 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWeb-BO: towards increased accessibility of Bayesian optimisation (BO) for chemistry
Austin M. Mroz *ab,
Piotr N. Tokaa,
Ehecatl Antonio del Río Chanonac and
Kim E. Jelfs*a
*ab,
Piotr N. Tokaa,
Ehecatl Antonio del Río Chanonac and
Kim E. Jelfs*a
aDepartment of Chemistry, Imperial College London, White City Campus, W12 0BZ, UK. E-mail: a.mroz@imperial.ac.uk; k.jelfs@imperial.ac.uk
bI-X Centre for AI in Science, Imperial College London, White City Campus, W12 0BZ, UK
cDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, SW7 2AZ, UK
First published on 30th July 2024
Abstract
Historically, the chemical discovery process has predominantly been a matter of trial-and-improvement, where small modifications are made to a chemical system, guided by chemical knowledge, with the aim of optimising towards a target property or combination of properties. While a trial-and-improvement approach is frequently successful, especially when assisted by the help of serendipity, the approach is incredibly time- and resource-intensive. Complicating this further, the available chemical space that could, in theory, be explored is remarkably vast. As we are faced with near infinite possibilities and limited resources, we require improved search methods to effectively move towards desired optima, e.g. chemical systems exhibiting a target property, or several desired properties. Bayesian optimisation (BO) has recently gained significant traction in chemistry, where within the BO framework, prior knowledge is used to inform and guide the search process to optimise towards desired chemical targets, e.g. optimal reaction conditions to maximise yield, or optimal catalyst exhibiting improved catalytic activity. While powerful, implementing BO algorithms in practice is largely limited to interfacing via various APIs – requiring advanced coding experience and bespoke scripts for each optimisation task. Further, it is challenging to seamlessly link these with electronic lab notebooks via a graphical user interface (GUI). Ultimately, this limits the accessibility of BO algorithms. Here, we present Web-BO, a GUI to support BO for chemical optimisation tasks. We demonstrate its performance using an open source dataset and associated emulator, and link the platform with an existing electronic lab notebook, datalab. By providing a GUI-based BO service, we hope to improve the accessibility of data-driven optimisation tools in chemistry; https://suprashare.rcs.ic.ac.uk/web-bo/.
1 Introduction
Decisions lie at the core of data-driven discovery. Within any step of a discovery process, whether searching for a new drug molecule, optimising material properties for a target application, or identifying new reactivities and synthetic routes, we are faced with choices. These choices range from identifying which experimental conditions to run a reaction in next, to determining whether to perform a simulation rather than experiment to save experimental cost and resources. Each of these decisions is associated with a cost; and we, as chemists, operate under some budget, which can take several forms, including: (i) available consumables, (ii) instrument time, (iii) high-performing computing resources, and so on. Historically, chemical intuition via a trial-and-improvement approach has guided our decision making in new discoveries. Yet, this traditional approach is very resource- and time-intensive. Indeed, it is estimated to take an average of 20 years to realize a new material at the industrial level.1,2 Such long timescales for discovery are problematic considering the gravity of the current challenges facing humanity.The traditional trial-and-improvement approach to discovery has been accelerated by significant advances in experimental hardware, including robotics and automation,3 which increases the rate and scale at which experiments may be performed. While powerful, the scale at which we are able to perform experiments is minimal compared to the search space that we are faced with. Indeed, it is estimated that there are between 1023 and 1060 hypothetical small (drug-like) molecules.4 Notably, this estimate does not include synthetic considerations. Chemical intuition guiding testing choices, where one factor/variable is changed at a time, has found success historically;5 however, this approach rarely yields optimal parameters, incorporates researcher bias, and typically requires a large number of experiments to identify subtle trends.6 Design of Experiments (DoE) is a statistical approach that screens multiple variables in parallel to gain a better understanding of the design space. In this way, DoE elucidates the interaction of a large number of experimental variables from comparably few experiments.7,8 Yet, DoE lacks the ability to effectively explore design spaces.9 Indeed, we require informed search techniques that consider factors not originally present in the initial model assumptions, to efficiently and effectively optimise towards desired features.
Recently, Bayesian optimisation (BO) has shown significant promise for chemical applications, from reaction optimisation9–11 to chemical and materials design,12–15 among others.16–18 BO's foundation on Bayesian principles allows more effective and efficient identification of optimal setup/parameters by incorporating accumulated measurements in a dynamic experimental planning workflow. General BO formulations feature several steps: (i) collate initial sample of data points, (ii) fit a probabilistic predictive model (termed surrogate model) to this data, (iii) predict performance of potential design alternatives, (iv) optimise over these alternatives through an acquisition function. It is the acquisition function that is used to determine the most promising points to evaluate; this is achieved by balancing exploration (sampling in data scarce regions) with exploitation (sampling in regions most likely to yield high objective values). Beyond that, there are many more complex forms of BO including; (i) multi-objective BO tasks, where several parameters are optimised at once,19 and (ii) multi-fidelity BO tasks, where cost and accuracy are balanced by taking advantage of varying approximations to the objective function (e.g. calculating vs. experimentally measuring a property of interest), and (iii) high-throughput (batch) BO where several possible solutions are suggested to be evaluated in parallel.20
To facilitate efforts to use BO in chemistry, bespoke BO packages have been introduced to help translate chemical problems to the BO formulation, including: Gauche,21 which provides an interface for applying Gaussian processes (GPs) to chemistry; GRYFFIN,22,23 which provides a platform to perform BO for chemical optimisation tasks over varying chemical landscapes; EDBO+,24 which offers a web application to facilitate BO for chemical tasks; and OLYMPUS,25 which offers a framework specifically to aid benchmarking optimisation algorithms. While these packages present specific tools to aid optimisation, there exist larger platforms to facilitate experimental planning and optimisation task implementations, including BayBE,26 software recently released by Merck that offers a back-end to support BO in chemistry. Yet, each of these solutions are not easily accessible to non-experts in coding and may not be easily integrated by experimentalists via GUI-based electronic lab notebooks (ELNs). ELNs provide a digitized platform for experimental procedures, results, and notes – allowing for ease of data distribution, processing, and storage.27 Additional resources are necessary to bridge the gap between data-driven optimisation techniques, namely BO, and non-computational experts.
Here, we present Web-BO, a web application offering a graphical user interface (GUI) to facilitate the application of BO to a wide array of chemical problems (Fig. 1). Web-BO is a modular platform that is easily integrated into existing ELN frameworks, and can be used as a standalone database and optimiser for chemical tasks. All of the data is stored in a searchable SQL database in an intuitive and future-proof form, enabling efficient benchmarking of various data-driven optimisation algorithms. Lastly, with Web-BO no coding experience is necessary to interface with and apply BO algorithms to chemical optimisation applications. Here, we describe the basic working interface and organization of Web-BO (Section 2.2) and demonstrate its application and interface with an existing ELN (Section 2.3).
 | ||
| Fig. 1 Users (green) interact with Web-BO in two capacities, defining the optimisation task, and performing the measurements. Web-BO offers data storage (yellow) is a SQL database, or by integrating with an electronic lab notebook (ELN). Bayesian optimisation (BO) is facilitated by solvers on the back-end, which take the current dataset, update the surrogate model, and select the next experiment (purple). | ||
2 Results and discussion
Web-BO offers an interactive GUI for applying BO to chemical problems. With our particular focus on increased accessibility here, we also linked Web-BO to an existing open-source electronic lab notebook (ELN), datalab.28 datalab is a recently developed ELN consisting of a Flask-based python web server paired with an intuitive GUI for efficient and effective data storage and maintenance across research group(s). In the remainder of this section we describe the basic version of the BO framework (Section 2.1), the organization and use of Web-BO (Section 2.2), and demonstrate the utility of Web-BO for a reaction optimisation case study (Section 2.3).2.1 Bayesian optimisation overview
In this section, we offer a brief introduction to BO and its basic formulation; for a more detailed description, we direct readers to ref. 13, 29 and 30.BO is a model-based, derivative-free optimisation method that affords efficient optimisation of black-box functions that are expensive to evaluate. Within the context of chemistry, objective functions and subsequent function evaluations can take on a variety of forms. For example, one may wish to identify optimal reaction conditions to maximise yield; here, the black-box function inputs are the reaction conditions and the function is evaluated by performing the experiment and measuring the yield. As in this example, solving directly to find the global optimal value is infeasible because the form of the objective function is unknown and function evaluations are expensive. Therefore, instead an iterative procedure is implemented where the black-box function (f) is sampled in an informed manner, as shown in Algorithm 1.29 Next, while the remaining budget (for example financial or number of experiments that can be practically carried out) is greater than the expended resources, the next data-point (x) to be sampled is determined based on the optimisation policy. The suggested experiment is then performed to yield the objective function measurement (y), and the dataset (![[scr D, script letter D]](https://www.rsc.org/images/entities/char_e523.gif) ) is updated with the results. This procedure is repeated until the budget is expired or user-defined optimisation criteria are met.
) is updated with the results. This procedure is repeated until the budget is expired or user-defined optimisation criteria are met.

To demonstrate the procedure outlined in Algorithm 1, let us consider an unknown, real-valued, objective function (f), which is defined over some real-space domain (χ). Our objective is to identify the globally maximal value, f*, and associated point x*,
 | (1) |
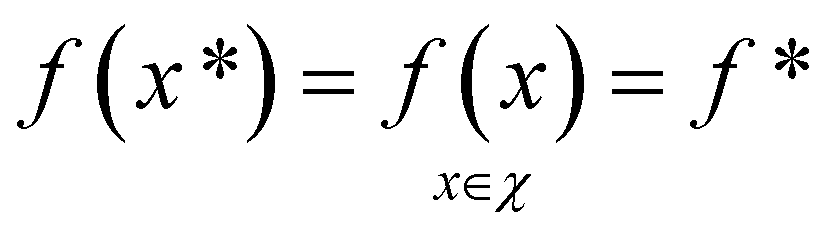 | (2) |
The objective function value of each point within the parameter space is then able to be predicted by the surrogate model and presented as a mean value with an associated uncertainty. The possible set of predictions and associated uncertainties are ranked by fitting an acquisition function. The acquisition function balances exploitation (sampling areas in the parameter space where uncertainty is small) with exploration (sampling areas in the parameter space where uncertainty is large). From this scoring function, the next set of experiments is suggested. The suggested measurements are then performed, and the dataset is updated with the new observations.
