
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral spin-crossover complexes based on an enantiopure Schiff base ligand with three chiral carbon centers†
Alejandro
Regueiro
,
Víctor
García-López
 ,
Alicia
Forment-Aliaga
and
Miguel
Clemente-León
*
,
Alicia
Forment-Aliaga
and
Miguel
Clemente-León
*
Instituto de Ciencia Molecular (ICMol), Universidad de Valencia, Catedrático José Beltrán 2, 46980 Paterna, Spain. E-mail: miguel.clemente@uv.es
First published on 3rd June 2024
Abstract
The preparation of Fe(II) complexes combining monodentate NCX− (X = S or Se) and the tetradentate Schiff base chiral ligands RR-L1 and SS-L1 = (RR- or SS-L1 = 1R,2R or 1S,2S)-N1,N2-bis(pyridin-2-ylmethylen)cyclohexane-1,2-diamine in acetone results in an unexpected reaction. Thus, four enantiomerically pure compounds of formulas [Fe(RR-S-L2)(NCX)2] and [Fe(SS-R-L2)(NCX)2] (X = S or Se) are formed by the new asymmetrical ligand L2. In L2, one acetone solvent molecule is incorporated into the ligand forming a bond with the C atom of one of the two CN imine groups of L1, which is transformed into an amine (Mannich reaction). This reaction is diastereoselective as the incorporation of acetone leads to an asymmetric C adjacent to the NH group with opposite chirality S- or R- to that of the cyclohexane carbons (RR- or SS-, respectively). Therefore, L2 contains three C chiral centers. Structural and magnetic characterization of these compounds demonstrates that they show in the bulk a gradual spin-crossover behavior and LIESST effect. Interestingly, the presence of an intramolecular hydrogen bond between the integrated acetone molecule and the NH group can trigger a secondary stimuli-responsive behavior in the system. Therefore, by changing the solvent polarity, the color of the complex in solution can be easily tuned.
Introduction
Spin-crossover (SCO) complexes undergo an entropy driven spin state change between the low-spin (LS) and high-spin (HS) states which can be triggered by a variety of external stimuli such as temperature, pressure, analytes, or electric fields.1 The incorporation of chirality to SCO compounds can afford additional functionalities such as nonlinear optical and dielectric properties, which can take advantage of their switching capabilities. Indeed, chiral SCO compounds have been proposed for magneto-optical and spintronic applications.2 The strategies to prepare chiral SCO compounds are based on the spontaneous resolution of achiral precursors3 and the use of chiral counteranions4 or ligands.5 Among them, Fe(II) SCO complexes having the general formula [Fe(L)n(NCX)2] (L = bidentate, n = 2, or tetradentate, n = 1, chiral Schiff base ligand; X = S, Se or BH3) have afforded several examples of chiral SCO complexes.3b,6–8 In this work, we have tried to extend this family of chiral SCO complexes to the tetradentate ligands (1R,2R and 1S,2S)-N1,N2-bis(pyridin-2-ylmethylen)cyclohexane-1,2-diamine (RR-and SS-L1, see Scheme 1), which contain two imine groups and two chiral centers, in combination with NCS− and NCSe− anions. In a previous work, Tao et al. used a similar strategy to prepare chiral SCO complexes with a related ligand containing two amine groups of formula [Fe(RR- or SS-L3)(NCSe)2] (L3 = 1R,2R and 1S,2S)-N1,N2-bis(pyridin-2-ylmethyl)cyclohexane-1,2-diamine, (see Scheme 1), which showed in the bulk a complete thermal and photo-induced SCO with Light-Induced Excited Spin-State Trapping (LIESST) and reverse-LIESST effects.7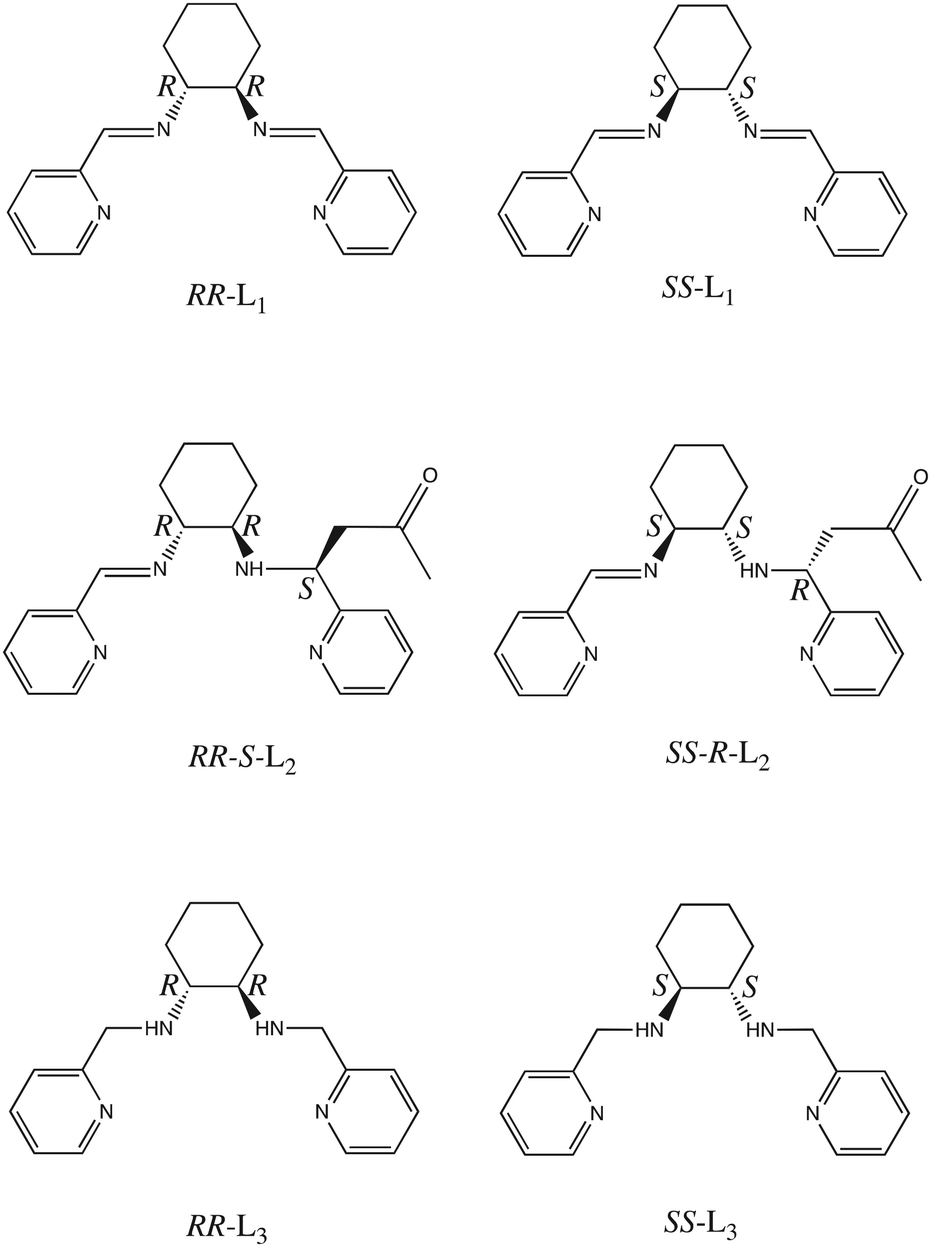 | ||
| Scheme 1 Molecular structures of the chiral ligands RR-L1, SS-L1, RR-S-L2, SS-R-L2, RR-L3 and SS-L3. | ||
