
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The wondrous world of ABX3 molecular perovskites†‡
Silva M.
Kronawitter
 and
Gregor
Kieslich
*
and
Gregor
Kieslich
*
Department of Chemistry, TUM School of Natural Sciences, Technical University of Munich, Lichtenbergstraße 4, 85748 Garching, Germany. E-mail: gregor.kieslich@tum.de
First published on 9th September 2024
Abstract
The substitution of atoms with molecular building blocks to form hybrid organic–inorganic networks has been an important research theme for several decades. ABX3 molecular perovskites (MolPs) are a subclass of hybrid networks, adopting the perovskite structure with cationic and anionic molecules on the A-site and X-site. MolPs such as ((CH3)2NH2)Zn(HCOO)3 or ((n-C3H7)4N)Mn(C2N3)3 show a range of fascinating structure–chemical properties, including temperature-driven phase transitions that include a change of polarity as interesting for ferroelectrics, pressure-driven order–disorder phase transitions as interesting for barocaloric solid-state refrigeration, and most recently, melting-behaviour before decomposition with subsequent glass formation after cooling. In this feature article, we take a more personal perspective, overviewing the field's current state and outlining future directions. We start by comparing the MolPs’ structural chemistry with their inorganic parents, a comparison that helps us identify opportunities for material design. After discussing the MolPs’ potential as barocalorics, ferroelectrics, and in the area of glasses, we outline some challenges that lie ahead. Beyond their relevance as a hybrid analogue of inorganic perovskites, we find that MolPs' chemical parameter space provides exciting opportunities for systematically developing design guidelines for functional materials.
 Silva M. Kronawitter | Silva M. Kronawitter received her bachelor's degree in Chemistry from the University of Regensburg in 2018. She then pursued her master's degree at the Technical University of Munich, completing it in 2019. In January 2021, Silva began her PhD studies in the Structural Chemistry of Functional Materials research group at the Technical University of Munich, under the supervision of Dr. Gregor Kieslich. Her research primarily focuses on the rational design and stimuli-responsiveness of molecular perovskites through exploring molecular substitution approaches. |
 Gregor Kieslich | Gregor Kieslich is a chemist who completed his PhD in solid-state chemistry at Mainz under W. Tremel in 2013. Afterwards, he moved to Cambridge in 2014. There, he worked until 2016 with A. K. Cheetham, starting to appreciate the beauty of hybrid compounds. Since 2017, he has been leading a research team at the Technical University of Munich. His work is centred around the structural chemistry of functional materials, with a healthy disregard for classical subject boundaries. Gregor has received several awards and fellowships, including acceptance into the Heisenberg programme of the DFG (2023) and the Dozentenpreis from the Fonds of the Chemical Industry (2022). |
Introduction
The targeted development of functional materials is an ongoing effort in applied solid-state chemistry and materials science. When casting material functionality as the specific material responsiveness in a useful context, the challenge shifts to understanding and designing a material's macroscopic responsiveness to external stimuli such as temperature, pressure, and electric fields, amongst others. In this pursuit, using organic and inorganic moieties for building up structures – making the material a hybrid – has developed into a similarly exciting and important research direction.1–5 Examples of whole material families derived by this approach are metal–organic frameworks and hybrid organic–inorganic perovskites (HOIPs). Both nicely show that using molecules as building blocks unlocks an unlimited chemical scope for synthesising materials with optimised and new properties, including metal–organic frameworks with chemically engineered pore-space6 and HOIPs with optimised photovoltaic properties, to name only two prominent examples.7,8Intriguingly, many hybrid compounds have a structurally related, inorganic parent. Examples are zeolitic imidazolate frameworks,9 diamondoid metal–organic frameworks,5 the entire family of HOIPs4 and Ruddlesden Popper-type layered HOIPs.10,11 This relation does not come by chance,12 and historically, inorganic network structures served as inspiration for designing hybrids.12 In the search for crystal chemistry principles that guide materials synthesis, the existence of well-studied inorganic parent materials holds exciting opportunities. On the one hand, and starting from established structure–chemical concepts, this link can highlight new design opportunities that come with the simple use of molecules. On the other hand, this link can pinpoint where established principles reach their limitations, motivating the development of improved or new structure–chemical concepts. Therefore, we see this parent-child relation as a cornerstone for efficiently harnessing the chemical space as available for synthesising hybrid materials.
In this context, materials with the iconic ABX3 perovskite structure motif13,14 have a unique role, and both inorganic perovskites and HOIPs are era-defining material families. For inorganic perovskites, the piezoelectric and ferroelectric properties of BaTiO3,15,16 PbZr1−xTixO317 and PbMg1−xNbxO318 shall be mentioned as early milestones,19 and the compositional variability of the perovskite structure soon led to the discovery of ferromagnetic SrRuO3,20 non-linear optical KNbO3,21 antiferromagnetic KNiF3,22,23 multiferroic BiMnO324 and superconductive YBa2Cu3O7 amongst many other examples.25,26 The report of the hybrid organic–inorganic (CH3NH3)PbI3 in 1978 marks the next important point,27,28 although its potential in the context of photovoltaics was not recognised until 2009.29 Since then, lead halide-based HOIPs with A+ = (CH3NH3)+, ((NH2)2CH)+, (CH7N2)+ and (C2H6N)+ have led to a shift in paradigm for obtaining high-performance semiconductors for photovoltaics, photodetectors and light emitting diodes.30–33
In HOIPs, the X-anion is typically a halide, making the 3D [BX3]− network purely inorganic. This is different from what we here refer to as molecular perovskites (MolPs). In these, the A-cation, e.g. dimethylammonium ((CH3)2NH2)+, guanidinium ((NH2)3C)+, tripropylmethylammonium ((n-C3H7)3(CH3)N)+ or tetramethylammonium ((CH3)4N)+, and the X-anion, e.g. formate (HCOO)−, hypophosphite (H2POO)−, dicyanamide (C2N3)− or azide (N3)−, are molecules and form perovskite-type materials such as ((CH3)2NH2)Zn(HCOO)3,34 ((NH2)3C)Mn(H2POO)3,35 ((n-C3H7)3(CH3)N)Ni(C2N3)336 or ((CH3)4N)Mn(N3)3,37 see Fig. 1. Therefore, molecular in MolPs serves to highlight the use of molecules on the A- and X-site. In MolPs, the B-cation and X-anion form a negatively charged, 3D coordination network. All B-cations are octahedrally coordinated by the coordinating atoms of the X-anion and linked to their six neighbouring B-cations via six molecular X-site bridges. The A-cation sits in the void of the ReO3-type coordination network for charge balance and, additionally, can interact with the network via hydrogen bonds.
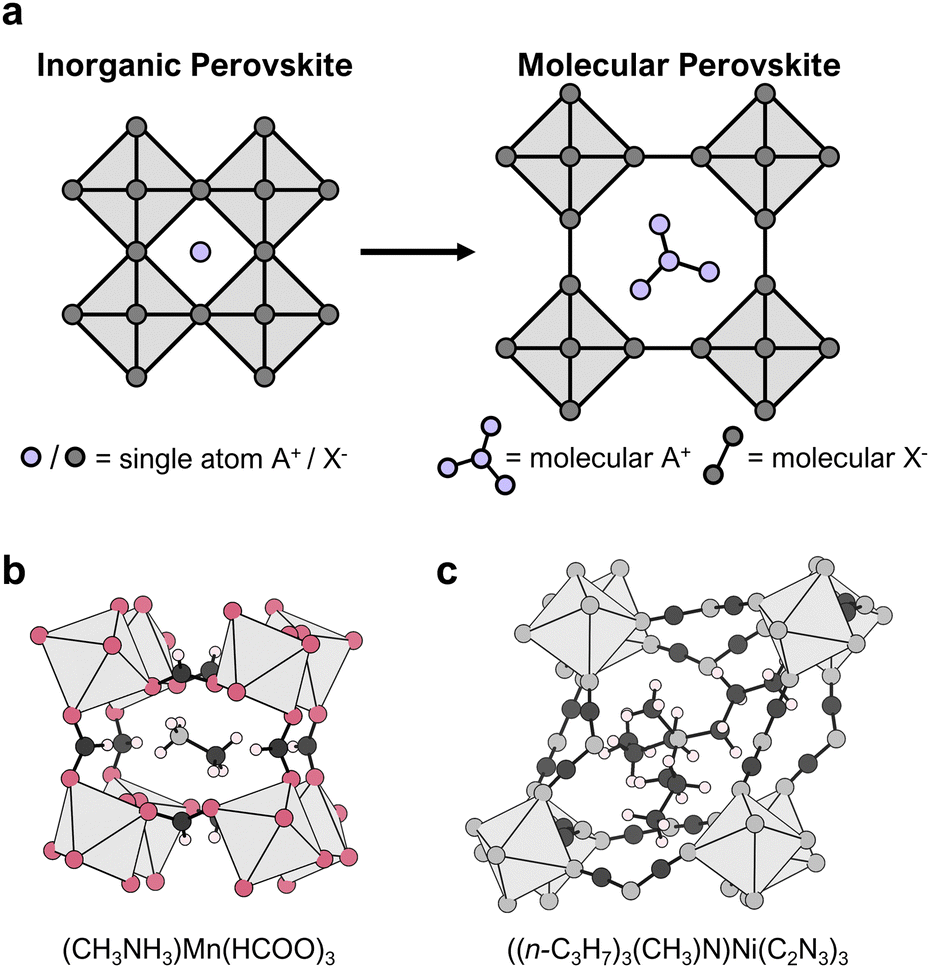 | ||
| Fig. 1 (a) 2D representation of the ABX3 perovskite building principle, emphasising the difference between inorganic perovskites and MolPs, where single atoms on the A- and X-site are substituted by molecular ions. In (b) and (c), two representative MolPs are shown, (b) (CH3NH3)Mn(HCOO)3 (CCDC number: 234550; colour code: Mn – light grey, O – red, N – grey, C – dark grey and H – light pink) and (c) ((n-C3H7)3(CH3)N)Ni(C2N3)3 (CCDC number: 2068844; colour code: Ni – light grey, C – dark grey, N – grey and H – light pink). | ||
The bond type in the 3D BX3 network of MolPs makes them insulators, and therefore, the interest of the material science community focuses on different aspects. For instance, some MolPs show pressure-driven phase transitions with large entropy changes, which renders them interesting for next-generation cooling approaches based on the barocaloric effect.38,39 Likewise, and very similar to their inorganic parents, the right combination of framework distortions breaks inversion symmetry, raising interest in the context of (improper) ferroelectrics and multiferroics.40 Additionally, their modular crystal structure enables the systematic study of how small chemical changes impact such properties and for developing knowledge-informed design principles for functional MolPs. This combination of fundamental structural chemistry and applied solid-state chemistry has initially sparked our interest and inspiration.
