
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multimodal imaging of a liver-on-a-chip model using labelled and label-free optical microscopy techniques†
Jan
Majer‡
 ab,
Aneesh
Alex‡
cd,
Jindou
Shi
def,
Eric J.
Chaney
de,
Prabuddha
Mukherjee
de,
Darold R.
Spillman
Jr.
deg,
Marina
Marjanovic
degh,
Carla F.
Newman
a,
Reid M.
Groseclose
c,
Peter D.
Watson
*b,
Stephen A.
Boppart
*defghi and
Steve R.
Hood
*ad
ab,
Aneesh
Alex‡
cd,
Jindou
Shi
def,
Eric J.
Chaney
de,
Prabuddha
Mukherjee
de,
Darold R.
Spillman
Jr.
deg,
Marina
Marjanovic
degh,
Carla F.
Newman
a,
Reid M.
Groseclose
c,
Peter D.
Watson
*b,
Stephen A.
Boppart
*defghi and
Steve R.
Hood
*ad
aPre-Clinical Sciences, Research Technologies, GSK, Stevenage, UK. E-mail: steve.r.hood@gsk.com
bSchool of Biosciences, Cardiff University, Cardiff, UK. E-mail: watsonpd@cardiff.ac.uk
cPre-Clinical Sciences, Research Technologies, GSK, Collegeville, PA, USA
dGSK Center for Optical Molecular Imaging, University of Illinois Urbana-Champaign, Urbana, IL, USA. E-mail: boppart@illinois.edu
eBeckman Institute for Advanced Science and Technology, University of Illinois Urbana-Champaign, Urbana, IL, USA
fDepartment of Electrical and Computer Engineering, University of Illinois Urbana-Champaign, Urbana, IL, USA
gNIH/NIBIB P41 Center for Label-Free Imaging and Multiscale Biophotonics (CLIMB), University of Illinois Urbana-Champaign, Urbana, IL, USA
hDepartment of Bioengineering, University of Illinois Urbana-Champaign, Urbana, IL, USA
iInterdisciplinary Health Sciences Institute, University of Illinois Urbana-Champaign, Urbana, IL, USA
First published on 11th September 2024
Abstract
A liver-on-a-chip model is an advanced complex in vitro model (CIVM) that incorporates different cell types and extracellular matrix to mimic the microenvironment of the human liver in a laboratory setting. Given the heterogenous and complex nature of liver-on-a-chip models, brightfield and fluorescence-based imaging techniques are widely utilized for assessing the changes occurring in these models with different treatment and environmental conditions. However, the utilization of optical microscopy techniques for structural and functional evaluation of the liver CIVMs have been limited by the reduced light penetration depth and lack of 3D information obtained using these imaging techniques. In this study, the potential of both labelled as well as label-free multimodal optical imaging techniques for visualization and characterization of the cellular and sub-cellular features of a liver-on-a-chip model was investigated. (1) Cellular uptake and distribution of Alexa 488 (A488)-labelled non-targeted and targeted antisense oligonucleotides (ASO and ASO-GalNAc) in the liver-on-a-chip model was determined using multiphoton microscopy. (2) Hyperspectral stimulated Raman scattering (SRS) microscopy of the C–H region was used to determine the heterogeneity of chemical composition of circular and cuboidal hepatocytes in the liver-on-a-chip model in a label-free manner. Additionally, the spatial overlap between the intracellular localization of ASO and lipid droplets was explored using simultaneous hyperspectral SRS and fluorescence microscopy. (3) The capability of light sheet fluorescence microscopy (LSFM) for full-depth 3D visualization of sub-cellular distribution of A488-ASO and cellular phenotypes in the liver-on-a-chip model was demonstrated. In summary, multimodal optical microscopy is a promising platform that can be utilized for visualization and quantification of 3D cellular organization, drug distribution and functional changes occurring in liver-on-a-chip models, and can provide valuable insights into liver biology and drug uptake mechanisms by enabling better characterization of these liver models.
Introduction
Primary human hepatocytes (PHH) are considered the gold standard for hepatic in vitro cell culture models due to the preservation of the metabolic pathways and functionality of the human liver in those cell types.1 One of the main challenges for in vitro human hepatic models in liver disease research and pharmacological studies is to ensure the stability and maintenance of the differentiated state of hepatocytes for several days or even weeks. It has been shown that 3D PHH cultures preserve the physiological hepatic function better than 2D cultures.2 Liver-on-a-chip models represent advanced 3D systems that simulate the intricate physiology of the human liver within a laboratory setting.3,4 Hepatic complex in vitro models (CIVMs) can incorporate various cell types, including PHH, Kupffer cells (KC), and hepatic stellate cells, along with extracellular matrix, to mimic the microenvironment of the liver. PHHs and KCs represent ∼60% and 15% of all liver cells, respectively. Unlike PHHs, KCs are nonparenchymal cells that reside exclusively in liver, where they are the necessary forefront of pathogen and injury response.5,6 Hepatic CIVMs are a powerful tool for studying liver diseases, drug metabolism, toxicity testing and development of therapeutic drugs. By replicating key aspects of the liver function, such as drug metabolism, bile production and immune responses in a controlled in vitro environment, these models provide a cost-efficient, robust, and human biology-relevant ethical alternative to traditional in vivo animal testing that can accelerate drug discovery and development.7,8 Additionally, CIVMs enable development of personalized medicine approaches by using patient-derived cells to tailor treatments for individual patients.9 As liver CIVMs continue to evolve, they hold great promise for advancing our understanding of liver biology and improving healthcare outcomes.Currently, studies using commercially available liver-on-a-chip models rely on analysis of albumin, urea, lactate dehydrogenase or other soluble biomarkers from cellular media for assessing cell health and functionality.10,11 Several assays for measuring metabolic, transcriptomic and phenotypic changes in these models have been developed as well.3,5 These biomarker measurements determine the ability of a liver CIVM to recreate the liver microenvironment in vitro and its potential to be used as a tool for gaining deeper insights into liver disease mechanisms, drug metabolism and drug-induced toxicity.12 Given the heterogenous and complex nature of the liver-on-a-chip models, brightfield or fluorescence imaging techniques are often employed to capture the changes occurring in these models with different media, treatment or environmental conditions.5 These techniques do not provide information on the 3D organization or functional changes occurring in the liver-on-a-chip models. To visualize 3D morphological architecture of the liver-on-a-chip models, confocal fluorescence imaging systems are utilized.5,13 However, limited optical penetration depth achieved using these optical imaging techniques is a major constraint in the structural and functional evaluation of these typically thick and complex liver-on-a-chip CIVMs. Advancements in optical microscopy techniques that can help to overcome the current challenges in visualization and characterization of liver CIVMs are crucial for enhancing our ability to investigate liver biology and drug treatment effects in these complex in vitro systems effectively.
This work describes the utilization of both labelled and label-free optical micro(spectro)scopy techniques for studying a liver-on-a-chip model. The liver-on-a-chip CIVM used in this study is a 3D perfused microphysiological system (MPS), which has been described previously by Kostrzewski et al.11 This in vitro liver model is capable of maintaining co-cultures of metabolically active PHH and KC for extended periods compared to 2D monocultures or spheroid cultures.14,15 Multiphoton microscopy (MPM) techniques such as simultaneous label-free autofluorescence multiharmonic (SLAM) microscopy16,17 and stimulated Raman scattering (SRS)18 microscopy offers several advantages for visualizing and characterizing this liver-on-a-chip platform. One key advantage of MPM techniques is its ability to penetrate deeper into thick and dense biological samples, such as the CIVMs, while minimizing photodamage to the cells by utilizing longer wavelength near-infrared pulsed laser beams.19 This enables researchers to visualize cellular interactions and structures in three dimensions with high spatial resolution, providing valuable insights into the complex microenvironment of the liver CIVM deep beneath its surface. SLAM microscopy is a novel MPM technique that performs simultaneous imaging of endogenous fluorophores such as nicotinamide adenine dinucleotide (phosphate) (NAD(P)H) from three-photon excited autofluorescence (3PF), flavin adenine dinucleotide (FAD) from two-photon excited autofluorescence (2PF), non-centrosymmetric structures such as collagen fibers from second harmonic generation (SHG) and structural interfaces such as cellular membranes, lipid droplets, and extracellular vesicles from third harmonic generation (THG).16,17 SRS microscopy is a coherent Raman imaging technique that reveals the vibrational properties of a sample with high chemical specificity at a sub-cellular resolution. SRS has been widely applied in cell classification,20 lipid distribution,21 and drug detection studies.22 However, utilization of these advanced optical imaging techniques for visualization and characterization of CIVMs have been limited so far. The enhancement in depth of penetration and complementary information on both structural and functional features of CIVMs obtained using the advanced MPM techniques have the potential to provide detailed insights into pharmacokinetic/pharmacodynamic (PK/PD) studies conducted using these CIVMs.
In this study, we demonstrate the benefits of MPM techniques to characterize microstructural features of a liver-on-a-chip model, which contained a co-culture of PHH and KC (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio). The aim of this study was to examine the effect of tagging an antisense oligonucleotide (ASO) with N-acetylgalactosamine (GalNAc) on the cellular uptake and intracellular localization of ASO. ASOs are a class of highly specific therapeutics, which use the cell's own gene regulatory machinery to alter the cellular gene expression. They rely on the complementary Watson–Crick base pairing to hybridize with their RNA targets.23 This mechanism of action allows to tailor the ASO to a specific gene by altering the nucleotide sequence. Hence ASOs have the potential to deliver new drugs to patients sooner and at lower costs in contrast with the market-dominating small molecule therapeutics, where challenging and multi-iterative molecular design strategies are required for each new therapeutic target. This was reflected in the list of FDA approvals in 2023, which included four novel therapeutic oligonucleotides,24 which was ∼20% of oligonucleotide therapeutics available on the market suggesting further market growth can be expected.
:1 ratio). The aim of this study was to examine the effect of tagging an antisense oligonucleotide (ASO) with N-acetylgalactosamine (GalNAc) on the cellular uptake and intracellular localization of ASO. ASOs are a class of highly specific therapeutics, which use the cell's own gene regulatory machinery to alter the cellular gene expression. They rely on the complementary Watson–Crick base pairing to hybridize with their RNA targets.23 This mechanism of action allows to tailor the ASO to a specific gene by altering the nucleotide sequence. Hence ASOs have the potential to deliver new drugs to patients sooner and at lower costs in contrast with the market-dominating small molecule therapeutics, where challenging and multi-iterative molecular design strategies are required for each new therapeutic target. This was reflected in the list of FDA approvals in 2023, which included four novel therapeutic oligonucleotides,24 which was ∼20% of oligonucleotide therapeutics available on the market suggesting further market growth can be expected.