The reaction of Fe(II) with RR- and SS-L1 and NCX− in acetone to prepare analogous compounds reported in this work results in an unexpected reaction. Thus, in the obtained complexes, one acetone solvent molecule is incorporated into the ligand forming a bond with the C atom of one of the two CN imine double bonds of L1, which is transformed into an amine. This leads in the compounds [Fe(RR-S-L2)(NCX)2] and [Fe(SS-R-L2)(NCX)2] (X = S or Se) to the asymmetrical β-amino ketone ligands RR-S- and SS-R-L2 with three chiral centers (see Scheme 1). Interestingly, an internal hydrogen bond opens the possibility to tune the molecular absorption of these systems. The structural, spectroscopic and magnetic characterization of these enantiomerically pure compounds is reported in this work.
Results and discussion
Synthesis
The chiral diimine ligands RR-L1 and SS-L1 were prepared by condensation of one equivalent of (S,S or R,R)-1,2-diaminocyclohexane and two equivalents of 2-pyridine carboxaldehyde in toluene.9 Slow diffusion of an aqueous solution of FeSO4 with ascorbic acid (to avoid FeII oxidation) and a solution containing a mixture of RR- or SS-L1 and KSCN or KSeCN in acetone at 5 °C (see Experimental section) leads to crystals of [Fe(RR-S- or SS-R-L2)(NCX)2] (X = S or Se). In these complexes, RR- and SS-L1 reacted with one acetone solvent molecule through last steps of a Mannich reaction.10 This resulted in the reaction of the C of one of the two imine bonds with acetone leading to the asymmetric RR-S- or SS-R-L2 ligands, which contain one amine and one imine in the crystals of [Fe(RR-S- or SS-R-L2)(NCX)2] (X = S or Se). A possible mechanism for this reaction is the activation of the imine as electrophile by the initial protonation or coordination to the metallic center and its nucleophilic attack by the enolate form of the acetone. The tautomeric form of the carbonyl compound can be favored by the acid pH of the aqueous solution (pH ∼ 2.8). This type of reaction has been used to prepare β-aminocarbonyl compounds,11 however, to fully understand the reaction mechanism, additional studies would be necessary. On the other hand, this reaction is diastereoselective. Thus, only one diasteroisomer is formed, in which the chirality of the new C stereocenter produced by the nucleophilic attack of the CH3 group of the acetone molecule to the C of the imine group is the opposite to that of the two chiral centers of the cyclohexane ring as shown by the crystal structure and circular dichroism (see below).The relevance of the formation of the amine group and incorporation of acetone to improve ligand flexibility and stabilize the final complex was confirmed by testing other solvents different than acetone, i.e.: ethanol and toluene, which did not lead to crystals. Furthermore, several attempts to extend this reaction to analogous Schiff base derivatives with substituents in the 5th position of the pyridine ring (MeO or Cl) or to other metals (Zn) did not allow the growth of crystals. On the other hand, the same results were obtained in the absence of ascorbic acid working in the N2 atmosphere of a glove box to rule out a reaction between ascorbic acid and the ligand as the origin of the unexpected reaction. Interestingly, without the addition of the ascorbic acid, the pH of the Fe(II) solution is still acid (pH = 3) while in a Zn(II) solution pH increases considerably (pH = 5), which could hinder the Zn complex formation. Finally, analogous synthetic method using equimolar mixtures of RR-L1 and SS-L1 ligands lead to the growth of separate crystals of [Fe(RR-S- and SS-R-L2)(NCX)2] in contrast to that observed with L3 in which a different phase with a racemic mixture of the enantiomeric complexes was obtained.7 This confirms the stereoselectivity of the reaction in agreement to that expected for the Mannich reaction.11
Powder X-ray diffraction (PXRD) patterns (see Fig. S1†) and elemental analysis (see Experimental section) confirm the purity of these compounds. Thermogravimetric analysis suggest that the complexes are stable until 470 K since significant weigh loss is not detected below this temperature (see Fig. S2†).
Structure
The crystal structures of [Fe(RR-S- or SS-R-L2)(NCX)2] (X = S or Se) were solved by single crystal X-ray diffraction from 90 K to 400 K. They crystallize in the non-centrosymmetric monoclinic P21 space group (see Tables S1–S4†). The asymmetric unit of all these structures contains one crystallographically independent neutral [Fe(RR-S- or SS-R-L2)(NCX)2] (X = S or Se) complex. Fe(II) is coordinated by the four N of the tetradentate RR-S- or SS-R-L2 ligand and the two N from NCS− or NCSe− in cis configuration leading to a distorted octahedral geometry (see Fig. 1, S3 and S4†). The L2 ligand presents three asymmetric C centers: the two C centers from the 1,2-diaminocyclohexane precursor, which keep the same configuration of the L1 precursor (RR- or SS-), and the C atom from the CN imine group bonded to the CH2 group from the acetone molecule, which shows opposite chirality to that of the two other chiral centers of the ligand (S- or R-, respectively). Interestingly, they contain enantiopure forms of the Λ-[Fe(RR-S-L2)(NCX)2] and Δ-[Fe(SS-R-L2)(NCX)2] complexes. The absolute configurations of these compounds were determined unambiguously using single crystal X-ray diffraction with Flack parameters close to 0 (see Tables S1–S4†). Therefore, these chiral ligands containing three asymmetric C enables spontaneous resolution of the Δ and Λ enantiomers.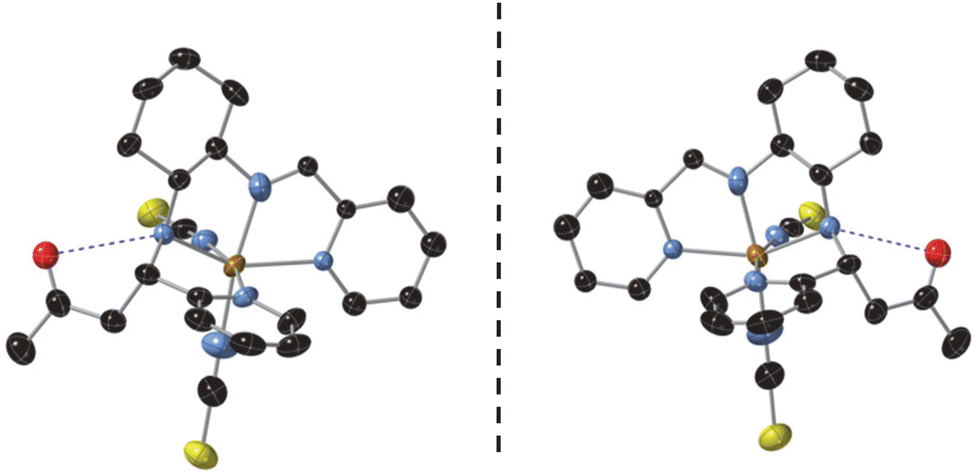 | ||
| Fig. 1 Asymmetric unit of [Fe(RR-S-L2)(NCS)2] (left) and [Fe(SS-R-L2)(NCS)2] (right) at 90 K. Fe (brown), S (yellow), C (black), N (blue) and O (red). Hydrogen and disordered atoms have been omitted for clarity. Hydrogen bonds are drawn as blue dashed lines. | ||
The average Fe–N bond lengths of [Fe(RR-S- or SS-R-L2)(NCSe)2] at 120 K (1.969(7) Å for RR-S- and 1.963(10) Å for SS-R-) are typical of the LS state. At higher temperatures, there is a gradual increase of the average Fe–N distances to reach maximum values at 400 K (2.145(17) Å for RR-S- and 2.14(4) Å for SS-R-), which are close to the expected ones for the HS state. Therefore, these gradual changes in metal–ligand distances suggest an also gradual SCO in a wide temperature for the two NCSe− enantiomorphs (see Table S5†). The average Fe–N bond lengths of the two NCS− enantiomorphs at similar temperatures are higher as expected for the lower ligand field of NCS− with respect to NCSe−. Thus, [Fe(RR-S- or SS-R-L2)(NCS)2] show average Fe–N distances of 2.061(5) Å for RR-S- and 2.053(7) Å for SS-R- at 90 K (see Table S5†), which are indicative of a mixture of LS and HS states. These distances increase with the temperature to reach maximum values of 2.163(7) Å for RR-S- at 300 K and 2.165(11) Å for SS-R- at 400 K typical of a HS state (see Table S5†). These changes in metal–ligand distances and unit cell volumes indicate a gradual and incomplete SCO (see Fig. S5†). At the same time, the four compounds exhibit an increase in the octahedral distortion parameters Σ and Θ, similar to that of related compounds (see Table S5†). Finally, the phenyl ring attached to the imine group of the two NCS− enantiomorphs presents a disorder, which was modelled with two possible configurations with occupancies of 0.5. This disorder is not observed in the two NCSe− enantiomorphs. There is one intramolecular hydrogen-bond involving NH from the amine group and O from the incorporated acetone molecule (see Fig. 1). This hydrogen-bond could play a role in the unexpected reaction of the ligand in the complex as it could be responsible in solution of the close contact between the acetone solvent molecules and the protonated imine bonds. At the same time, it could contribute to the stabilization of one of the two possible diasteroisomers.