In this feature article, we survey important structure–chemical properties of MolPs. By drawing on research highlights from us and others, we provide a more personal reflection on how the field developed over the past decade, focusing on the interplay between composition, structure and properties. We close this feature article with an outlook, discussing some remaining milestones, including the development of synthetic guidelines for MolPs with targeted properties.
Structure–chemical properties
Historical remarks
The first report of MolPs dates back to 1973,41 but the relation to their inorganic parents was not mentioned until 2005.34,42 MolPs that were reported between 1973–2000 are based on relatively small X-site anions such as (HCOO)− and (N3)−. By establishing the diffusion-based room temperature crystallisation method, these early works laid the basis for all subsequent reports. Until today, the mild-solution approach in its variants is the prevailing synthetic approach. Starting from approximately 2003, research turned to larger X-site anions such as (C2N3)− and [M(CN)2]−. These larger anions increase the size of the ReO3-type network, enabling the use of larger and chemically more complex A-cations.43–45 A literature survey from us shows that MolPs encompass over 160 members, see Fig. 2 and Table S1 in the ESI.‡ Among all members, formate- and dicyanamide-based MolPs are the largest subclasses. They include the series of multiferroic ((CH3)2NH2)B(HCOO)334,40 and barocaloric ((n-C3H7)4N)B(C2N3)346,47 (B2+ = Mn2+, Fe2+, Co2+, etc.) and most of our understanding of structure–property relations has developed around these two families.48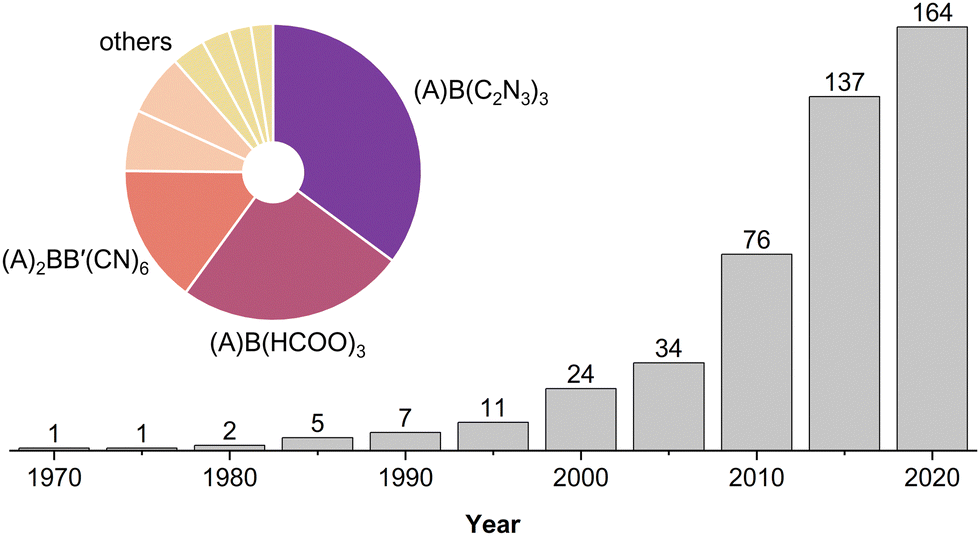 | ||
| Fig. 2 Evolution of ABX3 MolPs over time, with the accumulated number of MolPs given as a small number above the bars. The pie chart illustrates that the families of (A)B(C2N3)3 (purple), (A)B(HCOO)3 (red), and (A)2BB′(CN)6 (dark orange) are the largest. Summarised as others are the families of (A)B(N3)3 and (A)B(H2POO)3 (both light orange) and (A)2BB′(SCN)6, (A)B(ClO4)3, (A)B(BM4)3 and (A)B(M(CN)2)3 (all yellow). | ||
In total, over 50 different A-site cations have been employed. Based on the relation between [BX3]−-cage size and the available space for the A-cation, an increased chemical space is available when larger X-anions are used. Therefore, it can be approximated that the number of MolPs for a certain subclass scales with the [BX3]−-cage size. Looking at Fig. 2 and Table S1 (ESI‡), this is true for (C2N3)−, while only a few candidates for X− = [M(CN)2]− (M+ = Au+ and Ag+) have been reported. This implicates much room for the discovery of new compounds. Similar arguments can be made for the X-site families (A)2BB′(CN)6, (A)B(N3)3, (A)B(M(CN)2)3, (A)B(BM4)3 (M = F and H), (A)2BB′(SCN)6, (A)B(ClO4)3 and (A)B(H2POO)3 where commercially available A-cations have dominantly been used.
More than 50 years after the first report, MolPs have developed into a chemically rich material class, and it can be anticipated that many more MolPs will be discovered. Yet, the chemical space seems unlimited. Therefore, for streamlining the synthesis of new MolPs with interesting properties, synthetic guidelines coupled with concepts that facilitate the discovery of interesting and potentially functional properties, such as multiferroic behaviour, unusual temperature and pressure responses and large barocaloric effects, are required.
Stability criteria: tolerance factor as a synthetic guideline
Before discussing the role of molecular A- and X-site anions on the MolPs’ crystal chemistry, we shall first address the question of which A–B–X permutation can be expected to crystallise in a perovskite structure. In this context, the Goldschmidt's tolerance factor (α) is a central concept which has initially been developed for inorganic perovskites in 1926.49 For a given A–B–X combination, α can be calculated using the ionic radii of A, B and X, see eqn (1).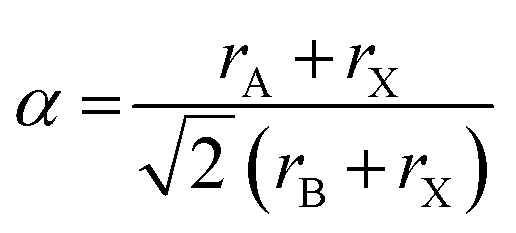 | (1) |
In 2014, we have extended this concept to HOIPs and MolPs.51 For calculating the sizes of molecular ions, A-site cations are approximated as rigid spheres with an effective radius rA,eff. Similarly, X-site anions are described as rigid cylinders with an effective radius rX,eff and an effective height hX,eff. For the B-site, the ionic radii from Shannon are used.52 This adaption modifies eqn (1) to eqn (2).53,54
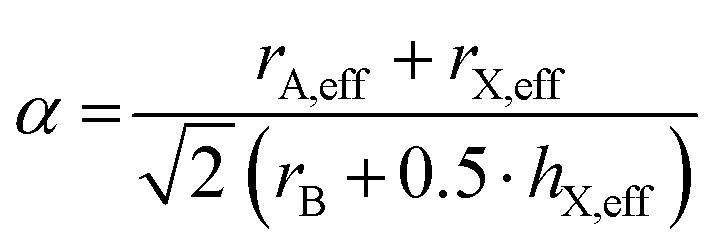 | (2) |
Describing a molecule as a sphere is a simplification that comes with the cost of accuracy. Nonetheless, there are numerous compound series where their stability can be rationalised with the tolerance factor argument alone.55 For instance, ((n-C2H5)4P)Mn(C2N3)356 and ((n-C3H7)4N)B(C2N3)3 (B2+ = Mn2+, Fe2+, Co2+ and Ni2+)57,58 both form perovskites, while the use of larger A-site cations to form ((n-C4H9)4N)B(C2N3)3 and ((n-C5H11)4N)B(C2N3)3 (B2+ = Mn2+, Fe2+ and Co2+)57,59 leads to different structure types. Likewise, a range of benzyltrialkylammonium A-site species shows that lower dimensional network structures are formed when the size of the A-site cation mismatches. ((C7H7)(CH3)3N)+ proved to be too small,60,61 while the larger cations ((C7H7)(C2H5)3N)+ and ((C7H7)(C4H9)3N)+ template the formation of perovskite-type ((C7H7)(C2H5)3N)B(C2N3)3 (B2+ = Mn2+ and Fe2+) and ((C7H7)(C4H9)3N)B(C2N3)3 (B2+ = Mn2+ and Co2+).44 Another example is (C10H20N)Cd(C2N3)3, which showcases the impact of the B-site metal's size. For B2+ = Cd2+, the [Cd(C2N3)3]− network is large enough for hosting (C10H20N)+ and a perovskite-type structure is formed. The network's size is significantly decreased for smaller metals such as B2+ = Mn2+, Co2+ and Ni2+, and diamond-like network structures are observed instead.43 Similar conclusions can be drawn for (NH4)B(HCOO)3, where ABX3 combinations with B2+ = Mn2+ and Cd2+ form perovskite structures,62,63 while B2+ = Cu2+ and Zn2+ are too small.64,65 Therefore, the tolerance factor sharpens our understanding of the role of the individual components’ sizes and has guided the interpretation of many research works in the context of MolPs43,46,55,58,66 and HOIPs54,67–69 over the last decade.
Looking at a more quantitative analysis which involves the calculation of individual αs, the absence of a concise and easy-to-adapt approach for extracting rA,eff for molecular cations from crystal structures has arguably hindered more rigorous bookkeeping. Over the last decade, several approaches were put forward to improve the A-site cation's size and shape description, as well as introducing anion dependent ionic radii for the metals.70,71 Focusing on the arguably most important work in this context, it was suggested to calculate the A-site cation's volume based on the isosurface of the molecule's electron density, where the geometric input comes from crystallographic data.48 This approach capitalises on the availability of free-of-charge analysis tools such as the CrystalExplorer software72 and improves the comparability of αs as calculated in different groups. By applying this methodology, it was shown that dicyanamide-based MolPs exhibit αs between 0.85 and 0.925, with only a few wrongly predicted examples.73 Elegantly, the A-site cation's globularity as a proxy of asphericity is calculated in parallel, which describes the characteristic shape of the molecular cations, with small globularity values indicating a strong asphericity. When interpreting a MolP as a coordination network that is templated by the A-cation, the importance of the A-site cation's size and globularity in dictating framework distortions is evident, although it has received only a little attention so far.48,74
Altogether, the tolerance factor is a good example of a concept that was initially developed in the context of inorganic condensed matter research and that was subsequently expanded to hybrid materials. Until today, it has helped to rationalise broader trends, guiding us and others on a day-to-day basis in the selection of potential A-site cations for the synthesis of new MolPs.
Framework distortions: octahedral tilts and shifts
We now discuss some of the structure–chemical ramifications of using molecules on the A- and X-site. Our starting point is again the family of inorganic perovskites where structural distortions can be categorised as B-site displacements, octahedral distortions and tilting of neighbouring octahedra.75,76 Here, we put the focus on framework distortions, which involve the tilting of corner-shared octahedra, see Fig. 3.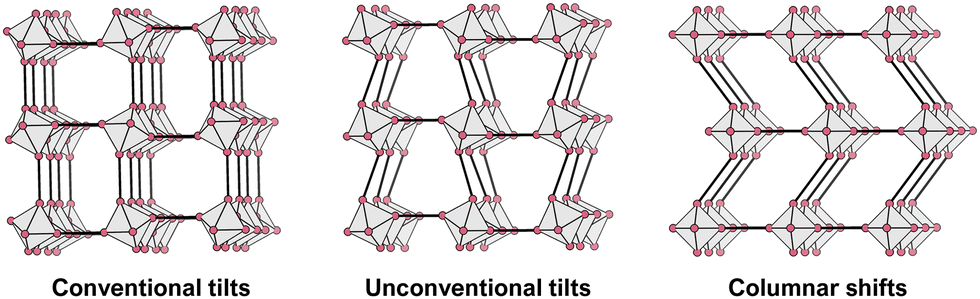 | ||
| Fig. 3 2D schematic of framework distortions occurring in MolPs. Conventional tilts can occur in perovskite oxides and MolPs, while unconventional tilts and columnar shifts also exist for MolPs. An atom-based connectivity, as observed in oxide perovskites, makes tilting of octahedra in the same direction and columnar shifts impossible. In other words, unconventional tilts and columnar shifts originate in the linker-based connectivity of neighbouring octahedra, and more generally, framework-type materials with polyatomic bridges can be expected to show a larger variety of framework distortions when compared to inorganic analogues. | ||
Concerted octahedral tilts distort the structure away from the cubic parent where no tilts exist. The occurrence of tilts is facilitated when α is smaller than 1, i.e. the size mismatch between A, B and X is compensated by framework distortions. This indicates that tilts and their amplitudes are susceptible to chemical composition, providing access to framework distortions through synthesis. This approach is known as tilt-engineering,77 can be used to break inversion symmetry and is, therefore, interesting for ferroelectrics and multiferroics. When considering tilts with a repeat unit of two octahedra in one direction, only a defined number of tilts is possible. These were initially categorised by M. Glazer in 1972 and are frequently referred to as Glazer tilts.75,78,79 Later on, group theory was applied to describe the active tilt distortions via their irreducible representation (irreps) with respect to their high-symmetry parent structure.75,80
For MolPs, the simple use of molecular X-site anions lifts some geometric constraints, thereby unlocking new distortions in the 3D network, see Fig. 3. In the so-called unconventional tilts, neighbouring octahedra tilt in the same direction.81 Additionally, columns and layers of octahedra can translate with respect to neighbouring layers and columns, a distortion that has been termed columnar shifts.82 In total, the possible set of framework distortions for MolPs encompasses conventional and unconventional tilts and columnar shifts.83 Notably, distortions with a repeat pattern of two octahedra can be described through their irreps with respect to a hypothetical parent, high-symmetry structure. For instance, ((CH3)4N)Cd(N3)384 and (C2H7N2])Mn(HCOO)385 are two examples where the active distortions are conventional out of phase octahedral tilts that transform with  as the primary order parameter. It should not go unmentioned, however, that examples with more complex repeat patterns exist where such a description is not meaningful anymore, including compounds such as (H2dabco)CsCl3 ((H2dabco)2+ = diprotonated 1,4-diazabicyclo[2.2.2]octane)86 and ((CH3)2NH2)Mn(H2POO)3.66
as the primary order parameter. It should not go unmentioned, however, that examples with more complex repeat patterns exist where such a description is not meaningful anymore, including compounds such as (H2dabco)CsCl3 ((H2dabco)2+ = diprotonated 1,4-diazabicyclo[2.2.2]octane)86 and ((CH3)2NH2)Mn(H2POO)3.66
Therefore, one important consequence of using molecular X-site ions is the increased number of available framework distortions, which strengthens tilt-engineering as a concept. In 2018, the theoretical groundwork was laid for how to break inversion symmetry through the targeted combination of distortion modes.38 Here, the group theoretical description of active distortions has proved immensely useful. An instructive example is (NH4)Cd(HCOO)3,63 which crystallises in the polar space-group Pna21. In total, three distortions are required to account for the compound's space-group symmetry: a planar shift which is described by the irrep 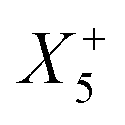 , an unconventional tilt that transforms as
, an unconventional tilt that transforms as 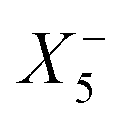 , and a conventional in-phase tilting with the irrep
, and a conventional in-phase tilting with the irrep  . Interestingly, the combination of shift distortions
. Interestingly, the combination of shift distortions 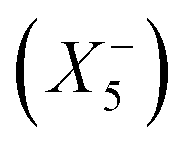 and unconventional tilts
and unconventional tilts 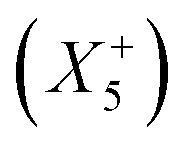 is already sufficient to break inversion symmetry. Therefore, the search for polar materials and potentially improper ferroelectrics translates to the question of how to design distortion modes via composition. Elegantly, irreps are also suitable for describing Jahn–Teller distortions, quadrupolar and dipolar A-site order. Together with framework distortions, a rich playground exists for designing distortion schemes and therewith polar materials. Yet, our understanding of the relation between composition and the active framework distortions is only developing, and so is our capability of chemical engineering targeted distortion schemes and their interplay. To fill this gap can be considered a milestone in the field.
is already sufficient to break inversion symmetry. Therefore, the search for polar materials and potentially improper ferroelectrics translates to the question of how to design distortion modes via composition. Elegantly, irreps are also suitable for describing Jahn–Teller distortions, quadrupolar and dipolar A-site order. Together with framework distortions, a rich playground exists for designing distortion schemes and therewith polar materials. Yet, our understanding of the relation between composition and the active framework distortions is only developing, and so is our capability of chemical engineering targeted distortion schemes and their interplay. To fill this gap can be considered a milestone in the field.
An overview study from 2020 has analysed distortion modes of over 70 MolPs with X-site anions such as (HCOO)−, (H2POO)−, (N3)−, (C2N3)− and [M(CN)2]−, revealing a relation between tilts and shifts and the used X-site anion.83 The linker geometry has been suggested as an important criterion, with bent X-site anions showing a tendency for conventional tilting, while linear X-site molecules such as (N3)− facilitate unconventional tilting. Columnar shifts are typically found in large [BX3]− network sizes such as ((n-C4H9)3(CH3)N)Mn(C2N3)387 and (((C6H5)3P)2N)Mn(Au(CN)2)3.81 Our future understanding of the link between composition and active distortion schemes in MolPs is linked to the rigorous report of distortion schemes when reporting new MolPs. The availability of free and user-friendly softwares88 is similarly important in this context, including their improvement towards batch analysis options and straightforward how-to instructions so that the barrier for novices for doing such an analysis is manageable.
Tilt and shift polymorphism in MolPs
We now turn our attention to a new form of polymorphism, which has its origin in the structure–chemical peculiarities of MolPs. Briefly, it should be iterated that temperature and pressure dependent polymorphism can play a defining role for a material's functionality. Likewise, pharmaceutics and mineralogy have taught us about the importance of metastable polymorphs that, compared to the thermodynamic stable phase, can show different physicochemical properties.89,90 MolPs have been reported exhibiting both stimuli-driven polymorphism and metastable polymorphic forms. In this part, the focus is put on metastable polymorphs while temperature- and pressure-driven phase transitions are discussed in the context of ferroelectrics and barocalorics.In 2021, our group discovered that depending on the synthetic conditions, two MolP-type polymorphs of ((n-C3H7)3(CH3)N)B(C2N3)3 (B2+ = Mn2+, Co2+ and Ni2+) can be obtained that are bench stable under ambient conditions.36 The polymorphs differ in the binding modes of the molecular anion (C2N3)− to the B-site metal, which results in different Glazer tilt systems. The different binding modes can be considered as conformational isomers and can be categorised after the anti–syn terminology as known from organic chemistry. Therefore, parallels can be drawn to conformational polymorphism found in molecular crystals,91 and this type of polymorphism is only possible for MolPs. We framed this phenomenon as tilt and shift polymorphism.
The metastable form of ((n-C3H7)3(CH3)N)B(C2N3)3 (B2+ = Mn2+, Co2+ and Ni2+) can be irreversibly transformed into the thermodynamic stable polymorph by temperature treatment. Therefore, the presence of tilt and shift polymorphism is detectable in calorimetry as an irreversible event. Previously, irreversible temperature induced heat signals have also been observed for ((C5H10)2N)Cd(C2N3)343 and ((C3H7)3N(C4H9))Mn(C2N3)3,92 which are likely related to tilt and shift polymorphism. Since all known examples involve relatively large X-site anions such as (H2POO)− and (C2N3)−,35,56,60,87 we propose that X-site anions with conformational degrees of freedom that allow variations of binding arrangements, torsion angles and distortions of the molecules with low energy differences promote the existence of tilt and shift polymorphism. The same train of thoughts can be applied to large A-site cations, e.g. tripropylmethyl-,36 benzyltriethyl-,44 tributylmethyl-ammonium,87 tetraethyl-56 and tributylmethyl-phosphonium,56 where different conformers can be expected to be relatively close in energy.
The existence of tilt and shift polymorphism is a direct manifestation of using molecular X-site anions. So far, only a limited number of cases are known which is related to difficulties in the targeted synthesis of single crystals of both polymorphs suitable for structural characterisation. We believe, however, that with further efforts in the synthesis of new MolPs and developments around tilt-engineering, new examples will be discovered soon.
The complexity of (molecular) perovskites
By now it is evident that MolPs exhibit a larger chemical space for their synthesis when compared to their inorganic parents. In the next paragraph, we address the question if this additional chemical space is related to an increase in their crystal structure's complexity. As we will see, answering this question fulfils curiosity, sharpens our intuition of their crystal chemistry, and might have consequences for the thermodynamic assessment of order–disorder phase transitions in MolPs in the future.The applied scheme for assessing the complexity was already put forward in 2012 by S. Krivovichev.93 In short, the more information that is required to describe a crystal structure, the more complex it is. Elegantly, this definition of a crystal structure's complexity transfers the challenge of assessing complexity to the question of how much information is contained in a given crystallographic information file, which can be tackled via information theory, see eqn (3) for the adapted Shannon's formula.93,94
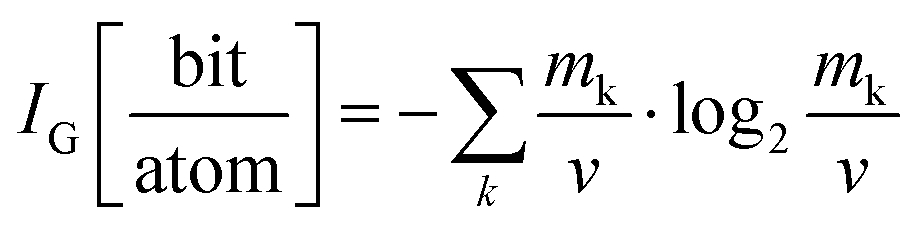 | (3) |
In 2022, we have performed a complexity analysis of 178 different perovskites, including inorganic perovskites, HOIPs and MolPs.97 A trend of increasing complexity, i.e. increasing IG, has been observed as a function of increasing B–B distance, which is taken as a proxy for the cage size, see Fig. 4. Given that a larger ReO3-type network requires a larger A-site cation with more carbon atoms, this finding is intuitive and agrees with chemical intuition. At this point, it is impossible to distinguish between a logarithmic or linear relationship trend; however, considering that the relative complexity increase is smaller when adding one atom to a complex structure compared to adding one atom to a less complex structure, we anticipate a logarithmic trend developing over time.
 | ||
| Fig. 4 Shown is the information content Ig,conf as calculated based on a *.cif file as a function of median B–B distance for a series of compounds that crystallise in the perovskite structure, see inset for colour code. The highlighted outliers are (H2dabco)CsCl3 (#), ((CH3)2NH2)Mn(H2POO)3 (*) and (PPN)Ni(Au(CN)2)3 (§) and are discussed in the text. Reproduced with permission from ref. 97. Copyright 2022, Royal Society of Chemistry. | ||
On a more practical aspect of such an analysis, the method is suitable for flagging outliers that exhibit structural anomalies. For instance, and as discussed above, (H2dabco)CsCl3 and ((CH3)2NH2)Mn(H2POO)3 exhibit complex octahedral distortion pattern, and accordingly, their complexities are higher than expected based on the median of their respective subclass, see Fig. 4. Likewise, (PPN)Ni(Au(CN)2)3 ((PPN)+ = bis(triphenylphosphine)iminium) is observed to have a relatively low complexity. When analysing the crystal structure, unusually large thermal ellipsoids point at some problems related to the crystal structure solution, which are absent for the more complex and most likely isostructural compound (PPN)Cd(Au(CN)2)3. Therefore, the method can serve as a tool for identifying interesting structure–chemical features and flag problems related to the underlying crystallography.
Looking forward, a relation between a crystal's structure information content and its configurational entropy has been suggested, which is most exciting for closing ties to applied properties.98 Currently, there are open questions related to the final formulas, but by now, it is clear that we should distinguish between the configurational entropy of a crystal structure and a disordered part, where the latter is best described as the entropy of mixing.99 It will be exciting to see how far this approach will bring us in the quantitative analysis of the configurational entropy,100 opening many exciting avenues, including the analysis of phase transitions, the evaluation of the thermodynamic stabilities of polymorphs and even a configurational analysis of ion diffusion pathways in solid-state electrolytes to name only a few.
MolPs and beyond: towards AB2X6 materials
The use of molecular X-site anions and A-site cations introduces new structure–chemical features, and it has proved useful to cast these as new structural degrees of freedom. For instance, in this picture, unconventional octahedral tilts relate to new rotational degrees of freedom and columnar shifts to new translational degrees of freedom. As recently noted by A. L. Goodwin and H. B. Boström,101 these structural degrees of freedom can be seen as structural elements and, when combined, have the potential to spark the development of new types of functional materials and properties, much like the discovery of new chemical elements enriched chemistry. This metaphor beautifully captures the opportunities of designing materials when using molecular building blocks and can be applied not only to MolPs but also to coordination networks more generally.Staying in this picture, the discovery of new structural degrees of freedom strengthens the overall concept. The use of molecular building blocks on the A-site opens the chemical toolbox for the targeted integration of new structural degrees of freedom. In 2022, we succeeded in introducing the spatial orientation of divalent A2+ cations within a ReO3-type [Mn(C2N3)3]− network as a new degree of freedom which is geometric in nature.102 Compared to A1+B2+X1−3 MolPs, an A2+ cation with two spatially separated charges is used, e.g. ((R)3N(CH2)nN(R′)3)2+ (R,R′ = alkyl chains), leading to the general formula A2+B2+2X1−6. The cationic charge centres of the A2+ cation are located in neighbouring [Mn(C2N3)3]− cages, forming a MolP where A+ cations are bridged by a –(CH2)n– chain. It was observed that depending on the –(CH2)n– chain length, different order patterns form, such as a herringbone-type pattern for n = 4 and a head-to-tail pattern for n = 5. Interestingly, a relation between pressure and temperature-responsiveness and the order pattern has been observed. For instance, the material with the herringbone-type order pattern exhibits negative linear compressibility, which is absent for the material with the head-to-tail ordering. Thus, the newly geometric degree of freedom couples to external stimuli, although it should be noted that the underlying crystal chemistry is a bit more complex, relying on an interplay between the A2+ cations and the 3D network.
This work showcases that MolPs and more general ReO3-type network structures,4,103 still provide a large chemical scope. Especially for synthetic chemists, the use of chemically tailored A-site cations, i.e. through simple organic chemistry based on substitution of amines, provides much room, which we see as key for learning of how to control distortion modes in MolPs and how to integrate additional functions in the future.
Towards applied properties
This section will discuss ferroelectric MolPs, their potential in solid-state refrigeration and their glass-forming properties. We will draw comparisons to the above-mentioned structure–chemical features, which will help pinpointing future opportunities within the respective fields as well as identifying open gaps that need to be filled to move MolPs closer to application.Ferroelectric MolPs
Ferroelectricity is the presence of a switchable electric polarization in a solid-state compound with inorganic perovskites such as BaTiO3 and PZT (PbZrxTi1−xO3) as commercially important examples. Since all ferroelectrics are also pyro- and piezoelectric, they cover a wide range of technologically relevant applications.104,105 A prerequisite for a material to be ferroelectric is the absence of inversion symmetry and the existence of a polar axis; in other words, designing a ferroelectric is bound to our capability to break inversion symmetry by targeting structural distortions via composition. For oxide-based perovskites, this has proved more difficult as initially anticipiated.106The situation is slightly different in MolPs, where additional distortion modes are available, that, when coupled in the right way,38 can lead to improper ferroelectric behaviour.107 (NH4)Cd(HCOO)3 was the aforementioned example, and in a similar way, polarity of ((NH2)3C)Cu(HCOO)3,108 ((n-C3H7)(C2H5)3P)Mn(C2N3)3,109 (C4H10N)Cd(H2POO)3110 and ((CH3OCH2)(C2H5)3P)Mn(C2N3)3109 can be explained. Therefore, there is on-going effort in learning of how to control distortion modes and their amplitudes,111 where exploiting organic chemistry related to A-site cations is the most intuitive starting point for steering framework distortions.
Looking at more qualitative synthetic strategies, parallels can be drawn to the field of molecular materials, where a few phenomenological principles exist that lead to polar compounds.112,113 MolPs and closely related materials can benefit from such principles due to the chemical freedom as provided by the A-site cation. Examples are the hybrid organic–inorganic hexagonal perovskites A′MnCl3 and ACdBr3, where the substitution of A+ = ((CH3)4N)+ with less symmetric cations such as A+ = ((CH3)3NCH2Cl)+ or A+′ = ((CH3)3NCH2Br)+ led to large spontaneous polarization.114,115 Likewise, this concept can be used to increase the polarization of an already polar MolPs, which was introduced as molecular tailoring for metal-free ferroelectric perovskites.112 The change in polarization in MolPs when going from ((C2H5)4P)Mn(C2N3)3 to ((n-C4H9)(CH3)P)Mn(C2N3)3 can thus be rationalised among other examples.56
So tailored chemical modifications of the A-site cation bring many opportunities in making MolPs polar. The synthesis of A-site solid solutions provides another handle that can even lead to polar materials despite both endmembers being non-polar.116 In this regard, we can note that step by step, the community is getting closer in the rational synthesis of polar MolPs; yet, it is a challenging and multidisciplinary task. Here, we would like to stress the importance of subject boundary-crossing works related to the conceptual understanding that drives ferroelectricity in individual MolPs based on conceptual toy-models and in-depth analysis of distortion modes.38,117 We believe these are key in the development and, when synergistically combined with synthetic work, will soon bring more fascinating breakthroughs to fruition.118
Melting and glass-forming MolPs
The discovery of meltable, glass-forming MolPs was only in 2021.119 Conceptually, MolP forming glasses such as ((n-C3H7)4N)B(C2N3)3 (B2+ = Mn2+, Co2+ and Fe2+),119 ((n-C4H9)3(CH3)N)B(C2N3)3 (B2+ = Mn2+, Fe2+, Co2+ and Ni2+)87 and ((n-C3H7)3(n-C4H7)N)Mn(C2N3)3,92 are a subclass of glass-forming coordination polymers.120–122 Although this is still a quite young area, several glass-forming coordination polymers are known today, such as 3D network materials including zeolitic imidazolate frameworks123,124 and carboxylate-based metal–organic frameworks,125,126 and coordination polymers with extended networks in 2D and 1D such gold thiolate-based127 and Zn-phosphate-based128,129 coordination polymers. The synthetic challenge for designing meltable coordination polymers lies in engineering the melting temperature Tm so that Tm drops below the decomposition temperature Td.122,130,131 Given the thermal liability of molecular fragments, melting is still considered an atypical behaviour for most coordination polymers. After melting, glass formation occurs during cooling.Regarding MolPs, melting has only been reported for a few members of the (A)B(C2N3)3 family.132 This highlights the role of the (C2N3)− ligand, which, as a relatively large ligand, allows for integrating larger quaternary ammonium species such as ((n-C3H7)3(n-C4H9)N)+, ((n-C3H7)4N)+ and ((n-C4H9)3(CH3)N)+ that push Tm below Tdvia larger entropic contributions. The size effect of the A-site cation on Tm has been investigated through A-site substitution in a (A)B(C2N3)3 series, where a size increase, i.e. by an increase of the alkyl chains from ((n-C3H7)4N)+ < ((n-C4H9)4N)+ < ((n-C5H11)4N)+, decreases Tm; however, the structure of the parent phase also changed, making it difficult to isolate the effect of the A-site cation on Tm.59 In addition, the impact of the divalent metal has been explored, showing a decrease of Tm along the series Mn2+ > Fe2+ > Co2+.59,119 We would like to note that this trend is not observed for the meltable series of ((n-C4H9)3(CH3)N)B(C2N3)3 with B2+ = Mn2+, Fe2+, Co2+ and Ni2+,87 where the highest Tm is observed for ((n-C4H9)3(CH3)N)Ni(C2N3)3. It seems that argumentations based on ligand-field stabilisation energy and the hard soft acid base concept must be carefully evaluated. Further studies are required to understand the detailed role of B2+, with opportunities for fine-tuning Tm.
More recently, our group has turned towards engineering properties of MolP-based glasses with focus on Tmvia the use of modifiers.92 This was inspired by work on inorganic glasses where modifiers are established for tuning glass properties.133 In our work, we showcased how a suitable modifier, e.g. Li(C2N3), lowers Tm of ((n-C3H7)3(n-C4H9)N)Mn(C2N3)3 through the formation of an eutectic mixture. This has opened the gap between Tm and Td, suppressing partial decomposition of individual molecular components. Such a partial decomposition is a parasitic process that occurs during melting when Tm and Td are too close. Additionally, slow cooling of the mixture enables a scalable glass formation compared to more established melt quenching methods, e.g. thermogravimetric analysis-differential scanning calorimetry-based routines, where typically only little amounts are obtained.
First principles of dicyanamide-based glasses that relate Tm to crystal chemical principles appeared, including parallels drawn to ionic liquids by us and others.59,92 There are yet open questions to tackle, including the question of how to integrate functionality into MolP-derived134 and, more generally, coordination polymer-derived glasses. Overall, the discovery of coordination polymer-based glasses is fascinating, filling a long-standing gap between inorganic glasses, metal-based glasses, and polymer-based glasses, with many unknown opportunities ahead.
Barocaloric cooling with MolPs
A regularly observed feature of MolPs is the A-site order–disorder phase transition behaviour as a response to temperature or pressure variation. In 2017, the pressure-driven phase transition in ((n-C3H7)4N)Mn(C2N3)3 has been recognised as a functional response in the context of solid-state refrigeration based on the barocaloric effect.46 In short, in barocalorics, a pressure-driven phase transition is used to run a thermodynamic cycle, with the pressure dependent isothermal entropy change as a major performance criteria.135 Materials with giant caloric effects, i.e. ΔS > 10 J K−1 kg−1, offer potential as an alternative cooling technology. Having in mind that commonly applied refrigerants are green-house gases, the opportunity of a new, greenhouse-gas free refrigerating technology based on the barocaloric effect is intriguing.136Solid-state refrigeration based on the barocaloric effect is a young area. Material testing is still a core task, and the identification of design guidelines for materials with optimised barocaloric properties is a key milestone.137 This is, however, a multidimensional challenge: in addition to performance-related criteria such as isothermal entropy change, barocaloric coefficient, small hysteresis, and low operating pressure, a good barocaloric also checks with more applied criteria such as suitable thermal conductivity, high elemental abundance, low toxicity and long material lifetime to name only the most important points.
Some examples of MolPs with a giant barocaloric response are ((n-C3H7)4N)B(C2N3)3 (B2+ = Mn2+ and Cd2+)46,47 and ((CH3)4N)B(N3)3 (B2+ = Mn2+ and Cd2+).84,138,139 Compared to other state-of-the-art barocalorics such as neopentylglycol ((CH3)2C(CH2OH)2),140–142n-alkanes143,144 or molecular Fe(II) spin-crossover compounds ([FeL2][BF4]2 with L = 2,6-di(pyrazol-1-yl)pyridine),145 which require high driving pressures (p > 1000 bar),137 MolPs generate giant isothermal entropy changes at already small working pressures such as p < 70 bar.46 We would like to note that the ((CH3)2NH2)Mg(HCOO)3 presents one example which shows a giant barocaloric effect (ΔS ∼ 39 J K−1 kg−1) at a relatively large pressure, i.e. p < 2000 bar;146 however, when using the barocaloric strength as a benchmark, i.e. ΔS/Δp, ((n-C3H7)4N)Mn(C2N3)3 outperforms many other competing barocalorics.48
Strikingly, first reports have appeared based on knowledge-informed material optimisation. For instance, after recognising the importance of alkane-chain disorder as source of entropy changes,143 alkylammonium cations with relatively large chains such as A+ = (C10H21NH3)+ and (C9H19NH3)+ have been incorporated into a 2D layered metal-halide perovskite, overall resulting in what is considered a colossal barocaloric effect with ΔS > 200 J K−1 kg−1 at p < 300 bar.147 This example showcases the value of design principles and fosters material class independent, critical evaluation of structural features that lead to a large barocaloric performance.
In this context, we see the role of MolPs as a material platform for systematically testing working hypotheses of how to address barocaloric properties via compositional and structural changes. For instance, the phase transition temperature (Ttrs) has been found to be sensitive to the choice of the B2+ with Ttrs increasing along the B-site metal series (B2+ = Mn2+ > Co2+ > Ni2+).36 A similar tunability has been reported for the compressibility, which is intuitively coupled to the barocaloric coefficient (dTc/dp).142,148 We anticipate that more of such reports are coming soon, step-by-step teaching us of how to design high-performance barocalorics for replacing established refrigeration technologies.
Perspective and conclusion
To conclude this feature paper, so far, we have mentioned the key structure–chemical properties and principles of MolPs, and by discussing three application-oriented areas, we addressed the question of how such principles can become useful in the context of applied solid-state chemistry. We want to close this feature article by mentioning two important points that, in our opinion, will be important in shaping the future of MolPs as a material class.MolPs as material family – expansion?
The majority of reported MolPs rely on commercially available A-site cations, providing much room for new MolPs based on tailor-made A-site cations. The above statistical analysis shows that we only scratched on the surface of MolPs based on X-site anions such as [Au(CN)2]− or (N3)−. Therefore, there is great scope for the discovery of many new MolPs without even having considered the use of larger yet unknown X-site anions, the synthesis of solid–solution for fine-tuning properties111,149–151 and the study of the underlying defect chemistry.152,153In going forward, it is important to ask if the discovery of new MolPs is equal to expanding our understanding of their crystal chemistry, and if it is possible to facilitate the synthesis of MolPs with interesting physicochemical properties? We think this is true if synthetic efforts go parallel with the continuous application and improvement of intuitive principles and descriptive approaches such as the tolerance factor concept, its data analysis-derived analogue67 or the distortion mode analysis.
When A-site cations become larger, they exhibit a larger number of conformers that differ in energy, size and shape. Including such considerations into the synthetic guidelines, such as the tolerance factor, seems fruitful, and the required workflow for efficiently analysing the A-cation's conformational energy landscape has recently been developed.154 From our current understanding, the A-site cations must match in size and shape with B and X so that the MolP forms. Moreover, we recently observed that the combination of size and shape plays a large role in dictating distortion amplitudes,111 providing a new perspective in tuning these. Therefore, exploiting the A-site cation's chemistry, size and shape provides chemical access to tilt distortion amplitudes and arguably their nature. Additionally, and as emphasised above, interesting physicochemical properties and phenomena rely on an interplay of various structure–chemical aspects where the A-site exhibits an important templating role. Therefore, from our current understanding, it is this unique feature – the molecular nature of the A-site cation – that should be put in the centre of synthetic efforts as an approach for controlling the crystal chemistry of MolPs and, therewith, their properties.
MolPs in material science – functionality ?
So, what lies beyond the fascinating analogies that can be drawn between MolPs and their inorganic parents? The future relevance of MolPs as a material class is closely coupled to the question of identifying material functionality that is, at least to some extent, unique to MolPs.The most intuitive approach in this regard is the above-mentioned goal of controlling framework distortions for incorporating improper ferroelectric behaviour. As we already know how to tune distortion mode amplitudes,111 knowledge that may be used to increase the polarization of an existing polar MolP, we may have achieved step two before step one. Given the many synthetic opportunities that we have, it seems only a matter of time until we know how to engineer distortion modes via A-site chemistry in MolPs.
Within the area of barocalorics, MolPs have proved as interesting model candidates, especially when including layered systems such as (C10H21NH3)2MnCl4.147,155 For the barocaloric ((n-C3H7)4N)Mn(C2N3)3, we can observe that the order–disorder phase transition is not only related to the A-site cation as observed for many MolPs, but that significant disorder is also occurring in the 3D [Mn(C2N3)3]− network.46 In other words, and drawing comparisons to the plastic crystal neopentylglycol as benchmark material,141,142 ((n-C3H7)4N)Mn(C2N3)3 can be considered a plastic network, where the A-site and the 3D network contribute to the phase transition thermodynamics. A similar behaviour is observed for some of the azides, such as ((CH3)4N)Mn(N3)3 and related compounds.37,156 Therefore, if we can understand and subsequently design contributions from the 3D network to the phase transition energetics, MolPs might indeed become promising contenders as barocaloric working media.39
As a last point, we would like to mention the potential of meltable MolPs. In the attempt to bring functionality in the glass product, the use of functional modifiers or functional building blocks in the parent crystalline MolP phase seems most promising. These could be chiral A-site cations157 or redox-chemistry accessible transition metals.134 The emerging principles and the knowledge that we have from inorganic glasses provide a flying start for establishing and rationalising composition–property relations in a short term. Notably, for a potential functional coordination polymer-based glass, the parent phase, i.e. in our case a MolP, only plays a subsidiary role, and the individual building blocks are more important; however, the modular building principle of MolPs enables to systematically explore the impact of composition and the individual components’ role on the melting process while excluding a large impact of the starting phase structure. Overall, we expect that more meltable MolPs will be discovered over time, and we are convinced that they can play a key role in establishing principles of meltable coordination polymers and in bringing functions into coordination polymer-based glasses.
