
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMononuclear Fe(III) complexes with 2,4-dichloro-6-((quinoline-8-ylimino)methyl)phenolate: synthesis, structure, and magnetic behavior†
Ah Rim
Jeong
a,
Si Ra
Park
b,
Jong Won
Shin
*a,
Jihyun
Kim
b,
Ryuya
Tokunaga
c,
Shinya
Hayami
 c and
Kil Sik
Min
*b
c and
Kil Sik
Min
*b
aDepartment of Chemistry, Kyungpook National University, Daegu 41566, Republic of Korea. E-mail: jwshin@kisti.re.kr
bDepartment of Chemistry Education, Kyungpook National University, Daegu 41566, Republic of Korea. E-mail: minks@knu.ac.kr
cDepartment of Chemistry, Kumamoto University, Kumamoto 860-8555, Japan
First published on 19th March 2024
Abstract
Three Fe(III)-based coordination complexes [Fe(dqmp)2](NO3)·H2O (1), [Fe(dqmp)2](BF4)·2CH3COCH3 (2), and [Fe(dqmp)2](ClO4) (3) were synthesized from Fe(NO3)3·9H2O/Fe(ClO4)3·xH2O, NaBF4, and 2,4-dichloro-6-((quinoline-8-ylimino)methyl)phenol (Hdqmp) in methanol/acetone and characterized. The structures of complexes 1–3 were determined via single-crystal X-ray crystallography at 100 K and room temperature, and their magnetic properties in the solid and solution forms were investigated. All complexes showed meridional structures with two tridentate dqmp− ligands coordinated with Fe(III) cations. In the solid state, complex 1 showed an abrupt and complete spin crossover at 225 K, whereas complexes 2 and 3 exhibited an incomplete spin crossover at 135 and 150 K, respectively. In a dimethylformamide solution, the complexes showed counterion-dependent spin transitions. In contrast to the solid state, in solution, complex 1 did not exhibit complete spin crossover. However, complexes 2 and 3 showed more complete spin transitions in solutions than in the solid state. The relaxation times, T1 and T2, for 1 and 2 were determined and both increased with temperature from 220 to 380 K. The T1 of 1 was larger than that of 2 at 380 K, and the T1 values were larger than the T2 values.
Introduction
Spin crossover (SCO) is a magnetic behavior generally exhibited by d4–d7 transition metal ions. It is a transition from high-spin to low-spin or vice versa induced by external stimuli such as temperature, light, and pressure. SCO has been extensively studied because of its potential applications in molecular sensors, molecular switches, displays, and contrast agents.1 Fe(III) complexes with d5 electron configurations are excellent candidates for SCO behavior because Fe(III) ions exhibit a large difference in the magnetic moment between high-spin (HS; S = 5/2) and low-spin (LS; S = 1/2) states and are rather stable in air.2 In fact, the air stability of Fe(III) ions enables their use in diverse magnetic applications such as magnetic sensors and contrast agents.3In this context, Fe(III) complexes based on N-(8-quinolinyl)salicylaldimine (Hqsal) have shown very prominent SCO behaviors and have been extensively investigated to reveal significant SCO phenomena.4 The Hqsal ligand has a rigid and fixed geometry in a meridional fashion as a tridentate (Scheme 1). Interestingly, the ligand can form bis-type metal complexes and induce excellent SCO behavior in Fe(III) complexes. These complexes can be potentially stable and show SCO even in solutions. Harding et al. have reported a Fe(III) complex, [Fe(qsal-I)2]OTf, with two qsal-I ligands (Hqsal-I = 5-iodo-N-(8-quinolyl)salicylaldimine), which first showed abrupt complete SCO at 228 K with 8 K hysteresis.5 The cooperativity of hysteresis was attributed to the extensive π–π stacking and C–H⋯O/F hydrogen bonding. Mercuri et al. reported a Fe(III) dimeric complex [Fe(qsal-5,7-Cl2)(NCS)(MeO)]2 containing four qsal-5,7-Cl2, two NCS−, and two MeO− species, which exists as a monomeric structure in solution.6 This qsal-type complex showed a strong antiferromagnetic interaction through coordinated methoxide ions (J = −21 K). In contrast to monomeric qsal-type complexes, no SCO behavior was observed in this complex. Wang et al. reported four Fe(III) complexes, [Fe(qsal-4-F)2]Y (Y = NO3−, PF6−, BF4−, or OTf−), with a new 4-fluoro-N-(8-quinolyl)salicylaldimine (Hqsal-4-F) ligand.7 These complexes are monomeric species and exhibit SCO behaviors depending on the counter anions. For example, in the case of NO3−, two-step symmetry-breaking SCO was observed, whereas in the case of PF6−, a two-step gradual SCO was observed. Moreover, [Fe(qsal-4-F)2]BF4 underwent a gradual SCO, whereas [Fe(qsal-4-F)2]OTf almost entirely retained the HS state. These results indicate that the position of the substituent and the anions play critical roles in the SCO behavior of complexes.
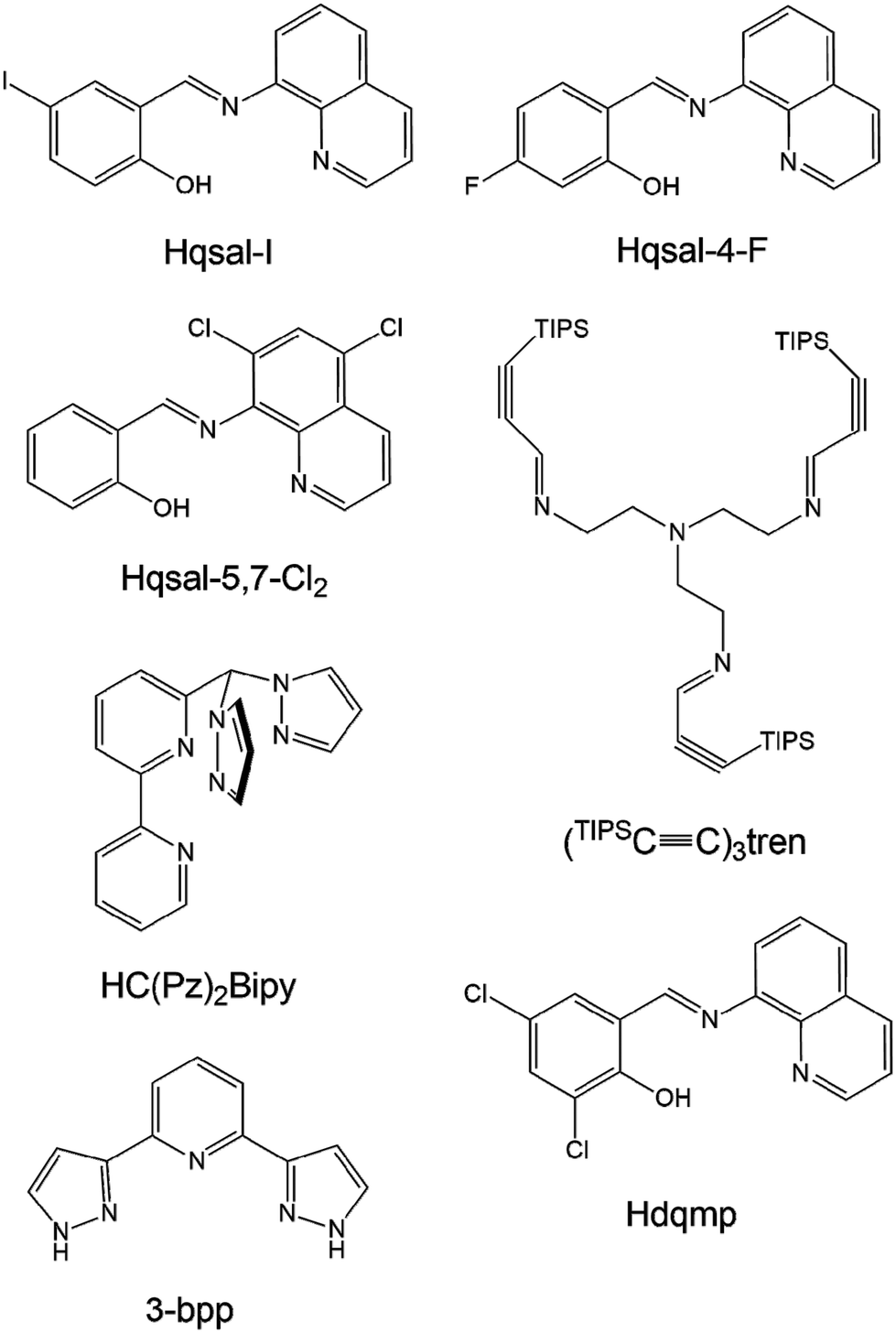 | ||
| Scheme 1 Structures and abbreviations of multidentate ligands mentioned in the Introduction of this study. | ||
Livesay and Shores reported a monomeric complex, [((TIPSC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)3tren)Fe(OTf)2] (Scheme 1), which is in the HS state at all temperatures in solids and solutions (i.e., cyclopentanone, methanol, and dichloromethane).8 However, it shows incomplete SCO with a T1/2 value of 171 K in acetone, which is attributed to triflate dissociation upon decreasing temperature. Herres-Pawlis et al. reported bis(pyrazolyl)bipyridinylmethane Fe(II) complexes that exhibit various SCO behaviors in different solvents.9 In acetonitrile or benzonitrile, the environment of the complex ion changed from N5O to N6, causing a temperature-dependent SCO behavior, which was different from the HS state of the solid sample. Barrett and Halcrow reported [Fe(3-bpp)2]X2 (3-bpp = 2,6-dipyrazol-3-ylpyridine), which exhibits versatile SCO behaviors at 259 K for X = BPh4− and 277 K for X = Br− in acetone–water solvent.10 The T1/2 values of the salts of five anions exhibit a clear trend: BPh4− ≈ BF4− > CF3SO3− > NO3− > Br−.
C)3tren)Fe(OTf)2] (Scheme 1), which is in the HS state at all temperatures in solids and solutions (i.e., cyclopentanone, methanol, and dichloromethane).8 However, it shows incomplete SCO with a T1/2 value of 171 K in acetone, which is attributed to triflate dissociation upon decreasing temperature. Herres-Pawlis et al. reported bis(pyrazolyl)bipyridinylmethane Fe(II) complexes that exhibit various SCO behaviors in different solvents.9 In acetonitrile or benzonitrile, the environment of the complex ion changed from N5O to N6, causing a temperature-dependent SCO behavior, which was different from the HS state of the solid sample. Barrett and Halcrow reported [Fe(3-bpp)2]X2 (3-bpp = 2,6-dipyrazol-3-ylpyridine), which exhibits versatile SCO behaviors at 259 K for X = BPh4− and 277 K for X = Br− in acetone–water solvent.10 The T1/2 values of the salts of five anions exhibit a clear trend: BPh4− ≈ BF4− > CF3SO3− > NO3− > Br−.
In the present study, the effects of the functional group of qsal ligands and anions on spin transition were investigated. We aimed to determine the changes in spin transition in solution through nuclear magnetic resonance (NMR) and confirm the possibility of the application of the prepared complexes as MRI contrast agents. Herein, we report three Fe(III) monomeric complexes, [Fe(dqmp)2](NO3)·H2O (1), [Fe(dqmp)2](BF4)·2CH3COCH3 (2), and [Fe(dqmp)2](ClO4) (3), which were prepared in methanol/acetone from Fe(III) salt and 2,4-dichloro-6-((quinoline-8-ylimino)methyl)phenol (Hdqmp) in air. These complexes exhibited octahedral monomeric structures and SCO in solid and solution states depending on the counteranion.
Experimental
General
All chemicals used in the synthesis were of reagent grade and used without further purification. Hdqmp was prepared as previously reported.11 Infrared spectra were recorded using a Thermo Fisher Scientific Nicolet iS5 spectrophotometer (±1 cm−1) with a KBr disk. Elemental analyses were performed using a Fisons/Carlo Erba EA1108 instrument in air. UV–vis absorption spectra were measured using a SCINCO S-2100 spectrophotometer. Magnetic susceptibilities were measured in an applied field of 5000 Oe between 5 and 300/400 K on a Quantum Design MPMS superconducting quantum interference device (SQUID) magnetometer. Diamagnetic corrections were made [417.78 (1), 407.20 (2), and 415.20 × 10−6 (3) emu mol−1] using Pascal's constants.12Syntheses of the compounds
X-ray crystallographic data collection and refinement
Single-crystal diffraction data of 1–3 were collected using their crystals at low and room temperatures, i.e., 100(2) and 298(2) K for 1 and 2, and 100(2) and 293(2) K for 3. The crystal diffraction data obtained at the low temperature were designated as 1a, 2a, and 3a and 1b, 2b, and 3b at room temperature, respectively. The data were recorded using synchrotron radiation (λ = 0.70000 and 0.63000 Å) on an ADSC Quantum-210 detector at 2D SMC with a silicon (111) double-crystal monochromator at the Pohang Accelerator Laboratory, Korea. The ADSC Q210 ADX program13 was used for data collection (detector distance: 62 mm, omega scan; Δω = 1°, exposure time: 3 s per frame), and HKL3000sm (Ver. 703r)14 was used for cell refinement, reduction, and absorption correction. The crystal structures of 1a, 1b, 2a, 2b, 3a, and 3b were solved by direct methods15 and refined via full-matrix least-squares refinement using the SHELXL-2018 computer program.16 The positions of all nonhydrogen atoms were refined using anisotropic displacement factors. All hydrogen atoms were placed using a riding model, and their positions were constrained relative to their parent atoms using the appropriate HFIX command in SHELXL-2018, except for the hydrogen atoms of water molecules. During the refinements of the structures (2b, 3a, and 3b), electron density peaks were located and believed to be highly disordered solvent molecules. All attempts to model the solvent molecules were unsuccessful, and they were removed using the SQUEEZE routine from PLATON software.17 The SQUEEZE routine indicated that there were large solvent cavities and electrons: 495 Å3 and 115 for 2b, 700 Å3 and 188 for 3a, and 837 Å3 and 198 for 3b, per unit cell. In the final cycles of refinement, the contribution of electrons to the electron density was removed from the observed data. In 2b, 3a, and 3b, the density, the F(000) value, the molecular weight and the formula were given without taking into account the results obtained with SQUEEZE. In addition, in the structure of 3, the ClO4− ion is disordered at 100 K. Thus, the oxygen atoms of 3a were treated with 1.00 sof for one O atom and 0.75 sof for 4 O atoms. Unlike in 3a, the perchlorate ion in 3b at 293 K is well refined. The crystallographic data and the results of refinements of 1a, 1b, 2a, 2b, 3a, and 3b are summarized in Table S1.†NMR measurements
A series of 1D 1H NMR spectra were obtained using a Varian Unity INOVA NMR spectrometer (Korea Basic Science Institute, Western Seoul Center). All spectra were acquired using a 500 MHz NMR (11.7 T) spectrometer within a temperature range of 220–380 K. The Evans NMR spectra were recorded using custom-made NMR tubes with a pure solvent in the inner tube and the sample in the outer tube. For Evans NMR, the pure inner solvent residue was used as a reference. For the calculation of the magnetic data, the following equation was used: χg = (3Δf)/(4πmf), where Δf is the difference in chemical shift between residual proton signals [Hz], f is the spectrometer frequency [Hz], and m is the mass concentration of the paramagnetic sample [g cm−3]. Additionally, χM was obtained by multiplying χg by the molar mass. Finally, we used the following equation to calculate the χM values: χM = (477Δδ)/(2 × 106M), where χM is molar susceptibility [cm3 mol−1], Δδ is frequency shift [ppm], and M is molar concentration [mol L−1].1g,18 The NMR spectra for determining relaxation time were measured at the KAIST Analysis Center for Research Advancement using a 400 MHz (9.4 T) Agilent DD2 NMR spectrometer (Agilent Technologies Inc., Santa Clara, CA). The proton T1 relaxation rates of dimethylformamide (DMF) in the presence of either 1 or 2 were determined using the inversion recovery method with 12 delays. For T2 measurements, the Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence was utilized, acquiring a total of 10–12 data points. Measurements were conducted at four temperatures: 220, 300, 340, and 380 K. Throughout the process, a recycle delay of 1 s was used. Raw data were processed and subjected to Fourier transformation using VnmrJ software (Agilent Technologies). Subsequently, peak integration and T1 and T2 curve fittings were performed using both VnmrJ software and Matlab (MathWorks, Natick, MA).Results and discussion
Synthesis and characterization
Dark-brown complexes [Fe(dqmp)2](NO3)·H2O (1) and [Fe(dqmp)2](ClO4) (3) were synthesized in yields of 42% and 53%, respectively, from a solution of Fe(NO3)3·9H2O/Fe(ClO4)3·5H2O and Hdqmp at room temperature in MeOH/acetone. At the same time, dark-brown [Fe(dqmp)2](BF4)·2CH3COCH3 (2) was prepared in high yield (70%) from Fe(NO3)3·9H2O, NaBF4, and Hdqmp at room temperature in MeOH/acetone. The prepared complexes were coordinated with two dqmp− ligands, causing distorted octahedral cationic species. However, the counterions of complexes 1, 2, and 3 were different: NO3−, BF4−, and ClO4−, respectively. The compositions of 1–3 were determined via elemental analyses, infrared spectroscopy, and single-crystal X-ray diffraction. The IR spectra of the complexes showed a common strong ν(CN) band at ca. 1604 ± 2 cm−1 which was assigned to the imine group of the dqmp− ligand. This band was consistent with coordination to the FeIII center, as it shifted by ca. 17 cm−1 to lower energy compared with the free ligand Hdqmp. Stretches for the anions, NO3−, BF4−, and ClO4−, were also observed in the IR spectra, that is, strong bands of the counterions were noted at 1384 (NO3−), 1056 (BF4−), and 1089 cm−1 (ClO4−).19Description of the crystal structures
The crystal structures of monomeric complexes [1a, 1b, 2a, 2b, 3a, and 3b] were determined by X-ray crystallography at low temperature (100 K, a) and at room temperature (298/293 K, b); the crystallographic data are summarized in Table S1,† and selected bond lengths are provided in Table 1. All complexes include isostructural monomeric [FeIII(dqmp)2]+ cation with two tridentate dqmp− ligands that coordinated meridionally to each other, that is, two phenolate (and quinolones) groups were cis relative to each other and two imine nitrogen atoms were in a trans configuration. Complexes 1–3 contained NO3−, BF4−, and ClO4− anions, respectively. In addition, complexes 1 and 3 crystallized with water molecules as a lattice solvent, whereas complex 2 crystallized with acetone molecules. ORTEP drawings of 1a, 2a, and 3a are shown in Fig. 1.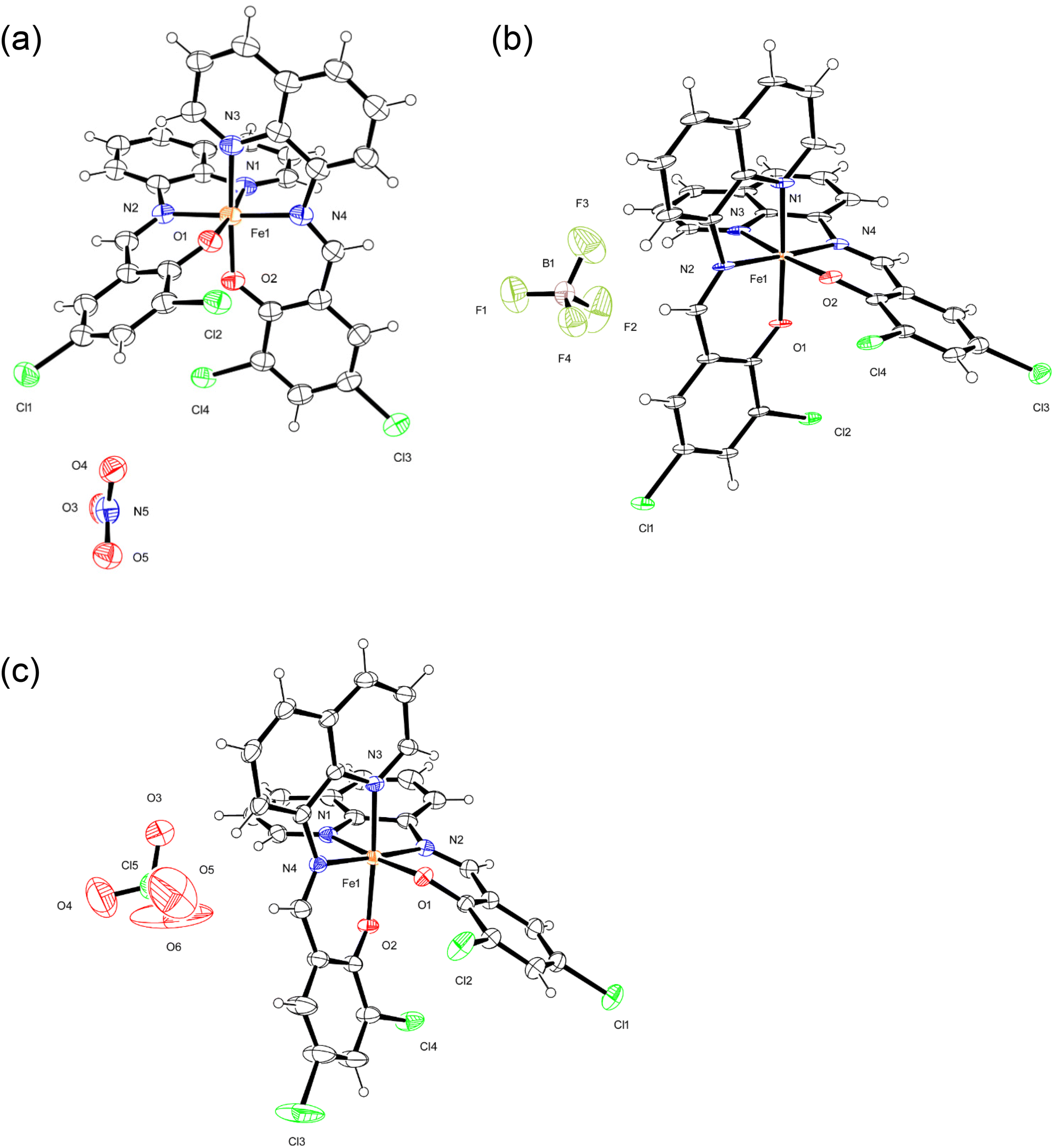 | ||
| Fig. 1 ORTEP views of 1a (a), 2a (b), and 3a (c). The atoms are represented by 40% (1a and 2a) and 30% (3a) probable thermal ellipsoids. Solvent molecules were omitted for clarity. | ||
| 1a | 1b | 2a | 2b | 3a | 3b | |
|---|---|---|---|---|---|---|
| Fe1–N1 | 1.994(3) | 2.194(3) | 1.978(2) | 2.014(3) | 1.972(2) | 1.986(3) |
| Fe1–N2 | 1.919(3) | 2.093(3) | 1.933(3) | 1.942(4) | 1.935(2) | 1.938(3) |
| Fe1–N3 | 1.976(3) | 2.124(3) | 1.984(2) | 2.019(4) | 1.981(2) | 1.974(3) |
| Fe1–N4 | 1.939(3) | 2.138(3) | 1.939(3) | 1.950(4) | 1.933(2) | 1.940(3) |
| Fe1–O1 | 1.874(2) | 1.923(2) | 1.879(2) | 1.882(3) | 1.870(2) | 1.880(2) |
| Fe1–O2 | 1.881(2) | 1.910(3) | 1.867(2) | 1.873(3) | 1.876(2) | 1.870(2) |
At 298 K, the average Fe–N and Fe–O bond lengths of 1 were 2.137(2) and 1.919(2) Å, respectively, which are typical of [Fe(qsal-X)2]+ in the HS state.7,20 At 100 K, the average Fe–N and Fe–O bond lengths were 1.957(2) and 1.878(1) Å, respectively, which are consistent with those of a LS FeIII center.21 The changes in the average bond lengths between the structures at 298 and 100 K were ΔFe–N = 0.180 Å and ΔFe–O = 0.041 Å. These values are comparable with those reported for [Fe(qsal)2]I3 and [Fe(qsal-OMe)2]Cl2·MeOH·0.5H2O and indicate a complete SCO.22 Different from the structure of 1, the average Fe–N and Fe–O bond lengths of 2 were slightly changed depending on the temperature, that is, the changes in the average bond lengths between the structures at 298 and 100 K were ΔFe–N = 0.016 Å and ΔFe–O = 0.005 Å, which is indicative of an incomplete SCO. Based on the average Fe–N and Fe–O bond lengths, the spin state of 2 was very close to those of an LS FeIII center in the [Fe(qsal-X)2]+ species.7,20,21 Likewise, the average Fe–N and Fe–O bond lengths of 3 were very slightly changed due to temperature, that is, the changes in the average bond lengths between the structures at 298 and 100 K were ΔFe–N = 0.006 Å and ΔFe–O = 0.002 Å, and indicated an incomplete SCO. In this case, the spin state of 3 was nearly consistent with those of the LS FeIII centers.20,21
At 100 K, the average bite distances and angles of the five-membered chelate rings were 2.587(3) Å and 82.73(8)° for 1, 2.595(3) Å and 82.97(9)° for 2, and 2.583(2) Å and 82.69(7)° for 3, respectively. These are almost identical within the estimated standard deviations. This means the structures of 1–3 at 100 K were quite similar. At room temperature, the average bite distances and angles of the five-membered chelate rings were 2.646(3) Å and 76.47(8)° for 1, 2.594(2) Å and 81.80(7)° for 2, and 2.585(2) Å and 82.56(7)° for 3, respectively. The distance and angle of 1 were quite different from those of 2 and 3. This can be attributed to the high spin state of 1 at room temperature and the low spin states of 2 and 3 at room temperature. Additionally, we calculated the octahedral distortion parameters Σ and Φ of 1a–3b.23 The Σ and Φ values were 43.1 and 123.7° for 1a, and 76.1 and 275.8° for 1b, respectively. From the big difference of the Σ and Φ values between 1a and 1b, we have observed the structural change due to the spin transition. However, complexes 2 and 3 did not exhibit any significant changes for the Σ and Φ values (42.0 and 124.3° for 2a, 41.3 and 129.0° for 2b, 41.2 and 121.0° for 3a, and 41.7 and 123.8° for 3b, respectively).23 These values are attributed to the very small transformation of the spin states.
All aromatic groups (i.e., quinoline and benzene) of 1a and 2a were involved in π–π interactions with the adjacent aromatic groups of the neighboring Fe(III) complex. Owing to these π–π interactions, all Fe(III) complex ions were linked in a two-dimensional network extending along the a(b + c) plane (Fig. 2 and 3). A ladder-like 1D chain structure was formed along axis a, due to the π–π interactions between the quinoline and benzene groups. In 1a, the shortest centroid⋯centroid distance between two quinolines was 3.73 Å, and that between quinoline and benzene was 3.94 Å. Furthermore, the π–π interactions between the ladder-like 1D chains existed through two quinoline groups (centroid⋯centroid distance 3.47 Å), forming a 2D sheet structure (Fig. 2). In 2a, the shortest centroid⋯centroid distance between two quinolines was 3.70 Å, and that between quinoline and benzene was 3.77 Å. Furthermore, the π–π interactions between the ladder-like 1D chains existed through two quinoline groups (centroid⋯centroid distance, 3.62 Å), inducing a 2D sheet structure (Fig. 3). In contrast to 1a, within the ladder-like 1D chain of 1b, the shortest centroid⋯centroid distance between two quinolines was 3.67 Å, and that between quinoline and benzene was 4.38 Å. Between the ladder-like 1D chains, the centroid⋯centroid distance was 3.59 Å through two quinoline groups. Contrary to 2a, within a ladder-like 1D chain of 2b, the shortest centroid⋯centroid distance between the two quinolines was 3.77 Å and that between quinoline and benzene was 3.87 Å. Between the ladder-like 1D chains, the centroid⋯centroid distance through two quinoline groups was 3.71 Å.
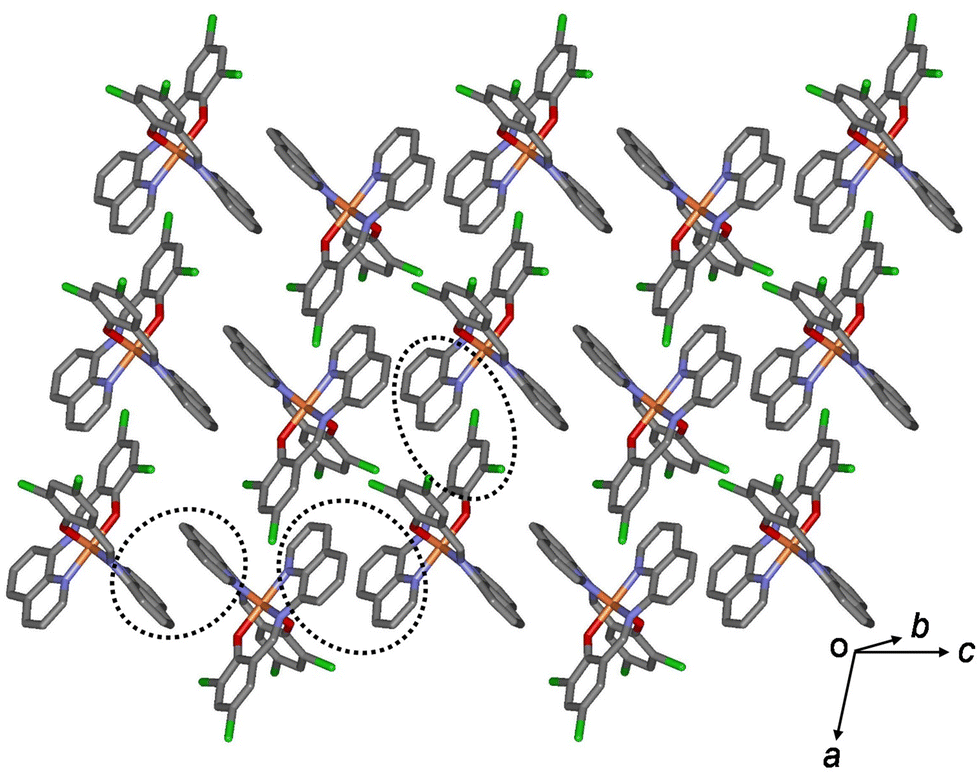 | ||
| Fig. 2 Schematic of 1a, showing an extended 2D layered structure formed via π–π interactions (dotted circles). | ||
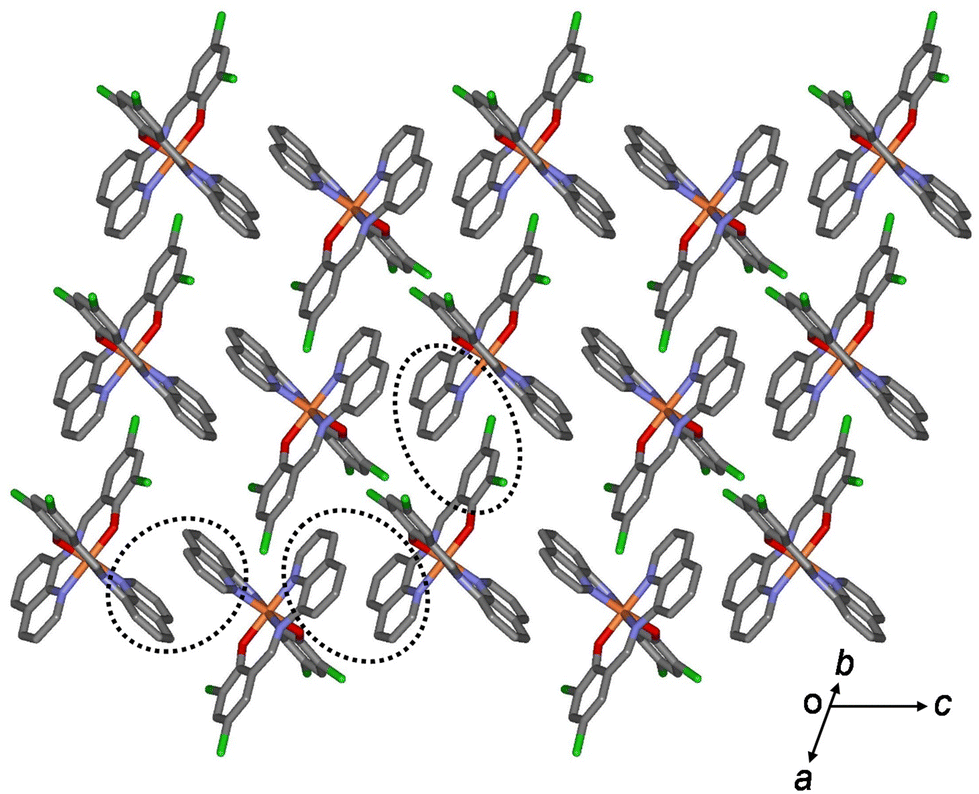 | ||
| Fig. 3 Schematic of 2a, showing an extended 2D layered structure formed via π–π interactions (dotted circles). | ||
In the case of 3a, owing to these π–π interactions, all Fe(III) complex ions were linked together, forming a two-dimensional network extending along the bc plane (Fig. 4). A 1D chain structure was formed along the c axis because of the π–π interactions between the quinoline groups. The shortest centroid⋯centroid distances of 3a between the two quinolines were 3.52 and 3.66 Å, respectively. Furthermore, the π–π interactions between the 1D chains existed through the quinoline and benzene groups (centroid⋯centroid distance, 4.29 Å), inducing a 2D sheet structure. In contrast to 3a, in a 1D chain of 3b, the shortest centroid⋯centroid distances between the two quinolines were 3.66 and 3.57 Å, respectively. Between the 1D chains, the centroid⋯centroid distance was 4.33 Å through the quinoline and benzene groups. As mentioned above, the shortest centroid⋯centroid distances between two quinolines or between quinoline and benzene decreased in the ladder-like 1D chains in the order of complexes 1 > 2 > 3. This means that they were densely stacked in the order of 1, 2, and 3, and can be supported by suggesting that spin transition is difficult to occur in the order.
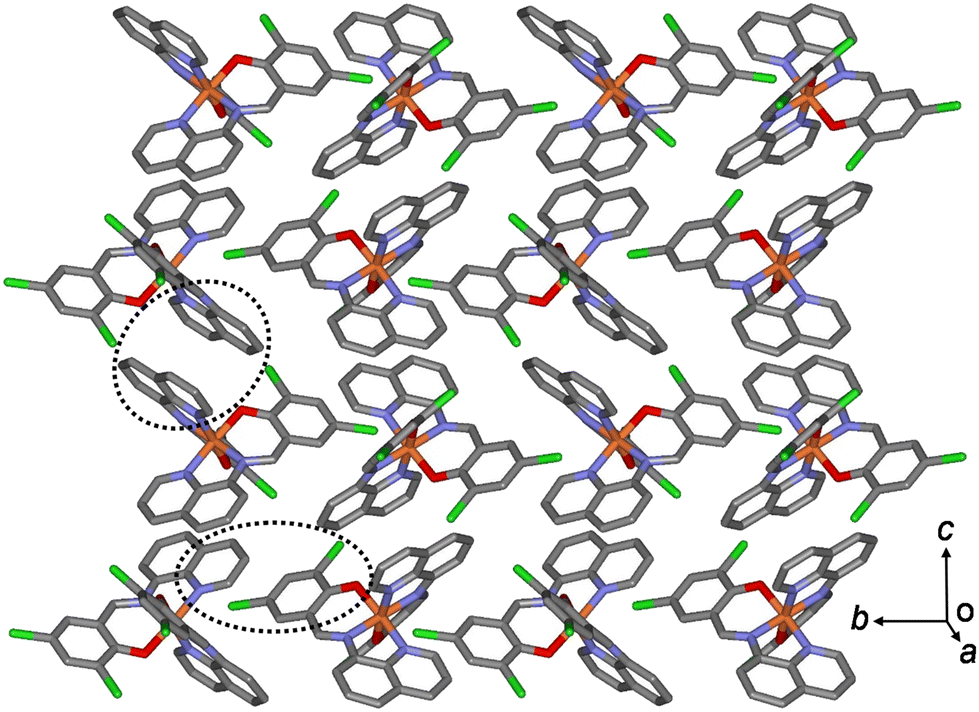 | ||
| Fig. 4 Schematic of 3b, showing an extended 2D layered structure formed via π–π interactions (dotted circles). | ||
Magnetic properties of solids 1–3
Magnetic susceptibility, χ, of polycrystalline samples 1–3 at 2–300 K was measured at 5000 Oe on a SQUID magnetometer. At 300 K, the χMT value of 1 was ca. 4.09 emu K mol−1 and was almost constant until 224 K, which is the expected value for the HS Fe(III) ions (S = 5/2). Subsequently, χMT abruptly decreased to 0.463 emu K mol−1 at 204 K, indicating an SCO behavior (Fig. 5).24 With further cooling, the χMT value very slightly decreased to 0.428 emu K mol−1 at 5 K, which is the expected value for the LS Fe(III) ion (S = 1/2).25 With an increase in temperature from 5 to 300 K in the second run, χMT showed a similar trend to that in the first run, with a small hysteresis. From 5 to 213 K, the χMT value gradually increased from 0.436 to 0.481 emu K mol−1, followed by an abrupt increase to 3.85 emu K mol−1 at 241 K, indicating an SCO, and then further increased to 4.04 emu K mol−1 at 290 K. Thus, the critical temperatures during heating showed a thermal hysteresis with a width of approximately 9 K at 225 K. Interestingly, this complex shows spin transition that can be clearly stabilized in both HS and LS states.1i,2c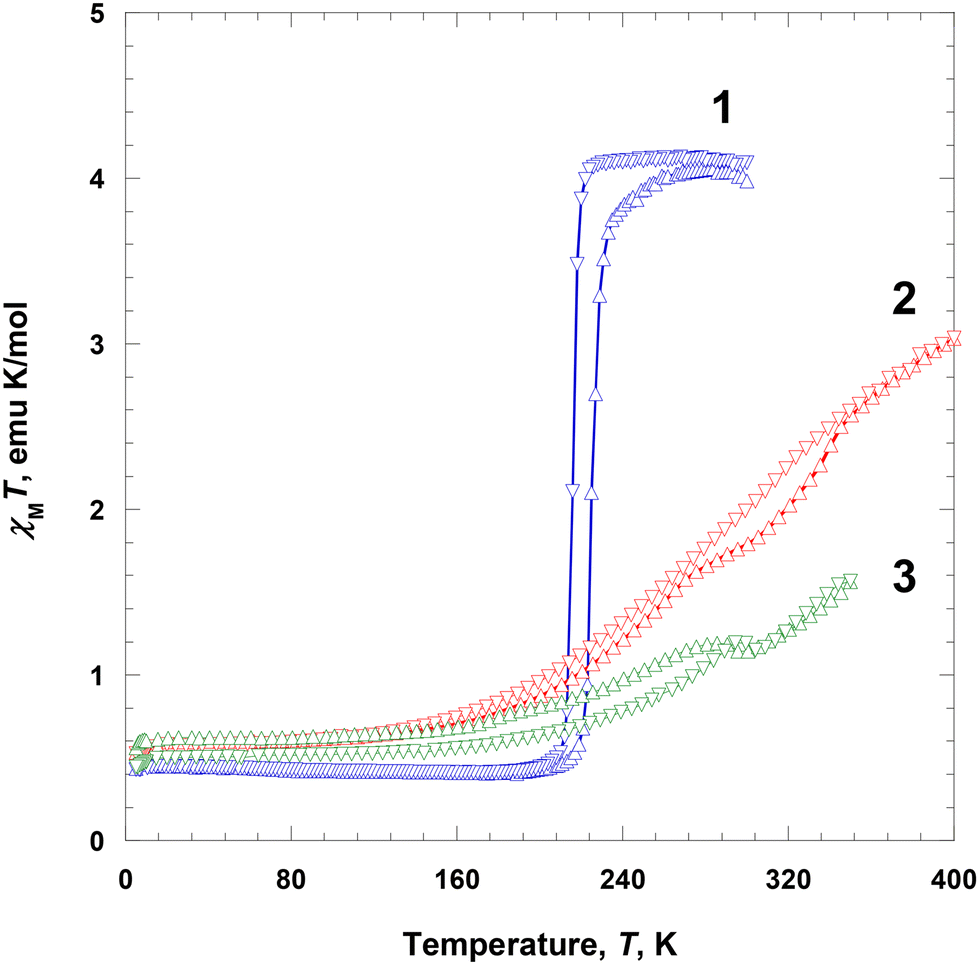 | ||
| Fig. 5 Magnetic susceptibility data, displayed as the χMT product for 1–3 at 5000 Oe during heating (Δ) and cooling (∇). | ||
The χMT value of 2 at 5 K was 0.531 emu K mol−1 and was nearly constant until 135 K (0.658 emu K mol−1), which is the expected value for the LS Fe(III) ions (S = 1/2). Subsequently, it rapidly increased to 3.03 emu K mol−1 at 400 K, indicating an SCO (Fig. 5).24 The χMT value at 400 K was significantly lower than the expected spin-only value of 4.38 emu K mol−1, indicating that the magnetic spin state at 400 K did not fully recover to the HS state of the Fe(III) ions (ca. 80% high spin).20b With a decrease in temperature from 400 to 5 K in the second run, χMT showed a similar trend to that in the first run, with a small hysteresis. With cooling from 400 to 135 K, the χMT value gradually decreased from 3.03 to 0.666 emu K mol−1; with further cooling to 5 K, it decreased only slightly to 0.525 emu K mol−1, which is the expected value for the LS Fe(III) ions (S = 1/2). In this case, the LS state at 5 K is fully recovered. In the region of SCO, there is a very small thermal hysteresis.
The χMT value of 3 at 5 K was 0.560 emu K mol−1 and was nearly constant until 150 K (0.670 emu K mol−1), which is the expected value for the LS Fe(III) ions (S = 1/2), followed by a prominent increase to 1.19 emu K mol−1 at 285 K, indicating an SCO (Fig. 5).24 Between 285 and 300 K, a phase transition occurred with a very small decrease in χMT. Above 300 K, the χMT value continuously increased with temperature until 350 K (1.56 emu K mol−1). This indicates that the magnetic spin state at 350 K did not fully recover to the HS state of the Fe(III) ions (ca. 50% HS).20b With a decrease in temperature from 350 to 305 K in the second run, χMT showed a similar trend to that in the first run. Specifically, between 350 and 305 K, χMT gradually decreased from 1.57 to 1.15 emu K mol−1, followed by a slight increase to 1.19 emu K mol−1 at 295 K. It then gradually decreased to 0.544 emu K mol−1 until 150 K, and remained almost constant until 5 K (0.544 emu K mol−1), ultimately taking the expected value for the LS Fe(III) ions (S = 1/2).24 The χMT curves obtained during heating and cooling exhibited a similar pattern; however, below 300 K, there was a small difference in magnetic moments, which was attributed to an annealing effect during heating.
In the crystalline solids of 1–3, the completeness of spin transition in 1–3 decreased in the order of NO3− > BF4− > ClO4−, which was attributed to the size of the counter anions. With respect to the counter anions, the SCO behaviors can be related to the shape and size of each anion. Nitrate ion is planar and shows hydrogen bonds (C–H⋯O) that extend in three directions in 1, whereas BF4−/ClO4− extends in four directions in 2 and 3, respectively, and are tightly connected by hydrogen bonds (C–H⋯F/O). Thus, when it is spherical and has a large volume like BF4− or ClO4−, the Fe(III) species in the solid state can be disturbed to induce spin transition due to the increase of the volume of the Fe(III) ion. Finally, an SCO is more difficult to occur in 2 and 3 than in 1.
Magnetic properties of the solutions of 1–3
Magnetic susceptibilities of the solution samples of 1–3 were measured in a DMF-d7 solvent using an NMR spectrometer at several different temperatures during heating from 220 to 380 K (Fig. 6). The effective magnetic moment, μeff(T) [=(8χMT)1/2], was calculated using χM obtained using the Evans method (Tables S2–S4 and Fig. S1–S3†).18 At 220 K, the μeff(T) value of 1 was ca. 2.38μB, which is slightly higher than that of the LS Fe(III) ion (S = 1/2). With the increase in temperature from 220 to 380 K, the effective moment gradually increased to 4.28μB. This indicates that the magnetic spin states at 380 K did not fully recover to the HS state of the Fe(III) ions (ca. 68% HS, assuming μeff(T) = 5.92μB).26 This behavior is rather different from that in the solid state. Interestingly, 1 showed a complete SCO in the solid state, whereas the solution of 1 did not show a complete SCO, that is, the magnetic moment of 1 differed between the solid and solution forms. This means that the SCO behavior is related to crystal packing and solvent effects.27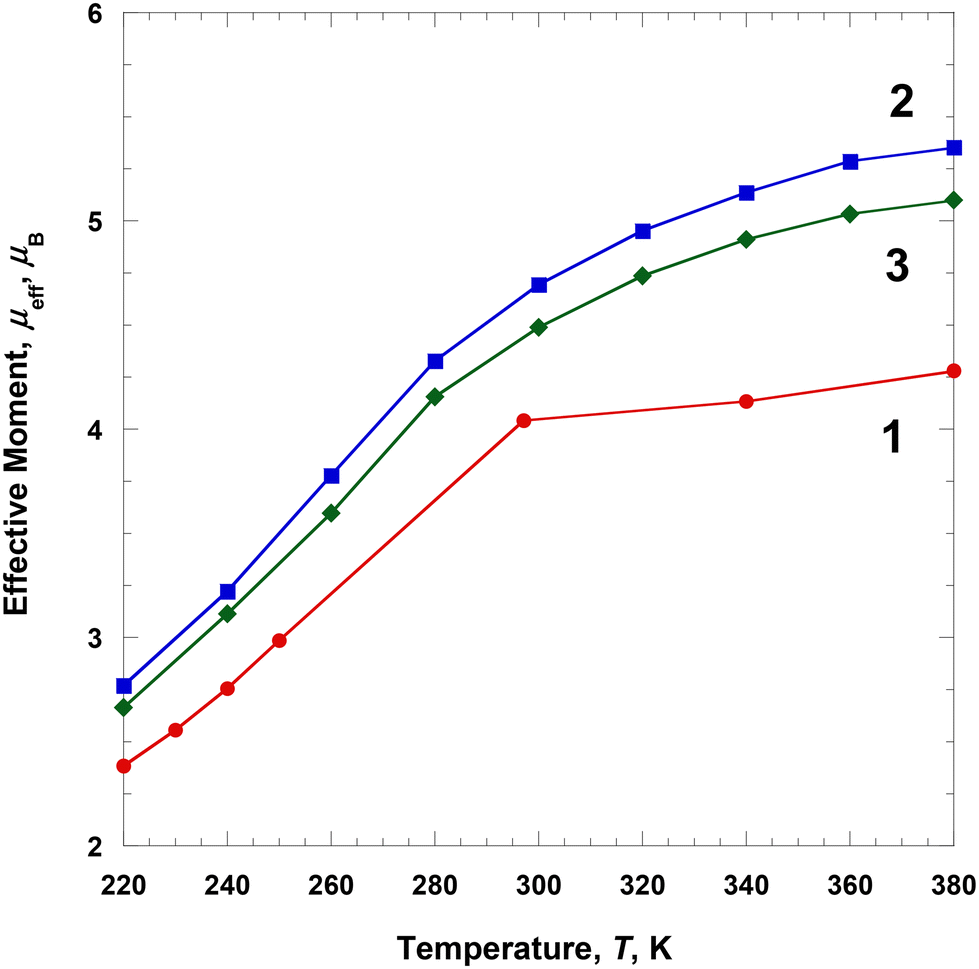 | ||
| Fig. 6 Temperature dependence of μeff for 1–3 dissolved in DMF-d7. | ||
At 220 K, the μeff(T) value of 2 was ca. 2.77μB, which is higher than that of the LS Fe(III) ions (S = 1/2) and close to the S = 1 spin state.25 With the increase in temperature from 220 to 380 K, the effective moment gradually increased to 5.35μB. This indicates that the magnetic spin states at 380 K did not fully recover to the HS state of the Fe(III) ions (ca. 90% HS, assuming μeff(T) = 5.92μB).26 In contrast to solution 1, complex 2 showed a clearer SCO as the temperature increased to 380 K, that is, the SCO of solution 2 was different from that of the solid state 2. At 220 K, the μeff(T) value of 3 was ca. 2.66μB, which is higher than that of the LS Fe(III) ions (S = 1/2) and close to the S = 1 spin state.25 With an increase in temperature from 220 to 380 K, the effective moment gradually increased to 5.10μB. This indicates that the magnetic spin states at 380 K did not fully recover to the HS state of the Fe(III) ions (ca. 84% HS, assuming μeff(T) = 5.92μB).26 These results are very similar to those of solution 2 and differ from those of solid 3. In the case of solution 3, some interactions related to Fe(III) cation, ClO4− anion, and DMF solvent significantly affected the SCO behavior.
In solutions 1–3, all complexes existed as monomeric units without any interactions between their complex cations. We expected that the complex cations would interact with the DMF solvent and their counteranions in solution. Surprisingly, the complexes showed an SCO behavior in solution in the measured temperature range. Although complexes 2 and 3 exhibited an incomplete SCO in the solid state, their SCOs were observed very distinctly in solutions. All complexes exhibited clear SCO behaviors in solution and were expected to relate to weak interactions by the counter ions. The obtained results suggest that the counterions have a more significant effect on SCO than π–π interactions.
Proton relaxation times of 1 and 2
Complexes 1 and 2 were dissolved in DMF-d7, and the T1 and T2 relaxation times of the prepared solutions were measured through NMR at ca. 2.90 ppm (Fig. 7). The longitudinal proton relaxation time (T1) for 1 and 2 was determined at 9.4 T (400 MHz) and 220, 300, 340, and 380 K (Fig. S4 and S6†). The transverse proton relaxation times (T2) were measured using a multiecho CPMG pulse sequence (Fig. S5 and S7†). At 220 K, complex 1 showed the T1 and T2 relaxation times of 0.11 and 0.047 s, respectively, and the relaxation times gradually increased with a temperature up to 380 K. At 380 K, the T1 and T2 relaxation times were 0.31 and 0.21 s, respectively. Thus, from 220 to 380 K, T1 and T2 increased by ca. 2.8 and 4.5 times, respectively. This means that the ratio of the HS state was proportional to T1 and T2. In the case of complex 2, at 220 K, the T1 and T2 relaxation times were 0.12 and 0.061 s, respectively, and an increase in temperature up to 380 K. At 380 K, the T1 and T2 relaxation times were 0.27 and 0.19 s, respectively. Thus, from 220 to 380 K, T1 and T2 increased by ca. 2.3 and 3.1 times, respectively. In 1 and 2, T1 was larger than T2, and both times increased with temperature because of the increase in magnetic moment. T1 and T2 depended on the magnetic moments and could potentially be adjusted to MRI contrast agents. The large difference in the T1/T2 relaxation times between low and high temperatures can considerably affect contrast in MRI.28 It is known that weighted MRI images depend on T1 and T2. Thus, owing to their temperature-dependent magnetic moments, 1 and 2 may be applied in MRI for diagnostic purposes.29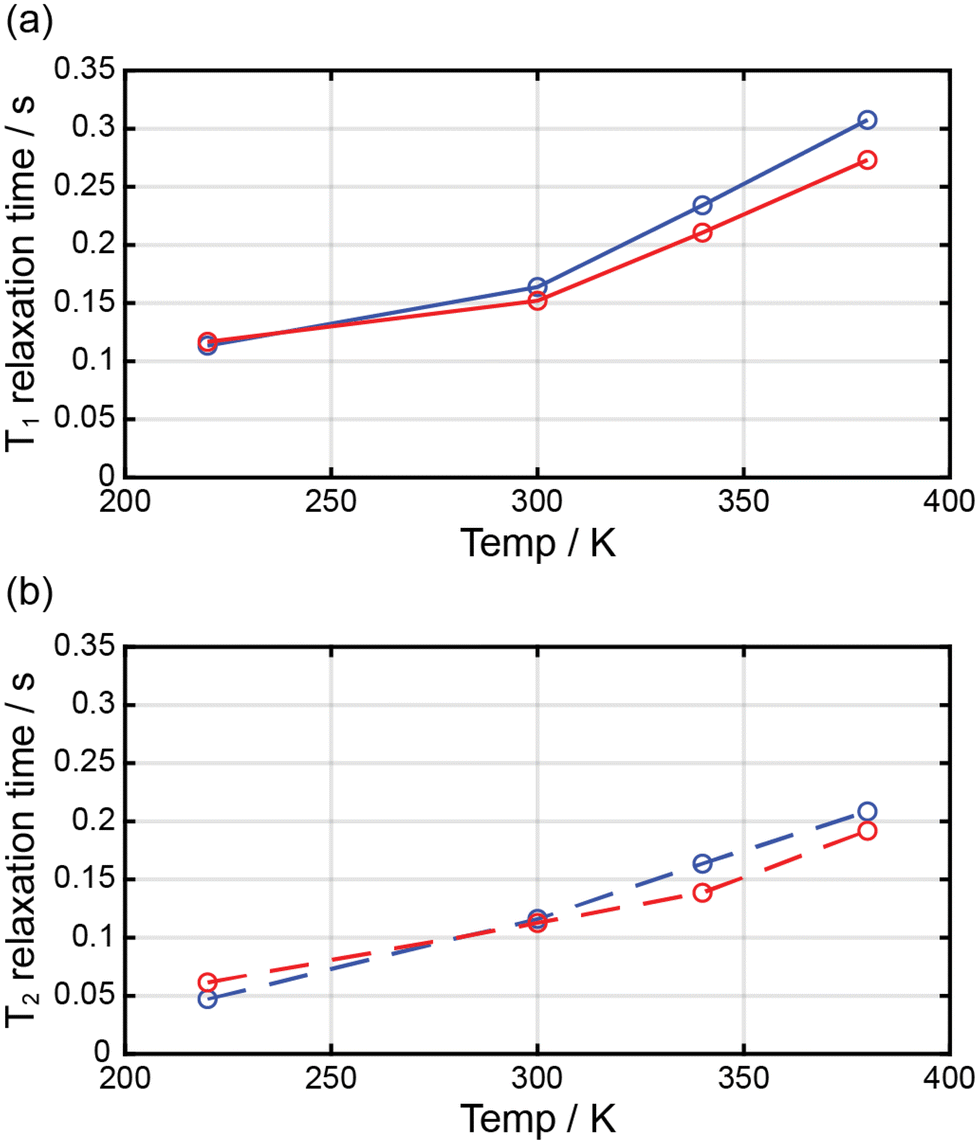 | ||
| Fig. 7 Temperature dependence of the relaxation times for 1 (blue circle) and 2 (red circle) dissolved in DMF-d7. | ||
Conclusions
We synthesized a series of mononuclear Fe(III) complexes with two tridentate ligands (dqmp−) and different counteranions; these complexes exhibited SCO behaviors in the solid and solution states. In all three complexes, two dqmp− ligands were meridionally coordinated in a tridentate manner with one uncoordinated counterion. Their Fe(III) cationic species were isomorphous, although the counterions were different. The solid structures exhibited strong π–π interactions between cationic units. Interestingly, the solid sample with NO3− anions showed a very abrupt SCO around 225 K; however, the complexes with BF4− and ClO4− anions exhibited an incomplete SCO in the solid state. Remarkably, in solutions, Fe(III) complexes with BF4− and ClO4− anions showed a clearer SCO than that with NO3− anions. Furthermore, the T1 and T2 relaxation times of the complexes with NO3− and BF4− depended on the temperature and were proportional to their magnetic moments in solution form. Owing to this behavior, the prepared complexes may potentially be applied as contrast agents in MRI; however, their solubility problems would have to be resolved. In future work, these SCO complexes should be modified and studied to obtain water-soluble species.Author contributions
Ah Rim Jeong: data curation, investigation, and visualization. Si Ra Park: data curation and investigation. Jong Won Shin: data curation, investigation, and writing – review & editing. Jihyun Kim: data curation and investigation. Ryuya Tokunaga: data curation and investigation. Shinya Hayami: investigation and methodology. Kil Sik Min: funding acquisition, project administration, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program (No. 2021R1F1A1062108) and under the framework of international cooperation program (No. 2021K2A9A2A08000136) through the National Research Foundation of Korea (NRF). X-ray crystallography at the PLS-II 2D-SMC beamline was supported in part by MEST and POSTECH.References
- (a) Y.-S. Meng and T. Liu, Acc. Chem. Res., 2019, 52, 1369–1379 CrossRef CAS PubMed; (b) L. T. Birchall, A. T. Raja, L. Jackson and H. J. Shepherd, Cryst. Growth Des., 2023, 23, 1768–1774 CrossRef CAS; (c) X. Li, D. Zhang, Y. Qian, W. Liu, C. Mathonière, R. Clérac and X. Bao, J. Am. Chem. Soc., 2023, 145, 9564–9570 CrossRef CAS PubMed; (d) Y.-C. Sun, F.-L. Chen, K.-J. Wang, Y. Zhao, H.-Y. Wei and X.-Y. Wang, Inorg. Chem., 2023, 62, 14863–14872 CrossRef CAS PubMed; (e) T. Kanetomo, K. Yokoyama, Y. Suzuki, H. Ida, A. Okazawa and M. Enomoto, Dalton Trans., 2023, 52, 12496–12503 RSC; (f) H.-D. Li, S.-G. Wu and M.-L. Tong, Chem. Commun., 2023, 59, 6159–6170 RSC; (g) A. Tsukiashi, K. S. Min, H. Terasawa, S. Yoshinaga, M. Takeda, R. Ohtani, M. Nakamura, L. F. Lindoy and S. Hayami, Chem. Lett., 2018, 47, 598–600 CrossRef CAS; (h) I.-R. Jeon, O. Jeannin, R. Clérac, M. Rouzières and M. Fourmigue, Chem. Commun., 2017, 53, 4989–4992 RSC; (i) T. Fujinami, M. Koike, N. Matsumoto, Y. Sunatsuki, A. Okazawa and N. Kojima, Inorg. Chem., 2014, 53, 2254–2259 CrossRef CAS PubMed; (j) S. Sundaresan and S. Brooker, Inorg. Chem., 2023, 62, 12192–12202 CrossRef CAS PubMed; (k) K. Lee, J. Park, I. Song and S. M. Yoon, Bull. Korean Chem. Soc., 2021, 42, 1170–1183 CrossRef CAS.
- (a) A. Miyawaki, K. Eda, T. Mochida, T. Sakurai, H. Ohta, T. Nakajima and K. Takahashi, Inorg. Chem., 2021, 60, 12735–12739 CrossRef CAS PubMed; (b) Z.-Y. Li, H. Ohtsu, T. Kojima, J.-W. Dai, T. Yoshida, B. K. Breedlove, W.-X. Zhang, H. Iguchi, O. Sato, M. Kawano and M. Yamashita, Angew. Chem., Int. Ed., 2016, 55, 5184–5189 CrossRef CAS PubMed; (c) K. Dankhoff and B. Weber, Dalton Trans., 2019, 48, 15376–15380 RSC.
- (a) L. Palagi, E. Di Gregorio, D. Costanzo, R. Stefania, C. Cavallotti, M. Capozza, S. Aime and E. Gianolio, J. Am. Chem. Soc., 2021, 143, 14178–14188 CrossRef CAS PubMed; (b) J. Xie, A. Haeckel, R. Hauptmann, I. P. Ray, C. Limberg, N. Kulak, B. Hamm and E. Schellenberger, Magn. Reson. Med., 2021, 85, 3370–3382 CrossRef CAS PubMed; (c) M. Lewkowitz, J. Adams, N. S. Sullivan, P. Wang, M. Shatruk, V. Zapf and A. S. Arvij, Sci. Rep., 2023, 13, 2769 CrossRef CAS PubMed.
- (a) M. Nihei, T. Shiga, Y. Maeda and H. Oshio, Coord. Chem. Rev., 2007, 251, 2606–2621 CrossRef CAS; (b) M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC; (c) D. J. Harding, P. Harding and W. Phonsri, Coord. Chem. Rev., 2016, 313, 38–61 CrossRef CAS.
- D. J. Harding, W. Phonsri, P. Harding, I. A. Gass, K. S. Murray, B. Moubaraki, J. D. Cashion, L. Liu and S. G. Telferd, Chem. Commun., 2013, 49, 6340–6342 RSC.
- S. A. Sahadevan, E. Cadoni, N. Monni, C. S. de Pipaón, J.-R. G. Mascaros, A. Abhervé, N. Avarvari, L. Marchiò, M. Arca and M. L. Mercuri, Cryst. Growth Des., 2018, 18, 4187–4199 CrossRef.
- H.-J. Sheng, C.-C. Xia, X.-Y. Zhang, C.-C. Zhang, W.-J. Ji, Y. Zhao and X.-Y. Wang, Inorg. Chem., 2022, 61, 12726–12735 CrossRef CAS PubMed.
- B. N. Livesay and M. P. Shores, Inorg. Chem., 2021, 60, 15445–15455 CrossRef CAS PubMed.
- K. Keisers, H. M. Hüppe, L. Iffland-Mühlhaus, A. Hoffmann, C. Göbel, U.-P. Apfel, B. Weber and S. Herres-Pawlis, Inorg. Chem., 2020, 59, 15343–15354 CrossRef CAS PubMed.
- S. A. Barrett and M. A. Halcrow, RSC Adv., 2014, 4, 11240 RSC.
- (a) K. Takano, M. Takahashi, T. Fukushima, M. Takezaki, T. Tominaga, H. Akashi, H. Takagi and T. Shibahara, Bull. Chem. Soc. Jpn., 2012, 85, 1210–1221 CrossRef CAS; (b) D. Chakraborty, D. Mandal, V. Ramkumar, V. Subramanian and J. V. Sundar, Polymer, 2015, 56, 157–170 CrossRef CAS.
- G. A. Bain and G. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- A. J. Arvai and C. Nielsen, ADSC Quantum-210 ADX Program, Area Detector System Corporation, Poway, CA, USA, 1983 Search PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, ed. C. W. Carter Jr. and R. M. Sweet, Academic Press, New York, 1997, vol. 276, part A, p. 307 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, PLATON program, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- (a) K. De Buysser, G. G. Herman, E. Bruneel, S. Hoste and I. Van Driessche, Chem. Phys., 2005, 315, 286–292 CrossRef CAS; (b) E. M. Schubert, J. Chem. Educ., 1992, 69, 62 CrossRef CAS; D. F. Evans, J. Chem. Soc., 1959, 2003–2005 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley and Sons, Inc., New Jersey, 6th edn, 2009, part B, pp. 84–120 Search PubMed.
- (a) B. J. C. Vieira, J. C. Dias, I. C. Santos, L. C. J. Pereira, V. da Gama and J. C. Waerenborgh, Inorg. Chem., 2015, 54, 1354–1362 CrossRef CAS PubMed; (b) R. Díaz-Torres, W. Phonsri, K. S. Murray, L. Liu, M. Ahmed, S. M. Neville, P. Harding and D. J. Harding, Inorg. Chem., 2020, 59, 13784–13791 CrossRef PubMed.
- (a) A. Tsukiashi, M. Nakaya, F. Kobayashi, R. Ohtani, M. Nakamura, J. M. Harrowfield, Y. Kim and S. Hayami, Inorg. Chem., 2018, 57, 2834–2842 CrossRef CAS PubMed; (b) J. J. Whittaker, P. Harding, J. K. Clegg and D. J. Harding, Cryst. Growth Des., 2020, 20, 7006–7011 CrossRef CAS.
- (a) K. Takahashi, T. Sato, H. Mori, H. Tajima and O. Sato, Phys. B, 2010, 405, S65–S68 CrossRef CAS; (b) D. J. Harding, D. Sertphon, P. Harding, K. S. Murray, B. Moubaraki, J. D. Cashion and H. Adams, Chem. – Eur. J., 2013, 19, 1082–1090 CrossRef CAS PubMed.
- N. Ortega-Villar, A. L. Thompson, M. C. Muñoz, V. M. Ugalde-Saldívar, A. E. Goeta, R. Moreno-Esparza and J. A. Real, Chem. – Eur. J., 2005, 11, 5721–5734 CrossRef CAS PubMed.
- (a) P.-C. Yang, K.-P. Yu, C.-T. Hsieh, J. Zou, C.-T. Fang, H.-K. Liu, C.-W. Pao, L. Deng, M.-J. Cheng and C.-Y. Lin, Chem. Sci., 2022, 13, 9637–9643 RSC; (b) O. Kahn, Molecular Magnetism, VCH, New York, 1993, p. 26 Search PubMed.
- P. B. Tsitovich, F. Gendron, A. Y. Nazarenko, B. N. Livesay, A. P. Lopez, M. P. Shores, J. Autschbach and J. R. Morrow, Inorg. Chem., 2018, 57, 8364–8374 CrossRef CAS PubMed.
- (a) J. W. Shin, A. R. Jeong, S. Jeoung, H. R. Moon, Y. Komatsumaru, S. Hayami, D. Moon and K. S. Min, Chem. Commun., 2018, 54, 4262–4265 RSC; (b) K. S. Min, K. Swierczek, A. G. DiPasquale, A. L. Rheingold, W. M. Reiff, A. M. Arif and J. S. Miller, Chem. Commun., 2008, 317–319 RSC; (c) M. A. Blagov, N. G. Spitsyna, N. S. Ovanesyan, A. S. Lobach, L. V. Zorina, S. V. Simonov, K. V. Zakharov and A. N. Vasiliev, Dalton Trans., 2023, 52, 1806–1819 RSC; (d) Z.-J. Ouyang, X.-Y. Mo, J.-Q. Ye, X.-X. Yu, S.-Y. Huang, X.-L. Liu, W.-B. Chen, S. Gao and W. Dong, Dalton Trans., 2021, 50, 5960–5967 RSC.
- D. Y. Aleshin, I. Nikovskiy, V. V. Novikov, A. V. Polezhaev, E. K. Melnikova and Y. V. Nelyubina, ACS Omega, 2021, 6, 33111–33121 CrossRef CAS PubMed.
- A. Tsukiashi, K. S. Min, H. Kitayama, H. Terasawa, S. Yoshinaga, M. Takeda, L. F. Lindoy and S. Hayami, Sci. Rep., 2018, 8, 14911 CrossRef PubMed.
- (a) M. Yu, B. S. Bouley, D. Xie and E. L. Que, Dalton Trans., 2019, 48, 9337–9341 RSC; (b) D. Maheshwaran, T. Nagendraraj, T. S. Balaji, G. Kumaresan, S. S. Kumaran and R. Mayilmurugan, Dalton Trans., 2020, 49, 14680–14689 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2002656–2002661. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04385a |
| This journal is © The Royal Society of Chemistry 2024 |