Neighboring [Fe(RR- or SS-L2)(NCSe)2] complexes are connected to each other through weak interactions involving the pyridine ring attached to the amine group with the CH3 from acetone and the CH2 from cyclohexane of two neighboring molecules (CH–π interactions), thus, leading to a double chain of complexes along the b axis (see Fig. 2). In the two NCS− enantiomers, analogous double chains are observed (see Fig. S6†). The presence of bulky CH3COCH2 groups prevents the formation of NH⋯Se interactions observed in the complexes with L3 ligands.7 This lack of strong intermolecular interactions could be responsible of the non-cooperative gradual spin-crossover found in these compounds in contrast to the more abrupt spin transition of [Fe(RR- and SS-L3)(NCSe)2].7 A similar trend has been observed in other bis-(thiocyanato) Fe(II) complexes of [4-X-N-(phenyl(pyridin-2-yl)methylene)aniline] (X = Cl, Br, CH3).12
 | ||
| Fig. 2 Double chains of complexes in the structure of [Fe(RR-S-L2)(NCS)2] (top) and [Fe(SS-R-L2)(NCS)2] (bottom) at 90 K linked through intermolecular interactions (red dashed lines). Fe (brown), S (yellow), C (black), N (blue) and O (red). | ||
UV-vis and circular dichroism (CD) spectroscopy
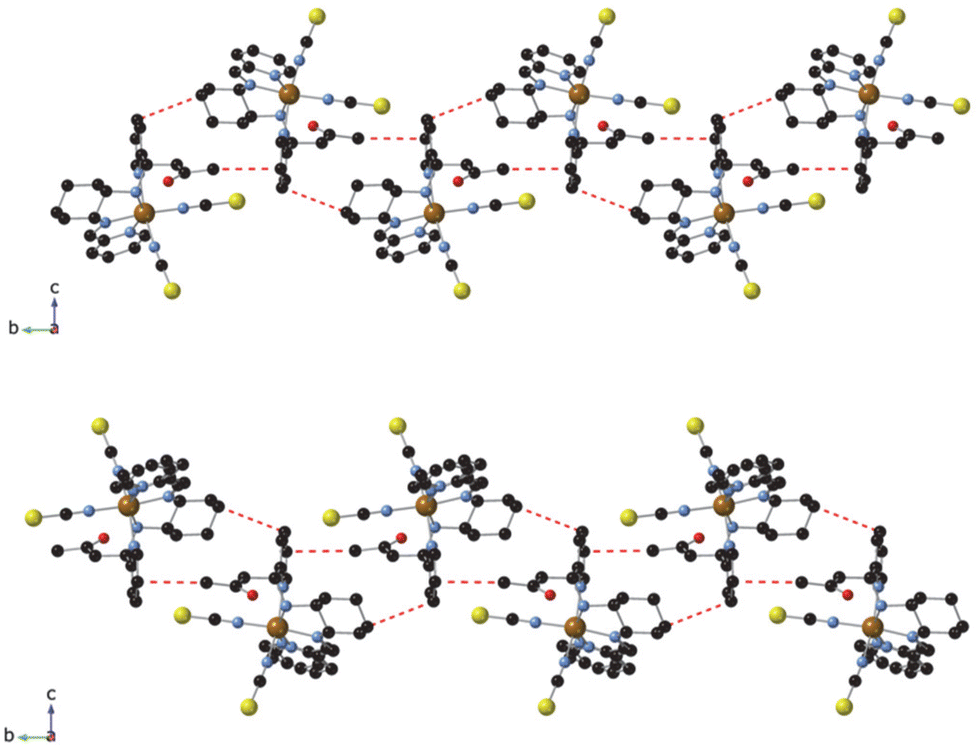
The chirality of the four complexes was confirmed by CD spectroscopy. Thus, CD spectra in CHCl3 of the two enantiomers of [Fe(RR-S- or SS-R-L2)(NCX)2] are mirror images of each other (see Fig. 3 and S7†). These CD spectra are analogous to those of [Fe(RR- or SS-L3)(NCSe)2] reported in the literature, in which the high anisotropy factors in the 250–340 nm region suggest that π–π* transitions of the ligand contribute to the optical activity.7 | ||
| Fig. 3 CD spectra of [Fe(RR-S- or SS-R-L2)(NCSe)2] (top) and [Fe(RR-S- or SS-R-L2)(NCS)2] (bottom) in CHCl3. | ||
Remarkably, during solubility tests in different solvents, we observed different colours of the complexes when using CHCl3 (blue solution) or CH3CN (violet solution) as solvents. UV-vis spectra of [Fe(RR-S- or SS-R-L2)(NCX)2] in CHCl3 and MeCN reflect this observation (see Fig. 4 and S8†). The intense bands in the UV region can be assigned to π–π* transitions, while the two bands at 373 and 567 nm (MeCN) and 385 and 608 nm (CHCl3) for X = Se and at 390 and 570 nm (MeCN) and 412 and 614 nm (CHCl3) for X = S can be assigned to a metal-to-ligand charge transfer (MLCT) by analogy to related compounds.13,14 The relatively low ε values of these bands (103 M−1 cm−1) are indicative of a predominant HS state,14 although additional measurements are needed to clarify this point such as NMR spectroscopy using Evans’ method. Solid-state UV-vis spectra of the four compounds show broader bands at 390 and 580 nm. This is caused by scattering and absorption flattening due to an inhomogeneous particle size distribution (see Fig. S9†). An interesting aspect of the observed solvent effect is that it can unlock a secondary stimuli-responsive behavior in the system. Therefore, by changing the nature of the solvent, the color of the complex in solution can be easily tuned, which suggests possible applications in sensing.
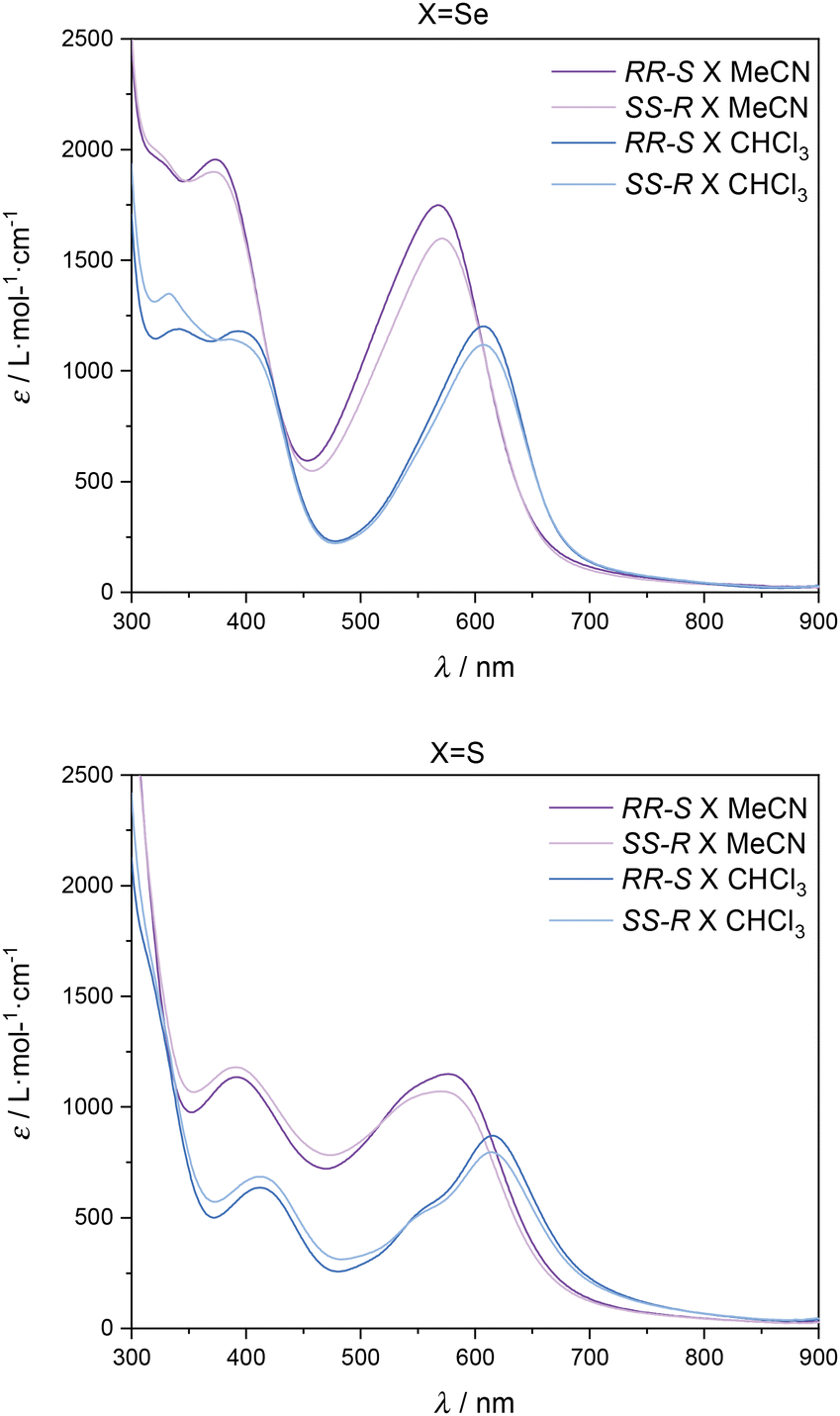 | ||
| Fig. 4 UV-vis spectra of CHCl3 and MeCN solutions of [Fe(RR-S- or SS-R-L2)(NCSe)2] (top) and [Fe(RR-S- or SS-R-L2)(NCS)2] (bottom). | ||
Magnetic properties
The product of the molar magnetic susceptibility times the temperature (χMT) of the four compounds is shown in Fig. 5. The magnetic properties of the two enantiomers of [Fe(RR-S- or SS-R-L2)(NCSe)2] and [Fe(RR-S- or SS-R-L2)(NCS)2] are almost identical. They present the same behavior in the heating and cooling modes from 5 to 400 K in agreement with the absence of solvent molecules in the structures. Furthermore, they show very gradual SCOs as expected for the lack of strong intermolecular interactions in agreement with temperature dependent structural characterization results. Thus, χMT of [Fe(RR-S- or SS-R-L2)(NCS)2] decreases from 3.5 cm3 K mol−1 at 400 K (close to the expected value for 100% HS) to 1.3 cm3 K mol−1 at 80 K. Below this temperature, χMT reaches a plateau with an abrupt decrease below 20 K due to zero-field splitting. This compound exhibits then an incomplete SCO from the fully HS state at 400 K to ∼37% of HS fraction below 80 K considering 3.5 cm3 K mol−1 as 100% HS. On the other hand, χMT of [Fe(RR-S- or SS-R-L2)(NCSe)2] shows a gradual decrease from 2.8 to 0.3 cm3 K mol−1 in the 400–150 K temperature range. Therefore, lower χMT values in the same range of temperatures are found for [Fe(RR-S- or SS-R-L2)(NCSe)2] enantiomers in agreement with the shorter Fe–N distances found in their structures and higher ligand field of NCSe− ligand. In this case, a LS state with a residual ∼9% HS fraction is obtained below 150 K, while fully HS state is not reached even at 400 K (∼80% of HS fraction at this temperature considering 3.5 cm3 K mol−1 as 100% HS). The T1/2, defined as the temperature at which the HS and LS fractions are the same, is 110 K for [Fe(RR-S- or SS-R-L2)(NCS)2] enantiomers and 260 K for [Fe(RR-S- or SS-R-L2)(NCSe)2] enantiomers.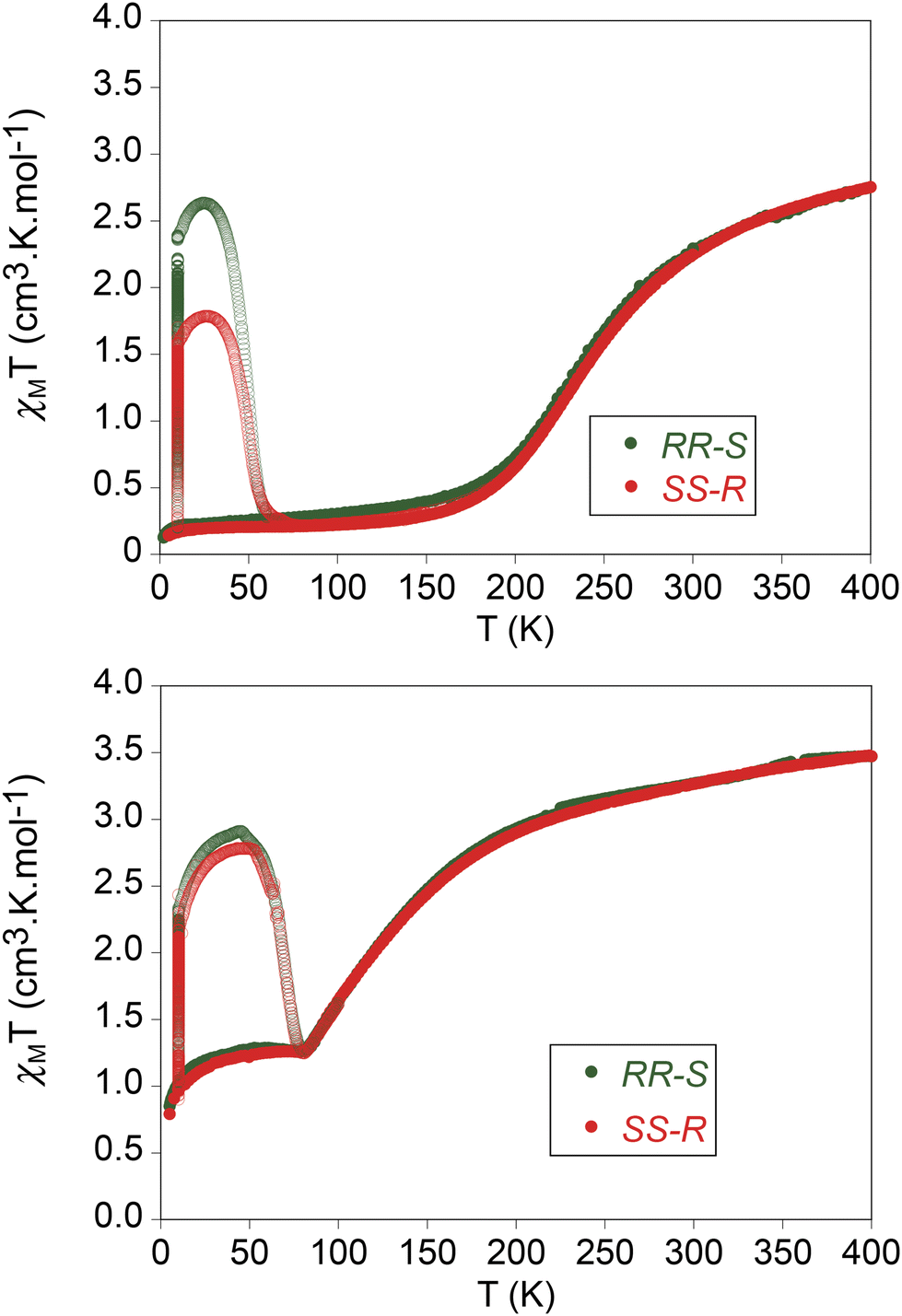 | ||
| Fig. 5 Thermal variation of χMT for [Fe(RR-S-L2)(NCSe)2] (green circles) and [Fe(SS-R-L2)(NCSe)2] (red circles) (top), [Fe(RR-S-L2)(NCS)2] (green circles) and [Fe(SS-R-L2)(NCS)2] (red circles) (bottom). Full circles: data recorded without irradiation; empty circles: data recorded after irradiation at 10 K. | ||
[Fe(RR-S- or SS-R-L2)(NCS)2] and [Fe(RR-S- or SS-R-L2)(NCSe)2] were irradiated at 10 K at 808 nm. A drastic increase of the magnetic signal was observed for the four compounds (see Fig. 5). After several hours of irradiation at 10 K (see Fig. S10†), the irradiation was switched off and the temperature was increased at a scan-rate of 0.3 K min−1 (the so-called T(LIESST) experiment). In the 10–50 K temperature range, an increase of χMT was observed with maximum values of 2.6 cm3 K mol−1 ([Fe(RR-S-L2)(NCSe)2]), 1.8 cm3 K mol−1 ([Fe(SS-R-L2)(NCSe)2]), 2.9 cm3 K mol−1 ([Fe(RR-S-L2)(NCS)2]) and 2.8 cm3 K mol−1 ([Fe(SS-R-L2)(NCS)2]) consistent with maximum LS to HS photoconversions close to 80% of the LS centers at 10 K. The different photoconversion of the RR-S- or SS-R- compounds could be attributed to sample preparation factors, different crystal size or irradiation time (see Fig. S10† and associated text). The T(LIESST), defined as the minimum of the derivative of χMT after irradiation with temperature, is 70 K for [Fe(RR-S- or SS-R-L2)(NCS)2] and 50 K for [Fe(RR-S- or SS-R-L2)(NCSe)2] in line with that obtained for other bis-(thiocyanato) or bis-(selenocyanato) Fe(II) complexes.7,12 In contrast to [Fe(RR- or SS-L3)(NCSe)2] compounds irradiation with 808 nm induced an increase of the signal instead of the reverse-LIESST effect observed for those compounds.7