One of the major hurdles in therapeutic ASO development is to deliver oligonucleotides in sufficient concentration to the target tissue and in the cells of interest. Chemical changes to the ASO polymer backbone as well as novel formulations and functional group conjugation to oligonucleotide ends were introduced to improve the delivery efficiency. In liver, ASO is taken up at a higher rate by nonparenchymal cells (sinusoidal endothelial cells, Kupffer cells) instead of hepatocytes.25 To target hepatocytes, GalNAc (targeting ligand) is conjugated to an ASO, which is specifically recognized by asialoglycoprotein receptor (ASGPR) present on hepatocyte cell membranes.26 While GalNAc modification of an ASO was previously reported to improve the ASO uptake in murine hepatocytes in 2D culture,27 and in murine tissues,28 it is unknown whether CIVMs containing a co-culture of PHH and KC will exhibit a similar ASO uptake enhancement. We investigated the cellular uptake and distribution of Alexa 488 (A488)-labelled ASOs (non-targeted ASO and ASO-GalNAc) in the liver-on-a-chip CIVM using multiphoton imaging techniques. Moreover, light sheet fluorescent microscopy (LSFM) was used to further understand the ASO distribution in the liver-on-a-chip model by utilizing the fast-scanning capability of the LSFM, and its ability to move and illuminate the sample at various angles.
Materials and methods
Liver-on-a-chip model
The liver-on-a-chip model used in this study is an array of open-well bioreactors, which housed collagen-coated scaffolds with a porous filter base, a pneumatic micropump and a reservoir that ensured a constant flow of nutrients mimicking the liver microenvironment (Fig. 1A).11 PHH and KC were obtained from Life Technologies (Thermo Fisher Scientific, USA) and BioIVT (USA), respectively. The human biological samples were sourced ethically, and their research use was in accord with the terms of the informed consents under an IRB/EC approved protocol. Cell culture work and ASO treatment of cells was conducted on the PhysioMimix system (CN Bio Innovations, UK). Cells were seeded onto scaffolds at 6 × 105 cells for PHH and 6 × 104 cells for KC in 1.6 mL of seeding media in the MPS on day 0. Seeding media consisted of advanced Dulbecco's Modified Eagle Medium (DMEM; 12491-015, Life Technologies, UK) with cocktail A and fetal bovine serum (FBS): plating supplement pack CM-3000 (Life Technologies, UK) and maintenance media consisted of William's medium E (WEM; A12176-01, Gibco, Thermo Fisher Scientific, US) with cocktail B: maintenance supplement pack CM4000 (Life Technologies, UK) and 100 nM hydrocortisone. Cells were incubated for multiple days to sufficiently cover the scaffold and form microtissue-like structures along the edges of the scaffold pores. An oxygen gradient was created across the microtissues as culture medium flowed through the tissue bed, mimicking the oxygen gradient in in vivo liver sinusoids.29 Cells were incubated for 4 days with a change of media after the first 24 h interval and on day 4. On day 7, DMEM was exchanged, and samples were dosed with different concentrations of A488-labelled non-targeted ASO and ASO-GalNAc (2 μM, 10 μM, 50 μM; Ionis, USA), respectively. ASO was diluted in WEM maintenance media to create a specific concentration in the full working volume of each well (1.8 mL). During the experiment, the cells seeded on the MPS scaffolds were maintained at a flow rate of 1 μL s−1. This controlled flow of ASO-containing media through the scaffold pores of the MPS scaffold helped to expose all seeded cells to a similar concentration of ASO. The ASO targeted the gene MALAT1 – a highly conserved nuclear long non-coding RNA. Samples were fixed with 4% paraformaldehyde (PFA) at 1 h, 4 h, 24 h, and 48 h time points post ASO treatment. Liver-on-a-chip scaffolds were removed from the MPS, washed, and stored in phosphate buffer saline (PBS). | ||
| Fig. 1 Brightfield and single-photon fluorescence microscopy of a liver-on-a-chip scaffold. A) and B) Schematics of a liver PhysioMimix perfused system and liver-on-a-chip scaffold, respectively. C) Brightfield image of a porous quarter scaffold seeded with primary human hepatocytes and Kupffer cells. D) A close-up brightfield image showing the cells residing on the scaffold surface and inside the scaffold pores. E) and F) Representative confocal single-photon excitation fluorescence images showing distribution of ASO signal intensity in cells on the scaffold surface (left column) and at 28.5 μm depth inside the scaffold pores (right column) shown at different magnifications, respectively. Scale bars = D and E) 200 μm, F) 50 μm. | ||
Spheroid cell culture
HepG2 cells were cultured in 60 mm Petri dishes and incubated at 37 °C in a 5% CO2 atmosphere. Cells were cultured in a high glucose Dulbecco's Modified Eagle Medium supplemented with GlutaMAX™ (Gibco, UK) and with 10% (v/v) fetal bovine serum (Gibco, UK). HepG2 spheroids were formed on an ultra-low binding 96-U-shaped well plate by seeding 400 cells per well, which were incubated for 5 days. Afterwards, cells were treated with ASO (2 μM) and fixed with PFA at 1 h time point post treatment.Multiphoton imaging platform
The femtosecond laser source used in the SLAM imaging system had a central wavelength of 1040 nm with 10 MHz pulse repetition rate (FemtoTrain, Spectra Physics, Milpitas, United States). The design details of the SLAM system were described previously.30 Briefly, the 1040 nm laser output was used to pump a nonlinear photonic crystal fiber (PCF, LMA-PM-15, NKT Photonics, Denmark). The PCF generated broadband pulses spanning from 870 nm to 1300 nm, which were subsequently tailored to a bandwidth from 990 nm to 1120 nm (130 nm bandwidth) by a pulse shaper to enable generation of 2PF, SHG, 3PF and THG signals simultaneously (Fig. 2A). All the nonlinear signals from the sample were acquired using a lab-built multimodal multiphoton imaging system set up as an inverted microscope, and the emitted signals were spectrally split into 4 different detection channels. The first detection channel captured the THG signal directed towards the photomultiplier tube (H7421-40, Hamamatsu, Japan) using a 405 nm dichroic beam splitter (Di02-R405-25 × 36, Semrock) and a 370 nm (bandwidth: 36 nm) bandpass filter (FF01-370/36-25, Semrock). The second detection channel collected the 3PF signal from NAD(P)H autofluorescence over the spectral window from 405–504 nm (FF01-451/106-25, Semrock). The third detection channel detected the SHG signal emitted between 542 nm and 566 nm using a bandpass filter (FF-01-554/23-25, Semrock) and a dichroic beam splitter edged at 593 nm (FF593-Di03-25 × 36, Semrock). The fourth detection channel primarily acquired the 2PF signal from FAD autofluorescence within the range from 593 nm to 700 nm excited by the 1040 nm portion of the broadband pulse.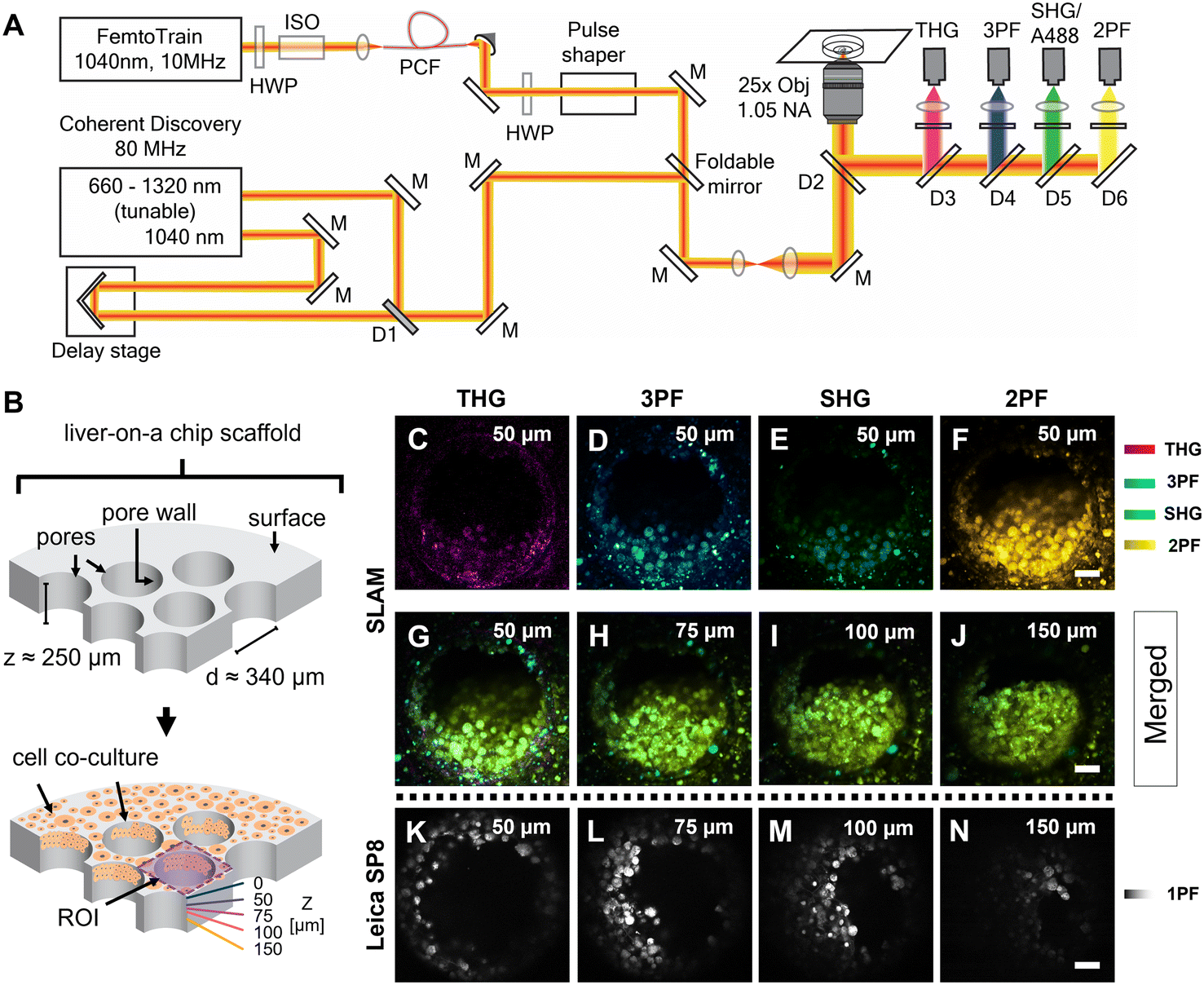 | ||
| Fig. 2 SLAM imaging of an untreated control liver-on-a-chip scaffold pores. A) Schematic of the custom-built inverted multiphoton imaging system used for imaging liver-on-a-chip scaffolds. The imaging platform has two laser sources: 1) a 10 MHz FemtoTrain laser with a central wavelength of 1040 nm for SLAM imaging and 2) an 80 MHz Coherent discovery laser source. B) Schematic of the liver-on-a-chip MPS scaffold. ROI – region of interest. C–F) Representative SLAM images obtained from one of the pores of a control untreated liver-on-a-chip scaffold at a depth of 50 μm below the scaffold surface. G–J) Merged SLAM images obtained from depths corresponding to 50 μm, 75 μm, 100 μm and 150 μm below the scaffold surface, respectively. 2PF – two photon fluorescence, SHG – second harmonic generation, 3PF – three photon fluorescence and THG – third harmonic generation. K–N) Comparative A488 single photon fluorescence images acquired with the Leica SP8 confocal microscope. 1PF – single photon fluorescence. Scale bars = 50 μm. | ||
In addition to capturing SLAM images of the liver-on-a-chip model, an 80 MHz Coherent discovery laser source with a synchronized dual output was used for multiphoton imaging of the A488-labelled samples. Both 800 nm (tunable output) and 1040 nm (fixed output) were simultaneously used for the achieving two-photon excitation of A488 dye. The laser beam was coupled into the custom-built inverted microscope designed for the SLAM system using a foldable mirror (Fig. 2A). A 25×, 1.05 NA objective (Olympus XLPLN25XWMP2) was used for focusing the laser beam onto the sample, and the fluorescence signal was collected in epifluorescence (epi) mode. Signal from the A488 dye was collected in the third detection channel (542–566 nm) corresponding to the SHG detection channel of the SLAM set up.