This brings us to our final thought. Due to the relation between MolPs to inorganic perovskites, MolPs will maintain a material class of large interest. It is also clear that advances related to the discovery of functional MolPs are a multidisciplinary research challenge, crossing synthetic organic chemistry, inorganic structural science and physicochemical chemistry, see Fig. 5. The height of relevance of MolPs as an independent material family, however, will be closely bound to the discovery of new and improved functionalities that have the potential to reignite the fascination of a wider materials science community.
 | ||
| Fig. 5 An artistic illustration highlighting the multidisciplinary research topics related to MolPs. | ||
Author contributions
S. M. Kronawitter and G. Kieslich contributed to the conceptualisation, review design, writing and editing.Data availability
This is a feature article and consequently, no original new research data is being reported; however, in the article we perform a literature survey. The result of the survey is given in the attached ESI‡ as tabulated data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M. K. and G. K. acknowledge support from the DFG through research grants (450070835) and the Heisenberg program (524525093).Notes and references
- C. L. Hobday and G. Kieslich, Dalton Trans., 2021, 50, 3759–3768 RSC.
- A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780–4795 RSC.
- O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS.
- W. Li, Z. Wang, F. Deschler, S. Gao, R. H. Friend and A. K. Cheetham, Nat. Rev. Mater., 2017, 2 CAS.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 CrossRef CAS.
- R.-B. Lin, Z. Zhang and B. Chen, Acc. Chem. Res., 2021, 54, 3362–3376 CrossRef CAS PubMed.
- B. Park, B. Philippe, S. M. Jain, X. Zhang, T. Edvinsson, H. Rensmo, B. Zietz and G. Boschloo, J. Mater. Chem. A, 2015, 3, 21760–21771 RSC.
- J. Albero, A. M. Asiri and H. García, J. Mater. Chem. A, 2016, 4, 4353–4364 RSC.
- D. W. Lewis, A. R. Ruiz-Salvador, A. Gómez, L. M. Rodriguez-Albelo, F.-X. Coudert, B. Slater, A. K. Cheetham and C. Mellot-Draznieks, CrystEngComm, 2009, 11, 2272 RSC.
- X. Gao, X. Zhang, W. Yin, H. Wang, Y. Hu, Q. Zhang, Z. Shi, V. L. Colvin, W. W. Yu and Y. Zhang, Adv. Sci., 2019, 6, 1900941 CrossRef CAS PubMed.
- D. B. Mitzi, C. A. Feild, W. T. A. Harrison and A. M. Guloy, Nature, 1994, 369, 467–469 CrossRef CAS.
- R. Robson, Dalton Trans., 2008, 5113–5131 RSC.
- G. Rose, Ann. Phys., 1839, 124, 551–573 CrossRef.
- St. v Náray-Szabó, Naturwissenschaften, 1943, 31, 202–203 CrossRef.
- A. von Hippel, Rev. Mod. Phys., 1950, 22, 221–237 CrossRef CAS.
- H. D. Megaw, Nature, 1945, 155, 484–485 CAS.
- B. Jaffe, R. S. Roth and S. Marzullo, J. Appl. Phys., 1954, 25, 809–810 CrossRef CAS.
- G. A. Smolenskii and A. I. Agranovskaya, Sov. Phys. Tech. Phys., 1958, 28, 1380–1389 Search PubMed.
- A. R. Chakhmouradian and P. M. Woodward, Phys. Chem. Miner., 2014, 41, 387–391 CrossRef CAS.
- A. Callaghan, C. W. Moeller and R. Ward, Inorg. Chem., 1966, 5, 1572–1576 CrossRef CAS.
- T. Fukuda and Y. Uematsu, Jpn. J. Appl. Phys., 1972, 11, 163–169 CrossRef CAS.
- H. Yamaguchi, K. Katsumata, M. Hagiwara, M. Tokunaga, H. L. Liu, A. Zibold, D. B. Tanner and Y. J. Wang, Phys. Rev. B, 1999, 59, 6021–6023 CrossRef CAS.
- M. Safa and B. K. Tanner, Phys. B + C, 1977, 86–88, 1347–1348 CrossRef.
- A. Moreira dos Santos, S. Parashar, A. R. Raju, Y. S. Zhao, A. K. Cheetham and C. Rao, Solid State Commun., 2002, 122, 49–52 CrossRef CAS.
- M. K. Wu, J. R. Ashburn, C. J. Torng, P. H. Hor, R. L. Meng, L. Gao, Z. J. Huang, Y. Q. Wang and C. W. Chu, Phys. Rev. Lett., 1987, 58, 908–910 CrossRef CAS PubMed.
- F. S. Galasso, Perovskites and high Tc superconductors, Gordon and Breach Science Publishers, New York, 1990 Search PubMed.
- D. Weber, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1978, 33, 1443–1445 CrossRef.
- D. Weber, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1978, 33, 862–865 CrossRef.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- Z. Liang, Y. Zhang, H. Xu, W. Chen, B. Liu, J. Zhang, H. Zhang, Z. Wang, D.-H. Kang, J. Zeng, X. Gao, Q. Wang, H. Hu, H. Zhou, X. Cai, X. Tian, P. Reiss, B. Xu, T. Kirchartz, Z. Xiao, S. Dai, N.-G. Park, J. Ye and X. Pan, Nature, 2023, 624, 557–563 CrossRef CAS PubMed.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- M. M
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) czka, M. Ptak, A. Ggor, D. Stefańska, J. K. Zarȩba and A. Sieradzki, Chem. Mater., 2020, 32, 1667–1673 CrossRef.
czka, M. Ptak, A. Ggor, D. Stefańska, J. K. Zarȩba and A. Sieradzki, Chem. Mater., 2020, 32, 1667–1673 CrossRef. - H. R. Petrosova, O. I. Kucheriv, S. Shova and I. A. Gural'skiy, Chem. Commun., 2022, 58, 5745–5748 RSC.
- P. Jain, N. S. Dalal, B. H. Toby, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2008, 130, 10450–10451 CrossRef CAS PubMed.
- Y. Wu, S. Shaker, F. Brivio, R. Murugavel, P. D. Bristowe and A. K. Cheetham, J. Am. Chem. Soc., 2017, 139, 16999–17002 CrossRef CAS PubMed.
- S. Burger, S. Grover, K. T. Butler, H. L. B. Boström, R. Grau-Crespo and G. Kieslich, Mater. Horiz., 2021, 8, 2444–2450 RSC.
- X.-H. Zhao, X.-C. Huang, S.-L. Zhang, D. Shao, H.-Y. Wei and X.-Y. Wang, J. Am. Chem. Soc., 2013, 135, 16006–16009 CrossRef CAS PubMed.
- H. L. B. Boström, M. S. Senn and A. L. Goodwin, Nat. Commun., 2018, 9, 2380 CrossRef PubMed.
- J. M. Bermúdez-García, M. Sánchez-Andújar and M. A. Señarís-Rodríguez, J. Phys. Chem. Lett., 2017, 8, 4419–4423 CrossRef PubMed.
- P. Jain, V. Ramachandran, R. J. Clark, H. D. Zhou, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625–13627 CrossRef CAS PubMed.
- E. Sletten and L. H. Jensen, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 1752–1756 CrossRef CAS.
- H. F. Clausen, R. D. Poulsen, A. D. Bond, M.-A. S. Chevallier and B. B. Iversen, J. Solid State Chem., 2005, 178, 3342–3351 CrossRef CAS.
- S. Burger, S. Kronawitter, H. L. B. Boström, J. K. Zaręba and G. Kieslich, Dalton Trans., 2020, 49, 10740–10744 RSC.
- M.-L. Tong, J. Ru, Y.-M. Wu, X.-M. Chen, H.-C. Chang, K. Mochizuki and S. Kitagawa, New J. Chem., 2003, 27, 779–782 RSC.
- P. M. van der Werff, E. Martínez-Ferrero, S. R. Batten, P. Jensen, C. Ruiz-Pérez, M. Almeida, J. C. Waerenborgh, J. D. Cashion, B. Moubaraki, J. R. Galán-Mascarós, J. M. Martínez-Agudo, E. Coronado and K. S. Murray, Dalton Trans., 2005, 285–290 RSC.
- J. M. Bermúdez-García, M. Sánchez-Andújar, S. Castro-García, J. López-Beceiro, R. Artiaga and M. A. Señarís-Rodríguez, Nat. Commun., 2017, 8, 15715 CrossRef PubMed.
- J. M. Bermúdez-García, S. Yáñez-Vilar, A. García-Fernández, M. Sánchez-Andújar, S. Castro-García, J. López-Beceiro, R. Artiaga, M. Dilshad, X. Moya and M. A. Señarís-Rodríguez, J. Mater. Chem. C, 2018, 6, 9867–9874 RSC.
- J. García-Ben, L. N. McHugh, T. D. Bennett and J. M. Bermúdez-García, Coord. Chem. Rev., 2022, 455, 214337 CrossRef.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- W. B. Jensen, J. Chem. Educ., 2010, 87, 587–588 CrossRef CAS.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2014, 5, 4712–4715 RSC.
- R. D. Shannon, Acta Cryst. A, 1976, 32, 751–767 CrossRef.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2015, 6, 3430–3433 RSC.
- M. Becker, T. Klüner and M. Wark, Dalton Trans., 2017, 46, 3500–3509 RSC.
- S. Burger, M. G. Ehrenreich and G. Kieslich, J. Mater. Chem. A, 2018, 6, 21785–21793 RSC.
- Y.-Q. Wu, J.-Y. Zhang, X. He, Z.-X. Wang, H.-L. Cai and M.-X. Li, Cryst. Growth Des., 2021, 21, 6245–6253 CrossRef CAS.
- J. A. Schlueter, J. L. Manson and U. Geiser, Inorg. Chem., 2005, 44, 3194–3202 CrossRef CAS PubMed.
- J. M. Bermúdez-García, M. Sánchez-Andújar, S. Yáñez-Vilar, S. Castro-García, R. Artiaga, J. López-Beceiro, L. Botana, A. Alegría and M. A. Señarís-Rodríguez, J. Mater. Chem. C, 2016, 4, 4889–4898 RSC.
- B. K. Shaw, C. Castillo-Blas, M. F. Thorne, M. L. Ríos Gómez, T. Forrest, M. D. Lopez, P. A. Chater, L. N. McHugh, D. A. Keen and T. D. Bennett, Chem. Sci., 2022, 13, 2033–2042 RSC.
- M. Mączka, A. Gągor, J. K. Zaręba, M. Trzebiatowska, D. Stefańska, E. Kucharska, J. Hanuza, N. Pałka, E. Czerwińska and A. Sieradzki, Dalton Trans., 2021, 50, 10580–10592 RSC.
- M. Mączka, A. Gągor, A. Stroppa, J. N. Gonçalves, J. K. Zaręba, D. Stefańska, A. Pikul, M. Drozd and A. Sieradzki, J. Mater. Chem. C, 2020, 8, 11735–11747 RSC.
- J.-Q. Liu, J. Wu, J. Wang, L. Lu, C. Daiguebonne, G. Calvez, O. Guillou, H. Sakiyama, N. S. Weng and M. Zeller, RSC Adv., 2014, 4, 20605 RSC.
- L. C. Gómez-Aguirre, B. Pato-Doldán, A. Stroppa, S. Yáñez-Vilar, L. Bayarjargal, B. Winkler, S. Castro-García, J. Mira, M. Sánchez-Andújar and M. A. Señarís-Rodríguez, Inorg. Chem., 2015, 54, 2109–2116 CrossRef PubMed.
- P. Peksa, J. K. Zarȩba, M. Ptak, M. Mczka, A. Ggor, S. Pawlus and A. Sieradzki, J. Phys. Chem. C, 2020, 124, 18714–18723 CrossRef CAS.
- G.-C. Xu, X.-M. Ma, L. Zhang, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2010, 132, 9588–9590 CrossRef CAS PubMed.
- Y. Wu, T. Binford, J. A. Hill, S. Shaker, J. Wang and A. K. Cheetham, Chem. Commun., 2018, 54, 3751–3754 RSC.
- C. J. Bartel, C. Sutton, B. R. Goldsmith, R. Ouyang, C. B. Musgrave, L. M. Ghiringhelli and M. Scheffler, Sci. Adv., 2019, 5, eaav0693 CrossRef CAS PubMed.
- Q. Sun and W.-J. Yin, J. Am. Chem. Soc., 2017, 139, 14905–14908 CrossRef CAS PubMed.
- X. Cai, Y. Zhang, Z. Shi, Y. Chen, Y. Xia, A. Yu, Y. Xu, F. Xie, H. Shao, H. Zhu, D. Fu, Y. Zhan and H. Zhang, Adv. Sci., 2022, 9, e2103648 CrossRef PubMed.
- S. Gholipour, A. M. Ali, J.-P. Correa-Baena, S.-H. Turren-Cruz, F. Tajabadi, W. Tress, N. Taghavinia, M. Grätzel, A. Abate, F. de Angelis, C. A. Gaggioli, E. Mosconi, A. Hagfeldt and M. Saliba, Adv. Mater., 2017, 29 Search PubMed.
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- J. García-Ben, A. García-Fernández, P. Dafonte-Rodríguez, I. Delgado-Ferreiro, U. B. Cappel, S. Castro-García, M. Sánchez-Andújar, J. M. Bermúdez-García and M. A. Señarís-Rodríguez, J. Solid State Chem., 2022, 316, 123635 CrossRef.
- I. E. Collings, J. A. Hill, A. B. Cairns, R. I. Cooper, A. L. Thompson, J. E. Parker, C. C. Tang and A. L. Goodwin, Dalton Trans., 2016, 45, 4169–4178 RSC.
- P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 32–43 CrossRef.
- P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 44–66 CrossRef.
- M. J. Pitcher, P. Mandal, M. S. Dyer, J. Alaria, P. Borisov, H. Niu, J. B. Claridge and M. J. Rosseinsky, Science, 2015, 347, 420–424 CrossRef CAS PubMed.
- A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3384–3392 CrossRef CAS.
- A. M. Glazer, Phase Transit., 2011, 84, 405–420 CrossRef CAS.
- C. J. Howard and H. T. Stokes, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 782–789 CrossRef.
- J. A. Hill, A. L. Thompson and A. L. Goodwin, J. Am. Chem. Soc., 2016, 138, 5886–5896 CrossRef CAS PubMed.
- H. L. B. Boström, J. A. Hill and A. L. Goodwin, Phys. Chem. Chem. Phys., 2016, 18, 31881–31894 RSC.
- H. L. B. Boström, CrystEngComm, 2020, 22, 961–968 RSC.
- Z.-Y. Du, Y.-P. Zhao, W.-X. Zhang, H.-L. Zhou, C.-T. He, W. Xue, B.-Y. Wang and X.-M. Chen, Chem. Commun., 2014, 50, 1989–1991 RSC.
- M. Mączka, J. Janczak, M. Trzebiatowska, A. Sieradzki, S. Pawlus and A. Pikul, Dalton Trans., 2017, 46, 8476–8485 RSC.
- L. A. Paton and W. T. A. Harrison, Angew. Chem., Int. Ed., 2010, 49, 7684–7687 CrossRef CAS PubMed.
- M. Mączka, A. Gągor, M. Ptak, D. Stefańska, L. Macalik, A. Pikul and A. Sieradzki, Dalton Trans., 2019, 48, 13006–13016 RSC.
- B. J. Campbell, H. T. Stokes, D. E. Tanner and D. M. Hatch, J. Appl. Crystallogr., 2006, 39, 607–614 CrossRef CAS.
- K. Simkiss, Nature, 1964, 201, 492–493 CrossRef CAS.
- J.-P. Brog, C.-L. Chanez, A. Crochet and K. M. Fromm, RSC Adv., 2013, 3, 16905 RSC.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170–2191 CrossRef CAS PubMed.
- S. M. Kronawitter, S. A. Hallweger, J. Meyer, C. Pedri, S. Burger, A. Alhadid, S. Henke and G. Kieslich, APL Mater., 2023, 11, 31119 CrossRef CAS.
- S. Krivovichev, Acta Cryst. A, 2012, 68, 393–398 CrossRef CAS.
- S. V. Krivovichev, Angew. Chem., Int. Ed., 2014, 53, 654–661 CrossRef CAS PubMed.
- W. Hornfeck, Z. Kristallogr. - Cryst. Mater., 2022, 237, 127–134 CrossRef CAS.
- D. A. Banaru, W. Hornfeck, S. M. Aksenov and A. M. Banaru, CrystEngComm, 2023, 25, 2144–2158 RSC.
- S. A. Hallweger, C. Kaußler and G. Kieslich, Phys. Chem. Chem. Phys., 2022, 24, 9196–9202 RSC.
- S. V. Krivovichev, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 274–276 CrossRef CAS PubMed.
- S. V. Krivovichev, V. G. Krivovichev, R. M. Hazen, S. M. Aksenov, M. S. Avdontceva, A. M. Banaru, L. A. Gorelova, R. M. Ismagilova, I. V. Kornyakov, I. V. Kuporev, S. M. Morrison, T. L. Panikorovskii and G. L. Starova, Mineral. Mag., 2022, 86, 183–204 CrossRef.
- A. E. Phillips and H. C. Walker, J. Phys. Energy, 2024, 6, 11001 CrossRef.
- H. L. B. Boström and A. L. Goodwin, Acc. Chem. Res., 2021, 54, 1288–1297 CrossRef PubMed.
- S. Burger, K. Hemmer, D. C. Mayer, P. Vervoorts, D. Daisenberger, J. K. Zaręba and G. Kieslich, Adv. Funct. Mater., 2022, 32, 2205343 CrossRef CAS.
- H. A. Evans, Y. Wu, R. Seshadri and A. K. Cheetham, Nat. Rev. Mater., 2020, 5, 196–213 CrossRef CAS PubMed.
- J. F. Scott, Science, 2007, 315, 954–959 CrossRef CAS PubMed.
- M. E. Lines and A. M. Glass, Principles and applications of ferroelectrics and related materials, Clarendon Press, Oxford, 2001 Search PubMed.
- N. A. Benedek and C. J. Fennie, J. Phys. Chem. C, 2013, 117, 13339–13349 CrossRef CAS.
- A. P. Levanyuk and D. G. Sannikov, Sov. Phys. Usp., 1974, 17, 199–214 CrossRef.
- K.-L. Hu, M. Kurmoo, Z. Wang and S. Gao, Chem. – Eur. J., 2009, 15, 12050–12064 CrossRef CAS PubMed.
- F.-J. Geng, L. Zhou, P.-P. Shi, X.-L. Wang, X. Zheng, Y. Zhang, D.-W. Fu and Q. Ye, J. Mater. Chem. C, 2017, 5, 1529–1536 RSC.
- M. Mączka, A. Gągor, D. Stefańska, J. K. Zaręba and A. Pikul, Dalton Trans., 2022, 51, 9094–9102 RSC.
- S. M. Kronawitter, S. Park, S. A. Hallweger, E. Myatt, J. Pitcairn, M. J. Cliffe, D. Daisenberger, M. Drees and G. Kieslich, Mater. Adv., 2024, 5, 6440–6445 RSC.
- H.-Y. Liu, H.-Y. Zhang, X.-G. Chen and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 15205–15218 CrossRef CAS PubMed.
- H.-Y. Zhang, Y.-Y. Tang, P.-P. Shi and R.-G. Xiong, Acc. Chem. Res., 2019, 52, 1928–1938 CrossRef CAS PubMed.
- Y.-M. You, W.-Q. Liao, D. Zhao, H.-Y. Ye, Y. Zhang, Q. Zhou, X. Niu, J. Wang, P.-F. Li, D.-W. Fu, Z. Wang, S. Gao, K. Yang, J.-M. Liu, J. Li, Y. Yan and R.-G. Xiong, Science, 2017, 357, 306–309 CrossRef CAS PubMed.