Conclusions
The use of a chiral tetradentate ligand with two imine groups in combination with NCS− and NCSe− leads to a family of chiral SCO compounds in which acetone, used as the reaction media, plays a non-innocent role, giving rise to an unexpected reaction. Thus, a new C–C bond is formed between an acetone molecule and the C of the imino group creating a new asymmetric ligand. This causes the reduction from imino to amino group. It seems that this reaction with acetone and the flexibility afforded by the amine group is crucial for the obtention of a crystalline compound since the use of other ligands with two imine groups not undergoing this reaction did not allow the growth of crystals. The presence of the acetone molecule in the structure leads to weaker intermolecular interactions and more isolated Fe(II) complexes that lead to a more gradual SCO in the bulk than that observed in related complexes with two amine groups. However, the incorporation of this acetone molecule could represent an advantage with respect to previous compounds. We have to take into account that the interaction with the solvent can modify the intramolecular hydrogen bond between the incorporated acetone molecule and the NH groups directly coordinated to Fe(II). This can result in alterations in the ligand field, the spin state, and the color of the complex. Preliminary characterization by UV-vis spectroscopy suggests that this could be possible. An additional advantage of the presence of acetone in these complexes could be related to the sensing of chiral molecules capable to interact with the new stereocenter formed by the incorporation of acetone.Experimental
Synthesis
RR-L1 and SS-L1 were prepared according to the literature.9 The other reagents and solvents were obtained from commercial sources and used without further purification.Synthesis of [Fe(RR-S- or SS-R-L2)(NCS)2] and [Fe(RR-S- or SS-R-L2)(NCSe)2]
[Fe(RR-S- or SS-R-L2)(NCX)2] (X = S or Se) were obtained through a liquid-to-liquid diffusion method. First, 3 mL aqueous solution of FeSO4·7H2O (0.3 mmol, 83.4 mg) and a few mg of ascorbic acid were added to the bottom of the diffusion tube. A water–acetone (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution, as an intermediate section, and 3 mL acetone solution of KNCSe (0.6 mmol, 86.4 mg) or KNCS (0.6 mmol, 58.3 mg) and the corresponding chiral ligand (0.3 mmol, 88.8 mg of RR- or SS-L1), as top layer, were added. The tube was sealed and kept at 5 °C. After three weeks blue needle crystals were obtained. Elemental analysis suggests the absorption of 1 to 3 water molecules. Elemental analysis calcd (%) for C23H26FeN6OSe2·2H2O: C, 42.35; H, 4.64; N, 12.88 and C23H26FeN6OSe2·H2O: C, 43.55; H, 4.45; N, 13.25; found for [Fe(RR-S-L2)(NCSe)2]: C, 42.23; H, 4.10; N, 12.55. Elemental analysis found for [Fe(SS-R-L2)(NCSe)2]: C, 43.06; H, 4.23; N, 12.74. Elemental analysis calcd (%) for C23H26FeN6OS2: C, 52.87; H, 5.02; N, 16.09; S, 12.28. Elemental analysis calcd (%) for C23H26FeN6OS2·3H2O: C, 47.92; H, 5.59; N, 14.58; S, 11.12. Elemental analysis found for [Fe(RR-S-L2)(NCS)2]: C, 47.54; H, 4.63; N, 14.06; S, 10.77. Elemental analysis found for [Fe(SS-R-L2)(NCS)2]: C, 52.25; H, 5.10; N, 15.47; S, 11.89.
:1) solution, as an intermediate section, and 3 mL acetone solution of KNCSe (0.6 mmol, 86.4 mg) or KNCS (0.6 mmol, 58.3 mg) and the corresponding chiral ligand (0.3 mmol, 88.8 mg of RR- or SS-L1), as top layer, were added. The tube was sealed and kept at 5 °C. After three weeks blue needle crystals were obtained. Elemental analysis suggests the absorption of 1 to 3 water molecules. Elemental analysis calcd (%) for C23H26FeN6OSe2·2H2O: C, 42.35; H, 4.64; N, 12.88 and C23H26FeN6OSe2·H2O: C, 43.55; H, 4.45; N, 13.25; found for [Fe(RR-S-L2)(NCSe)2]: C, 42.23; H, 4.10; N, 12.55. Elemental analysis found for [Fe(SS-R-L2)(NCSe)2]: C, 43.06; H, 4.23; N, 12.74. Elemental analysis calcd (%) for C23H26FeN6OS2: C, 52.87; H, 5.02; N, 16.09; S, 12.28. Elemental analysis calcd (%) for C23H26FeN6OS2·3H2O: C, 47.92; H, 5.59; N, 14.58; S, 11.12. Elemental analysis found for [Fe(RR-S-L2)(NCS)2]: C, 47.54; H, 4.63; N, 14.06; S, 10.77. Elemental analysis found for [Fe(SS-R-L2)(NCS)2]: C, 52.25; H, 5.10; N, 15.47; S, 11.89.
Structural resolution
Single crystals of all complexes were mounted on a glass fibre using a viscous hydrocarbon oil to coat the crystal and then transferred directly to the cold nitrogen stream for data collection. X-ray data were collected at different temperatures on a Supernova diffractometer equipped with a graphite-monochromated Enhance (Mo) X-ray Source (λ = 0.71073 Å). The program CrysAlisPro, Oxford Diffraction Ltd, was used for unit cell determinations and data reduction. Empirical absorption correction was performed using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm. The structures were solved with the ShelXT structure solution program15 and refined with the SHELXL-2013 program,16 using Olex2.17 Non-hydrogen atoms were refined anisotropically, and hydrogen atoms were placed in calculated positions refined using idealized geometries (riding model) and assigned fixed isotropic displacement parameters. Crystallographic data are summarized in Tables S1–S4.† The disorder of the pyridine ring attached to the imine group in the structures of [Fe(RR-S- or SS-R-L2)(NCS)2] at 300 K, [Fe(RR-S-L2)(NCS)2] at 90, 150, 250 and 300 K and [Fe(SS-R-L2)(NCS)2] at 90, 130 and 200 K was solved with two possible configurations with occupancies of 0.5. In these cases, it was not possible to refine anistropically all the disordered atoms. Furthermore, AFIX constraints and DFIX restraints were used to fix the geometry of part of these disordered fragments. CCDC 2343602–2343619 contain the supplementary crystallographic data for this paper.† For X-ray powder pattern, a 0.7 mm glass capillary was filled with a polycrystalline sample of de complexes and mounted and aligned on an Empyrean PANalytical powder diffractometer, using CuKα radiation (λ = 1.54177 Å). A total of 3 scans were collected at room temperature in the 2θ range 5–40°.Physical characterization
The Fe/S and Fe/Se ratios were measured with a Philips ESEM X230 scanning electron microscope equipped with an EDAX DX-4 microsonde. Elemental analyses (C, H, S and N) were performed with a CE Instruments EA 1110 CHNS Elemental analyzer. CD spectra were measured in a Jasco J-1500 Circular Dichroism spectrophotometer. UV-vis spectra were measured in a Jasco V-670 spectrophotometer. Magnetic measurements were performed with a Quantum Design MPMS-XL-5 SQUID magnetometer with a applied magnetic fields of 0.1 or 0.5 T. LIESST measurements were performed irradiating with a MDL-H-808-5W system from Microbeam (λ = 808 nm, optical power 8.5 mW cm−2) coupled via an optical fiber to the cavity of the SQUID magnetometer. It was verified that irradiation resulted in no significant change in magnetic response due to heating of the sample. The photomagnetic samples consisted of a thin layer of compound whose weight was corrected by comparison of a thermal spin crossover curve with that of a more accurately weighted sample of the same compound.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Spanish MCIN (Grant PID2020-117264GB-I00 funded by MCIN/AEI/10.13039/501100011033 and Unidad de Excelencia María de Maeztu CEX2019-000919-M), the Generalitat Valenciana PO FEDER program INFRA GV 2020, IDIFEDER/2020/063 and IDIFEDER/2021/075, and H2020 program (ERC2021-COG-10104342). AR thanks the Spanish MCIN FPI predoctoral fellowship (PRE2021-098327). This study forms part of the Advanced Materials programme and was supported by MCIN with funding from the European Union NextGenerationEU (PRTR-C17.I1) and by Generalitat Valenciana (MFA/2022/050). We all thank J. M. Martínez-Agudo and G. Agustí for magnetic measurements.References
- (a) Spin Crossover in Transition Metal Compounds, Topics in Current Chemistry, ed. P. Gütlich and H. A. Goodwin, Springer Verlag, Berlin-Heidelberg-New York, 2004, vol. 233–235 Search PubMed; (b) Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, UK, 2013 Search PubMed.
- K. Senthil Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176 CrossRef CAS.
- (a) S. Ohkoshi, S. Takano, K. Imoto, M. Yoshikiyo, A. Namai and H. Tokoro, Nat. Photonics, 2014, 8, 65 CrossRef CAS; (b) C. Bartual-Murgui, L. Piñeiro-López, F. J. Valverde-Muñoz, M. C. Muñoz, M. Seredyuk and J. A. Real, Inorg. Chem., 2017, 56, 13535 CrossRef CAS PubMed; (c) T. Charytanowicz, K. Dziedzic-Kocurek, K. Kumar, S. I. Ohkoshi, S. Chorazy and B. Sieklucka, Cryst. Growth Des., 2023, 23, 4052 CrossRef CAS; (d) C. T. Kelly, R. Jordan, S. Felton, H. Müller-Bunz and G. G. Morgan, Chem. – Eur. J., 2023, 2023, e202300275 CrossRef PubMed.
- (a) I. A. Gural'skiy, V. A. Reshetnikov, A. Szebesczyk, E. Gumienna-Kontecka, A. I. Marynin, S. I. Shylin, V. Ksenofontov and I. O. Fritsky, J. Mater. Chem. C, 2015, 3, 4737 RSC; (b) V. Jornet-Mollá, Y. Duan, C. Giménez-Saiz, Y. Y. Tang, P. F. Li, F. M. Romero and R. G. Xiong, Angew. Chem., Int. Ed., 2017, 56, 14052 CrossRef PubMed; (c) A. Naim, Y. Bouhadja, M. Cortijo, E. Duverger-Nédellec, H. D. Flack, E. Freysz, P. Guionneau, A. Iazzolino, A. O. Hamouda, P. Rosa, O. Stefańczyk, A. Valentín-Pérez and M. Zeggar, Inorg. Chem., 2018, 57, 14501 CrossRef CAS PubMed; (d) V. B. Jakobsen, L. O'Brien, G. Novitchi, H. Müller-Bunz, A. L. Barra and G. G. Morgan, Eur. J. Inorg. Chem., 2019, 2019, 4405 CrossRef CAS.
- (a) R. T. Acha and M. Pilkington, CrystEngComm, 2015, 17, 8897 RSC; (b) L. F. Qin, C. Y. Pang, W.-K. Han, F.-L. Zhang, L. Tian, Z.-G. Gu, X. Ren and Z. Li, CrystEngComm, 2015, 17, 7956 RSC; (c) L. F. Qin, C.-Y. Pang, W. K. Han, F. L. Zhang, L. Tian, Z. G. Gu, X. Ren and Z. Li, Dalton Trans., 2016, 45, 7340 RSC; (d) D. H. Ren, D. Qiu, C. Y. Pang, Z. Li and Z. G. Gu, Chem. Commun., 2015, 51, 788 RSC; (e) J. Ru, F. Yu, P. P. Shi, C. Q. Jiao, C. H. Li, R. G. Xiong, T. Liu, M. Kurmoo and J. L. Zuo, Eur. J. Inorg. Chem., 2017, 2017, 3144 CrossRef CAS; (f) K. E. Burrows, S. E. McGrath, R. Kulmaczewski, O. Cespedes, S. A. Barrett and M. A. Halcrow, Chem. – Eur. J., 2017, 23, 9067 CrossRef CAS PubMed; (g) N. Suryadevara, A. Pausch, E. Moreno-Pineda, A. Mizuno, J. Bürck, A. Baksi, T. Hochdörffer, I. Šalitroš, A. S. Ulrich, M. M. Kappes, V. Schünemann, W. Klopper and M. Ruben, Chem. – Eur. J., 2021, 27, 15171 CrossRef CAS PubMed.
- Y. Sekimoto, M. R. Karim, N. Saigo, R. Ohtani, M. Nakamura and S. Hayami, Eur. J. Inorg. Chem., 2017, 2017, 1049 CrossRef CAS.
- T. T. Ma, X. P. Sun, Z. S. Yao and J. Tao, Inorg. Chem. Front., 2020, 7, 1196 RSC.
- X. H. Zhao, Y. F. Deng, J. Q. Huang, M. Liu and Y. Z. Zhang, Inorg. Chem. Front., 2024, 11, 808 RSC.
- Y. Zhang, L. Xiang, Q. Wang, X. F. Duan and G. Zi, Inorg. Chim. Acta, 2008, 361, 1246 CrossRef CAS.
- M. Arendt, B. Westermann and N. Risch, Angew. Chem., Int. Ed., 1998, 37, 1044 CrossRef.
- (a) B. Eftekhari-Sis, A. Abdollahifar, M. M. Hashemi and M. Zirak, Eur. J. Org. Chem., 2006, 5152 CrossRef CAS; (b) H. Wu, Y. Shen, L. Y. Fan, Y. Wan, P. Zhang, C. F. Chen and W. X. Wang, Tetrahedron, 2007, 63, 2404 CrossRef CAS.
- K. Bhar, W. Guo, M. Gonidec, V. Nikhil Raj M, S. Bhatt, F. Perdih, P. Guionneau, G. Chastanet and A. K. Sharma, Dalton Trans., 2022, 51, 9302 RSC.
- N. Ortega-Villar, V. M. Ugalde-Saldívar, M. C. Muñoz, L. A. Ortiz-Frade, J. A. Alvarado-Rodríguez, J. A. Real and R. Moreno-Esparza, Inorg. Chem., 2007, 46, 7285 CrossRef CAS PubMed.
- I. Prat, A. Company, T. Corona, T. Parella, X. Ribas and M. Costas, Inorg. Chem., 2013, 52, 9229 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2343602–2343619. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00924j |
| This journal is © The Royal Society of Chemistry 2024 |