Images were collected from stained samples to validate the cellular features observed in the liver-on-a-chip model. In order to chemically confirm that the ASO was localized the cell nuclei and membranes, the chip was stained with Hoechst 33342 and eosin. Hoechst and eosin stains were excited sequentially at 750 nm and 1040 nm, respectively. Fluorescence emission signal from Hoechst and eosin stains was detected in the 3PF and A488 channels, respectively.
SRS and fluorescence microscopy
An SP8 confocal microscope (Leica Microsystems, Germany) was coupled with a PicoEmerald integrated pulsed laser system (APE, Germany) with a fixed-wavelength Stokes beam at 1031.2 nm and an optical parametric oscillator-tuned pump beam capable of a range of ∼720–940 nm was utilized for this study. The SRS signal was acquired in transillumination mode, where the two beams were spatially and temporally overlapped and focused on a sample using HC FLUOTAR L 25×/0.95 NA Water and HC PL IRAPO 40×/1.10 NA Water CORR lenses (Leica Microsystems, Germany). The transmitted light was collected using an S1 1.4 NA oil immersion condenser (Leica Microsystems, Germany), and the stimulated Raman loss in the pump beam was detected using a photodiode. The photodiode signal was denoised in real time using a UHFLI 600 MHz lock-in amplifier (Zurich Instruments, Switzerland) coupled with the picoEmerald reference pump output. The time constant was fixed at equal to one-half of the pixel dwell time to allow sufficient lock-in integration time without losing the temporal resolution. Data were acquired at an 8-bit depth. Images were acquired by raster scanning the laser beams with a pixel size set to ∼190 nm. The laser powers of the pump and Stokes beams were attenuated to 15–75 mW and 30–150 mW, respectively, where the Stokes intensity was always twice as high as the pump intensity. Hyperspectral SRS images were acquired from 2788 cm−1 to 3059 cm−1 with an 8 cm−1 step size. Single CH3 (2945 cm−1) and CH2 (2866 cm−1) frequency images were acquired by tuning the pump wavelength to 791 nm and 796 nm, respectively.Single-photon excitation fluorescence was collected in parallel using a PMT epi-detector in tandem with the SRS data by exciting the A488 dye with a Leica TCS SP8 confocal microscope equipped with a continuous wave argon laser source (excitation 488 nm). The epifluorescence signal was collected with the same 25× lens used for the SRS image capture.
Hyperspectral SRS data were processed and analyzed with the Hyperspectral Image Analysis (HIA) tool.31 Prior to component extraction, data were first denoised using the built-in SVD algorithm. Individual components were extracted using the non-negative matrix factorization-based FSC3 algorithm,32 which provided the absolute concentration of individual components per pixel. The total number of components was set to six with an active spatial correlation constraint equal to 1 pixel to avoid extreme deviation between neighboring pixels and to mimic the effect of the limited spatial resolution of the microscope objective. Weighting was used to reduce local noise-dominated signal deviations and highlight localized chemical components. The weighting terms α, α′, and γ were set to 0.5, 0.5, and 1, respectively.
Light-sheet fluorescence microscopy
After fixation, the chip and spheroid samples were washed with PBS and incubated with cell mask 647 (1:500) or concavalin A (ConA) 571 nm (1:500), and Hoechst 33342 (1:5000) for 1 h. Subsequently, the scaffolds were thoroughly washed and cut into thin strips to fit into glass capillary sample holders and partially submerged in 1.5% (v/v) low-melting agarose solution. A Zeiss light sheet Z.1 microscope was used to acquire imaging data using the W Plan-Apochromat 20×/1.0 corr objective lens immersed in PBS. Hoechst, A488, ConA, and cell mask were excited with 405, 488, 561, and 638 nm continuous laser lines, respectively. The light sheet thickness was set to 4–6.6 μm. Images of 1920 × 1920 pixels with Z < 300 μm were collected at a 16-bit depth and subsequently processed with Arivis Vision4D (Carl Zeiss AG, Germany).
Cell segmentation and ASO quantification
The 3D cell segmentation was achieved by leveraging the Cellpose model.33 Using the pre-trained segmentation model, individual cells were segmented from the 3-D multiphoton image stacks. To quantify the cellular ASO uptake, the average fluorescence intensity measured in the A488 channel (third detection channel) was determined for each segmented cell. The statistical differences in the amount of drug uptake of non-targeted ASO and ASO-GalNAc were assessed using Mann–Whitney U tests.For quantification of non-targeted ASO and ASO-GalNAc specifically in the hepatocytes with in vivo relevant cuboidal morphology, images were collected from the selected pores of the scaffold identified based on the wide-area mosaic images obtained from the liver-on-a-chip scaffold. As shown in Fig. S3,† cuboidal hepatocytes were manually segmented using ImageJ software and the mean fluorescence intensity per cell detected in the A488 channel was calculated.
Results and discussion
Optical imaging of liver-on-a-chip model using single-photon fluorescence microscopy
PHH and KC were seeded on collagen-coated porous polystyrene scaffolds, which were embedded in a perfused MPS with a controlled flow of nutrients, mimicking human liver physiology (Fig. 1A and B). The scaffold (thickness ≈ 250 μm) was made of translucent polystyrene with closely dispersed pores (diameter ≈ 340 μm) (Fig. 1C), wherein cells were observed clearly adhering to the scaffold surface and the pore walls (Fig. 1D). The brightfield images demonstrate that the extent of microtissue formations and relative areas occupied by the cells varied between various pores in the scaffold.Prior to 4% PFA fixation, the cells shown in Fig. 1 were treated with 2 μM fluorescently labelled non-targeted ASO (A488-ASO) for 1 h. The non-targeted ASO was observed inside the cells on the scaffold surface and inside the pores (depth = 28.5 μm) with the single photon excitation fluorescence modality (Fig. 1E and F). However, the signal intensity of non-targeted ASO that originated from cells inside the pores were lower compared to the non-targeted ASO signal observed in the cells located on the scaffold surface. Since the cells inside the pores are exposed to a similar concentration of non-targeted ASO as the one on the scaffold surface, the reduction in fluorescence signal with increasing depth can be attributed to the scattering of excitation photons and the fluorescence photons originating from the focal point, which both travel through microtissue formations along the laser beam pathway. This was a major limitation for the single-photon fluorescence microscopy technique to investigate the microstructural features and non-targeted A488-ASO distribution across the depth of the liver-on-a-chip model. Advanced optical imaging techniques such as MPM techniques or LSFM that can provide deeper penetration into microtissue formations are needed to visualize and quantify the heterogeneity of ASO distribution in CIVMs.
Label-free imaging of liver-on-a-chip model using SLAM microscopy
Fixed liver-on-a-chip scaffolds were examined using a custom-built SLAM microscope to evaluate the capability of multiphoton imaging technology to visualize and characterize the microstructural details of the liver-on-a-chip model. Liver scaffolds were placed on a 35 mm glass-bottom dish, submerged in PBS, and placed flat in the sample holder, perpendicular to the objective lens and the optical pathway of the SLAM inverted microscope setup (Fig. 2A). Using the SLAM imaging set up, backscattered autofluorescence and multiharmonic signals were detected simultaneously from the selected regions of interest within a control untreated liver-on-a-chip scaffold.As indicated in Fig. 2B, pseudo-color-coded SLAM images obtained from regions of interest (ROIs) centered at scaffold pores at a depth of 50 μm below the scaffold surface are shown in Fig. 2C–F. In a SLAM microscope, morphological characteristics of the microtissue formations were mainly observed in the 2PF (FAD, yellow) and 3PF (NAD(P)H, cyan) detection channels.17 As expected, the boundary of the scaffold pore to which cells were attached and cellular interfaces were observed in the THG channel (Fig. 2C). Typically, the SHG channel of a SLAM microscope captures the signal originating from non-centrosymmetric structures such as collagen fibers in the context of animal and human biology.34 Instead, a small portion of the NAD(P)H and FAD autofluorescence was collected in the SHG channel at a depth of 50 μm inside the pores, since the detection window of this channel was centered at ∼550 nm (Fig. 2E). In the SLAM images obtained from the untreated liver-on-a-chip scaffold sample, the signal intensities detected in the 2PF and 3PF channels arising from the cellular autofluorescent components were stronger compared to the signal intensities detected in the THG and SHG channels. The signal intensities detected in the SLAM channels were contrast-enhanced to depict the complementary information observed in the respective channels. To compare the SLAM images obtained from different depths (50 μm, 75 μm, 100 μm and 150 μm), the absolute pixel intensities detected in each SLAM channel were pseudo-color-coded, and signal from individual channels were combined to produce the merged images corresponding to different depths (Fig. 2G–J). The merged images had a greenish-yellow appearance due to the dominance of 2PF (yellow) and 3PF (cyan) signals arising from the microtissue formations inside the pores observed at different depths. As shown in Fig. 2G–J, it was possible to visualize cellular features in the liver-on-a-chip scaffold pores up to a depth of 150 μm using SLAM microscopy. The reduction of the fluorescence signal intensities with increasing depth in the SLAM images was lower compared to the single photon fluorescence (1PF) images owing to the application of a longer wavelength excitation laser (Fig. 2K–N). Although 1PF signal was detected at a depth of 150 μm, this was only true for a few top-level cell layers, which were unobscured by the remaining microtissue, whereas the rest of the image was dominated by low intensity blurred signal or by background. Detecting clear A488 1PF signal at the 150 μm depth was achieved by imaging exclusively scaffold pores where the cells grow in an inverted conical shape along the longitudinal direction of the scaffold pore, which minimized the single photon scattering. These results demonstrate the capability and necessity of MPM techniques such as SLAM to visualize microstructural features from deeper regions of liver-on-a-chip models in a non-invasive and label-free manner.
