
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeted synthesis of gold nanorods and characterization of their tailored surface properties using optical and X-ray spectroscopy†
David G.
Schauer
 ab,
Jona
Bredehoeft
a,
Umar
Yunusa
b,
Ajith
Pattammattel
c,
Hans Jakob
Wörner
a and
Emily A.
Sprague-Klein
*b
ab,
Jona
Bredehoeft
a,
Umar
Yunusa
b,
Ajith
Pattammattel
c,
Hans Jakob
Wörner
a and
Emily A.
Sprague-Klein
*b
aETH Zurich, Dept. of Chemistry and Applied Biosciences, Laboratory of Physical Chemistry, Vladimir-Prelog-Weg 2 (HCI E 241), 8093 Zürich, Switzerland
bDepartment of Chemistry, Brown University, Providence, Rhode Island 02912, USA. E-mail: emily_sprague-klein@brown.edu
cNational Synchrotron Light Source II, Brookhaven National Laboratory, Upton, NY 11973, USA
First published on 24th September 2024
Abstract
In recent years, nanophotonics have had a transformative impact on harnessing energy, directing chemical reactions, and enabling novel molecular dynamics for thermodynamically intensive applications. Plasmonic nanoparticles have emerged as a tool for confining light on nanometer-length scales where regions of intense electromagnetic fields can be precisely tuned for controlled surface chemistry. We demonstrate a precision pH-driven synthesis of gold nanorods with optical resonance properties widely tunable across the near-infrared spectrum. Through controlled electrostatic interactions, we can perform selective adsorbate molecule attachment and monitor the surface transitions through spectroscopic techniques that include ground-state absorption spectrophotometry, two-dimensional X-ray absorption near-edge spectroscopy, Fourier-transform infrared spectroscopy, and surface-enhanced Raman spectroscopy. We elucidate the electronic, structural, and chemical factors that contribute to plasmon-molecule dynamics at the nanoscale with broad implications for the fields of energy, photonics, and bio-inspired materials.
1 Introduction
Gold nanoparticles (≤100 nm for one axis) have found a wide range of applications in diverse fields such as catalysis, sensing, electronics, and biomedicine. Spherical gold nanoparticles have traditionally been the most widely employed in various application domains. However, their plasmonic response is only limited to the visible region. Consequently, anisotropic nanostructures, such as gold nanorods (GNRs), have attracted a great deal of attention in recent years.1 This is because their plasmonic properties can be easily tuned by adjusting the aspect ratio of the structures. This tunability and the associated possibility of harnessing anisotropic plasmonic resonances have made GNRs indispensable for a variety of applications.2 For example, GNRs are utilized in biomedicine as mediators of photothermal treatment to target and destroy cancer cells, in radiation therapy, (cancer) cell imaging, or as a less toxic and more efficient drug delivery alternative (depending on the surfactant).3–11 They can also be used in displays, as encoders for security systems, as nanoantennas, or as resonant light-driven artificial biomotors.2,12–15Because of the structural anisotropy, GNRs possess both translational and rotational degrees of freedom resulting in a longitudinal (length) as well as a transverse (diameter) surface plasmon wavelength (LSPW, TSPW).11,16,17 Depending on the length-to-diameter aspect ratio, the GNRs exhibit unique absorption spectra indicated by a distinct surface plasmon resonance. Plasmons describe collective oscillations of the quasi-free electrons excited by the electric field of the incident light. Looking at GNRs with a size much smaller than the wavelength of the incident light, the plasmonic oscillations encompass the entire surface of the GNRs, then referred to as localized surface plasmons resonance (LSPR), as shown in Fig. 1. These charge oscillations induce a dipole negatively interfering with the incident electric field, leading toward the absorption of either the LSPW or TSPW in the Vis/NIR range. The LSPW can cover a broad spectrum in the Vis/NIR range (in this work, between ≈ 700–1100 nm). The shorter the length of the GNRs, the more blueshifted their LSPW.2,18–25
 | ||
| Fig. 1 Schematic representation of the collective electronic oscillations of (a) GNRs possessing both a transverse and longitudinal surface plasmon wavelength and (b) GNSs having a totally symmetric localized surface plasmon resonance. | ||
In order to control the length of the GNRs – and hence the LSPW – the pH, growth time, and volume of the solutions are among the critical parameters to be considered during the synthesis process. At present, there is a variety of methods for synthesizing GNRs using different techniques and chemicals. The method of seed-mediated growth can be applied to control the LSPW.26–31 In this method, adjusting the pH, growth and reaction time, chemicals used, and the upscaling factor have a significant impact on the shape, the geometry, and the size of the GNRs.22,32–35 Throughout this work, the seed-mediated growth method from Leng et al.36 was further developed by including a precise adjustment of the synthesis parameters mentioned above. This can lead to an unprecedented level of detail in fine-tuning the morphology of the GNRs and their optical properties.
Herein, we demonstrate a nanofabrication approach that allows us to reliably and reproducibly tune the LSPR to the photothermal region for controlled heating applications that are not hot electron-activated. This approach confers three main advantages: surface selectivity, tunability of the optical and electronic properties, and control of heat dissipation. An application where this approach is especially advantageous is when considering molecular analytes commonly employed in photodynamic therapy applications. Notably, a common therapeutic dye, methylene blue (MB), degrades under ultraviolet light and from hot electron excitation in solid-state materials. Therefore, utilizing LSPR scaffolding to enhance molecular dynamics and absorptive properties is beneficial.37 In this manuscript, we evaluate site-specific coordination of a target analyte molecule for bio-inspired complex materials and demonstrate the utility of a pH-mediated synthetic approach to create nanostructures with a high degree of uniformity, shape, and size control. The tunability of surfactant chemistry also enables preferential binding of blue dyes to the nanoparticle surface, which has important applications in phonon (heat) dissipation into condensed phase media.38–47
2. Experimental methods and characterization
2.1. Targeted growth and fine-tuning of noble gold nanostructures
Two batches of solutions were prepared for each experimental run to carry out the targeted synthesis: several growth solutions and one seed solution. About 100 mL stock solutions of the used chemicals in MilliQ water (18.2 MΩ cm at 25 °C) were prepared in order to ensure reproducibility across several experimental runs. The growth solutions were prepared using cetyltrimethylammonium bromide (CTAB, 0.20 M, 5.0 mL), hydrogen tetrachloroaurate(III) trihydrate (HAuCl4·3H2O, 5.0 × 10−3 M, 1.0 mL), hydroxylamine hydrochloride (NH2OH·HCl, 0.1 M, 1.0 mL), silver nitrate (AgNO3, 0.01 M, 0.10 mL) and sodium hydroxide (NaOH, 0.2 M, for pH increase), which were stored under constant stirring (300 rpm) in a 28 °C water bath.For obtaining the GNRs, the pH in the growth solution was set between ≈5.0–8.0 since a higher pH led to the formation of gold nanospheres (GNSs). To initiate the growth process, 15 L of the seed solution, consisting of CTAB (0.20 M, 5 mL), HAuCl4·3H2O (5.0 × 10−4 M, 5.0 mL) and freshly prepared (<30 min) ice-cold sodium borohydride (NaBH4, 0.01 M, 0.60 mL), were added into the growth solutions after precise adjustment of the pH, using a pH-meter (Metrohm 744 pH with a Hamilton SlimTriode electrode, Orion Star A211). After seeding, the growth solutions were kept in screwable amber glass vials (20 mL) in a 28 °C water bath throughout the whole growth process. A scheme of the synthesis process is shown in Fig. S1 in the ESI.† These CTAB-GNRs are stable for months in the water bath (excess CTAB will crystallize at room temperature). After 24–48 h, the growth process has ended naturally or can be terminated by centrifugation (5600 rpm, 15 mL conical falcon tubes, 5–10 min). A more detailed synthesis protocol can be found in the extended experimental section of the ESI.†
In order to investigate the different plasmon resonance responses and electrostatic interactions between the GNRs, the surfactant stabilizing the growth process and the attached dye molecules can be exchanged, as illustrated in Fig. 2a. Briefly, the CTAB-GNRs (OD ≈ 5) were centrifuged in 1.5 mL conical falcon tubes (6000 rpm, 30 min), and the supernatant was carefully removed, and the same volume of a 0.7 wt% poly(sodium 4-styrenesulfonate) (Na-PSS) dissolved in MilliQ-water was added. The suspension was put in a sonicator bath to redisperse the GNRs. Subsequently, the same process was repeated a second time using Na-PSS and another 1–2 times (depending on the concentration of the GNRs) using 0.7 wt% of sodium citrate (Na-CIT) dissolved in MilliQ-water. Both the PSS- and CIT-GNRs are stable at room temperature for several weeks and do not have to be stored in a water bath compared to CTAB-GNRs. Fig. 2b shows the resonance shift and the broadening of the absorption spectra using different surfactant-capped GNRs, and Fig. 2c a scanning and transmission electron microscope (SEM, TEM) image verifying the homogeneity and precise size distribution of the GNRs.48
 | ||
| Fig. 2 (a) A scheme of the surfactant-stabilized growth process resulting in CTAB-GNRs with a positive surface charge. Exchanging the surfactant using Na-PSS leads to a delocalized negative surface charge, which allows the final attachment of Na-CIT to the GNRs' surface, resulting in a negative surface charge. (b) The different absorption (resonance) spectra of CTAB-, PSS-, and CIT-GNRs. The inset shows the transmission/color of CTAB-GNRs (A), CTAB-GNRs + IC (B, C; different concentrations), and CIT-GNRs (D). (c) SEM and TEM image of CTAB-GNRs. | ||
Due to the positive/negative surface charge of the surfactant-capped GNRs, it is possible to attach different dye molecules to the GNRs' surface electrostatically. The following dyes were selected for this study: indigo carmine (IC), MB, and leucomethylene blue (LMB). IC (negative) is attached to the CTAB-GNRs, whereas LMB (neutral) and MB (positive) are attached to the CIT-GNRs. The dyes were prepared in 100 M stock solutions in MilliQ-water. For the CTAB-GNRs (OD ≈5), 1 mL of the solution was centrifuged (6000 rpm, 20 min), and ≥ 90% of the supernatant was carefully removed. The subsequent addition of the same volume of IC from its stock solution led to a color change from red/orange to purple/violet, as seen in Fig. 2b. For the CIT-GNRs (1 mL, OD ≈1), 50 L of LMB were added, and after ≈10 min 100 L of 10 wt% of polyvinylpyrrolidone (PVP) in MilliQ-water were added to prevent precipitation through agglomeration of the CIT-GNRs. The exact process was repeated for MB. The absorption spectra are displayed in Fig. 5. All these solutions are stable at room temperature for several weeks and were used for the subsequent characterizations.
2.2. UV/Vis/NIR absorption spectrophotometry
A Cary 60 spectrophotometer (Agilent Technologies, Santa Clara, CA) in dual-beam mode was used to scan the absorbance properties of the bare and functionalized gold nanostructures. A scan range of 200–1100 nm was used with a step size of 0.5 nm, an averaging time of 0.1 s, and a scan rate of 300 nm. The spectra were background-subtracted using solvent blanks performed in a 2 mm path-length quartz cuvette (Spectrocell).2.3. Nano-XANES imaging and spectroscopy
A focused high energy X-ray beam (<40 nm resolution) was used to image and obtain spectroscopic information on two differing sizes of CTAB-GNRs at the Au L3-edge (11918.7 eV)49 performed at beamline 3-ID Hard X-ray nanoprobe (HXN) of the National Synchrotron Light Source 2 (NSLS-II).50,51 The high photon flux (>108 photons s−1) allows for the detection of subtle changes in electronic information correlated to the nanoscale structures. An aliquot of concentrated sample (OD ≈ 10) was drop-casted onto silicon wafers and allowed to dry under ambient conditions before loading into a sealed sample chamber with a beryllium window. Once the sample was positioned at a 45° angle for two-dimensional fluorescence imaging, the chamber was pumped down to a pre-vacuum state and held under a helium atmosphere. Alignment and measurements were performed using Fresnel X-ray zone plates and a Si(111) double-crystal monochromator with a spectral resolution of 10−4 (dE/E). A scanning range of 2.5 × 2.5 m2 was used with a total of 64 energy points across an energy range of 11.880–12.000 keV (Fig. 3). The gold foil spectrum was measured in transmission mode with a large beam upstream to the microscope with a beam size of ≈ 2 × 0.5 mm2. The raw data and the corresponding first derivatives can be found in Fig. S5 in the ESI.† An overview of the signal-to-noise ratio and resolution, depending on the chosen size of the ROI on the two-dimensional X-ray fluorescence (2D-XRF) maps before processing the two-dimensional X-ray absorption near-edge structure (2D-XANES) spectra is displayed in Fig. S6 in the ESI.† Fig. S7 in the ESI† shows eleven accumulated spectra of the highest intensity regions (high concentration of GNRs) with an ROI of 5 × 5 px2, and the difference in the 2D-XANES spectra processed from these regions compared to the background (dark spots). Data was acquired using Bluesky software, and workup was performed using XMIDAS, xraylarch, Athena (Demeter), and MatLab.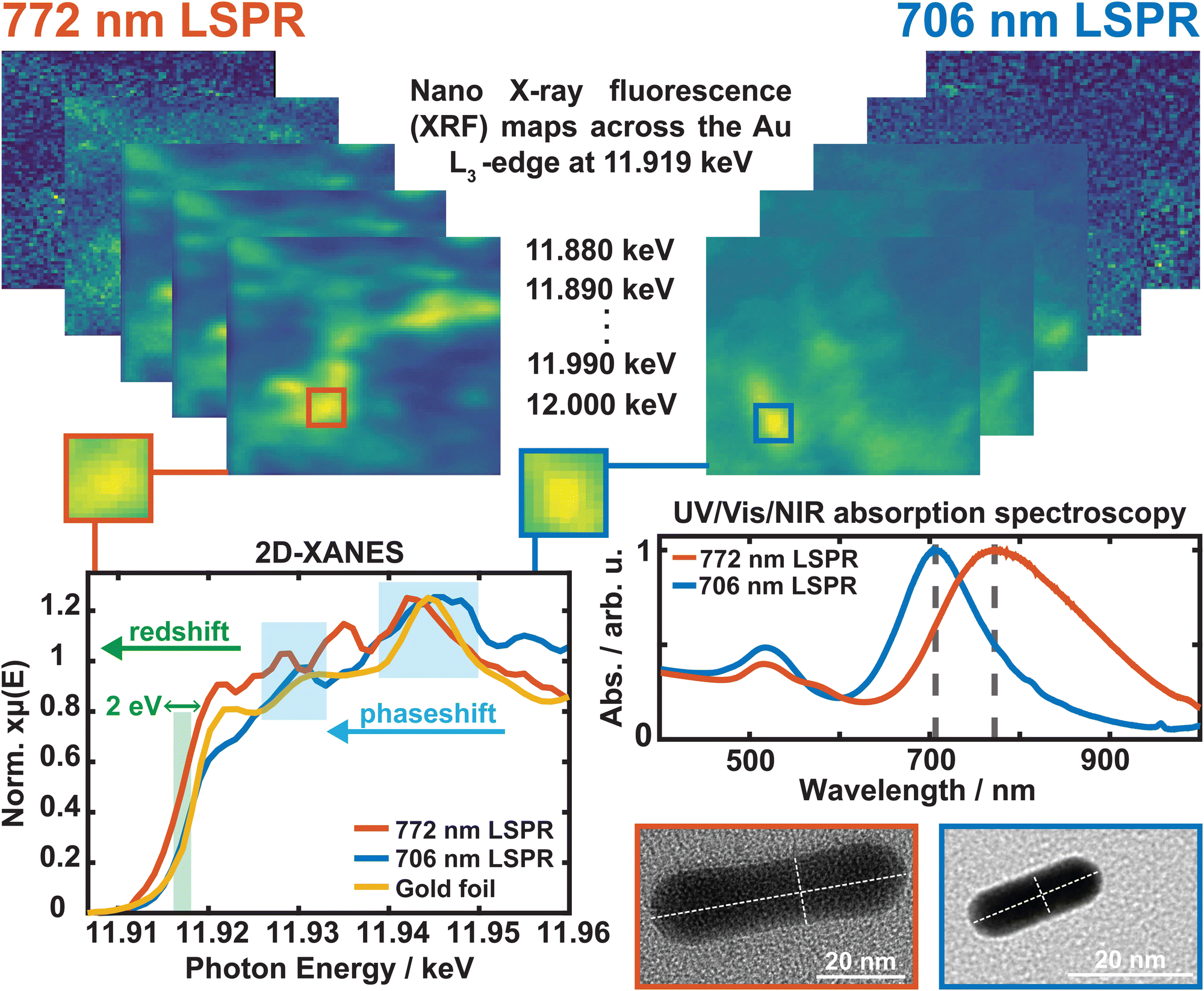 | ||
| Fig. 3 64 X-ray fluorescence (XRF) maps (2.5 × 2.5 m2) were recorded, stacked, and aligned, covering an energy range of 11.880–12.000 keV. The region with the highest intensity (GNRs, yellow) was chosen using an ROI of 5 × 5 px (200 × 200 nm2), and the 2D X-ray absorption near-edge structure (2D-XANES) spectra were processed for CTAB-GNRs possessing a 772 nm and 706 nm LSPW, and the gold foil. | ||
2.4. FTIR characterization of surfactant chemistry
Samples for the FTIR analysis were prepared by drop-casting an aliquot of concentrated GNR dispersion with the molecular adsorbates onto two different membranes (International Crystal Laboratories) consisting of either polyethylene or polytetrafluorethylen, allowing for a transmission range of 400–4000 cm−1. The spectra were then recorded using a Nicolet iS20 (Thermo Scientific) Fourier-transform infrared (FTIR) spectrometer (transmittance mode) with a resolution of 0.25 cm−1 and an acquisition of 64 scans per spectrum. The data was acquired and pre-processed using OMNIC software with workup and analysis performed in MatLab.2.5. SERS-based detection of molecular analytes
Samples for surface-enhanced Raman spectroscopy (SERS) measurements were prepared by drop-casting an aliquot of the mixture (dye + different surfactant-capped GNRs) to a glass microscope coverslip and allowed to dry under ambient conditions. A single wavelength excitation source (632.8 nm) was used to illuminate the sample in a confocal Raman imaging microscope (Witec Alpha 300) with a cone of collection determined by a 100× magnification objective detected in a 180° back-scattering geometry. The sample was positioned on a piezo-actuated stage with focusing in the z-direction and aligned to the 520 cm−1 peak in a silicon wafer. Data was acquired and pre-processed using Witec Project 5 software with workup and analysis performed in MatLab and Origin Pro.3 Results and discussion
One of the notable effects of the LSPR of GNRs is the extraordinary enhancement of the electric field around the surface, which results in dramatic absorption of specific wavelengths of incident light. The wavelengths transmitted by the GNRs determine the color of the nanoparticle suspension, as seen in Fig. 2b. Different geometries interact in a certain way with the electric field of the incident light, giving rise to a variety of unique dipole moments, generally described with the equation| p = ε0εmα|E0|, |
where ε(λ), εm and ε0 indicate the dielectric functions of the nanoparticle, its surrounding material, and the vacuum, R the radius of the sphere, and αo the spherical polarizability. In contrast, GNRs show two LSPRs, i.e., TSPW and LSPW, resulting in a separation of α

| panisot. = ε0εm(α‖|E0|‖ + α⊥|E0|⊥) |
where α‖,|E0|‖ describe the LSPW, and α⊥,|E0|⊥ the TSPW, L‖,⊥ the longitudinal and transverse axes (depending on the aspect ratio and geometry of the GNRs), and V the volume of the GNRs. Therefore, a clear separation of the LSPR response, especially in GNRs, can be observed, covering the UV/Vis (TSPW) and the Vis/NIR range (LSPW). As described by the Mie and Rayleigh theory, their dielectric functions determine the measured absorption cross sections (Cabs) for GNRs. The Cabs can be manipulated depending on the surfactant and ligands of individual GNRs, resulting in an altered LSPR response, which is the subject of this work.2,19,52,53 A scheme of the LSPW and the correlation to the lengths of the GNRs is illustrated in Fig. 4a.

 | ||
| Fig. 4 (a) Scheme of correlating different GNRs lengths and their LSPW. (b) Correlation between the pH of the solutions and the LSPW (◊) and TSPW (□) of the GNRs. A signature of GNSs (△) was also observed at basic pH values. The legend states the growth times, the assignment of the absorption peaks, and the upscaling of the total solution by a factor of 10. The error bars indicate the precision of the pH measurement. | ||
The absorption spectra of the GNRs discussed in this work can be found in Fig. S2–S4 in the ESI.† The peak associated with the TSPW is present at shorter wavelengths (≈500 nm), and the peak indicating the LSPW covers a spectrum between ≈ 700–1100 nm. A lower pH (≈5) led to a redshift (longer wavelengths), whereas an increase in pH (≈8.5) led to a blueshift (shorter wavelengths) of the LSPW. In order to illustrate this trend, all absorption peaks were precisely evaluated, using Voigt-fits (Fig. S4 in the ESI†), and plotted against their pH, as shown in Fig. 4b. At pH values greater than 7, the TSPW of the GNRs underwent a redshift and broadened spectrally. This change was either indicated by a small discontinuity of the curve up to the formation of a new third peak corresponding to the formation of isotropic particles, marked as “△” in Fig. 4b. As spheres only possess a single geometric parameter (diameter), they only show a single absorption peak in the spectra (≈550 nm), which neither corresponds to the LSPW nor the TSPW of the GNRs. As most samples contain traces of spheres, confirmed by SEM imaging, the Voigt model utilizes three distinct absorption peaks. The third peak corresponding to the spheres only became clearly visible at a basic pH, as shown in Fig. 4b.
When the GNRs were synthesized in the appropriate acidic pH regime, a clear correlation between the position of the LSPW and the growth time and the pH value was observed. The lower the pH, the more redshifted the LSPW and the greater the length of the GNRs. If the pH was adjusted to a more basic value, the LSPW became more blueshifted; hence, the GNRs' length would decrease. This is due to the increased reducing power of NH2OH·HCl at larger pH values,54,55 which leads to the formation of shorter GNRs in the growth process due to a faster consumption of the educts in the growth solution, leaving no sufficient time to form longer GNRs. The trend was no longer present for growth times shorter than one hour (yellow “◊”) since, at a lower pH, the LSPW was blueshifted. With an increasing pH (≥6.5), a trend comparable to that of GNRs with growth times longer than one hour was observed. This can be explained by the fact that at lower pH values (≈5), the reduction of HAuCl4·3H2O from Au+3 to Au0 takes longer (pH ≈ 2.7 of the growth solutions before adjustment). Adjusting the overall volume of the synthesis (upscaling with a factor of 10) while holding the pH, the concentration, and the growth time constant resulted in a blueshift of the LSPW, indicated as light-blue and -red “◊” in Fig. 4b. This trend could be explained by an inhomogeneous distribution of the seeds after the injection of the seed solution, leading to longer diffusion times and, thus, different reaction kinetics. This might be important to consider for large-scale applications of GNRs.
Fig. 5 shows the recorded UV/Vis/NIR spectra of the different surfactant-capped GNRs with the respective dyes (IC, LMB, MB) attached to their surface. CTAB-GNRs possess a bilayer of CTAB around the surface of the GNRs consisting of a hydrophobic (negative, carbon-tail) middle part and a hydrophilic (positive, nitrogen-head) end (Fig. 2a), leading towards an overall positive surface charge of the CTAB-GNRs. In comparison, PSS-GNRs exhibit a delocalized negative surface charge due to the benzene-π-system, making it easier to exchange the surfactant from CTAB to Na-PSS and subsequently to Na-CIT, having a negative surface charge, enabling the electrostatic attachment of differently charged dyes to the surfactant-capped GNRs. The negative IC binds with the CTAB-GNRs, whereas the neutrally charged LMB and the positively charged MB are attached to the CIT-GNRs.48 A clear shift of the LSPW (Δλ) can be observed in all the spectra (Fig. 5). This can be explained by the increased area over which the electron cloud is delocalized due to the attachment of the dye molecules. This increase in size leads to a decrease in the restoring force of the plasmonic electron cloud, which results in a redshift of the LSPR. This can be observed for the CIT-GNRs with MB and LMB attached to their surfaces, whereas the CTAB-GNR + IC follows an opposing trend (blueshift), possibly due to electron excess of the GNRs, when bound to CTAB.56
 | ||
| Fig. 5 Absorption spectra, showing the different surfactant-capped GNRs and the dyes, highlighting the shift in the individual spectra occurring due to the electrostatic interactions following the attachment of the dye to the surfactant-capped GNRs. | ||
Fig. 3 shows the nano-XANES mapping of CTAB-stabilized GNR oligomers with two different LSPRs. A 2D-XRF map was acquired at every X-ray absorption spectroscopy (XAS) monochromator position, yielding spatial resolution correlated to electronic information for the Au L3-absorption-edge dominated by 2p3/2 → 5d5/2,3/2 dipole allowed transitions. For the 772 nm LSPR, the brightest pixel region corresponds to the highest concentration of GNRs, giving a distinct 2D-XANES spectrum (red trace). Similarly, the nanorods with a 706 nm optical LSPR yield a 2D-XANES spectrum (blue trace) with an edge energy that overlaps nearly precisely with the gold foil standard (yellow trace). In contrast, a redshift of ≈2 eV (green box) is observed for GNRs with an optical LSPR of 772 nm. The electronic energetic shift of 2 eV in the 2D-XANES edge correlates to an observed optical shift of 66 nm, where additional phaseshifts are also observed in the oscillatory features past the white line energy (blue boxes). The magnitude of the LSPR redshift that we observe is much greater than ≈10 nm, signifying that the origin of this redshift is not due to subtle corner rounding or narrowing alone, as can be seen in previous studies on corner-sharpness-controlled gold nanoparticles.57 We therefore attribute the redshift to changes in GNR aspect ratio, electronic changes in the longitudinal LSPR mode, and ligand-induced electronic restructuring. Further correlation to structural and morphological information obtained by TEM imaging shows that the redshift can be ascribed primarily to a change in the oscillator strength of the longitudinal plasmon resonance going from a nanorod of dimensions ≈ 10 × 25 nm (706 nm, blue) to a nanorod of dimensions ≈20 × 60 nm (772 nm, red) resulting in a size-dependent shift in the gold d-band holes. This lowering in relative oscillator strength is also correlated with out-of-phase XANES amplitudes occurring at energies 11.935 keV and 11.930 keV, suggesting a shift in the 5d valence electron distribution across coordination sites ranging from Au0 ↔ Au−1 due to the increased electron-donating ability of the CH2 and CH3 groups of CTAB and a shift to more acidic growth solutions as the LSPR redshifts.58 The synthesis methodology included in this report uses the same chemical and surfactants from a prepared stock solution under identical conditions. Given the high fidelity of this one-pot synthesis protocol, we can draw comparisons in the LSPR redshift in the 772 nm and 706 nm GNR samples with only CTAB functionalization.
Fig. 6 displays the FTIR and SERS optical vibrational spectroscopy results following gold nanorod surface stabilization with CTAB and Na-CIT, after which the nanostructures underwent surface functionalization with blue dye adsorbates. In Fig. 6a, the 2915.90 cm−1 and 2849.01 cm−1 modes correspond to a symmetric and asymmetric stretching of the methyl and methylene groups in the CTAB surfactant. Vibrational modes in the mid-wavenumber region (1486.61 cm−1, 1472.38 cm−1, 1462.32 cm−1, 1430.86 cm−1, 1407 cm−1) correspond to asymmetric and symmetric bending of C–H bonds of the H3C–N+ moiety. Notably, modes in the fingerprint region exhibit higher peak amplitudes, suggesting a greater number of active sites for adsorbate molecule attachment. The modes include C–H bending motions that are typically not observed in pure CTAB (1012.20 cm−1 and 981.29 cm−1), C–N+ stretching motions (959.59 cm−1), and rocking modes of the CH2-chain (938 cm−1 and 908 cm−1). Fig. 6b shows the FTIR-detected CIT-GNRs following the surfactant exchange and surface stabilizing with citrate ions. The most prominent modes can be assigned to the symmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration (1584 cm−1) and the antisymmetric vibration of COO− (1400.73 cm−1). Both modes are shifted to higher relative wavenumbers (Δṽ > 5 cm−1) indicative of surface interactions and monodentate or bidentate binding to gold. High-wavenumber modes corresponding to O–H stretches are also present (2359 cm−1 and 2341 cm−1) where these are commonly seen in pure citrate but less often observed in citrate-GNRs. Vibrational modes below 1100 cm−1 correspond to symmetric and asymmetric stretches of C–O–C bonds.59–69
O vibration (1584 cm−1) and the antisymmetric vibration of COO− (1400.73 cm−1). Both modes are shifted to higher relative wavenumbers (Δṽ > 5 cm−1) indicative of surface interactions and monodentate or bidentate binding to gold. High-wavenumber modes corresponding to O–H stretches are also present (2359 cm−1 and 2341 cm−1) where these are commonly seen in pure citrate but less often observed in citrate-GNRs. Vibrational modes below 1100 cm−1 correspond to symmetric and asymmetric stretches of C–O–C bonds.59–69
 | ||
| Fig. 6 (a) and (b) FTIR spectra of CTAB-GNRs and CIT-GNRs. (c) SERS spectra of the different surfactant-capped GNRs (CTAB, CIT) with the dye molecules (IC, LMB, MB) electrostatically attached to their surfaces. | ||
SERS-detection of adsorbate molecules that are confined to nanometer regions between GNRs through electrostatic interactions are shown in Fig. 6c. IC displays prominent SERS modes at 1633.64 cm−1 (CO, CC stretches with in-plane C–H modes), 1622.96 cm−1, 1574.67 cm−1 (CC asymmetric stretches in the pyrrolidone ring combined with CO stretching), 1296.83 cm−1 (symmetric stretches of the SO3− group with C–C bending and N1–C2 stretching), 770.85 cm−1 (out-of-plane bending in the C–C and C–H bonds combined with pyrrolidone ring breathing), 676.01 cm−1 (out-of-plane N–H and C–C motion), 573.65 cm−1 (CC−CO−C bending), 551.74 cm−1 (CC−CO−C stretches), and 270.40 cm−1 (skeletal deformation modes combined with Au−O vibrations). Of interest are the absence of sulfonate modes with SERS transitions at 1360 cm−1, 1301 cm−1, 1141 cm−1, and 611 cm−1 which are suppressed likely due to symmetric electrostatic interactions at both ends of the anionic IC molecule with the cationic CTAB at the gold nanorod surfaces.