The BO formulation described here is the simplest, single-objective, optimisation problem; this is the current formulation that is supported by Web-BO. There are many more complex algorithms available, including algorithms where there are multiple objectives to optimise (multi-objective optimisation) and where function evaluations can be performed with methods of varying accuracy (multi-fidelity optimisation) implementations. The implementation of Web-BO would allow facile integration of multi-objective optimisation and multi-fidelity approaches in future releases.
2.2 Web-BO overview
At its core, Web-BO offers a GUI for BO application to chemical tasks. Fig. 2 outlines the basic structure and procedure provided by the Web-BO architecture as it relates to the BO solver back-end and integration with ELNs; this is distilled into five main steps:(1) Upload dataset: datasets are uploaded by the user in one of two supported formats: (i) csv file, or (ii) datalab collection. Table 1 presents a sample csv upload format; here, columns are variables and datapoints are rows.
| Solvent | Temperature | Pressure | Target |
|---|---|---|---|
| a | 25 | 1.0 | 2.34 |
| b | 45 | 2.3 | 4.56 |
| c | 55 | 2.3 | 10.33 |
(2) Define BO experiment: BO options are defined by the user via an interactive web form. This is comprised of three steps: (i) dataset selection, (ii) optimisation space definition, (iii) BO algorithm component selection (i.e. Gaussian process kernel, acquisition function, batch size, etc.). For example, in the case of the sample dataset presented in Table 1, ‘target’ is the optimisation objective, and the remaining variables (solvent, temperature, and pressure) define the optimisation space (e.g. we are changing those values to attempt to achieve a larger target).
(3) Make recommendation: recommendations for the next measurement(s) are made (currently by Web-BO using BayBE), which fits the surrogate model on the existing dataset, scoring the predictions using the acquisition function, and suggests the next candidate measurement(s) to take. Notably, the user may define how many experiments they would like to perform each iteration (batch size) (see Section 2.2.2). For example, the next candidate measurement to take in the sample dataset presented in Table 1 would consist of a solvent, temperature and pressure.
(4) Take measurements: measurements are acquired by the user; this may involve either laboratory work, simulations, or a combination of both.
(5) Update dataset: users update the dataset with measurements (e.g. new measurement(s) are appended to the end of the dataset) and the process is started over again, if the termination criteria have yet to be reached. In cases where the dataset is hosted by an ELN, changes to the dataset are reflected in the Web-BO database and the ELN database.
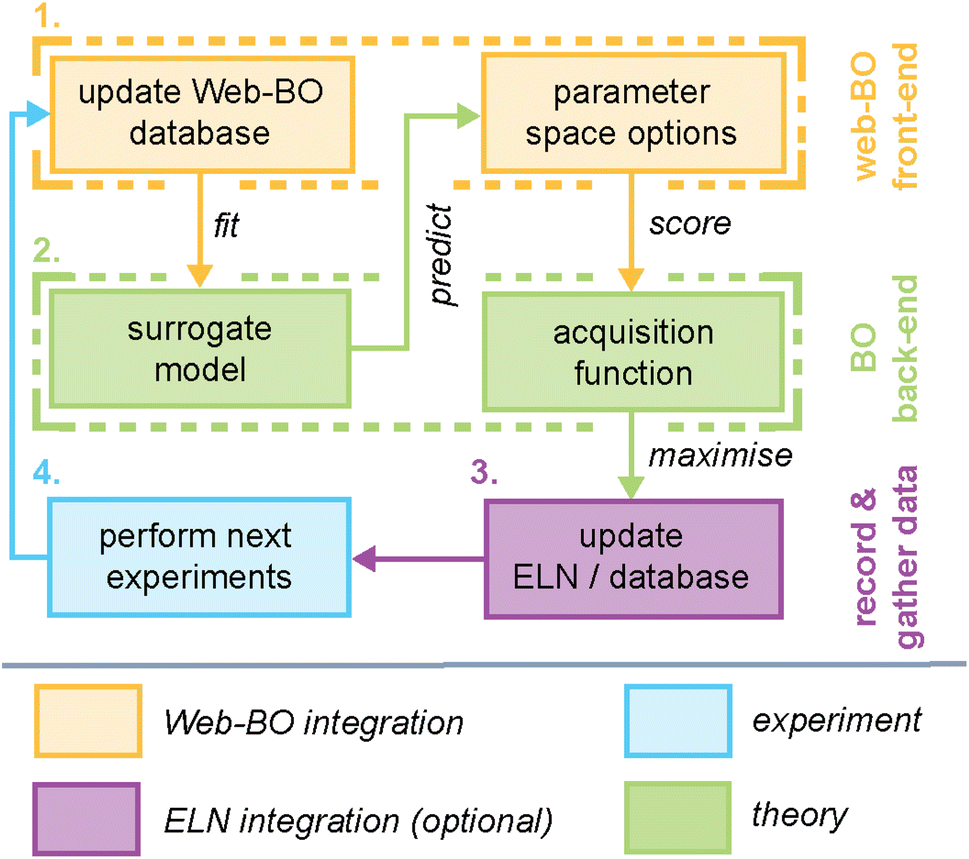 | ||
| Fig. 2 The role of Web-BO in a general Bayesian optimisation (BO) framework is highlighted in yellow. The back-end calculations are presented in green and the step involving experimentation is displayed in blue. While this is an iterative procedure, step numbers are added to illustrate the flow of initial cycles. | ||
Fig. 3 highlights features associated with each of the steps of the workflow provided by Web-BO. The iterative procedure described above and shown in Fig. 2 and 3 is afforded by the underlying database architecture, Fig. 4. Web-BO is structured such that datasets and optimisation experiments are stored independently; this allows multiple BO algorithms to be tested for one dataset, thereby enabling researchers to select the best-performing BO algorithm formulation for their specific task. Indeed, this is important considering that there is not a single ideal algorithm that is best suited to all (chemical) tasks.33 Next, we describe the dataset formats (Section 2.2.1) and BO formulation options (Section 2.2.2) supported by Web-BO.
 | ||
| Fig. 3 (a) The homepage displays all datasets and experiments in the database. The ‘Explore!’ button generates a sample optimisation task and dataset, “sample-Reizman-dataset”. (b) Datasets may be uploaded from datalab, which is the currently integrated electronic lab notebook (ELN). (c) Experiments are defined in several steps; here, two examples are shown: (i) dataset options are pre-populated from the database, (ii) the variable types for each parameter in the dataset must be defined. After the type is selected, the page dynamically queries for user input. Input validation is offered to ensure that the parameter space is accurately defined (i.e. format of categorical/chemical variable options are acceptable, and continuous/integer variable ranges fit within the constraints of the BO solver). (d) Running experiments involves generating new recommendations and: (i) adding measurements to the dataset. Recommendations are pre-populated in the form for ease of use, (ii) optimisation progress is visualised. | ||
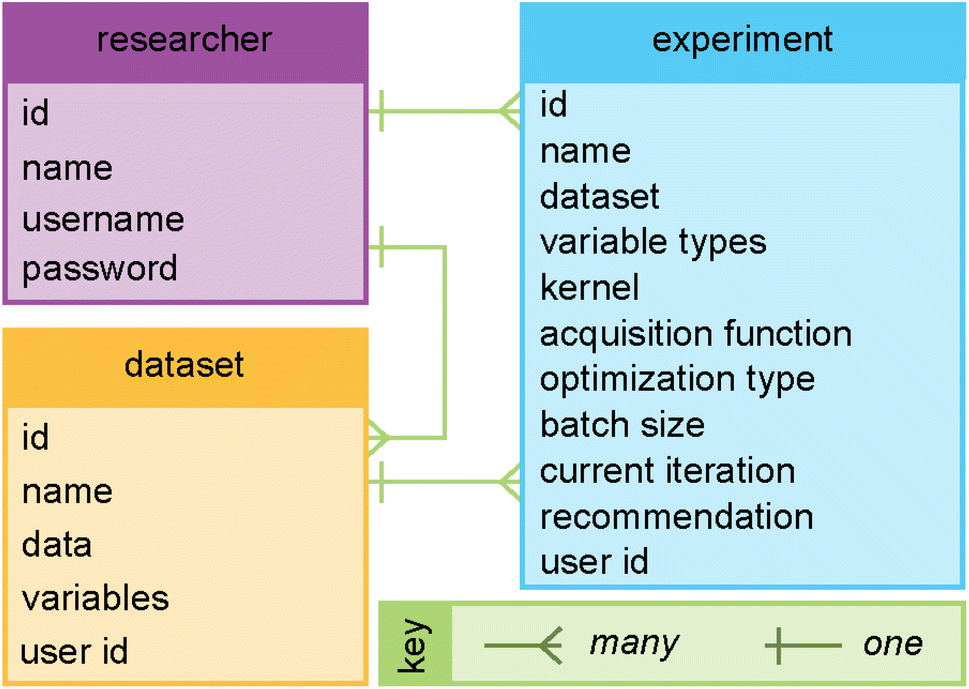 | ||
| Fig. 4 The entity relationship diagram for the database for Web-BO consists of three parts: (i) researcher, (ii) dataset, and (iii) experiment. The links between the entities denote the specific relationships; a single researcher can have many datasets and experiments, and a single dataset can be associated with many experiments. | ||
To support the use of ELNs in chemistry, Web-BO offers a modular platform that may be extended to support data structures offered by existing ELN platforms. Indeed, several ELN solutions specific to the chemical sciences have been introduced; including LabTrove,36,37 Indigo-ELN,38 OpenEnventory,39 Chemotion ELN,40 and datalab. Here, we demonstrate this utility by integrating datalab – a recent, open source addition to this space that supports a wide array of chemical applications. Datalab's modular architecture allows for customised organisation and data formats to be implemented. Fig. 3b outlines the information necessary to query a datalab instance using its API, including the API key, the domain name of the datalab repository, the variable names of the dataset, as well as two variables that describe the exact data to be collected (collection ID and block ID). The latter two are specific to the structure of datalab, they refer to the name of a collection of data in the repository and exact parts of the data to be collected, respectively.28
(1) Select dataset: dataset options are pre-populated from the datasets contained in the database.
(2) Define optimisation space: the type of variable needs to be defined for each optimisation parameter. The supported options are: (i) integer, where variables take on integer values between user-defined minimum and maximum values (e.g. number of units of a reagent); (ii) continuous, where variables represent measurements between user-defined minimum and maximum values (e.g. solvent ratio, temperature, pressure); (iii) categorical, where variables may be selected from a user-defined list of options (e.g. candidate solvents); and (iv) chemical, where variables are chemical structures represented as SMILES strings41 (e.g. reagents, products, etc.). When one of the variable types is selected, the user is queried for additional information. In the case of continuous and integer variables, the user is asked for miminum and maximum values. In the case of categorical and chemical values, the user is asked for a list of candidates to select from, which are uploaded as a csv file. Categorical candidates are one-hot encoded. Users are able to select from varying chemical encodings for chemical candidates, including Mordred, Morgan molecular fingerprints, and one-hot encoding. Web-BO offers the option to validate the optimisation space.
(3) Select BO components: there exist several parameters used to fully define the BO algorithm. First, the optimisation type describes whether the objective is to be minimised or maximised. The batch size describes how many experiments are performed in one iteration. The parameter to optimise is selected from a pre-populated dropdown menu. The GP kernel, which effectively indicates the similarity between two datapoints, is defined; options currently include Matern, and Tanimoto. Finally, the acquisition function is selected; options currently include Expected Improvement, and Probability of Improvement.
It is important to note that, while there are many surrogate models that may be implemented in BO, Web-BO currently only supports Gaussian processes (GPs) as surrogate models. GPs are non-parametric models that offer quantitative uncertainty, allow for exact Bayesian inference and are known to work well for sparse datasets.21,42 Due to these factors, they have demonstrated success across chemical applications.31 Presently, the back-end of Web-BO is supported by BayBE.26 In the future, we envisage integrating features that allow users to upload and integrate bespoke solvers, as well as offer support for alternative BO software platforms, including BOTorch,43 and BOFire.44
2.3 Case study
To facilitate ease of use and to help users acquaint themselves with the Web-BO framework, we offer a case study involving reaction condition optimization to maximize yield; the initial data and results are pre-loaded in the Data and Experiment databases for each new user account and accessed by selecting the ‘Explore!’ button on the homepage (Fig. 3a). Full demonstration of ELN integration requires a datalab account and webserver, detailed in Fig. 3b; therefore we offer detailed documentation on Github to support end users in this endeavour.35Determining the ideal reaction conditions (e.g. temperature, time, solvent ratio, etc.) that maximise yield, is a common optimisation task encountered in chemistry.45 Indeed, catalytic reaction optimisation is more complex; even for systems whose mechanism is well-understood, such as Suzuki–Miyaura cross-coupling reactions, selecting the ideal catalyst–ligand system for a particular reaction is nontrivial. This was recently highlighted by Reizman et al., who screened and optimised several Pd-catalyzed Suzuki–Miyaura cross-coupling reactions.46
The case study that is integrated in Web-BO concerns optimizing the coupling of 3-bromoquinoline with 3,5-dimethylisoxazole-4-boronic acid pinacol ester in the presence of 1,8-diazobicyclo[5.4.0]undec-7-ene (DBU) and THF/water, Fig. 5, originally presented by Reizman et al.46 The objective of this optimisation case study is to maximise the reaction yield, given five optimisation parameters, Fig. 5c. While the original study also optimised towards turnover number, to maintain simplicity in the case study, we elect to reformulate the optimisation task into a single-objective problem, where we want to maximise yield.
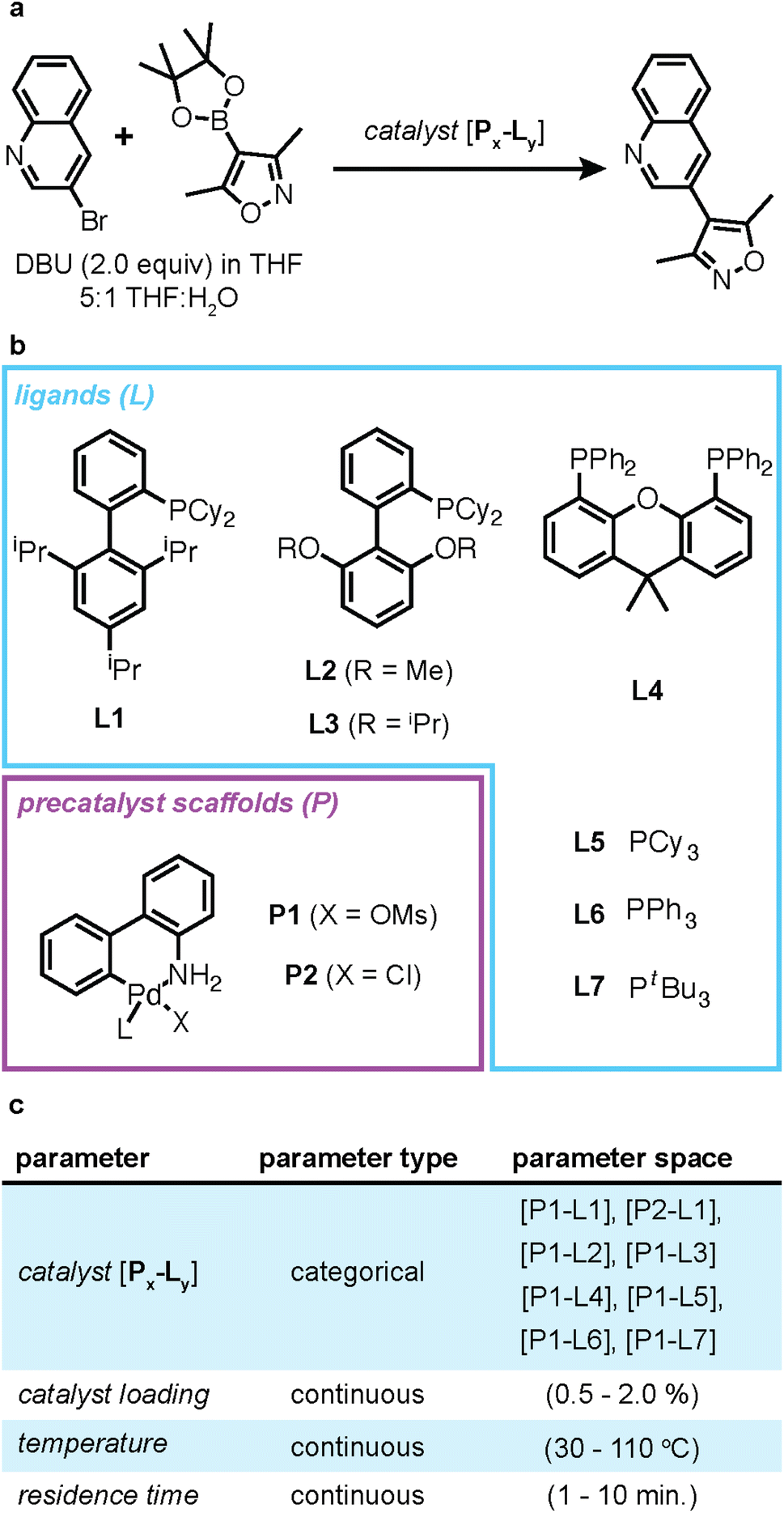 | ||
| Fig. 5 (a) Reaction scheme for the Suzuki–Miyaura cross-coupling of 3-bromoquinoline with 3,5-dimethylisoxazole-4-boronic acid pinacol ester in the presence of 1,8-diazobicyclo[5.4.0]undec-7-ene (DBU) and THF/water. (b) The precatalyst scaffolds (P) and ligands (L) that comprise the catalysts explored in this study. (c) The optimisation space for this case study is composed of four parameters. | ||
A critical step in the BO procedure involves performing the experiment (step 4 in Fig. 2). We rely on existing experimental emulators to facilitate this case study. Emulators are ML models trained on experimental data to reproduce chemical response surfaces; thus, instead of performing an experiment, we can query the model to predict the outcome. Here, we take advantage of the Suzuki–Miyaura cross-coupling emulator presented by Felton et al. when querying the objective function within the BO algorithm.47 Fig. 3 presents the main pages of Web-BO that are involved in setting up and running the case study. For a further demonstration, we encourage readers to engage with the video demonstration in the documentation.35
3 Conclusions and future outlooks
While chemical intuition is powerful and has led to significant advancements across disciplines, integrating data-driven decision making into the chemical design and optimisation process has demonstrated success covering a broad range of applications. Here, theory is used to suggest the next parameters for experimentation. BO is just one implementation of data-driven decision making – and has shown significant promise across chemical optimisation tasks. Yet, current implementations of this powerful tool require advanced coding knowledge. While coding has been increasingly integrated into chemical education, there still exists a need for tools that allow for alternative interaction and implementations of BO.Web-BO offers a modular GUI for exploration of BO application to chemical optimisation problems, enabling increased accessibility and ease of experimentation. There may be instances where researchers are unsure whether BO is the right algorithm for their task; in this case, Web-BO offers an intuitive platform with which to easily answer this question without the need to delve into the coding details. Indeed, this platform provides a visualisation of the steps necessary to develop a closed-loop workflow – where suggestions made by theory are directly sent to autonomous platforms for experimentation. Web-BO allows for the fact that closed-loop workflows are not always feasible and human interaction is required (human-in-the-loop);48 this is the solution provided by Web-BO.
Web-BO currently offers support for single-objective optimisation tasks using Gaussian processes (GPs) as the surrogate model. Indeed, there exist many other more complex BO formulations, including multi-objective BO (multiple parameters are optimised) and multi-fidelity BO (measurements possessing varying degrees of accuracy and cost may be performed), among others, which will be supported by Web-BO in the future. Further work will also integrate additional dataset upload methods, including support for additional ELNs and the ability to interface with MongoDB and SQL databases. Lastly, documentation for bespoke solver integration will be updated, allowing users to benchmark optimisation a range of conventional BO software packages and algorithms beyond BO.
Improving accessibility of BO for integration in chemical optimisation problems is paramount to realizing the full power of data-driven solutions to chemical challenges. Web-BO offers a step to realising this.
Author contributions
A. M. M. conceived and designed the project, developed the web application, and wrote the first draft of the manuscript. P. N. T. wrote the script to integrate the ELN. K. E. J. supervised and acquired funding. All authors contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Matthew Evans, one of the developers of datalab for insightful discussions and assistance in working with datalab's API. The authors would also like to thank Dr Diego Alonso Alvarez and Dr Lukas Turcani for their technical assistance in making Web-BO publicly available. A. M. M. is supported by the Eric and Wendy Schmidt AI in Science Postdoctoral Fellowship, a Schmidt Sciences program. K. E. J. acknowledges the European Research Council through Agreement No. 758370 (ERC-StG-PE5-CoMMaD) and the Royal Society for a University Research Fellowship. P. N. T. acknowledges the React CDT for funding (EP/S023232/1).Notes and references
- J. M. Cole, Acc. Chem. Res., 2020, 53, 599–610 CrossRef CAS PubMed.
- Z. Deng, V. Kumar, F. T. Bölle, F. Caro, A. A. Franco, I. E. Castelli, P. Canepa and Z. W. Seh, Energy Environ. Sci., 2022, 15, 579–594 RSC.
- R. L. Greenaway and K. E. Jelfs, Adv. Mater., 2021, 33, 2004831 CrossRef CAS PubMed.
- P. G. Polishchuk, T. I. Madzhidov and A. Varnek, J. Comput.-Aided Mol. Des., 2013, 27, 675–679 CrossRef CAS PubMed.
- D. W. Lendrem, B. C. Lendrem, D. Woods, R. Rowland-Jones, M. Burke, M. Chatfield, J. D. Isaacs and M. R. Owen, Drug Discovery Today, 2015, 20, 1365–1371 CrossRef PubMed.
- S. A. Weissman and N. G. Anderson, Org. Process Res. Dev., 2015, 19, 1605–1633 CrossRef CAS.
- Y. Zhang, D. W. Apley and W. Chen, Sci. Rep., 2020, 10, 4924 CrossRef CAS PubMed.
- P. M. Murray, F. Bellany, L. Benhamou, D.-K. Bučar, A. B. Tabor and T. D. Sheppard, Org. Biomol. Chem., 2016, 14, 2373–2384 RSC.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 CrossRef CAS PubMed.
- E. Braconi, Nat. Rev. Methods Primers, 2023, 3, 74 CrossRef CAS.
- C. J. Taylor, K. C. Felton, D. Wigh, M. I. Jeraal, R. Grainger, G. Chessari, C. N. Johnson and A. A. Lapkin, ACS Cent. Sci., 2023, 9, 957–968 CrossRef CAS PubMed.
- R.-R. Griffiths and J. M. Hernández-Lobato, Chem. Sci., 2020, 11, 577–586 RSC.
- A. Deshwal, C. M. Simon and J. R. Doppa, Mol. Syst. Des. Eng., 2021, 6, 1066–1086 RSC.
- S. Diwale, M. K. Eisner, C. Carpenter, W. Sun, G. C. Rutledge and R. D. Braatz, Mol. Syst. Des. Eng., 2022, 7, 622–636 RSC.
- Y. Jin and P. V. Kumar, Nanoscale, 2023, 15, 10975–10984 RSC.
- H. Takeda, H. Fukuda, K. Nakano, S. Hashimura, N. Tanibata, M. Nakayama, Y. Ono and T. Natori, Mater. Adv., 2022, 3, 8141–8148 RSC.
- A. M. K. Nambiar, C. P. Breen, T. Hart, T. Kulesza, T. F. Jamison and K. F. Jensen, ACS Cent. Sci., 2022, 8, 825–836 CrossRef CAS PubMed.
- T. Savage, N. Basha, J. McDonough, O. K. Matar and E. A. d. R. Chanona, Nat. Chem. Eng., 2023, 1, 522–531 CrossRef.
- A. K. Y. Low, F. Mekki-Berrada, A. Gupta, A. Ostudin, J. Xie, E. Vissol-Gaudin, Y.-F. Lim, Q. Li, Y. S. Ong, S. A. Khan and K. Hippalgaonkar, npj Comput. Mater., 2024, 10, 104 CrossRef CAS.
- J. Gonzalez, Z. Dai, P. Hennig and N. Lawrence, Proceedings of the 19th International Conference on Artificial Intelligence and Statistics, Cadiz, Spain, 2016, pp. 648–657 Search PubMed.
- R.-R. Griffiths, L. Klarner, H. B. Moss, A. Ravuri, S. Truong, S. Stanton, G. Tom, B. Rankovic, Y. Du, A. Jamasb, A. Deshwal, J. Schwartz, A. Tripp, G. Kell, S. Frieder, A. Bourached, A. Chan, J. Moss, C. Guo, J. Durholt, S. Chaurasia, F. Strieth-Kalthoff, A. A. Lee, B. Cheng, A. Aspuru-Guzik, P. Schwaller and J. Tang, GAUCHE: A Library for Gaussian Processes in Chemistry, 2023 Search PubMed.
- F. Häse, L. M. Roch, C. Kreisbeck and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 1134–1145 CrossRef PubMed.
- F. Häse, M. Aldeghi, R. J. Hickman, L. M. Roch and A. Aspuru-Guzik, Appl. Phys. Rev., 2021, 8, 031406 Search PubMed.
- J. A. Garrido Torres, S. H. Lau, P. Anchuri, J. M. Stevens, J. E. Tabora, J. Li, A. Borovika, R. P. Adams and A. G. Doyle, J. Am. Chem. Soc., 2022, 144(43), 19999–20007 CrossRef PubMed.
- F. Häse, M. Aldeghi, R. J. Hickman, L. M. Roch, M. Christensen, E. Liles, J. E. Hein and A. Aspuru-Guzik, Mach. Learn.: Sci. Technol., 2021, 2, 035021 Search PubMed.
- M. Fitzner, A. Šošić, A. Hopp and A. Lee, BayBE, https://github.com/emdgroup/baybe/ Search PubMed.
- S. G. Higgins, A. A. Nogiwa-Valdez and M. M. Stevens, Nat. Protoc., 2022, 17, 179–189 CrossRef CAS PubMed.
- M. L. Evans and J. D. Bocarsly, Datalab, https://github.com/the-grey-group/datalab Search PubMed.
- R. Garnett, Bayesian Optimization, Cambridge University Press, 2023 Search PubMed.
- B. Lei, T. Q. Kirk, A. Bhattacharya, D. Pati, X. Qian, R. Arroyave and B. K. Mallick, npj Comput. Mater., 2021, 7, 194 CrossRef.
- V. L. Deringer, A. P. Bartók, N. Bernstein, D. M. Wilkins, M. Ceriotti and G. Csányi, Chem. Rev., 2021, 121, 10073–10141 CrossRef CAS PubMed.
- N. Raimbault, A. Grisafi, M. Ceriotti and M. Rossi, New J. Phys., 2019, 21, 105001 CrossRef CAS.
- Q. Liang, A. E. Gongora, Z. Ren, A. Tiihonen, Z. Liu, S. Sun, J. R. Deneault, D. Bash, F. Mekki-Berrada, S. A. Khan, K. Hippalgaonkar, B. Maruyama, K. A. Brown, J. Fisher Iii and T. Buonassisi, npj Comput. Mater., 2021, 7, 188 CrossRef.
- A. M. Mroz, P. N. Toka and K. E. Jelfs, Web-BO, https://suprashare.rcs.ic.ac.uk/web-bo/ Search PubMed.
- A. M. Mroz, P. N. Toka and K. E. Jelfs, Web-BO, https://github.com/austin-mroz/webBO Search PubMed.
- A. E. Day, S. J. Coles, C. L. Bird, J. G. Frey, R. J. Whitby, V. E. Tkachenko and A. J. Williams, J. Chem. Inf. Model., 2015, 55, 501–509 CrossRef CAS PubMed.
- C. Willoughby, C. L. Bird, S. J. Coles and J. G. Frey, J. Chem. Inf. Model., 2014, 54, 3268–3283 CrossRef CAS PubMed.
- Indigo, https://github.com/epam/Indigo Search PubMed.
- F. Rudolphi and L. J. Goossen, J. Chem. Inf. Model., 2012, 52, 293–301 CrossRef CAS PubMed.
- P. Tremouilhac, A. Nguyen, Y.-C. Huang, S. Kotov, D. S. Lütjohann, F. Hübsch, N. Jung and S. Bräse, J. Cheminf., 2017, 9, 54 Search PubMed.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- M. M. Noack, G. S. Doerk, R. Li, J. K. Streit, R. A. Vaia, K. G. Yager and M. Fukuto, Sci. Rep., 2020, 10, 17663 CrossRef CAS PubMed.
- M. Balandat, B. Karrer, D. R. Jiang, S. Daulton, B. Letham, A. G. Wilson and E. Bakshy, Advances in Neural Information Processing Systems, 2020, ISSN 1049-5258 Search PubMed.
- BoFire, https://github.com/experimental-design/bofire Search PubMed.
- C. J. Taylor, A. Pomberger, K. C. Felton, R. Grainger, M. Barecka, T. W. Chamberlain, R. A. Bourne, C. N. Johnson and A. A. Lapkin, Chem. Rev., 2023, 123, 3089–3126 CrossRef CAS PubMed.
- B. J. Reizman, Y.-M. Wang, S. L. Buchwald and K. F. Jensen, React. Chem. Eng., 2016, 1, 658–666 RSC.
- K. C. Felton, J. G. Rittig and A. A. Lapkin, Chem.: Methods, 2021, 1, 116–122 CAS.
- M. Christensen, L. P. E. Yunker, P. Shiri, T. Zepel, P. L. Prieto, S. Grunert, F. Bork and J. E. Hein, Chem. Sci., 2021, 12, 15473–15490 RSC.
| This journal is © The Royal Society of Chemistry 2024 |