- W.-Q. Liao, Y.-Y. Tang, P.-F. Li, Y.-M. You and R.-G. Xiong, J. Am. Chem. Soc., 2018, 140, 3975–3980 CrossRef CAS PubMed.
- N. L. Evans, P. M. M. Thygesen, H. L. B. Boström, E. M. Reynolds, I. E. Collings, A. E. Phillips and A. L. Goodwin, J. Am. Chem. Soc., 2016, 138, 9393–9396 CrossRef CAS PubMed.
- D. J. W. Allen, N. C. Bristowe, A. L. Goodwin and H. H.-M. Yeung, J. Mater. Chem. C, 2021, 9, 2706–2711 RSC.
- H.-Y. Ye, Y.-Y. Tang, P.-F. Li, W.-Q. Liao, J.-X. Gao, X.-N. Hua, H. Cai, P.-P. Shi, Y.-M. You and R.-G. Xiong, Science, 2018, 361, 151–155 CrossRef CAS PubMed.
- B. K. Shaw, A. R. Hughes, M. Ducamp, S. Moss, A. Debnath, A. F. Sapnik, M. F. Thorne, L. N. McHugh, A. Pugliese, D. S. Keeble, P. Chater, J. M. Bermudez-Garcia, X. Moya, S. K. Saha, D. A. Keen, F.-X. Coudert, F. Blanc and T. D. Bennett, Nat. Chem., 2021, 13, 778–785 CrossRef CAS PubMed.
- T. D. Bennett and S. Horike, Nat. Rev. Mater., 2018, 3, 431–440 CrossRef.
- S. Horike, S. S. Nagarkar, T. Ogawa and S. Kitagawa, Angew. Chem., Int. Ed., 2020, 59, 6652–6664 CrossRef CAS PubMed.
- N. Ma and S. Horike, Chem. Rev., 2022, 122, 4163–4203 CrossRef CAS PubMed.
- T. D. Bennett, J.-C. Tan, Y. Yue, E. Baxter, C. Ducati, N. J. Terrill, H. H.-M. Yeung, Z. Zhou, W. Chen, S. Henke, A. K. Cheetham and G. N. Greaves, Nat. Commun., 2015, 6, 8079 CrossRef CAS PubMed.
- L. Frentzel-Beyme, M. Kloß, P. Kolodzeiski, R. Pallach and S. Henke, J. Am. Chem. Soc., 2019, 141, 12362–12371 CrossRef CAS PubMed.
- M. Kim, H.-S. Lee, D.-H. Seo, S. J. Cho, E.-C. Jeon and H. R. Moon, Nat. Commun., 2024, 15, 1174 CrossRef CAS PubMed.
- W. Xu, N. Hanikel, K. A. Lomachenko, C. Atzori, A. Lund, H. Lyu, Z. Zhou, C. A. Angell and O. M. Yaghi, Angew. Chem., Int. Ed., 2023, 62, e202300003 CrossRef CAS PubMed.
- S. Vaidya, O. Veselska, A. Zhadan, M. Diaz-Lopez, Y. Joly, P. Bordet, N. Guillou, C. Dujardin, G. Ledoux, F. Toche, R. Chiriac, A. Fateeva, S. Horike and A. Demessence, Chem. Sci., 2020, 11, 6815–6823 RSC.
- D. Umeyama, N. P. Funnell, M. J. Cliffe, J. A. Hill, A. L. Goodwin, Y. Hijikata, T. Itakura, T. Okubo, S. Horike and S. Kitagawa, Chem. Commun., 2015, 51, 12728–12731 RSC.
- D. Umeyama, S. Horike, M. Inukai, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2015, 137, 864–870 CrossRef CAS PubMed.
- C. Healy, K. M. Patil, B. H. Wilson, L. Hermanspahn, N. C. Harvey-Reid, B. I. Howard, C. Kleinjan, J. Kolien, F. Payet, S. G. Telfer, P. E. Kruger and T. D. Bennett, Coord. Chem. Rev., 2020, 419, 213388 CrossRef CAS.
- L. Feng, K.-Y. Wang, G. S. Day, M. R. Ryder and H.-C. Zhou, Chem. Rev., 2020, 120, 13087–13133 CrossRef CAS PubMed.
- C. Ye, L. N. McHugh, C. Chen, S. E. Dutton and T. D. Bennett, Angew. Chem., Int. Ed., 2023, 62, e202302406 CrossRef CAS PubMed.
- J. Lu, Z. Shan, J. Zhang, Y. Su, K. Yi, Y. Zhang and Q. Zheng, J. Non-Cryst. Solids: X, 2022, 16, 100125 CAS.
- S. Kosasang, N. Ma, S. Impeng, S. Bureekaew, Y. Namiki, M. Tsujimoto, T. Saothayanun, H. Yamada and S. Horike, J. Am. Chem. Soc., 2024, 146, 17793–17800 CrossRef CAS PubMed.
- X. Moya, S. Kar-Narayan and N. D. Mathur, Nat. Mater., 2014, 13, 439–450 CrossRef CAS PubMed.
- X. Moya and N. D. Mathur, Science, 2020, 370, 797–803 CrossRef CAS PubMed.
- D. Boldrin, Appl. Phys. Lett., 2021, 118 Search PubMed.
- J. Salgado-Beceiro, A. Nonato, R. X. Silva, A. García-Fernández, M. Sánchez-Andújar, S. Castro-García, E. Stern-Taulats, M. A. Señarís-Rodríguez, X. Moya and J. M. Bermúdez-García, Mater. Adv., 2020, 1, 3167–3170 RSC.
- R. X. Da Silva, C. W. de Araujo Paschoal, C. Costa Dos Santos, A. García-Fernández, J. Salgado-Beceiro, M. A. Señarís-Rodríguez, M. Sanchez-Andujar and A. Nonato Almeida de Abreu Silva, Molecules, 2020, 25, 4754 CrossRef CAS PubMed.
- A. Aznar, P. Lloveras, M. Barrio, P. Negrier, A. Planes, L. Mañosa, N. D. Mathur, X. Moya and J.-L. Tamarit, J. Mater. Chem. A, 2020, 8, 639–647 RSC.
- P. Lloveras, A. Aznar, M. Barrio, P. Negrier, C. Popescu, A. Planes, L. Mañosa, E. Stern-Taulats, A. Avramenko, N. D. Mathur, X. Moya and J.-L. Tamarit, Nat. Commun., 2019, 10, 1803 CrossRef CAS PubMed.
- B. Li, Y. Kawakita, S. Ohira-Kawamura, T. Sugahara, H. Wang, J. Wang, Y. Chen, S. I. Kawaguchi, S. Kawaguchi, K. Ohara, K. Li, D. Yu, R. Mole, T. Hattori, T. Kikuchi, S.-I. Yano, Z. Zhang, Z. Zhang, W. Ren, S. Lin, O. Sakata, K. Nakajima and Z. Zhang, Nature, 2019, 567, 506–510 CrossRef CAS PubMed.
- J. Lin, P. Tong, K. Zhang, K. Tao, W. Lu, X. Wang, X. Zhang, W. Song and Y. Sun, Nat. Commun., 2022, 13, 596 CrossRef CAS PubMed.
- C. M. Miliante, A. M. Christmann, R. P. Soares, J. R. Bocca, C. S. Alves, A. M. G. Carvalho and A. R. Muniz, J. Mater. Chem. A, 2022, 10, 8344–8355 RSC.
- S. P. Vallone, A. N. Tantillo, A. M. Dos Santos, J. J. Molaison, R. Kulmaczewski, A. Chapoy, P. Ahmadi, M. A. Halcrow and K. G. Sandeman, Adv. Mater., 2019, 31, e1807334 CrossRef PubMed.
- M. Szafrański, W.-J. Wei, Z.-M. Wang, W. Li and A. Katrusiak, APL Mater., 2018, 6, 100701 CrossRef.
- J. Seo, R. D. McGillicuddy, A. H. Slavney, S. Zhang, R. Ukani, A. A. Yakovenko, S.-L. Zheng and J. A. Mason, Nat. Commun., 2022, 13, 2536 CrossRef CAS PubMed.
- S. Grover, S. Burger, K. T. Butler, K. Hemmer, P. Vervoorts, G. Kieslich and R. Grau-Crespo, CrystEngComm, 2023, 25, 3439–3444 RSC.
- G. Kieslich, S. Kumagai, A. C. Forse, S. Sun, S. Henke, M. Yamashita, C. P. Grey and A. K. Cheetham, Chem. Sci., 2016, 7, 5108–5112 RSC.
- S. Chen, R. Shang, B.-W. Wang, Z.-M. Wang and S. Gao, Angew. Chem., Int. Ed., 2015, 54, 11093–11096 CrossRef CAS PubMed.
- L. He, P.-P. Shi, M.-M. Zhao, C.-M. Liu, W. Zhang and Q. Ye, Chem. Mater., 2021, 33, 799–805 CrossRef CAS.
- H. L. B. Boström, J. Bruckmoser and A. L. Goodwin, J. Am. Chem. Soc., 2019, 141, 17978–17982 CrossRef PubMed.
- H. L. B. Boström and G. Kieslich, J. Phys. Chem. C, 2021, 125, 1467–1471 CrossRef.
- A. M. Mroz, L. Turcani and K. E. Jelfs, Electron. Struct., 2023, 5, 45004 CrossRef.
- J. Li, M. Barrio, D. J. Dunstan, R. Dixey, X. Lou, J.-L. Tamarit, A. E. Phillips and P. Lloveras, Adv. Funct. Mater., 2021, 31 CrossRef.
- Z.-Y. Du, Y.-P. Zhao, C.-T. He, B.-Y. Wang, W. Xue, H.-L. Zhou, J. Bai, B. Huang, W.-X. Zhang and X.-M. Chen, Cryst. Growth Des., 2014, 14, 3903–3909 CrossRef CAS.
- Z.-B. Liu, L. He, P.-P. Shi, Q. Ye and D.-W. Fu, J. Phys. Chem. Lett., 2020, 11, 7960–7965 CrossRef CAS PubMed.
Footnotes |
| † The title is inspired by Mike Glazer's 2017 Bragg Lecture of the Royal Institution with the title “The Wondrous World of Perovskites”. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03833a |
| This journal is © The Royal Society of Chemistry 2024 |