MB displays normal vibrational modes at 1622 cm−1, 1433 cm−1, and 1405.53 cm−1 that are consistent with unbound surface-free molecules where structural motions correspond to skeletal bending at the C−N stretch and C−N ring stretch (1622.96 cm−1), asymmetric vibrations about the C−N stretch (1433.11 cm−1), and stretching about the C9−N10, C3−N2, C−N ring, and in-plane C−H bending motion (1405.53 cm−1). These C−N based modes are also the most intense peaks in the spectrum, which contrasts with previous studies of MB on silver nanocaps. A pronounced relative decrease in mode amplitude for the 400–1400 cm−1 region was observed. The relative peak amplitude changes can be ascribed to asymmetric electrostatic interactions between one end of the cationic MB molecule and the anionic citrate surface of the GNRs. All other modes shift when MB is adsorbed to GNRs with citrate surfactant functionalization. These shifts in SERS transitions include the C−N stretching mode (1217.67 cm−1), CH3 rocking motion with in-plane C−H bending (1157.68 cm−1), C−H out-of-plane bending (1126.03 cm−1), in-plane bending of C−H bonds (1041.88 cm−1), and skeletal deformations at the C−N−C moiety (479.28 cm−1). LMB was also used to functionalize the surfaces of GNRs, which shared many overlapping modes with MB. One of the more intense bands in LMB is centered at 1371 cm−1 corresponding to the CC ring stretch, less prevalent in our GNRs system. According to the SERS spectra, both LMB and MB share similar peak intensities, suggesting similar binding or attachment configurations to the GNRs. This observation of varied absorption dynamics is further supported by similar peak intensity shifts of the LSPR where the IC molecule induces the most pronounced shift followed by smaller effects in the MB and LMB surface attachment varieties.70–76
4. Conclusions
We report a precision pH-directed synthesis of GNRs functionalized with cationic CTAB surfactants with optical LSPR tunability of the longitudinal resonance throughout the near-infrared. Surfactant exchange using Na-PSS leads to a delocalized negative charge, allowing for the final attachment of Na-CIT, forming anionic surface charges. The tunable GNRs are probed by a nano-XANES technique that confers simultaneous high spatial and high electronic resolution to elucidate phase shifts due to d-band vacancies. Notably, a redshift in optical LSPR properties was observed to correlate with a redshift in the Au L3-edge due to the electron donating properties of the surfactant, which changes as the LSPW shifts. We also report a controlled adsorbate functionalization of the GNRs through electrostatic interactions between cationic–anionic species with the analyte molecules MB, IC, and LMB. The characterized electrostatic surface interactions and their tunability can confer significant advantages for applications that demand preferential binding to plasmonic nanostructures. In this way, we have demonstrated the precision synthesis of nanostructures with a high degree of uniformity, shape, and size with wide-ranging and essential applications to photonics, single molecule conductors, photodynamic therapy, and bioelectronics.Author contributions
Conceptualization: D. G. S., H. J. W., E. A. S. K. investigation: D.G. S., J. B., U. Y., A. P. data analysis & curation: D. G. S., J. B., A. P. visualization: D. G. S. funding acquisition: H. J. W., E. A. S. K. writing: D. G. S., J. B., U. Y., H. J. W., E. A. S. K.Conflicts of interest
There are no conflicts to declare.Data availability
Data is available for access at the HXN Beamline of NSLS-II at Brookhaven National Laboratory managed by the Department of Energy, at the Instrumentation for Molecular and Nanoscale Innovation (IMNI), the ScopeM facility at ETH Zurich and the Department of Chemistry at Brown University.Acknowledgements
The authors acknowledge A. Schneider, M. Seiler, M. Urban, and D. Zindel for technical support with mechanical, chemical, and electronic components for ongoing experiments, J. Trester and D. Matselyukh for their continuous support of the project, and N. Warren and A. Willemsen for insightful discussions. The ScopeM facility at ETH Zurich and the IMNI facility at Brown University are acknowledged for providing their instruments. The authors also acknowledge research support from Brown University startup funds.References
- J. Zheng, X. Cheng, H. Zhang, X. Bai, R. Ai, L. Shao and J. Wang, Chem. Rev., 2021, 121, 13342–13453 CrossRef CAS PubMed.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Maragò and M. A. Iatì, J. Phys.: Condens. Matter, 2017, 29, 203002 CrossRef PubMed.
- X. Wang, G. Li, Y. Ding and S. Sun, RSC Adv., 2014, 4, 30375–30383 RSC.
- H. Yang, H. He, Z. Tong, H. Xia, Z. Mao and C. Gao, J. Colloid Interface Sci., 2020, 565, 186–196 CrossRef CAS PubMed.
- C. Carnovale, G. Bryant, R. Shukla and V. Bansal, Prog. Mater. Sci., 2016, 83, 152–190 CrossRef CAS.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- L. Xie, X. Zhang, C. Chu, Y. Dong, T. Zhang, X. Li, G. Liu, W. Cai and S. Han, J. Nanobiotechnol., 2021, 19, 454 CrossRef CAS PubMed.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- R. A. Rippel and A. M. Seifalian, J. Nanosci. Nanotechnol., 2011, 11, 3740–3748 CrossRef CAS PubMed.
- J. Cao, T. Sun and K. T. Grattan, Sens. Actuators, B, 2014, 195, 332–351 CrossRef CAS.
- J. Pérez-Juste, I. Pastoriza-Santos, L. M. Liz-Marzán and P. Mulvaney, Coord. Chem. Rev., 2005, 249, 1870–1901 CrossRef.