Visualization and quantification of ASO uptake using multiphoton fluorescence microscopy
One of the promising applications of the liver-on-a-chip platforms is to facilitate the identification and development of drug compounds with optimal PK/PD properties, efficacy, and safety profiles. Currently, the major end point measurements of these in vitro assays include read outs of functionality biomarkers and profiling analysis.14 Imaging techniques provide valuable information on the spatial heterogeneity, drug treatment response and functional changes occurring in these liver-on-a-chip platforms. However, limited penetration depth and lack of 3-D information is a major restriction faced by the conventional optical imaging techniques for evaluating liver-on-a-chip models during PK/PD studies, and further advancements in imaging techniques that enable better characterization of the liver-on-a-chip models are needed.To that end, the custom-built multiphoton imaging platform was utilized to evaluate the cellular uptake and distribution of non-targeted ASO and ASO-GalNAc in the liver-on-a-chip scaffolds. Simultaneously synchronized laser excitation beams at 800 nm and 1040 nm were used to achieve efficient two-photon excitation of the fluorescent A488 label. The fluorescence signal emitted from A488 was collected in the third detection channel (Fig. 2A) and A488 fluorescence intensity was measured for liver-on-a-chip scaffolds treated with PBS only (control), non-targeted ASO and ASO-GalNAc, respectively (Fig. 3A–C). Cells detected in the pores under each of those treatment conditions were segmented along the depth of the pore using the Cellpose algorithm and color-coded based on the fluorescence intensity detected in the A488 channel (Movies M1–M3†). Among the two treatment groups, a higher level of A488 fluorescence intensity was detected in the images obtained from the liver-on-a-chip scaffolds treated with ASO-GalNAc group compared to the non-targeted ASO group at 1 h time point (n = 3/time point per group; Fig. S1†). This observed increase in uptake of ASO-GalNAc is consistent with the results obtained in with the results obtained in previous 2D hepatocyte monoculture studies.27 A non-uniform distribution of A488 signal was observed within each field of view in both treatment groups, indicating that there were cell–cell variations in non-targeted ASO and ASO-GalNAc uptake within the microtissue formations.
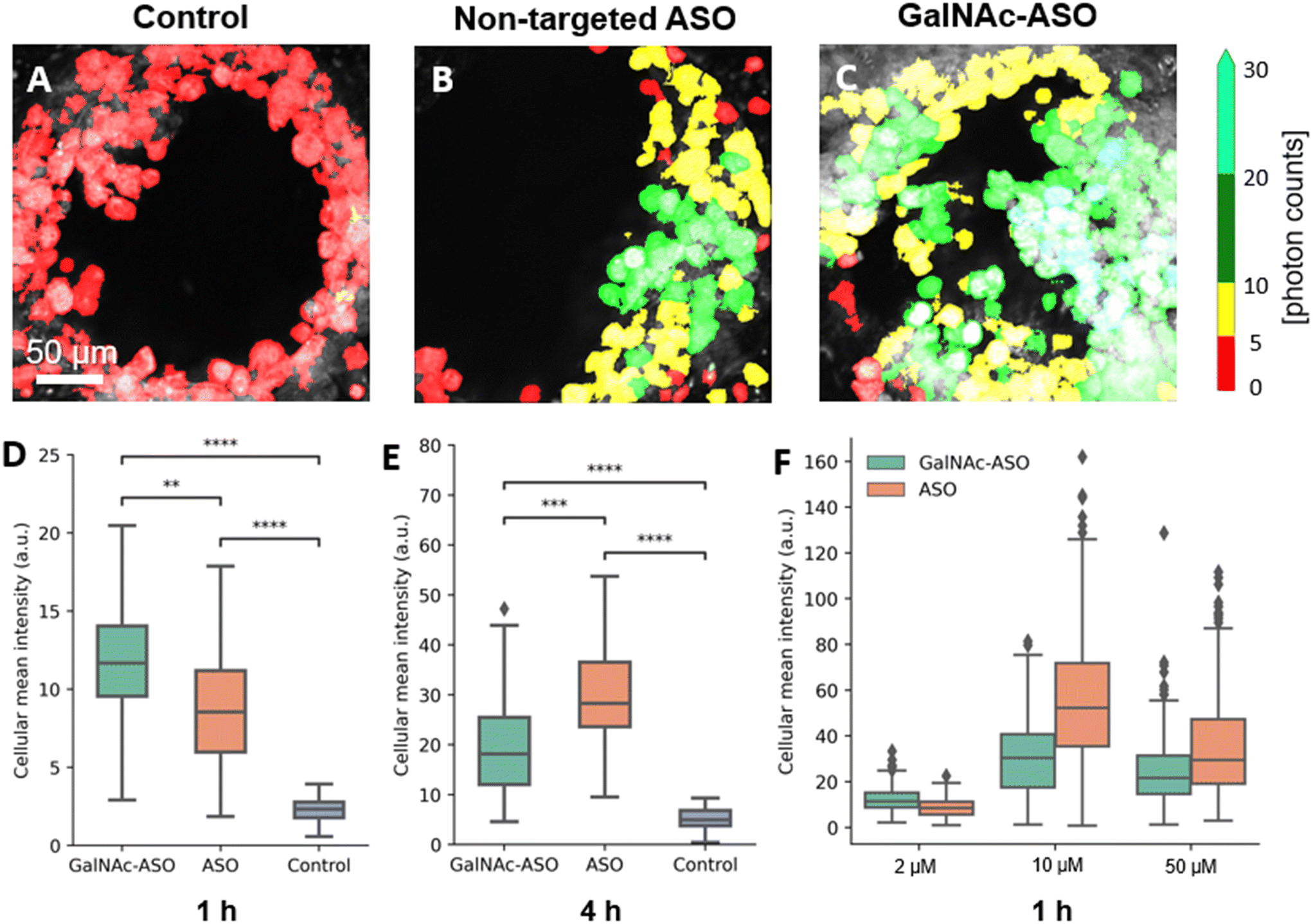 | ||
| Fig. 3 Visualization and quantification of non-targeted ASO and ASO-GalNAc in the liver-on-a-chip using multiphoton fluorescence microscopy. A–C) Representative false color-coded fluorescence intensity images obtained from one of the pores of the liver-on-a-chip scaffold belonging to control (treated with PBS only), non-targeted ASO and ASO-GalNAc groups, respectively, 1 h post-treatment at 2 μM concentration. D) and E) Quantification of ASO uptake (A488 fluorescence) in liver scaffolds dosed at 2 μM concentration in ASO-GalNAc, non-targeted ASO and control groups at 1 h and 4 h, respectively. F) Comparison of ASO-GalNAc and non-targeted ASO uptake following treatment with different concentrations at 1 h. **: P ≤ 0.001, ***: P ≤ 0.0001, ****: P ≤ 0.00001. Scale bar = 50 μm. | ||
The average value of A488 fluorescence intensity were measured from the liver-on-a-chip scaffolds dosed at 2 μM concentration under each treatment condition at 1 h and 4 h time points post-treatment (Fig. 3D and E). At 1 h time point, the average fluorescence intensity measured in the ASO-GalNAc group was significantly higher (P < 0.001) than in the non-targeted ASO group (Fig. 3D). However, the overall fluorescence intensity at the 4 h time point showed that non-targeted ASO uptake surpassed the ASO-GalNAc (P < 0.0001). Previous studies in monoculture primary mouse hepatocytes had reported a similar increased cellular uptake of ASO-GalNAc occurring during the first 2 h after the treatment in comparison to the non-targeted ASO group.27 The rapid uptake of ASO-GalNAc during the first hour post-treatment can be attributed to the high affinity of the GalNAc ligand with transmembrane ASGPR that is abundantly expressed on mammalian hepatocytes.26 The ASO-GalNAc-ASGPR complex is internalized into hepatocytes by clathrin-mediated endocytosis,35 which directs the cargo into endosomes, where the ligand–receptor complex is dissociated, the receptor is recycled, and majority of cargo is directed to the lysosomal pathway.36 Thus, the high affinity of GalNAc ligand with ASGPR facilitated efficient uptake of ASO-GalNAc into PHH during the first hour post-treatment, but was then slowed by lowered number of binding-available ASGPR on the cell surface. In contrast, ASO taken up into cells occurs via comparatively slower endocytic processes involving other membrane receptors such as scavenger receptors SR-AI/II.37 Although uptake occurs via less efficient pathways, it was observed that a significantly higher amount of non-targeted ASO was internalized into cells by 4 h time point (Fig. 3E), which suggested lower receptor saturation in this group.
To get a better understanding of the relationship between ASO availability and cellular uptake mechanisms, multiphoton fluorescence images were obtained from liver-on-a-chip scaffolds treated with different concentrations (2 μM, 10 μM and 50 μM) of non-targeted ASO and ASO-GalNAc at 1 h post-treatment (Fig. 3F). Interestingly, the average A488 fluorescence intensity was higher for the ASO-GalNAc group only in the lower concentration (2 μM) treatment condition. Both at 10 μM and 50 μM treatment conditions, non-targeted ASO group showed higher cellular uptake of ASO compared to the ASO-GalNAc group. These results were consistent with observations in previous studies that the cellular uptake of ASO-GalNAc via the ASGPR mediated pathway tend to reach a plateau 1–2 h post-treatment and at higher GalNAc concentrations.25,27 Although the uptake rate of the non-targeted ASO was less efficient than the ASO-GalNAc during the first hour post treatment, this group exhibited a steady increase in cellular uptake of ASO with time and concentration. This was likely due to the uptake being facilitated by a larger number of receptors on the cell surface compared to the GalNAc-binding ASGPR, which suggested the ASGPR-mediated endocytic pathway was saturated at high ASO-GalNAc concentrations (10 μM and 50 μM), likely due to the high binding affinity to GalNAc and insufficient receptor recycling rate.26 These results demonstrate the capability of MPM techniques to quantitatively measure ASO uptake and distribution in liver-on-a-chip samples at different ASO concentrations and its effect on cellular uptake. Further imaging studies measuring ASO uptake at different concentration levels and investigation of time required to reach saturation levels under each treatment condition are needed to obtain a deeper understanding of the ASO uptake mechanisms in the liver-on-a-chip model.
