-Vigne, Y. Ogawa, S. Molina-Boisseau, Y. Nishiyama, V. Meyer, M. Petit-Conil, K. Mazeau and L. Heux, Cellulose, 2017, 24, 3725–3739 CrossRef CAS.
- O. M. Terrett, J. J. Lyczakowski, L. Yu, D. Iuga, W. T. Franks, S. P. Brown, R. Dupree and P. Dupree, Nat. Commun., 2019, 10, 4978 CrossRef CAS PubMed.
- M. Lorenz, S. Sattler, M. Reza, A. Bismarck and E. Kontturi, Faraday Discuss., 2017, 202, 315–330 RSC.
- S. Eyley and W. Thielemans, Nanoscale, 2014, 6, 7764–7779 RSC.
- V. Arumughan, T. Nypelö, M. Hasani and A. Larsson, Adv. Colloid Interface Sci., 2021, 298 Search PubMed.
- C. Li, M. Akinc, J. Wiench, M. Pruski and C. H. Schilling, J. Am. Ceram. Soc., 2005, 88, 2762–2768 CrossRef CAS.
- C. Li and M. Akinc, J. Am. Ceram. Soc., 2005, 88, 1448–1454 CrossRef CAS.
- S. Çinar, L. Van Steenhuyse and M. Akinc, J. Am. Ceram. Soc., 2013, 96, 1077–1084 CrossRef.
- S. Çinar, D. D. Anderson and M. Akinc, J. Eur. Ceram. Soc., 2015, 35, 613–622 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00936j |
| ‡ These authors worked together on this publication and contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |