
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-induced valence tautomerism and polarization switching in mononuclear cobalt complexes with an enantiopure chiral ligand†
Wen-Huang
Xu
,
Yu-Bo
Huang
,
Wen-Wei
Zheng
,
Sheng-Qun
Su
,
Shinji
Kanegawa
 ,
Shu-Qi
Wu
* and
Osamu
Sato
*
,
Shu-Qi
Wu
* and
Osamu
Sato
*
Institute for Materials Chemistry and Engineering & IRCCS, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: sato@cm.kyushu-u.ac; jpwu.shuqi.152@m.kyushu-u.ac.jp
First published on 2nd January 2024
Abstract
Light-induced polarization switchable molecular materials have attracted attention for decades owing to their potential remote manipulation and ultrafast responsiveness. Here we report a valence tautomeric (VT) complex with an enantiopure chiral ligand. By a suitable choice of counter anions, a significant improvement in photoconversion has been demonstrated, leading to novel photo-responsive polarization switching materials.
Owing to the versatility of their switching properties, switchable molecular materials have become ideal candidates for energy storage and light-recording devices.1–4 Valence tautomeric (VT) compounds, which possess two nearly degenerate electronic states resulting from a charge transfer process between transition metal ions and redox-active ligands, exhibit desirable properties such as switchable valence, spin multiplicity, and polarization.4–17 The rich chemical tailorability in the molecular design and properties of VT compounds renders them highly attractive for the development of a wide variety of functional materials.5,13 Among a variety of external stimuli, light has received extensive research in recent years due to its fast response and remote control.18–25
In 2020, our group reported a cobalt mononuclear compound, [Co(phendiox)(rac-cth)]ClO4·0.5EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 (H2phendiox = 9,10-dihydroxyphenanthrene, rac-cth = racemic 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane), with a thermally induced polarization change during the VT process, owing to the change of its molecular dipole moment in a polar crystallographic space group. Upon temperature variations, the pyroelectric effect was observed in the same temperature range as with the VT process. However, the low light conversion efficiency (8%) and relatively fast relaxation dynamics at low temperatures precluded the direct observation of the light-induced polarization switching properties.
7 (H2phendiox = 9,10-dihydroxyphenanthrene, rac-cth = racemic 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane), with a thermally induced polarization change during the VT process, owing to the change of its molecular dipole moment in a polar crystallographic space group. Upon temperature variations, the pyroelectric effect was observed in the same temperature range as with the VT process. However, the low light conversion efficiency (8%) and relatively fast relaxation dynamics at low temperatures precluded the direct observation of the light-induced polarization switching properties.
Notably, various factors, such as counterions, solvents and crystal packing, play pivotal roles in modulating intermolecular interactions.26–30 These interactions, in turn, influence the electron transfer processes within VT compounds (Fig. 1).
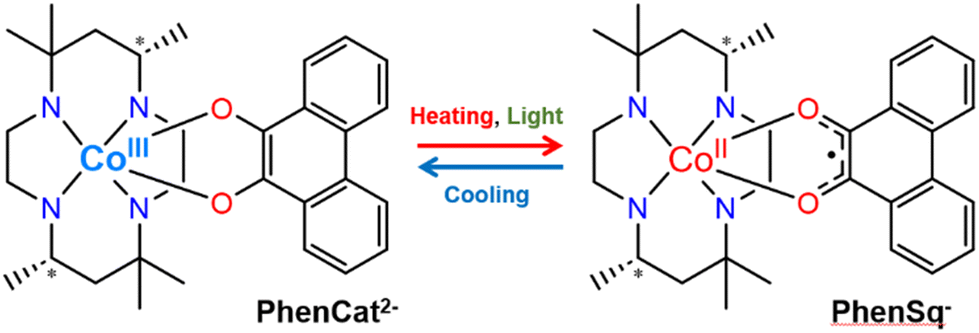 | ||
| Fig. 1 Electron transfer behaviour in [Co(phendiox)(SS-cth)]+ motifs. | ||
Extensive efforts involving the substitution of co-crystallized solvents and counterions were undertaken to enhance the photoconversion of [Co (phendiox)(rac-cth)]+. However, most of such compounds tend to crystallize in nonpolar space groups,31 and the molecular dipole moments cancel out, leading to a cancellation of molecular dipole moments and consequently, a lack of polarization change in the crystalline state.
Herein, we introduced an enantiopure chiral ligand as the strategy to break the crystallographic centrosymmetry, yielding VT compounds [Co(SS-cth)(phendiox)]PF6·0.5EtOH (1·0.5EtOH) and [Co(SS-cth)(phendiox)]AsF6·0.5EtOH (2·0.5EtOH). Notably, complex 2·0.5EtOH crystallizes in the polar P21 space group and exhibits the lowest VT transition temperature in this family of compounds. Correspondingly, it shows an improvement in the photoconversion properties; at 5 K, complex 2·0.5EtOH exhibits a conversion ratio of ca. 27% and an enhanced lifetime of electron-transferred metastable states of ca. 4000 seconds, which suggests that a significant polarization switching could be realized in these materials.
The direct current magnetic susceptibilities (χm) of 1·0.5EtOH and 2·0.5EtOH were measured in a 2000 Oe external magnetic field (Fig. 2). To prevent loss of co-crystallized ethanol molecules, the measurements of the pristine samples were performed from 300 to 5 K, followed by heating up to 400 K. The samples were then maintained at 400 K for an adequate duration of time, and another complete measurement cycle between 400 and 5 K was conducted to determine the magnetic properties of the desolvated samples.
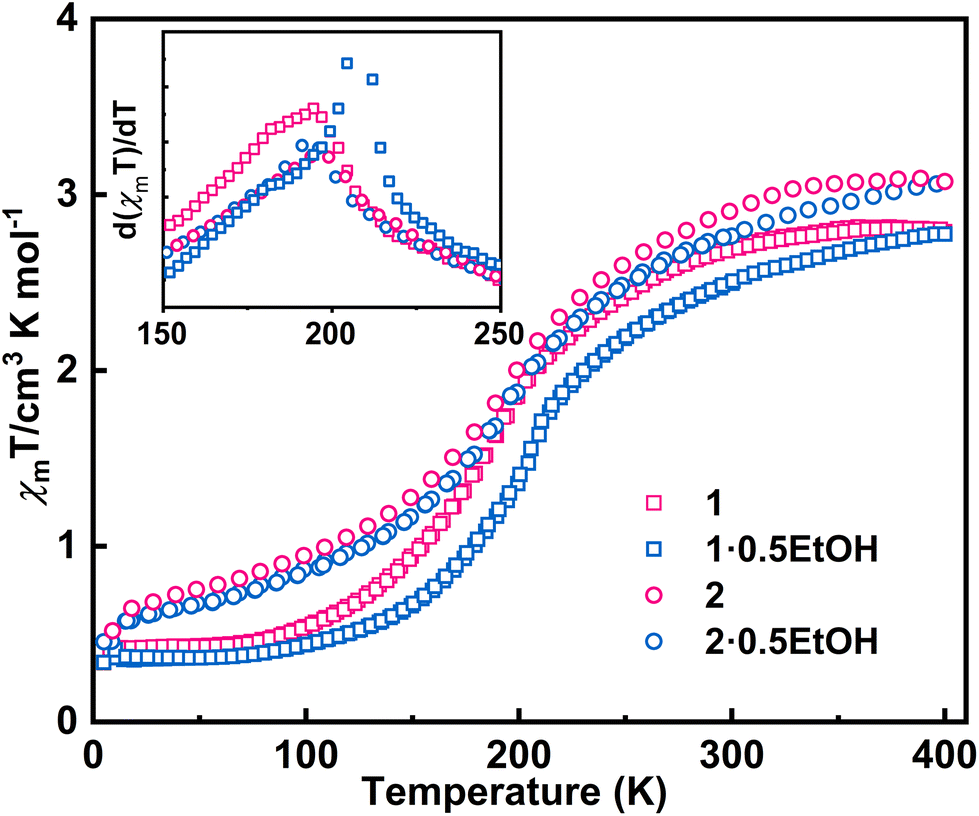 | ||
| Fig. 2 Magnetic properties of complexes 1 and 2. The temperature-dependent magnetic susceptibility was performed at a sweeping rate of 5 K min−1. The magnetic susceptibilities of compounds 1 and 2 are shown by open squares and circles, respectively. Magnetic properties before and after desolvation are marked in red and blue, respectively. Inset: First-order derivatives of the χmT product in the cooling process. | ||
These complexes exhibit similar magnetic behavior. The room-temperature χmT values of both complexes were slightly larger than the spin-only values of an uncoupled hs-CoII-semiquinonato (PhenSq−) pair (1: 2.75 cm3 K mol−1, 2: 3.0 cm3 K mol−1), indicating a predominant presence of this electronic configuration.7 Below 200 K, the χmT values dropped drastically to 0.5–0.7 cm3 K mol−1, corresponding to an increase in the population in the diamagnetic ls-CoIII-catecholato (PhenCat2−) ground state.32 Notably, the T1/2 (defined as the peak temperature of the first order derivative of χmT–T plots) of 2 (ca. 190 K) is slightly lower than that of 1 (ca. 200 K), suggesting the enhanced stability of high-spin species in the lattice of the hexafluoroarsenate salt, potentially attributable to the negative chemical pressure exerted by the AsF6− anion owing to its larger size compared to PF6−. During heating to 400 K, neither 1 nor 2 exhibit significant thermal hysteresis in χmT values. Upon solvent loss (Fig. S1†),33,34 the T1/2 for 1 and 2 decrease by 15 and 5 K, respectively. The solvent effect observed here closely resembles that of the previously reported complex, [Co(rac-cth)(phendiox)]ClO4·0.5EtOH. However, given that 1·0.5EtOH and 2·0.5EtOH exhibit significantly lower transition temperatures, an enhancement in the stability of the light-induced metastable high-spin state is anticipated, possibly leading to a better photoconversion performance.35
The X-ray single-crystal structures of 2·0.5EtOH at 78 and 300 K were obtained at the Spring-8 synchrotron facility. Crystallographic parameters and refinement parameters are summarized in ESI tables.† Different from its racemic counterpart in the C2/c space group, complex 2·0.5EtOH crystallized in the polar P21 space group.31,36 The use of chiral ligands plays a key role in the loss of the inversion symmetry in the crystal. In the molecular structure (Fig. 3), the Co center is octahedrally coordinated by four N atoms from the cth ligand and two O atoms from the phendiox ligand. At low temperature, two crystallographically independent molecular motifs could be found in the unit cell, distinguishable by the Co–O bond lengths; one exhibits lengths of 1.898 and 1.907 Å (α site), characteristic of the CoIII ion, whereas the other shows lengths of 2.017 and 1.999 Å (β site).
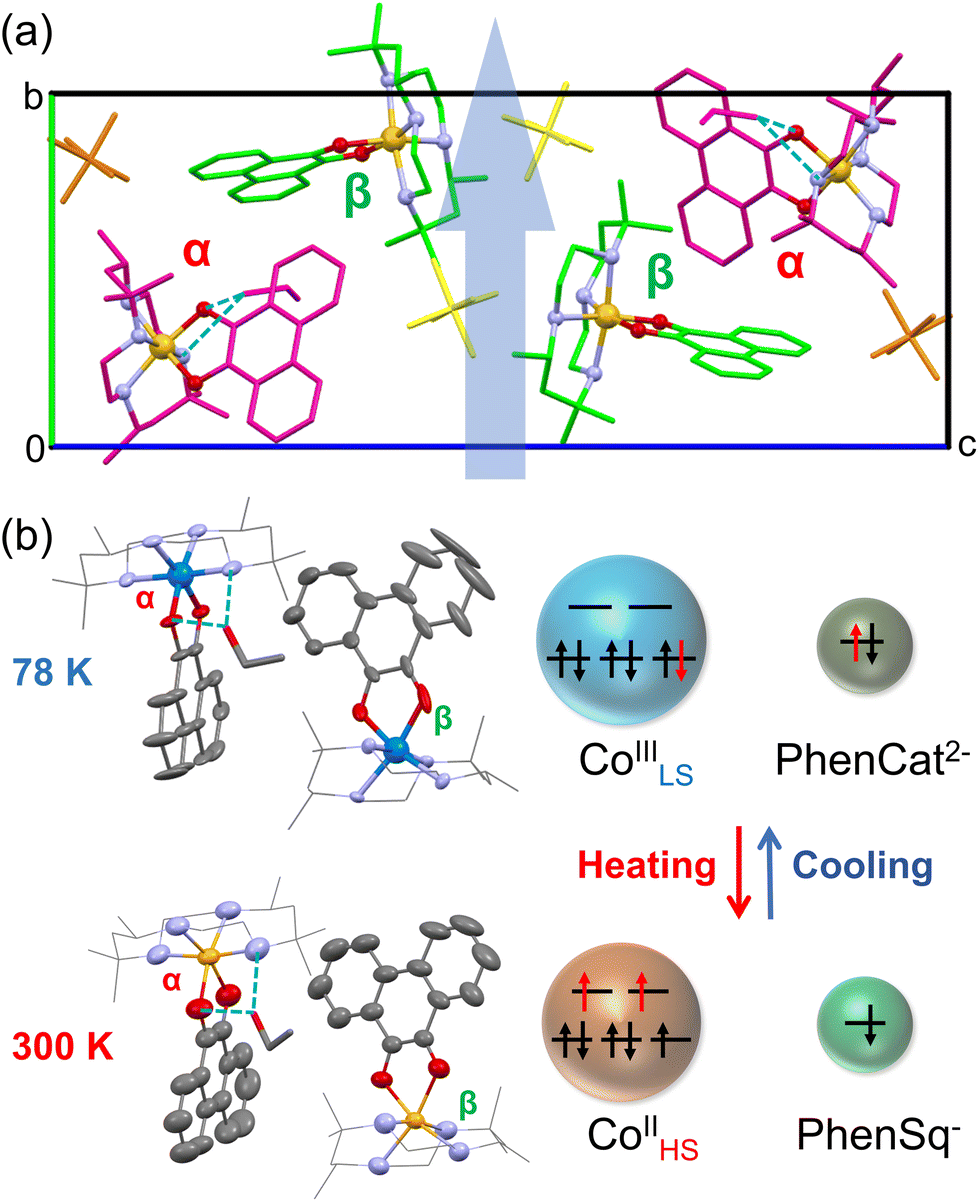 | ||
| Fig. 3 Crystal structure and packing of 2·0.5EtOH. (a) Molecular packing viewed along the a-axis. The Co is coordinated by four nitrogen atoms (in lilac) and two oxygen atoms (in red). Inequivalent motifs are labelled in red (α site) and green (β site), respectively. The polarization direction is indicated by a blue arrow. (b) Structure of the asymmetric unit in 2·0.5EtOH in the low temperature phase (upper) and the high temperature phase (lower). Counter ions and hydrogen atoms are omitted for clarity. | ||
The relatively longer Co–O bond lengths revealed an incomplete VT transition at β sites, which is consistent with the non-vanishing χmT value at this temperature. The difference in the completeness of the VT transition might stem from the distinct hydrogen-bonded structures at each site. Molecules at α sites show stronger hydrogen bond interactions, with an N–H⋯O distance of 3.019 Å and an O–H⋯O distance of 2.885 Å. In contrast, no significant hydrogen bonds form between ethanol and molecules at the β site. The different chemical environment leads to a different spin state at a low temperature. At 300 K, the Co–O bond lengths at both sites increased to the typical values of the CoII ion, ranging from 2.050 Å to 2.092 Å. Such changes in bond length are consistent with the usual VT behavior in the [Co(cth)(phendiox)]+ family.
Temperature-dependent infrared (IR) and UV-visible absorption spectra also confirmed the presence of electron transfer between metal and ligand sites of complex 2 (Fig. 4); the absorption peaks at approximately 1354 and 1378 cm−1, which decreased upon heating, correspond to the in-phase C–O vibration and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching in phenCat2−. The absorption feature at around 1600 cm−1 is identified as the C–C–C stretching mode, typical of the [CoIII(cth)(phencAT)]+ family.7 Meanwhile, the absorption peak at approximately 1458 cm−1 is ascribed to the C–O and CC stretching modes in phenSq−. Similarly, the characteristic π–π* transition band of phenCat2− centered at 520 nm increases with heating, confirming the electron transfer nature of the temperature-dependent electronic structures.
C stretching in phenCat2−. The absorption feature at around 1600 cm−1 is identified as the C–C–C stretching mode, typical of the [CoIII(cth)(phencAT)]+ family.7 Meanwhile, the absorption peak at approximately 1458 cm−1 is ascribed to the C–O and CC stretching modes in phenSq−. Similarly, the characteristic π–π* transition band of phenCat2− centered at 520 nm increases with heating, confirming the electron transfer nature of the temperature-dependent electronic structures.
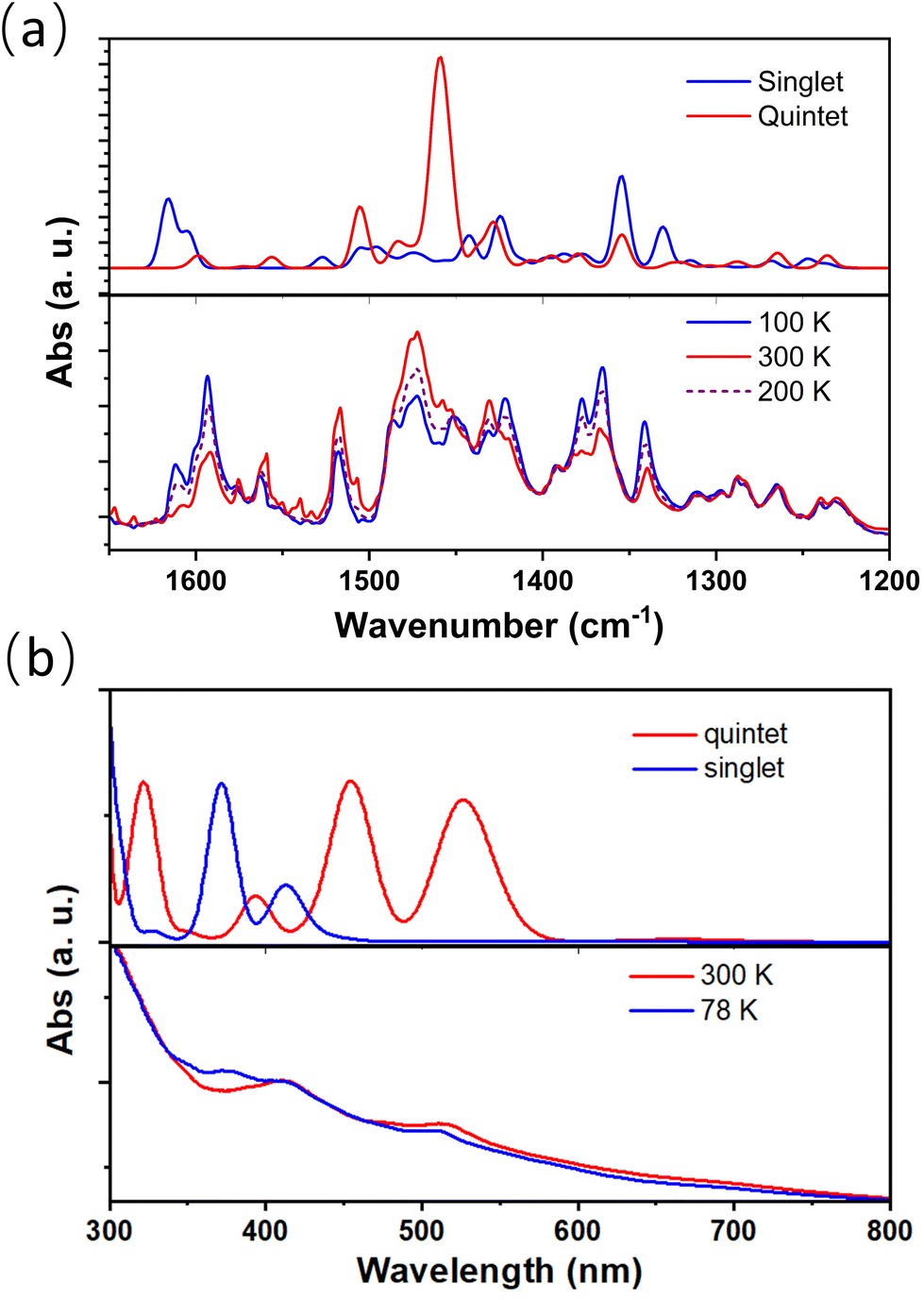 | ||
| Fig. 4 Experimental and calculated temperature-dependent IR and UV spectra of complex 2·0.5EtOH. (a) Calculated IR absorption for singlet and quintet states (upper), and temperature-dependent IR spectra recorded between 100 and 300 K (lower). (b) Calculated UV absorption spectra for singlet and quintet states (upper), and temperature-dependent UV spectra recorded between 78 and 300 K (lower). | ||
Based on the density functional theory (DFT) calculations reported previously,7 the [Co(cth)(phendiox)]+ motif possesses a permanent electric dipole moment of 13.26 Debye for a ls-CoIII-catecholato state and 7.47 Debye for a hs-CoII-semiquinonato state corresponding to the LT and HT phases, respectively. However, the change in net molecular dipole moments in a unit cell is largely cancelled owing to the approximately perpendicular alignment of the [Co(cth)(phendiox)]+ motif with respect to the polar b-axis. The dipole moment direction could be estimated from the O–Co–O bisection line, which forms angles of 100.15° and 89.75° with the b-axis for α and β molecules, respectively, at 300 K. At 78 K, these angles change slightly to 103.09° and 90.00°. Projecting the dipole moments to the polar axis, the change induced by the VT process is approximately 3.64 Debye per unit cell, corresponding to a polarization change of 353 nC cm−2.
Photomagnetic susceptibility measurements were performed using a 532 nm wavelength laser. An increase in χmT from 0.4 to 1.2 cm3 mol−1 K at 5 K indicated a photoinduced charge transfer process from ls-Co(III) to hs-Co(II). A gradual decrease in the χmT value upon increasing the temperature was then observed, followed by a collapse back to the normal (in the absence of photoirradiation) value at 53 K (Fig. 5). As the temperature increased, the curve coincided with the χmT vs. T plot of the bulk sample. The percentage of photoconversion was estimated to be nearly 27% using the reported high-temperature χmT value (approximately 3.0 cm3 mol−1 K) of the same family of compounds as the upper limit.
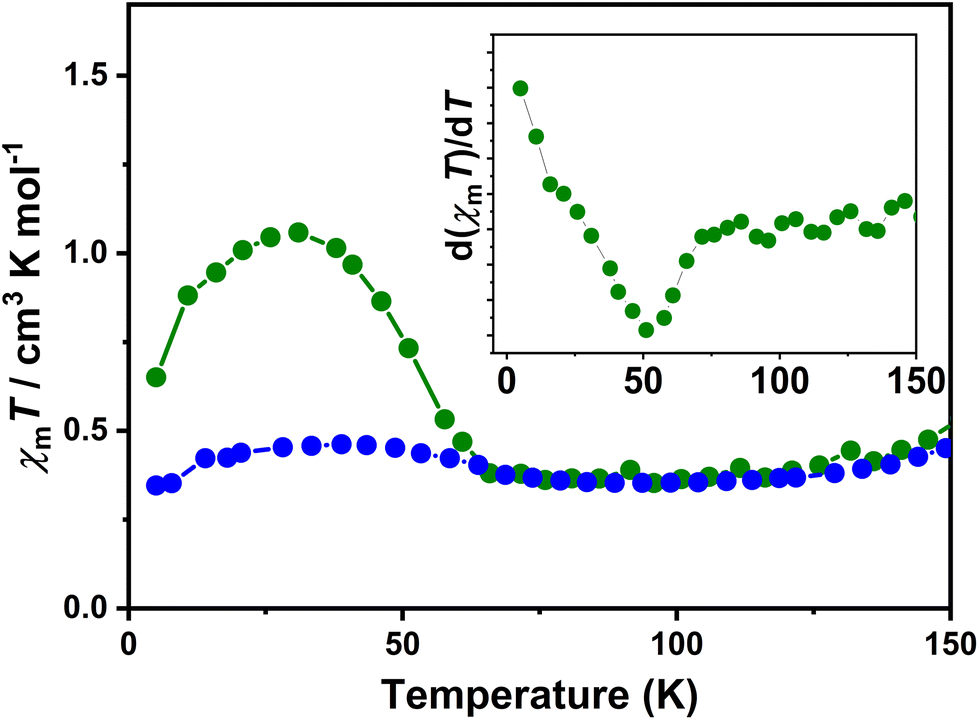 | ||
| Fig. 5 Temperature-dependent magnetic susceptibilities of complex 2·0.5EtOH after irradiation (green). Light-induced magnetization is performed by 532 nm irradiation. Magnetization of the bulk sample is shown in blue circles. Inset: First-order derivatives of the light induced χmT product. | ||
To further investigate the relaxation dynamics from the photoinduced metastable state, the time dependence of the magnetic susceptibility after irradiation was measured at different temperatures. The fraction of the metastable state as a function of time can be defined as
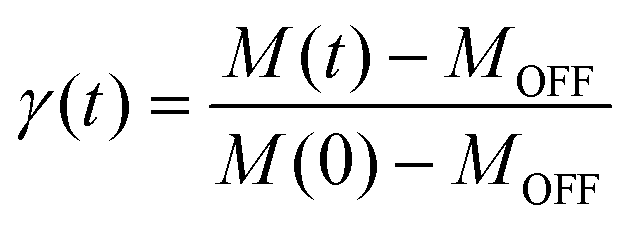 | (1) |
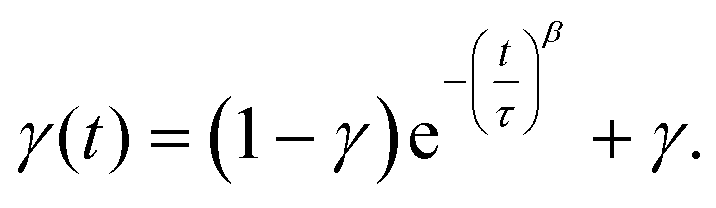 | (2) |
The parameter γ indicates the phenomenological HS fraction that persists in the infinite time limit, τ denotes the characteristic relaxation time and β indicates its distribution. Remarkably, the relaxation time for complex 2 reached ca. 4000 s at 5 K, which is over 3 times longer than that observed for [Co(rac-cth)(phendiox)]ClO4 and [Co(SS-cth)(phendiox)]PF6. This suggests the enhanced stability of the metastable state, which correlates with the substantial improvement of the photoconversion performance in this system (Fig. 6). Such a trend aligns with the established understanding that lower thermally-induced transition temperatures are indicative of slower relaxation times for the light-induced excited spin states within spin crossover systems.37 Furthermore, it is important to recognize that complex 2, with its polar crystal structure, allows for achieving a persistent polarization change through light irradiation by trapping the electron-transferred state. By assuming the Gaussian broadening of the pyroelectric coefficient during the relaxation process, we estimate its maximal value as 3.19 nC cm−2 K−1, which lies within the detectable range of a standard Keithley 6517B electrometer. However, the practical realization of such measurements poses significant challenges due to the extremely thin thickness of the crystals of complex 2, making experimental observations difficult to carry out.
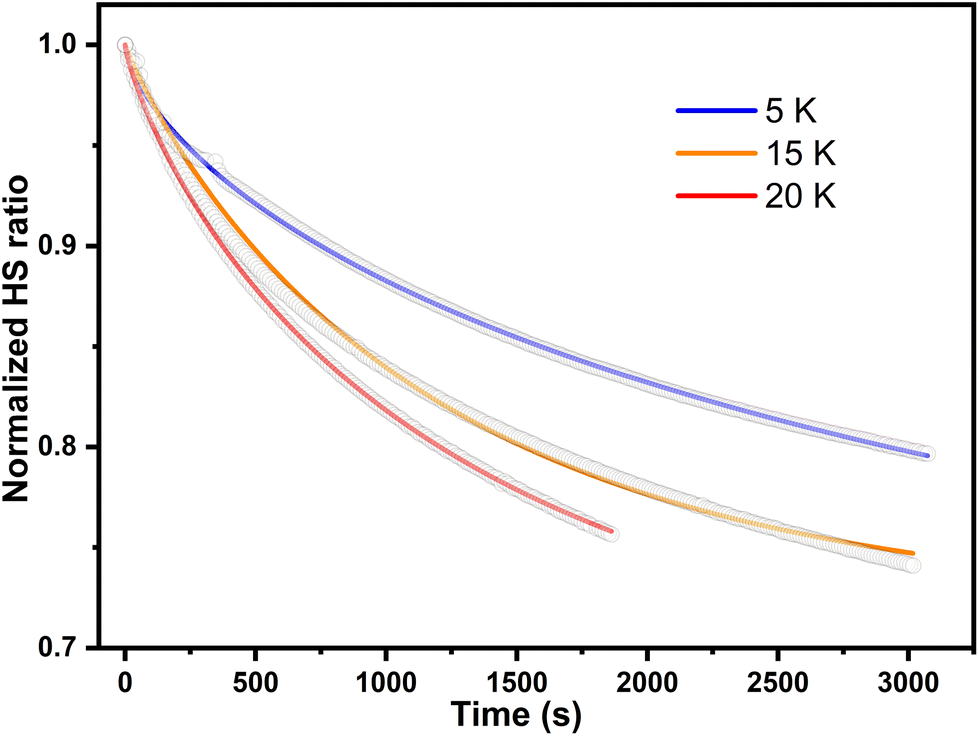 | ||
| Fig. 6 The relaxation plots of photoinduced magnetization of complex 2·0.5EtOH at 5, 15 and 20 K, which are shown in blue, orange, and red respectively. The wavenumber of excitation light is 532 nm. | ||
Conclusions
In this study, we introduced an enantiopure cth ligand to obtain two isostructural VT compounds crystallized in the polar P21 space group. By using the hexafluoroarsenate anion in complex 2, a lowering of VT transition temperatures by ca. 10 K has been observed, suggesting the improved stability of the hs-Co(II)-semiquinonato species in this lattice. Correspondingly, the light conversion efficiency and the lifetime of the metastable state were improved to approximately 27% and 4000 s at 5 K, respectively. As the electric dipole moments change substantially during the electron transfer process, light-induced polarization switching should be expected for this family of compounds. These results suggest the feasibility of achieving photoinduced persistent polarization by these strategies, which would lead to a promising outcome for applications in molecular electronics and optically rewritable information storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Wen-Huang Xu was supported by JST SPRING, Grant Number JPMJSP2136. Yu-Bo Huang thanks the China Scholarship Council for support. This work was supported by JSPS KAKENHI (21K05085, 22K14694, 20H00385 and 21K05086). This work was performed under the Cooperative Research Program of “Network Joint Research Center for Materials and Devices”. This work was supported by the MEXT Project of “Integrated Research Consortium on Chemical Sciences”. Shu-Qi Wu and Sheng-Qun Su are grateful for support from the Iketani Science and Technology Foundation (0341056A and 0351039A).References
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357–366 CrossRef CAS PubMed.
- M. D. Manrique-Juárez, S. Rat, L. Salmon, G. Molnár, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Coord. Chem. Rev., 2016, 308, 395–408 CrossRef.
- Y. Sekine, R. Akiyoshi and S. Hayami, Coord. Chem. Rev., 2022, 469, 214663 CrossRef CAS.
- G. K. Gransbury and C. Boskovic, Encyclopedia of Inorganic and Bioinorganic Chemistry, pp. 1–24. DOI:10.1002/9781119951438.eibc2785.
- S.-Q. Su, S.-Q. Wu, S. Kanegawa, K. Yamamoto and O. Sato, Chem. Sci., 2023, 14, 10631–10643 RSC.
- S.-Q. Wu, M. Liu, K. Gao, S. Kanegawa, Y. Horie, G. Aoyama, H. Okajima, A. Sakamoto, M. L. Baker, M. S. Huzan, P. Bencok, T. Abe, Y. Shiota, K. Yoshizawa, W. Xu, H.-Z. Kou and O. Sato, Nat. Commun., 2020, 11, 1992 CrossRef CAS PubMed.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704–705 CrossRef CAS PubMed.
- H. W. Liu, K. Matsuda, Z. Z. Gu, K. Takahashi, A. L. Cui, R. Nakajima, A. Fujishima and O. Sato, Phys. Rev. Lett., 2003, 90, 167403 CrossRef CAS PubMed.
- S. Kanegawa, Y. Shiota, S. Kang, K. Takahashi, H. Okajima, A. Sakamoto, T. Iwata, H. Kandori, K. Yoshizawa and O. Sato, J. Am. Chem. Soc., 2016, 138, 14170–14173 CrossRef CAS PubMed.
- F. Cheng, S. Wu, W. Zheng, S. Su, T. Nakanishi, W. Xu, P. Sadhukhan, H. Sejima, S. Ikenaga, K. Yamamoto, K. Gao, S. Kanegawa and O. Sato, Chem. – Eur. J., 2022, 28, e202202161 CrossRef CAS PubMed.
- J. Tao, H. Maruyama and O. Sato, J. Am. Chem. Soc., 2006, 128, 1790–1791 CrossRef CAS PubMed.
- A. Dei, D. Gatteschi, C. Sangregorio and L. Sorace, Acc. Chem. Res., 2004, 37, 827–835 CrossRef CAS PubMed.
- O. Drath and C. Boskovic, Coord. Chem. Rev., 2018, 375, 256–266 CrossRef CAS.
- J. Ferrando-Soria, J. Vallejo, M. Castellano, J. Martínez-Lillo, E. Pardo, J. Cano, I. Castro, F. Lloret, R. Ruiz-Garcia and M. Julve, Coord. Chem. Rev., 2017, 339, 17–103 CrossRef CAS.
- G. K. Gransbury, B. N. Livesay, J. T. Janetzki, M. A. Hay, R. W. Gable, M. P. Shores, A. Starikova and C. Boskovic, J. Am. Chem. Soc., 2020, 142, 10692–10704 CrossRef CAS PubMed.
- T. Tezgerevska, K. G. Alley and C. Boskovic, Coord. Chem. Rev., 2014, 268, 23–40 CrossRef CAS.
- P. Sadhukhan, S.-Q. Wu, S. Kanegawa, S.-Q. Su, X. Zhang, T. Nakanishi, J. I. Long, K. Gao, R. Shimada, H. Okajima, A. Sakamoto, J. G. Chiappella, M. S. Huzan, T. Kroll, D. Sokaras, M. L. Baker and O. Sato, Nat. Commun., 2023, 14, 3394 CrossRef CAS PubMed.
- S.-Q. Su, S.-Q. Wu, Y.-B. Huang, W.-H. Xu, K.-G. Gao, A. Okazawa, H. Okajima, A. Sakamoto, S. Kanegawa and O. Sato, Angew. Chem., Int. Ed., 2022, 61, e202208771 CrossRef CAS PubMed.
- A. Tashiro, S. Kanegawa, O. Sato and Y. Teki, Polyhedron, 2013, 66, 167–170 CrossRef CAS.
- T. J. Penfold, J. O. Johansson and J. Eng, Coord. Chem. Rev., 2023, 494, 215346 CrossRef CAS.
- A. Witt, F. W. Heinemann and M. M. Khusniyarov, Chem. Sci., 2015, 6, 4599–4609 RSC.
- H. Kuramochi, G. Aoyama, H. Okajima, A. Sakamoto, S. Kanegawa, O. Sato, S. Takeuchi and T. Tahara, Angew. Chem., Int. Ed., 2020, 59, 15865–15869 CrossRef CAS PubMed.
- R. Ash, K. Zhang and J. Vura-Weis, J. Chem. Phys., 2019, 151, 104201 CrossRef PubMed.
- O. Sato, A. Cui, R. Matsuda, J. Tao and S. Hayami, Acc. Chem. Res., 2007, 40, 361–369 CrossRef CAS PubMed.
- E. Evangelio, C. Rodriguez-Blanco, Y. Coppel, D. N. Hendrickson, J. P. Sutter, J. Campo and D. Ruiz-Molina, Solid State Sci., 2009, 11, 793–800 CrossRef CAS.
- Y. Mulyana, G. Poneti, B. Moubaraki, K. S. Murray, B. F. Abrahams, L. Sorace and C. Boskovic, Dalton Trans., 2010, 39, 4757–4767 RSC.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- A. Dei, A. Feis, G. Poneti and L. Sorace, Inorg. Chim. Acta, 2008, 361, 3842–3846 CrossRef CAS.
- G.-L. Li, S. Kanegawa, Z.-S. Yao, S.-Q. Su, S.-Q. Wu, Y.-G. Huang, S. Kang and O. Sato, Chem. – Eur. J., 2016, 22, 17130–17135 CrossRef CAS PubMed.
- A. Bencini, A. Caneschi, C. Carbonera, A. Dei, D. Gatteschi, R. Righini, C. Sangregorio and J. V. Slageren, J. Mol. Struct., 2003, 656, 141–154 CrossRef CAS.
- G. Poneti, M. Mannini, B. Cortigiani, L. Poggini, L. Sorace, E. Otero, P. Sainctavit, R. Sessoli and A. Dei, Inorg. Chem., 2013, 52, 11798–11805 CrossRef CAS PubMed.
- V. L. Nadurata and C. Boskovic, Inorg. Chem. Front., 2021, 8, 1840–1864 RSC.
- V. L. Nadurata, M. A. Hay, J. T. Janetzki, G. K. Gransbury and C. Boskovic, Dalton Trans., 2021, 50, 16631–16646 RSC.
- R. D. Schmidt, D. A. Shultz and J. D. Martin, Inorg. Chem., 2010, 49, 3162–3168 CrossRef CAS PubMed.
- A. Caneschi, A. Dei, F. Fabrizi de Biani, P. Gütlich, V. Ksenofontov, G. Levchenko, A. Hoefer and F. Renz, Chem. – Eur. J., 2001, 7, 3926–3930 CrossRef CAS PubMed.
- V. A. Money, C. Carbonera, J. Elhaïk, M. A. Halcrow, J. A. K. Howard and J.-F. Létard, Chem. – Eur. J., 2007, 13, 5503–5514 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2309543 and 2309544. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03915c |
| This journal is © The Royal Society of Chemistry 2024 |