Quantification of ASO uptake in cuboidal hepatocytes in the liver-on-a-chip model
In addition to quantifying the average ASO uptake in a liver-on-a-chip model at different time points post-treatment, MPM was used to investigate the spatial heterogeneity of the ASO uptake and 3D distribution within liver scaffolds. Primarily, cells exhibiting circular and cuboidal morphology were observed within the pores of the liver-on-a-chip scaffolds. A representative MPM image showing NAD(P)H autofluorescence detected in the second detection channel arising from circular and cuboidal cells at a depth of 30 μm inside the pore of a vehicle control (PBS only) liver-on-a-chip scaffold is shown in Fig. S2.† Under optimal microenvironmental conditions, PHH are known to exhibit cuboidal morphology with well-defined cell borders, and enhanced transporter and metabolic functions.38 To measure ASO uptake specifically in cuboidal hepatocytes, pores containing PHH with cuboidal morphology were identified by acquiring 2D wide-area mosaic images of the liver-on-a-chip scaffolds. A representative mosaic image obtained from ASO-GalNAc treated liver-on-a-chip scaffold is shown in Fig. 4A. The ASO-(GalNAc) was found to strongly localize in nuclei-like structures of the cells with additional high contrast fluorescence on the cell membrane and minimal signal detected in the cytoplasm (Fig. 4B). Hoechst and eosin staining was used to highlight the cellular morphology and confirm that the non-targeted ASO and ASO-GalNAc distribution in hepatocytes was primarily localized to the cell nuclei in the liver-on-a-chip scaffold (Fig. 4C).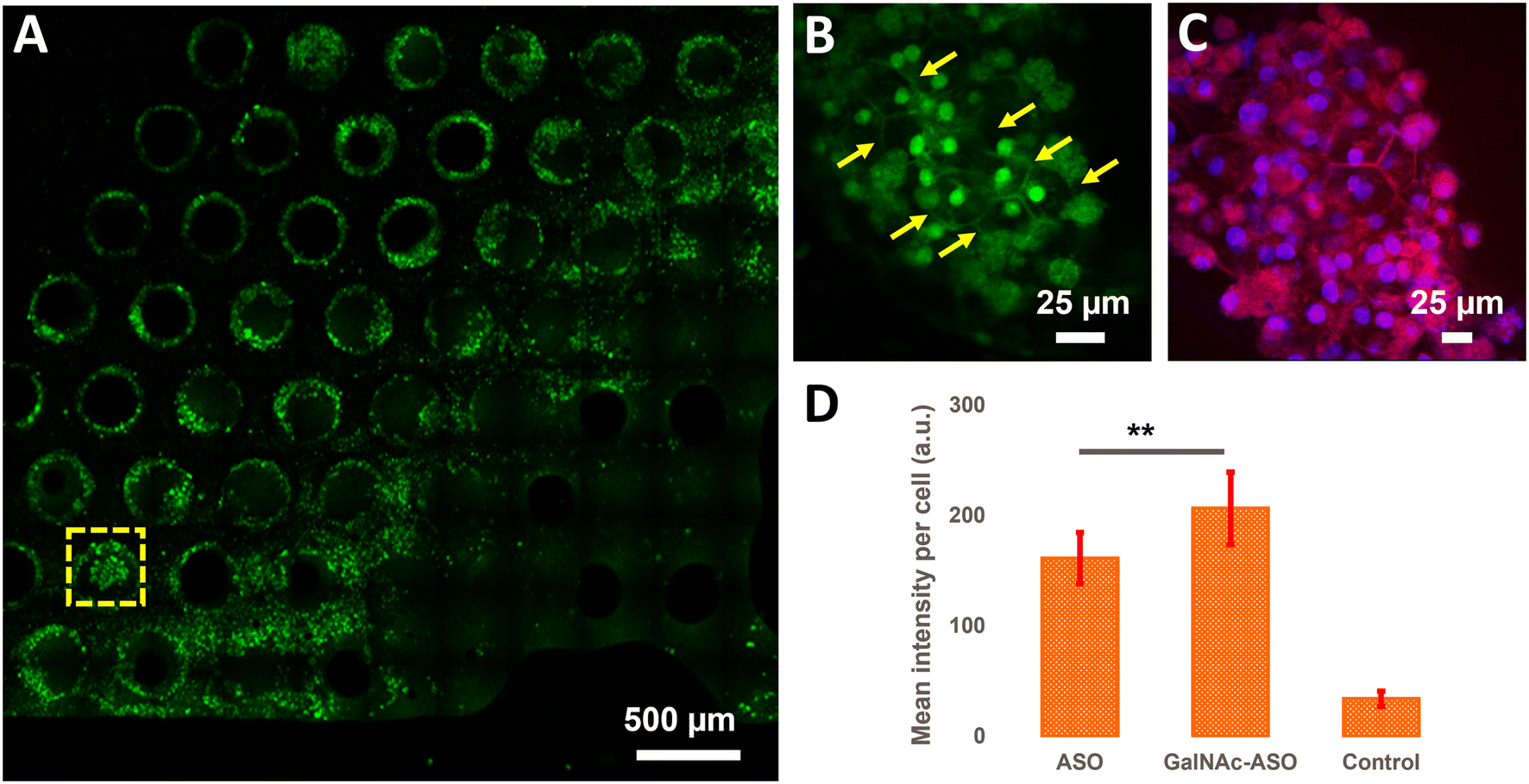 | ||
| Fig. 4 Visualization and quantification of ASO uptake in cuboidal hepatocytes in the liver-on-a-chip. A) A wide area 2D mosaic image of an ASO-GalNAc treated liver-on-a-chip scaffold at 2 μM concentration 1 h post-treatment. B) One of the pores containing cuboidal hepatocytes (yellow arrows) identified from the mosaic image. C) Image obtained from a liver-on-a-chip stained with Hoechst 33342 and eosin stains to confirm the nuclear (blue) and cell membrane (red) structures. D) Comparison of non-targeted ASO and ASO-GalNAc uptake in cuboidal hepatocytes 1 h post-treatment (N = 20 cuboidal cells/condition). **: P ≤ 0.01. Scale bars: A) 500 μm, B and C) 25 μm. | ||
Cellpose training and manual annotations of 3D data are still cumbersome and extremely labor-intensive.39 To quantify the cellular uptake of non-targeted ASO and ASO-GalNAc specifically in cuboidal hepatocytes, cuboidal cells were manually segmented (Fig. S3†; N = 20 cuboidal cells/condition) for both treatment conditions and the average fluorescence intensity per unit area in these cells were compared. At 1 h time point, significantly higher amount of ASO-GalNAc was detected in the cuboidal hepatocytes (N = 20 cuboidal cells/treatment condition) compared to non-targeted ASO (Fig. 4D). Similar to the trend observed in the average A488 fluorescence intensity measured from the liver-on-a-chip scaffolds at 1 h time point (Fig. 3D), ASO-GalNAc was taken up at a faster rate compared to non-targeted ASO in cuboidal hepatocytes as well during this time period. Thus, the differential uptake of ASO-GalNAc in cuboidal hepatocytes was confirmed, and these results demonstrate the potential of MPM techniques to visualize and quantify ASO uptake in a cell-type specific manner in liver-on-a-chip models.
The observed ASO localization in the nuclei of cuboidal hepatocytes was used to quantitatively prove the light penetration depth advantage of 2PF microscope systems over 1PF microscopes. In the 1PF confocal image dataset, the number of nuclear features detected with Cellpose segmentation model decreased immediately after focusing below the first layers of cells, and a maximum of three nuclei at low fluorescence intensities was found below approximately 35 μm per Z position. On the other hand, on average fourteen nuclei was identified from the 2PF dataset. This number dropped below ten at ∼60 μm, but ASO signal from the nuclei was measurable at 75 μm pore depth (Fig. S4†). In order to compare the fluorescence signal loss with depth between 1PF and 2PF image datasets, the average fluorescence intensity detected per nuclei per Z plane were determined over a depth of 0–75 μm. As shown in Fig. S4G and H,† the average fluorescence intensity per nuclei measured in the measured pore of the liver-on-a-chip model dropped faster with depth in the 1PF dataset compared to the 2PF dataset.
A majority of cuboidal hepatocytes was expected to be inside the scaffold pores, where the cells get mechanical cues and nutrients most resembling the sinusoidal environment of liver tissues. The deeper optical penetration into the liver-on-a-chip model achieved using MPM techniques is a crucial factor for ASO distribution and uptake studies in cuboidal hepatocytes in liver CIVMs given that differential intracellular distribution of ASOs between cuboidal and circular hepatocytes was observed.
Further studies using organ-on-a-chip models with different co-culture and environmental conditions are needed to gain deeper insights into the effect of cellular morphology and functionality on ASO uptake dynamics. Coupled together with other functional readouts, imaging studies using MPM techniques can be utilized to identify ASO candidates with optimal pharmacokinetic properties and obtain a better understanding of various biophysiological factors affecting ASO uptake in liver CIVMs.
Label-free cell classification of circular and cuboidal hepatocytes using SRS microscopy
Cell segmentation in 3D cell cultures is notoriously difficult. Moreover, finding an ideal cell type-specific fluorescent dye, which can be used to separate cell phenotypes, such as circular and cuboidal hepatocytes, can be an arduous task. As an alternative label-free approach, we explored hyperspectral SRS microscopy to examine whether the change in morphology between cuboidal and circular hepatocytes was accompanied by a change in chemical composition and chemical distribution in a PFA-fixed, ASO-untreated PHH monoculture grown in a liver-on-a-chip MPS. The liver scaffolds were examined in the C–H region of the Raman spectrum (2780–3050 cm−1), which encapsulates the diversity of CH, CH2, and CH3 molecular bonds.40The complex information embedded in the hyperspectral data cube containing voxels with spatial (XY) and vibrational (ω) information was first reduced in dimensionality to a 2D image by color-coding the vibrational information contained in the Raman spectrum, thus improving the interpretability of the data (Fig. 5A). The differently color-coded SRS information highlighted the cells in the sample and revealed a faint distinction between lipid droplets (white, <2900 cm−1) and cytoplasm (cyan, >2900 cm−1). A faint chemical contrast was observed between the cuboidal (yellow arrows) and circular PHHs (red arrows) in the compressed image shown as grey and blue cytoplasm hue. To improve the contrast between lipid and protein components in cells, stimulated Raman histology (SRH) workflow was applied. SRH has been proven to effectively replace hematoxylin and eosin staining with high accuracy by acquiring single images of CH2 and CH3 vibrational modes.41,42 The peaks at 2866 cm−1 and 2945 cm−1 roughly correspond to CH2 and CH3 modes, respectively. A separation between lipid rich and protein rich regions of cells was achieved by making a ratio |CH3–CH2| between the two images, which was overlayed on the CH2 (lipid) image following the method introduced by Freudiger et al.41 Indeed, the contrast previously observed in Fig. 5A was more pronounced in the SRH image (Fig. 5B), which showed a high lipid concentration (green signal) in circular hepatocytes. In contrast, the cytoplasm of the cuboidal hepatocytes appeared protein-rich (magenta), suggesting a contrast in cell physiology between circular and cuboidal hepatocytes.