- L. Shao, Z.-J. Yang, D. Andrén, P. Johansson and M. Käll, ACS Nano, 2015, 9, 12542–12551 CrossRef CAS PubMed.
- E. Rusak, I. Staude, M. Decker, J. Sautter, A. E. Miroshnichenko, D. A. Powell, D. N. Neshev and Y. S. Kivshar, Appl. Phys. Lett., 2014, 105, 221109 CrossRef.
- P. Biagioni, J.-S. Huang and B. Hecht, Rep. Prog. Phys., 2012, 75, 024402 CrossRef PubMed.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- Z. Jiang, G. Wen, Y. Luo, X. Zhang, Q. Liu and A. Liang, Sci. Rep., 2014, 4, 5323 CrossRef CAS PubMed.
- E. Hutter and J. H. Fendler, Adv. Mater., 2004, 16, 1685–1706 CrossRef CAS.
- S. A. Maier, Plasmonics: fundamentals and applications, Springer, New York, 2007 Search PubMed.
- C. Kittel, Introduction to solid state physics, Wiley, Hoboken, NJ, 8th edn, 2005 Search PubMed.
- U. Kreibig and M. Vollmer, Optical Properties of Metal Clusters, Springer Berlin Heidelberg, Berlin, Heidelberg, 1995, vol. 25 Search PubMed.
- W. Ni, X. Kou, Z. Yang and J. Wang, ACS Nano, 2008, 2, 677–686 CrossRef CAS PubMed.
- J. Zuloaga and P. Nordlander, Nano Lett., 2011, 11, 1280–1283 CrossRef CAS PubMed.
- J. M. Núñez-Leyva, E. S. Kolosovas-Machuca, J. Sánchez, E. Guevara, A. Cuadrado, J. Alda and F. J. González, Nanomaterials, 2021, 11, 1696 CrossRef PubMed.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS PubMed.
- A. Mehere and N. B. Chaure, Appl. Phys. A, 2020, 126, 662 CrossRef CAS.
- M.-Z. Wei, T.-S. Deng, Q. Zhang, Z. Cheng and S. Li, ACS Omega, 2021, 6, 9188–9195 CrossRef CAS PubMed.
- W. Zhou, Y. Hao, X. Wang, M. Li and S. Liang, Opt. Mater., 2023, 140, 113834 CrossRef CAS.
- X. Zhang, N. Tran, T. Egan, B. Sharma and G. Chen, J. Phys. Chem. C, 2021, 125, 13350–13360 CrossRef CAS.
- S. Jiang, F. Chen, X. Wen, C. Huang, Y. Xuan, H. Yao, Z. Wu, G. Huang and L. Meng, Inorg. Chem. Commun., 2023, 149, 110426 CrossRef CAS.
- Y. Guo, Q. Liu, A. Wei, S. Jiang, F. Chen, J. Huang, Y. He, G. Huang and Z. Wu, Dalton Trans., 2023, 52, 1052–1061 RSC.
- R. Gallagher, X. Zhang, A. Altomare, D. Lawrence, N. Shawver, N. Tran, M. Beazley and G. Chen, Nano Res., 2021, 14, 1167–1174 CrossRef CAS.
- J. Cheng, L. Ge, B. Xiong and Y. He, J. Chin. Chem. Soc., 2011, 58, 822–827 CrossRef CAS.
- J. O. Park, S.-H. Cho, D.-Y. Jeong, Y.-M. Kong and S. Y. Lee, Jpn. J. Appl. Phys., 2015, 54, 015001 CrossRef CAS.
- Q. Wei, J. Ji and J. Shen, J. Nanosci. Nanotechnol., 2008, 8, 5708–5714 CrossRef CAS.
- Y. Leng, X. Yin, F. Hu, Y. Zou, X. Xing, B. Li, Y. Guo, L. Ye and Z. Lu, RSC Adv., 2017, 7, 25469–25474 RSC.
- U. Yunusa, N. Warren, D. Schauer, P. Srivastava and E. Sprague-Klein, Nanoscale, 2024, 16, 5601–5612 RSC.
- J. Yu, C.-H. Hsu, C.-C. Huang and P.-Y. Chang, ACS Appl. Mater. Interfaces, 2015, 7, 432–441 CrossRef CAS.
- N. L. Warren, U. Yunusa, A. B. Singhal and E. A. Sprague-Klein, Chem. Phys. Rev., 2024, 5, 011307 CrossRef CAS.
- A. Taldaev, R. Terekhov, I. Nikitin, E. Melnik, V. Kuzina, M. Klochko, I. Reshetov, A. Shiryaev, V. Loschenov and G. Ramenskaya, Front. Pharmacol., 2023, 14, 1264961 CrossRef CAS.
- V. G. Rao, U. Aslam and S. Linic, J. Am. Chem. Soc., 2019, 141, 643–647 CrossRef CAS.
- M. N. Le, B. R. Wuertz, M. A. Biel, R. L. Thompson and F. G. Ondrey, Laryngoscope Invest. Otolaryngol., 2022, 7, 982–987 CrossRef.
- A. F. Dos Santos, L. F. Terra, R. A. M. Wailemann, T. C. Oliveira, V. D. M. Gomes, M. F. Mineiro, F. C. Meotti, A. Bruni-Cardoso, M. S. Baptista and L. Labriola, BMC Cancer, 2017, 17, 194 CrossRef.
- C. C. Carlin, A. X. Dai, A. Al-Zubeidi, E. M. Simmerman, H. Oh, N. Gross, S. A. Lee, S. Link, C. F. Landes, F. H. Da Jornada and J. A. Dionne, Chem. Phys. Rev., 2023, 4, 041309 CrossRef CAS.
- R. Boltes Cecatto, L. Siqueira De Magalhães, M. Fernanda Setúbal Destro Rodrigues, C. Pavani, A. Lino-dos Santos-Franco, M. Teixeira Gomes and D. Fátima Teixeira Silva, Photodiagn. Photodyn. Ther., 2020, 31, 101828 CrossRef CAS.
- C. Boerigter, R. Campana, M. Morabito and S. Linic, Nat. Commun., 2016, 7, 10545 CrossRef CAS.
- C. Boerigter, U. Aslam and S. Linic, ACS Nano, 2016, 10, 6108–6115 CrossRef CAS.
- J. G. Mehtala, D. Y. Zemlyanov, J. P. Max, N. Kadasala, S. Zhao and A. Wei, Langmuir, 2014, 30, 13727–13730 CrossRef CAS PubMed.
- J. A. Bearden and A. F. Burr, Rev. Mod. Phys., 1967, 39, 125–142 CrossRef CAS.
- A. Pattammattel, R. Tappero, M. Ge, Y. S. Chu, X. Huang, Y. Gao and H. Yan, Sci. Adv., 2020, 6, 7 Search PubMed.
- A. Pattammattel, R. Tappero, D. Gavrilov, H. Zhang, P. Aronstein, H. J. Forman, P. A. O'Day, H. Yan and Y. S. Chu, Metallomics, 2022, 14, 11 CrossRef PubMed.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409–453 Search PubMed.
- V. Myroshnychenko, J. Rodríguez-Fernández, I. Pastoriza-Santos, A. M. Funston, C. Novo, P. Mulvaney, L. M. Liz-Marzán and F. J. García De Abajo, Chem. Soc. Rev., 2008, 37, 1792 RSC.
- M. R. Rahman, F. S. Saleh, T. Okajima and T. Ohsaka, Langmuir, 2011, 27, 5126–5135 CrossRef CAS PubMed.
- X. Zou, E. Ying and S. Dong, J. Colloid Interface Sci., 2007, 306, 307–315 CrossRef CAS PubMed.
- D. Avsar, H. Ertuerk and M. P. Menguec, J. Quant. Spectrosc. Radiat. Transfer, 2020, 241, 106684 CrossRef CAS.
- J.-E. Park, Y. Lee and J.-M. Nam, Nano Lett., 2018, 18, 6475–6482 CrossRef CAS PubMed.
- L. Luo, J. Luo, H. Li, F. Ren, Y. Zhang, A. Liu, W.-X. Li and J. Zeng, Nat. Commun., 2021, 12, 1218 CrossRef CAS PubMed.
- A. Zoppi, S. Trigari, G. Margheri, M. Muniz-Miranda and E. Giorgetti, RSC Adv., 2015, 5, 8523–8532 RSC.
- R. Yuan, L. Gao, J. Liu, C. Tu, R. Tan and S. Xu, Eur. Polym. J., 2024, 203, 112644 CrossRef CAS.
- G. Su, C. Yang and J.-J. Zhu, Langmuir, 2015, 31, 817–823 CrossRef CAS PubMed.
- J. C. Mohan, G. Praveen, K. Chennazhi, R. Jayakumar and S. Nair, J. Exp. Nanosci., 2013, 8, 32–45 CrossRef CAS.
- R. Kurrey, M. K. Deb, K. Shrivas, B. R. Khalkho, J. Nirmalkar, D. Sinha and S. Jha, Anal. Bioanal. Chem., 2019, 411, 6943–6957 CrossRef CAS PubMed.
- H.-C. Kim, E. Kim, S. W. Jeong, T.-L. Ha, S.-I. Park, S. G. Lee, S. J. Lee and S. W. Lee, Nanoscale, 2015, 7, 16470–16480 RSC.
- B. R. Khalkho, M. K. Deb, R. Kurrey, B. Sahu, A. Saha, T. K. Patle, R. Chauhan and K. Shrivas, Spectrochim. Acta, Part A, 2022, 267, 120523 CrossRef CAS PubMed.
- A. Giri, A. Makhal, B. Ghosh, A. K. Raychaudhuri and S. K. Pal, Nanoscale, 2010, 2, 2704 RSC.
- M. M. Fathy, A. A. Elfiky, Y. S. Bashandy, M. M. Hamdy, A. M. Elgharib, I. M. Ibrahim, R. T. Kamal, A. S. Mohamed, A. M. Rashad, O. S. Ahmed, Y. Elkaramany, Y. S. Abdelaziz, F. G. Amin and J. I. Eid, Sci. Rep., 2023, 13, 2749 CrossRef CAS PubMed.
- R. Del Caño, J. M. Gisbert-González, J. González-Rodríguez, G. Sánchez-Obrero, R. Madueño, M. Blázquez and T. Pineda, Nanoscale, 2020, 12, 658–668 RSC.
- H. R. De Barros, L. Piovan, G. L. Sassaki, D. De Araujo Sabry, N. Mattoso, A. M. Nunes, M. R. Meneghetti and I. C. Riegel-Vidotti, Carbohydr. Polym., 2016, 152, 479–486 CrossRef CAS PubMed.
- I. Shadi, B. Chowdhry, M. Snowden and R. Withnall, Spectrochim. Acta, Part A, 2003, 59, 2201–2206 CrossRef CAS PubMed.
- N. Peica and W. Kiefer, J. Raman Spectrosc., 2008, 39, 47–60 CrossRef CAS.
- E. Papadopoulou, N. Gale, J. F. Thompson, T. A. Fleming, T. Brown and P. N. Bartlett, Chem. Sci., 2016, 7, 386–393 RSC.
- L. H. Oakley, D. M. Fabian, H. E. Mayhew, S. A. Svoboda and K. L. Wustholz, Anal. Chem., 2012, 84, 8006–8012 CrossRef CAS PubMed.
- R. P. Johnson, J. A. Richardson, T. Brown and P. N. Bartlett, J. Am. Chem. Soc., 2012, 134, 14099–14107 CrossRef CAS PubMed.
- S. Dutta Roy, M. Ghosh and J. Chowdhury, J. Phys. Chem. C, 2018, 122, 10981–10991 CrossRef CAS.
- J. Clayden, Chem. Commun., 2004, 127–135 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Extended description of the experimental methods and synthesis instructions. Overview of the fitting algorithm (Voigt-fits) applied to the absorption spectra of the GNRs to extract the peak positions. Raw data and acquisition using 2D X-ray absorption near-edge structure (2D-XANES) spectroscopy at the National Synchrotron Light Source II (NSLS-II) at Brookhaven National Laboratory (BNL). See DOI: https://doi.org/10.1039/d4cp01993h |
| This journal is © the Owner Societies 2024 |