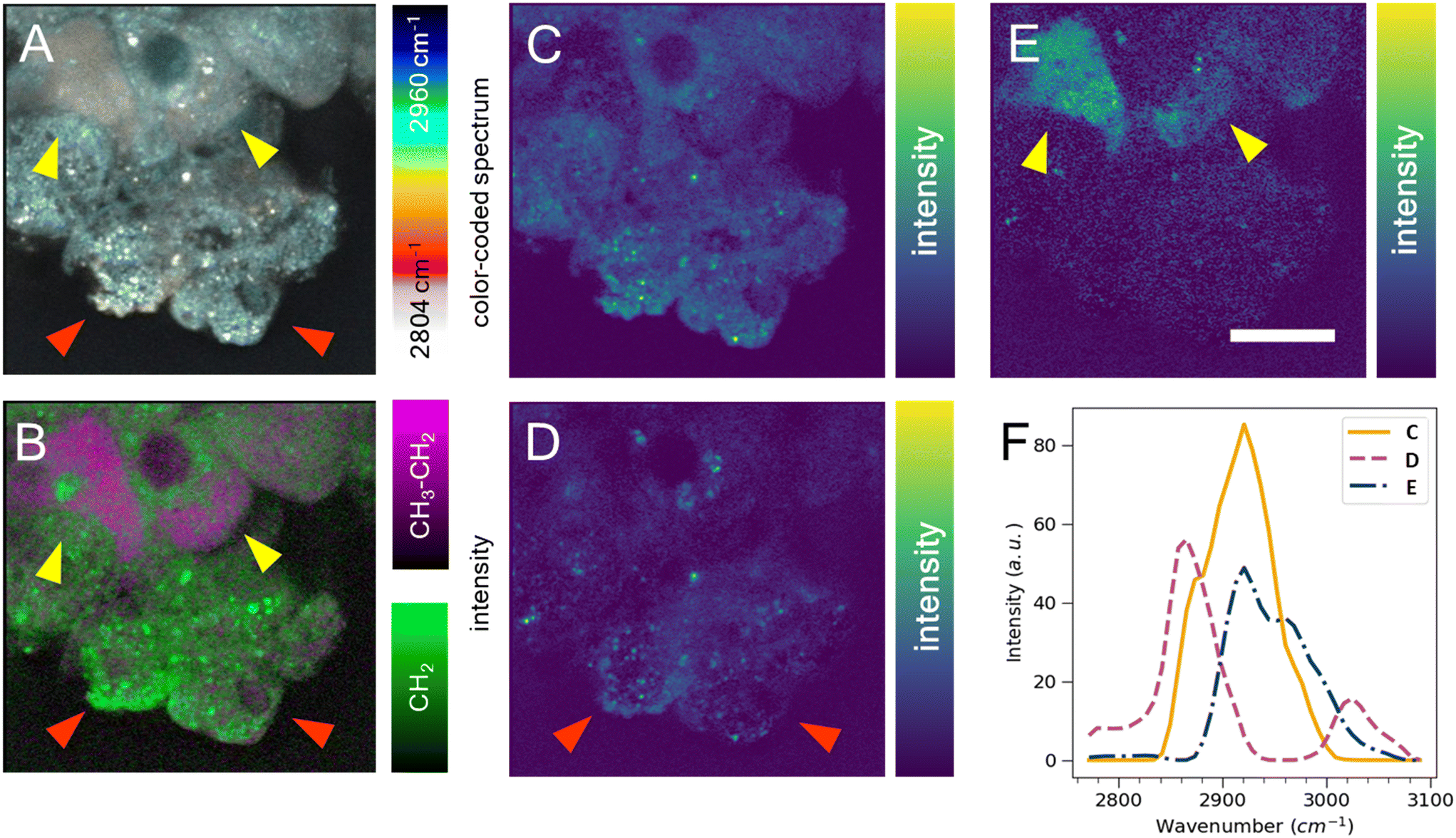 | ||
| Fig. 5 Heterogeneity of hepatocyte physiology with hyperspectral SRS in PHH in liver-on-a-chip microtissues. Hyperspectral SRS in the C–H region of Raman spectrum captures faint intercellular differences. A) Simplest dimensionality reduction by color-coding each spectral point. B) The image subtraction of SRS data cube slices captured at 2920 cm−1 and 2865 cm−1 (magenta) overlayed with the 2865 cm−1 image slice (green). Precise chemical component separation was achieved based on both spatial (C–E) and spectral (F) information with FSC3 analysis. C) The first FSC3 component highlighted the protein distribution. D) The second component highlighted the punctate distribution of putative lipids, which were found predominantly in circular cells (red arrows). E) The third component highlighted cytoplasm of cuboidal cells seen towards the top region of the image (yellow arrows). Scale bar = 10 μm. | ||
To better understand the chemical composition of the cytoplasm of cuboidal hepatocytes, the hyperspectral SRS data were analyzed using the FSC3 unsupervised non-negative matrix factorization algorithm,32 which resulted in the detection of chemical components that captured the putative protein component (Fig. 5C) and lipid components, which appeared as puncta throughout the circular cells (Fig. 5D). The cytoplasm of cuboidal hepatocytes was separated using the third FSC3 component with an improved signal contrast (yellow arrows in Fig. 5E), in comparison to the flattened and SRH images shown in Fig. 5A and B. Interestingly, the components associated with the cuboidal PHH cytoplasm was spatially homogeneous, which suggested it is freely distributed in the cell cytoplasm. The spectra of the individual FSC3 components revealed the approximate compositions of the CH2 and CH3 vibrational modes within the sample (Fig. 5F). The cuboidal cytoplasm spectral component shown in Fig. 5E consisted mostly of the asymmetric stretch vibrational mode, which overlapped with the protein CH3 signal. Although it does not directly establish that the third FSC3 component was protein-based, this observation in conjunction with its homogeneous distribution suggested that this component was primarily hydrophilic and not lipid-based. Moreover, this FSC3 component (Fig. 5E) was not observed in the perinuclear region, unlike the protein-associated component shown in Fig. 5C. Although the exact chemical composition cannot be determined from this spectral information, hyperspectral SRS results of the C–H region demonstrate the capability of this technique to be utilized as a powerful analytical method for phenotype separation of cell types within a liver-on-a-chip model in a label-free manner. Previous ASO uptake study in live hepatocytes by Mukherjee et al. showed that hepatocytes in simple non-confluent 2D cell culture remain in a circular shape.27 Therefore, the circular hepatocytes were considered to assume their shape when they are in an environment with low cell-to-cell contact. However, it is possible a small subpopulation of cells was detaching dead hepatocytes.
Subcellular localization of ASO and lipid droplets using simultaneous hyperspectral SRS and fluorescence microscopy
The SRS effect can generate the Raman effect with 108 times improved efficiency in comparison with spontaneous Raman systems, which can lead to highly contrasted hyperspectral data at high speed. Nevertheless, label-free detection of specific oligonucleotides in cells is not currently feasible owing to the low sensitivity, low analyte concentration and insufficient spectral contrast needed to measure the ASO of interest. However, the absence of fluorescence interference in SRS signal makes this technique well-suited for simultaneous label-free detection of cells and tracking of fluorescently labelled ASOs in the liver-on-a-chip samples. Combined fluorescence and SRS images were acquired at the interface between a scaffold pore and surface to obtain sufficient signal-to-noise ratio and reduce light scattering from deeper in the sample. The fluorescent signal of the A488 label revealed a heterogeneous distribution of ASO inside the cells (50 μM, 1 h) in the scaffold pores (Fig. 6A). The ASO localized into large spherical structures within cells, which were hypothesized to be the putative nuclei of cuboidal hepatocytes (white arrows) observed in the hyperspectral SRS images (Fig. 6A), whereas they appeared as dark circular regions in spectral components in the untreated control sample as shown in Fig. 5C. The dark feature of nuclei was attributed to a low concentration of lipids and proteins, which were present at higher concentration in cytoplasm. In circular hepatocytes (red arrow), the ASO was sequestered into putative cytoplasmic punctae. This disparity in the intracellular distribution of ASO observed between the cuboidal and circular hepatocytes point towards the importance of conducting drug uptake studies in CIVMs that better reflect the in vivo environment. By tuning the pump laser to 796 nm (2866 cm−1), CH2 vibrational mode was visualized in the sample (Fig. 6B). This image revealed both a homogeneous low-contrast signal inside the cells and high-contrast small lipid droplets, which were polarized in distribution towards the center of the pore and on the cell mass surface. As shown in Fig. 6C, a polystyrene scaffold pore wall was visualized by tuning the pump laser wavelength to 783.5 nm (3074 cm−1). Furthermore, small faint structures were observed inside the pore at the same frequency. It was hypothesized that these were either lipid components or debris overlapping with the vibrational frequency of polystyrene. No crosstalk was observed between the A488 fluorescence and SRS signals. Moreover, the spatial overlap between the punctate ASO and SRS CH2 signals was minimal, suggesting that there is no direct link between lipid deposits and sequestered ASO punctate trafficking inside the hepatocytes (Fig. 6D).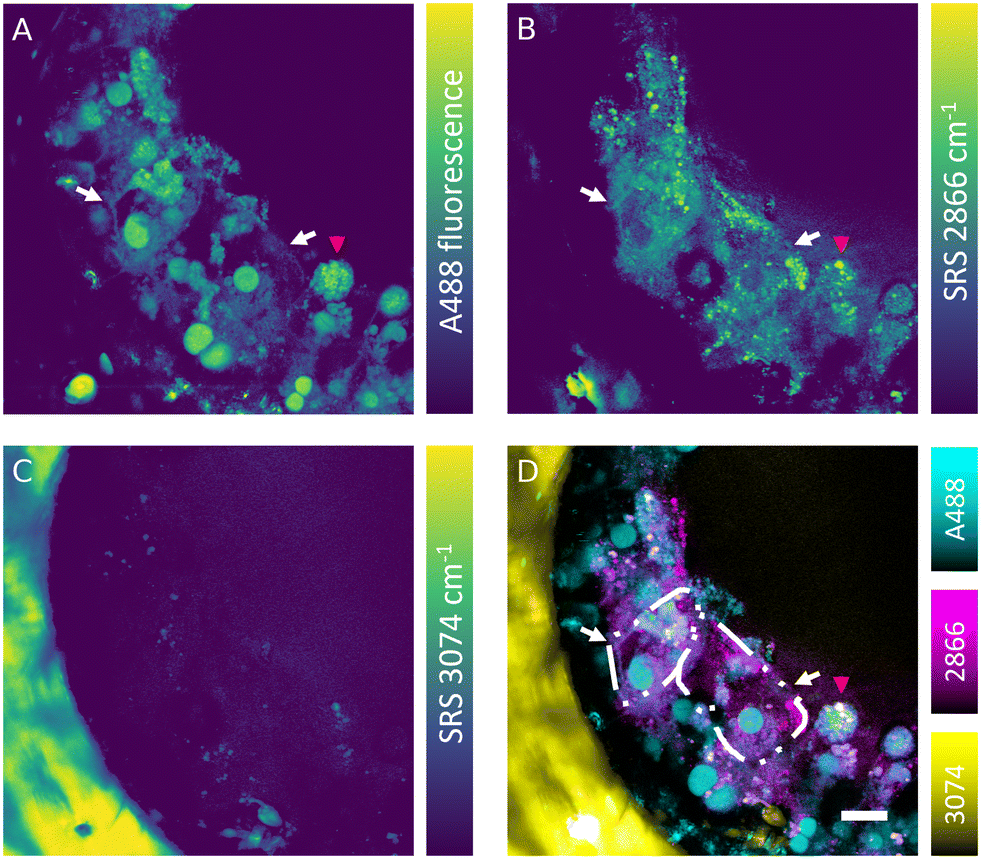 | ||
| Fig. 6 Simultaneous hyperspectral SRS and fluorescence microscopy of ASO treated liver-on-a-chip model (50 μM, 1 h. A) A488-labelled ASO was distributed in multiple cellular compartments and on cellular surfaces of cuboidal (white arrows) and circular (red arrow) hepatocytes. B) Single vibrational image of the CH2 highlighted predominantly lipid distribution in the cells. C) Vibrational mode corresponding to 3074 cm−1 highlighted the polystyrene scaffold wall. D) Overlay of fluorescence and SRS channels showing the distribution of lipids, polystyrene and the ASO. Cuboidal hepatocyte cell boundaries were highlighted with dash-dot-dot lines. Scale bar = 20 μm. | ||
Simultaneous SRS and fluorescence microscopy herein described offers a potential to track dynamics of fluorescently tagged drug and lipid distribution in biological systems without an introduction of further fluorescent labels in real time. These results suggest SRS can play a major role in the development of ASO delivery strategies for treating diseases such as the non-alcoholic fatty liver disease (NAFLD), where the potential of Raman in mapping the distribution and concentration of cholesterol and lipids during the disease progression has already been demonstrated.43
Enhanced 3D visualization of the ASO uptake in a liver-on-a-chip model using light sheet fluorescence microscopy
Although the advanced light penetration capability of the near-IR SRS and the MPM instrument enabled a visualization of cells localized deep within the scaffold pores (∼150 μm), strong scattering of light by microtissue formations in the scaffold pores prohibited visualization of the cellular features and organization in the entire pore. To address this restriction, the potential of LSFM was explored to perform deeper imaging of the liver scaffolds by exploiting its ability to collect light from the sample at a 90° angle. In combination with the LSFM ability to rotate the sample, the whole scaffold surface and pores walls were exposed to the microscope light path (Fig. S5†). LSFM images of the fixed liver-on-a-chip samples were collected in three channels. In addition to A488-labeled ASO (Fig. 7A, E and I), cell nuclei were stained with Hoechst (Fig. 7B, F and J) and cell membranes with cell mask A647 (Fig. 7C) or concanavalin A rhodamine (Fig. 7G and K). First, a maximum intensity projection of an overview scan of the unpermeabilized sample face was acquired (Fig. 7A–D), where two individual light sheet beams illuminated the front surface of the scaffold and rear part of the pore wall. The images confirmed a dense distribution of ASO-positive cells, as observed with previously described imaging modalities (Fig. 7A). The ASO distribution was confirmed to be highly accumulated in putative cell nuclei or cytoplasmic punctae. Unexpectedly, the Hoechst signal was observed in irregular shapes, which were considered to be cellular surfaces rather than round nuclei (Fig. 7B), which suggested either poor Hoechst permeability into samples or dying cells. Moreover, the cell mask fluorescence signal (Fig. 7C) was aggregated into clusters – likely, staining artefacts – and puncta within cells. However, the expected smooth membrane signal was observed. | ||
| Fig. 7 Light sheet fluorescence microscopy of the liver-on-a-chip scaffold pores. The A488-tagged ASO (A, E and I), the Hoechst-stained DNA (B, F and J) and cell surface stained with concavalin A (C, G and K) were imaged simultaneously in a single scaffold. A–D) The face and pore of the scaffold were visualized simultaneously (Z depth ≈ 400 μm). E–H) A single side of a scaffold pore highlighted the detailed distribution of ASO and in cells (Z depth ≈ 190 μm). Inset scale bar = 10 μm. I–L) Single plane image highlighted the fine structures of cuboidal cell walls. Green arrows show overlapping signal between cell nuclei and ASO. White and blue arrows point towards cuboidal and circular hepatocytes, respectively. | ||
Cell mask was replaced with concanavalin A (ConA), a membrane stain. The evaluation of fluorescence in HepG2 spheroids revealed the ConA highlighted cellular membranes with high contrast and that the Hoechst-stained nuclei with high specificity without labelling any other structures (Fig. S6†). Furthermore, upon treatment of a spheroid with ASO, the nuclear signal remained unchanged and the unlikely interaction between Hoechst and ASO was ruled out (Fig. S7†). The scaffolds were permeabilized to further improve staining of a sample. The 3D images were flattened using a maximum intensity projection (Fig. 7E–H). The overall intensity across all channels was found to be the highest in the microtissue near the scaffold pore edge and plateaued in the center of the pore. However, this most likely did not reflect the dye distribution and was most likely a result of adjacent microtissue interference in the center of the pore or the lack of it at the scaffold pore edge. The intensity fluctuation originating from light scattering are a well known issue in the light sheet community.44 The mutually exclusive ASO nuclear and cytoplasmic signals were consistent with previous findings and thus confirmed that the ASO distribution remained unaffected by the permeabilization step (Fig. 7E). Hoechst signal was clearly observed in the cell nuclei in the permeabilized sample, although a portion of the Hoechst signal was still observed in the cell cytoplasm (Fig. 7F). Nonetheless, the colocalization of the putative nuclear Hoechst and A488 signal was confirmed in part of the cell population, as shown in the image insets. Similarly, a clear spatial separation between the nuclear and punctate cytoplasmic ASO signals was observed in a separate cell population (Fig. 7H). MALAT1, the ASO target is a long non-coding RNA retained in nuclei45 and therefore the ASO must be delivered into nucleus for a successful MALAT1 gene suppression. Indeed, the colocalization of ASO and nucleus-specific Hoechst (binds to DNA double-strands) signal suggested the ASO is transported into nucleus. This was further confirmed by Buntz et al. in an earlier study, where upon microinjection, the ASO was passively transported into MCF-7 cell nuclei.46 Albeit MCF-7 are breast carcinoma cells, given the conserved nature of the MALAT1, the localization was expected to be nuclear-retained.
A single plane image from the Z-stack was used to observe the fluorescence distribution in the cells in detail (Fig. 7I–L). The cell boundaries in the single-plane image were captured in all channels, suggesting the potential capture of fluorescent markers on the cell surface. Owing to the visualization of cell membranes, the presence of two cell populations was suggested: cuboidal and spherical hepatocytes, which were previously found and separated using multiphoton and hyperspectral SRS techniques (Fig. 4 and 5), and were later observed with single photon fluorescence and SRS in tandem, with absence of ASO signal in the cytoplasm and high intensity of ASO signal in the nucleus (Fig. 6). The majority of the cells inside the scaffold pore had morphological features corresponding to the cuboidal hepatocytes, with a large extent of cell-to-cell contact as shown in Fig. 7L (white arrows). Circular hepatocytes were found on the surface of the cell mass inside the scaffold pores in low abundance with a low cell-to-cell contact (Fig. 7L, cyan arrow), which is a sign of improper cellular function, which can lead to eventual cell death.47 Finally, the distribution of the ConA-rhodamine label was assessed (Fig. 7G and K), which labelled the cell surface evenly in contrast with the cell mask. The single-plane image showed heterogeneous penetration of the ConA dye inside some of the cuboidal hepatic cells demonstrated by the complete cytoplasmic ConA signal, suggesting partial membrane permeabilization of the sample (Fig. 7K). The heterogeneity of cytoplasmic staining with ConA was tentatively associated with uneven Triton penetration during the permeabilization step possibly caused by hepatic cell secretion; although if any excretion indeed occurred, the layer of excreted matrix was likely very thin as it would have otherwise been detected with SRS. It was possible to visualize the entire scaffold pore using LSFM and this enabled imaging of deeper cellular features in the liver scaffolds compared to the MPM techniques investigated in this study. However, LSFM is dependent on the precision of the fluorescent labelling stains. As seen in the Hoechst fluorescent channel, membranous structures which were unlikely to be associated with DNA were also observed in that channel. With further optimization of staining methods, LSFM is capable of full-depth and large-scale sample imaging owing to its high-speed image capture and low fluorescence cytotoxicity.
The current study demonstrated the advantages of using LSFM for studying ASO uptake in liver-on-a-chip models. Further studies using LSFM to perform quantitative comparison of ASO uptake in cuboidal and circular hepatocytes are warranted to obtain a comprehensive understanding of ASO cellular uptake occurring in these liver CIVMs.
Conclusions
In this study, the capability of labelled and label-free multimodal optical imaging techniques for visualization and characterization of cellular and sub-cellular features in a liver-on-a-chip model was demonstrated. It was possible to visualize the cellular features of the microtissue formations in the pores of the liver-on-a-chip model up to a depth of 150 μm using SLAM microscopy in a non-invasive and label-free manner. To show the potential of MPM studies to support pharmacokinetic/pharmacodynamic studies, the non-uniform cellular uptake and distribution of non-targeted ASO and ASO-GalNAc in the liver-on-a-chip model was determined using a custom-built two-photon imaging platform. The differential uptake of non-targeted ASO and ASO-GalNAc was measured at different time points post-treatment and at varying concentrations. Furthermore, sub-cellular distribution of ASO was visualized and a significantly higher uptake of ASO-GalNAc was confirmed during the first hour post-treatment based on the MPM images. Furthermore, a full depth (∼250 μm) 3D visualization of the sub-cellular distribution of ASO and cellular phenotypes in the liver-on-a-chip scaffold pores was achieved using LSFM.SRS revealed that the cytoplasm of cuboidal hepatocytes appeared to be protein-rich in comparison to the lipid-rich cytoplasm of the circular hepatocytes indicating possible presence of lipid droplets and vesicles. Furthermore, cuboidal hepatocyte-specific spectral component was identified using the FSC3. In addition, the simultaneously acquired images of ASO (fluorescence) and lipid droplets (CH2, SRS) showed that the intracellular trafficking of lipids and endocytosed ASOs do not overlap.
Liver-on-a-chip MPS narrows the gap between in vitro assay and in vivo environment in a human liver by accurately mimicking the in vivo environment as shown by the large number of cuboidal hepatocytes which exhibited different chemical composition, morphology, and ASO distribution in contrast with the circular hepatocytes. In liver, hepatocyte morphology is strictly cuboidal. Therefore, data derived from non-cuboidal hepatocytes should be disregarded in order to accurately model the in vivo cell and tissue dynamics, either in a laboratory or computationally. Illustrating this, the study observed that the variations in hepatocyte morphology were found associated with a difference in the intracellular trafficking of ASOs.
Overall, this imaging study effectively integrated labelled and label-free multimodal microscopy techniques to investigate ASO cellular uptake and distribution in liver-on-a-chip CIVM by leveraging multiphoton fluorescence, coherent Raman microscopy and light sheet microscopy techniques. These advanced optical imaging methodologies have shown promising potential to provide deeper insights into therapeutic cargo delivery dynamics on a cellular level in liver CIVMs to support discovery and development of effective therapeutic ASOs.
Data availability
We appreciate the call for open data. However, given the proprietary nature of the collaboration between our institutions and the fact that the data is still subject to research, we can only share these upon a request and under an appropriate collaborative agreement.Conflicts of interest
S. A. B. received grant support from GSK related to the research described here. S. A. B. is co-founder of and holds equity interest in LiveBx, LLC, which specializes in consulting and building novel multimodal multiphoton imaging systems. J. M., A. A., C. N., R. G. and S. R. H. are employees of GSK. All other authors declare no competing interests.Acknowledgements
The authors thank CN Bio Innovations Ltd, Cambridge, UK for conducting the cell culture experiments and ASO dosing of the liver CIVM scaffolds. The authors gratefully acknowledge Cardiff University Bioimaging Hub Core Facility (RRID: SCR_022556), for their support and assistance in this work. This work was funded by European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 812992, and in part by GSK through the GSK Center for Optical Molecular Imaging at the Beckman Institute for Advanced Science and Technology, University of Illinois Urbana-Champaign.References
- K. Zeilinger, N. Freyer, G. Damm, D. Seehofer and F. Knöspel, Exp. Biol. Med., 2016, 241, 1684–1698 CrossRef CAS PubMed.
- L. Schyschka, J. J. M. Sánchez, Z. Wang, B. Burkhardt, U. Müller-Vieira, K. Zeilinger, A. Bachmann, S. Nadalin, G. Damm and A. K. Nussler, Arch. Toxicol., 2013, 87, 1581–1593 CrossRef CAS PubMed.
- S. Kaur, S. Kidambi, M. Ortega-Ribera, L. T. T. Thuy, N. Nieto, V. C. Cogger, W. F. Xie, F. Tacke and J. Gracia-Sancho, Cell. Mol. Gastroenterol. Hepatol., 2023, 15, 559–571 CrossRef CAS PubMed.
- S. Deguchi and K. Takayama, Inflammation Regener., 2022, 42, 62 CrossRef PubMed.
- T. Kostrzewski, S. Snow, A. L. Battle, S. Peel, Z. Ahmad, J. Basak, M. Surakala, A. Bornot, J. Lindgren, M. Ryaboshapkina, M. Clausen, D. Lindén, C. Maass, L. M. Young, A. Corrigan, L. Ewart and D. Hughes, Commun. Biol., 2021, 4, 1080 CrossRef CAS PubMed.
- J. Deng, W. Wei, Z. Chen, B. Lin, W. Zhao, Y. Luo and X. Zhang, Micromachines, 2019, 10, 676 CrossRef PubMed.
- J. E. Ekert, J. Deakyne, P. Pribul-Allen, R. Terry, C. Schofield, C. G. Jeong, J. Storey, L. Mohamet, J. Francis, A. Naidoo, A. Amador, J. L. Klein and W. Rowan, SLAS Discovery, 2020, 25, 1174–1190 CrossRef CAS PubMed.
- S. Y. Lee, D. Kim, S. H. Lee and J. H. Sung, APL Bioeng., 2021, 5, 041505 CrossRef CAS PubMed.
- R. Zitter, R. M. Chugh and S. Saha, Cancers, 2022, 14, 3006 CrossRef CAS PubMed.
- K. M. Bircsak, R. DeBiasio, M. Miedel, A. Alsebahi, R. Reddinger, A. Saleh, T. Shun, L. A. Vernetti and A. Gough, Toxicology, 2021, 450, 152667 CrossRef CAS PubMed.
- T. Kostrzewski, T. Cornforth, S. A. Snow, L. Ouro-Gnao, C. Rowe, E. M. Large and D. J. Hughes, World J. Gastroenterol., 2017, 23, 204–215 CrossRef PubMed.
- A. Rubiano, A. Indapurkar, R. Yokosawa, A. Miedzik, B. Rosenzweig, A. Arefin, C. M. Moulin, K. Dame, N. Hartman, D. A. Volpe, M. K. Matta, D. J. Hughes, D. G. Strauss, T. Kostrzewski and A. J. S. Ribeiro, Clin. Transl. Sci., 2021, 14, 1049–1061 CrossRef CAS PubMed.
- F. Lazzeri-Barcelo, N. Oliva-Vilarnau, M. Baniol, B. Leibiger, O. Bergmann, V. M. Lauschke, I. B. Leibiger, N. Moruzzi and P.-O. Berggren, Nat. Commun., 2024, 15, 767 CrossRef CAS PubMed.
- T. Kostrzewski, P. Maraver, L. Ouro-Gnao, A. Levi, S. Snow, A. Miedzik, K. Rombouts and D. Hughes, Hepatol. Commun., 2020, 4, 77–91 CrossRef CAS PubMed.
- A. Vivares, S. Salle-Lefort, C. Arabeyre-Fabre, R. Ngo, G. Penarier, M. Bremond, P. Moliner, J.-F. Gallas, G. Fabre and S. Klieber, Xenobiotica, 2015, 45, 29–44 CrossRef CAS PubMed.
- S. You, H. Tu, E. J. Chaney, Y. Sun, Y. Zhao, A. J. Bower, Y.-Z. Liu, M. Marjanovic, S. Sinha, Y. Pu and S. A. Boppart, Nat. Commun., 2018, 9, 2125 CrossRef PubMed.
- S. A. Boppart, S. You, L. Li, J. Chen and H. Tu, APL Photonics, 2019, 4, 100901 CrossRef PubMed.
- C. W. Freudiger, W. Min, B. G. Saar, S. Lu, G. R. Holtom, C. He, J. C. Tsai, J. X. Kang and X. S. Xie, Science, 2008, 322, 1857–1861 CrossRef CAS PubMed.
- K. König, J. Microsc., 2000, 200, 83–104 CrossRef PubMed.
- W. J. Tipping, L. T. Wilson, C. An, A. A. Leventi, A. W. Wark, C. Wetherill, N. C. O. Tomkinson, K. Faulds and D. Graham, Chem. Sci., 2022, 13, 3468–3476 RSC.
- K. Czamara, K. Majzner, A. Selmi, M. Baranska, Y. Ozaki and A. Kaczor, Sci. Rep., 2017, 7, 40889 CrossRef CAS PubMed.
- K. Sepp, M. Lee, M. T. J. Bluntzer, G. V. Helgason, A. N. Hulme and V. G. Brunton, J. Med. Chem., 2020, 63, 2028–2034 CrossRef CAS PubMed.
- T. C. Roberts, R. Langer and M. J. A. Wood, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- B. G. De La Torre and F. Albericio, Molecules, 2024, 29, 585 CrossRef CAS PubMed.
- C. M. Miller, M. Tanowitz, A. J. Donner, T. P. Prakash, E. E. Swayze, E. N. Harris and P. P. Seth, Nucleic Acid Ther., 2018, 28, 119–127 CrossRef CAS PubMed.
- T. P. Prakash, M. J. Graham, J. Yu, R. Carty, A. Low, A. Chappell, K. Schmidt, C. Zhao, M. Aghajan, H. F. Murray, S. Riney, S. L. Booten, S. F. Murray, H. Gaus, J. Crosby, W. F. Lima, S. Guo, B. P. Monia, E. E. Swayze and P. P. Seth, Nucleic Acids Res., 2014, 42, 8796–8807 CrossRef CAS PubMed.
- P. Mukherjee, E. Aksamitiene, A. Alex, J. Shi, K. Bera, C. Zhang, D. R. Spillman, M. Marjanovic, M. Fazio, P. P. Seth, K. Frazier, S. R. Hood and S. A. Boppart, Nucleic Acid Ther., 2022, 32, 163–176 CrossRef CAS PubMed.
- J. Scharner, S. Qi, F. Rigo, C. F. Bennett and A. R. Krainer, Mol. Ther.--Nucleic Acids, 2019, 16, 313–325 CrossRef CAS PubMed.
- K. Domansky, W. Inman, J. Serdy, A. Dash, M. H. M. Lim and L. G. Griffith, Lab Chip, 2010, 10, 51–58 RSC.
- J. H. Lee, J. Rico-Jimenez, C. Zhang, A. Alex, E. J. Chaney, R. Barkalifa, D. R. Spillman Jr, M. Marjanovic, Z. Arp, S. R. Hood and S. A. Boppart, Biomed. Opt. Express, 2019, 10, 5431–5444 CrossRef CAS PubMed.
- F. Masia, A. Karuna, P. Borri and W. Langbein, J. Raman Spectrosc., 2015, 46, 727–734 CrossRef CAS PubMed.
- F. Masia, A. Glen, P. Stephens, P. Borri and W. Langbein, Anal. Chem., 2013, 85, 10820–10828 CrossRef CAS PubMed.
- C. Stringer, T. Wang, M. Michaelos and M. Pachitariu, Nat. Methods, 2021, 18, 100–106 CrossRef CAS PubMed.
- X. Chen, O. Nadiarynkh, S. Plotnikov and P. J. Campagnola, Nat. Protoc., 2012, 7, 654–669 CrossRef CAS PubMed.
- A. A. D'Souza and P. V. Devarajan, J. Controlled Release, 2015, 203, 126–139 CrossRef PubMed.
- A. D. Springer and S. F. Dowdy, Nucleic Acid Ther., 2018, 28, 109–118 CrossRef CAS PubMed.
- C. M. Miller, A. J. Donner, E. E. Blank, A. W. Egger, B. M. Kellar, M. E. Østergaard, P. P. Seth and E. N. Harris, Nucleic Acids Res., 2016, 44, 2782–2794 CrossRef PubMed.
- J. L. Page, M. C. Johnson, K. M. Olsavsky, S. C. Strom, H. Zarbl and C. J. Omiecinski, Toxicol. Sci., 2007, 97, 384–397 CrossRef CAS PubMed.
- M. Eisenstein, Nature, 2023, 623, 1095–1097 CrossRef CAS PubMed.
- Y. Li, B. Shen, S. Li, Y. Zhao, J. Qu and L. Liu, Adv. Biol., 2021, 5, 2000184 CrossRef PubMed.
- C. W. Freudiger, R. Pfannl, D. A. Orringer, B. G. Saar, M. Ji, Q. Zeng, L. Ottoboni, W. Ying, C. Waeber, J. R. Sims, P. L. De Jager, O. Sagher, M. A. Philbert, X. Xu, S. Kesari, X. S. Xie and G. S. Young, Lab. Invest., 2012, 92, 1492–1502 CrossRef CAS PubMed.
- B. Sarri, F. Poizat, S. Heuke, J. Wojak, F. Franchi, F. Caillol, M. Giovannini and H. Rigneault, Biomed. Opt. Express, 2019, 10, 5378–5384 CrossRef CAS PubMed.
- F. X. Xu, G. N. Ioannou, S. P. Lee, C. Savard, C. L. Horn and D. Fu, J. Biomed. Opt., 2024, 29, 016008 Search PubMed.
- N. Yayon, A. Dudai, N. Vrieler, O. Amsalem, M. London and H. Soreq, Sci. Rep., 2018, 8, 4311 CrossRef PubMed.
- G. Arun, D. Aggarwal and D. L. Spector, Non-Coding RNA, 2020, 6, 22 CrossRef CAS PubMed.
- A. Buntz, T. Killian, D. Schmid, H. Seul, U. Brinkmann, J. Ravn, M. Lindholm, H. Knoetgen, V. Haucke and O. Mundigl, Nucleic Acids Res., 2019, 47, 953–969 CrossRef CAS PubMed.
- S. N. Bhatia, U. J. Balis, M. L. Yarmush and M. Toner, FASEB J., 1999, 13, 1883–1900 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00504j |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |