
Selective monoborylation of methane by metal–organic framework confined mononuclear pyridylimine-iridium(I) hydride†
Rahul
Kalita
a,
Manav
Chauhan
a,
Poorvi
Gupta
a,
Wahida
Begum
a,
Chhaya
Thadhani
a,
Biplab
Ghosh
b,
Balendra
 a,
Himani
Bisht
a and
Kuntal
Manna
*a
a,
Himani
Bisht
a and
Kuntal
Manna
*a
aDepartment of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110016, India. E-mail: kmanna@chemistry.iitd.ac.in
bBARC Beamlines Section, Indus-2, RRCAT, Indore 452013, India
First published on 29th May 2024
Abstract
Chemoselective monoborylation of methane in high yield is a grand challenge. We have developed a metal–organic framework confined pyridylimine-iridium hydride catalyst, which is efficient in methane C–H borylation using bis(pinacolato)diboron to afford methyl boronic acid pinacol ester in 98% GC-yield at 130 °C with a TON of 196. Mechanistic investigation suggests the oxidative addition of methane to IrIII(Bpin)2(H) species to form IrV(Bpin)2(CH3)(H)2 as the turnover limiting step.
Chemoselective C–H borylation of methane has drawn significant attention in recent years due to the abundance of methane as the low-cost carbon feedstock and the application of organoborane products as versatile synthetic intermediates in organic synthesis.1–3 However, the methane C–H bond activation under mild conditions is highly challenging owing to the intrinsic inertness stemming from the considerable C–H bond dissociation energy (104 kcal mol−1) with a large HOMO–LUMO gap, low acidity and polarization difficulty.4–7 In addition, methane borylation reactions typically suffer from poor selectivity originating from the over-borylation of methane C–H bonds, competitive solvent C–H borylation, and boron-byproduct formation. Furthermore, the mono-functionalized product is more reactive than methane itself, making the selective monoborylation of methane extremely challenging.
Early reports of developing homogeneous catalysts for methane borylation using bis(pinacolato)diboron (B2pin2) as the borylating agent were based on 1,10-phenanthroline- and diphosphine-ligated Ir, Rh and Ru complexes, which gave a mixture of monoborylated (CH3Bpin) and diborylated methane [CH2(Bpin)2] at 150 °C with selectivity spanning 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 31:1.8,9 Farha and co-workers elegantly employed a metal–organic framework (MOF) as the porous solid support with appropriate pore sizes,10–19 in which an embedded iridium(III) catalyst enables selective mono C–H borylation over multiborylation of methane within the confined reaction space via shape-selective catalysis, leading to the formation of CH3Bpin in 19.5% yield with a turnover number (TON)of67.20 Subsequently, a mono(phosphine) MOF-supported-Ir catalyst outperformed other Ir-catalysts based on chelating phenanthroline, bipyridine, and diphosphine ligands to give CH3Bpin with a TON of 127.21 Despite significant progress in developing catalysts for selective monoborylation of methane, these catalytic systems also generate a considerable amount of boron-byproducts such as HOBpin, pinBOBpin and Sol-Bpin during the borylation reactions.22–24 Herein, we report a MOF-confined mononuclear pyridylimine-ligated iridium(I)-hydride, which is a highly robust and active heterogeneous catalyst for chemoselective monoborylation of methane to afford CH3Bpin in a near-quantitative yield (Fig. 1).
:1 to 31:1.8,9 Farha and co-workers elegantly employed a metal–organic framework (MOF) as the porous solid support with appropriate pore sizes,10–19 in which an embedded iridium(III) catalyst enables selective mono C–H borylation over multiborylation of methane within the confined reaction space via shape-selective catalysis, leading to the formation of CH3Bpin in 19.5% yield with a turnover number (TON)of67.20 Subsequently, a mono(phosphine) MOF-supported-Ir catalyst outperformed other Ir-catalysts based on chelating phenanthroline, bipyridine, and diphosphine ligands to give CH3Bpin with a TON of 127.21 Despite significant progress in developing catalysts for selective monoborylation of methane, these catalytic systems also generate a considerable amount of boron-byproducts such as HOBpin, pinBOBpin and Sol-Bpin during the borylation reactions.22–24 Herein, we report a MOF-confined mononuclear pyridylimine-ligated iridium(I)-hydride, which is a highly robust and active heterogeneous catalyst for chemoselective monoborylation of methane to afford CH3Bpin in a near-quantitative yield (Fig. 1).
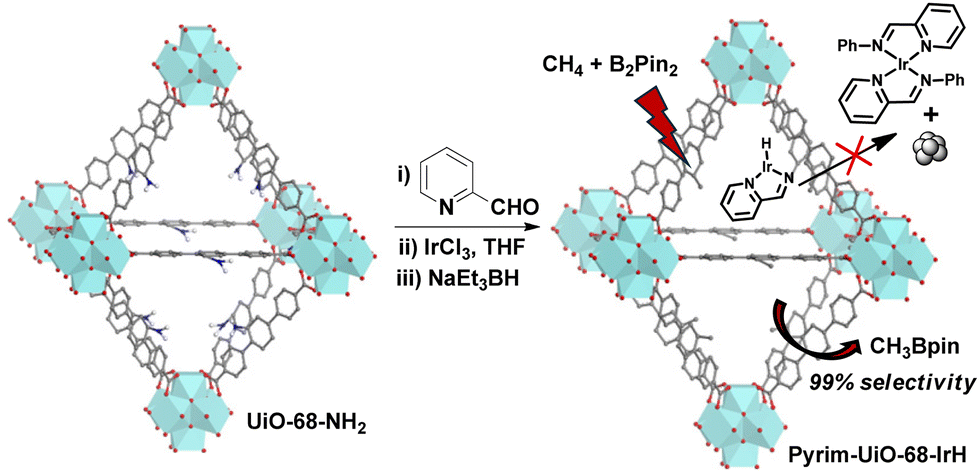 | ||
| Fig. 1 Synthesis of pyrim-UiO-IrH MOF via post-synthetic modification of pristine amino-functionalized UiO-68-MOF for methane monoborylation. | ||
Pyrim-UiO-IrH MOF has a UiO-68 topology,15,25 built from Zr6O4(OH)4 nodes and linear 2′-amino-[1,1′:4′,1′′-terphenyl]-4,4′′-dicarboxylate bridging linkers bearing pyridylimine-ligated iridium(I)-hydride species. Pyrim-UiO-IrH was synthesized by metalation of a pyridylimine-functionalized zirconium UiO-68-MOF (pyrim-UiO) with IrCl3·3H2O followed by treatment of NaEt3BH.26 The reaction of pyrim-UiO MOF and IrCl3·3H2O in THF formed pyrim-UiO-IrCl3 MOF bearing pyridylimine-ligated IrCl3 species at its linkers. The extended X-ray absorption fine structure (EXAFS) of pyrim-UiO-IrCl3 at the Ir L3-edge fitted well with the DFT-optimized structure of (pyrim)IrCl3(THF), indicating the presence of an octahedral IrIII-species, in which Ir3+ ion is coordinated two nitrogen atoms of pyridylimine, three chlorine atoms and a THF molecule (Fig. 2b). The treatment of pyrim-UiO-IrCl3 with NaEt3BH in THF at room temperature furnished pyrim-UiO-IrH. PXRD patterns of pyrim-UiO-IrH, simulated UiO-68 MOF, and pristine UiO-68-NH2 are comparable indicating that pyrim-UiO-IrH has a UiO-68 topology and that pristine MOF's structure and crystallinity were not altered during post-synthetic modifications (Fig. 2a).
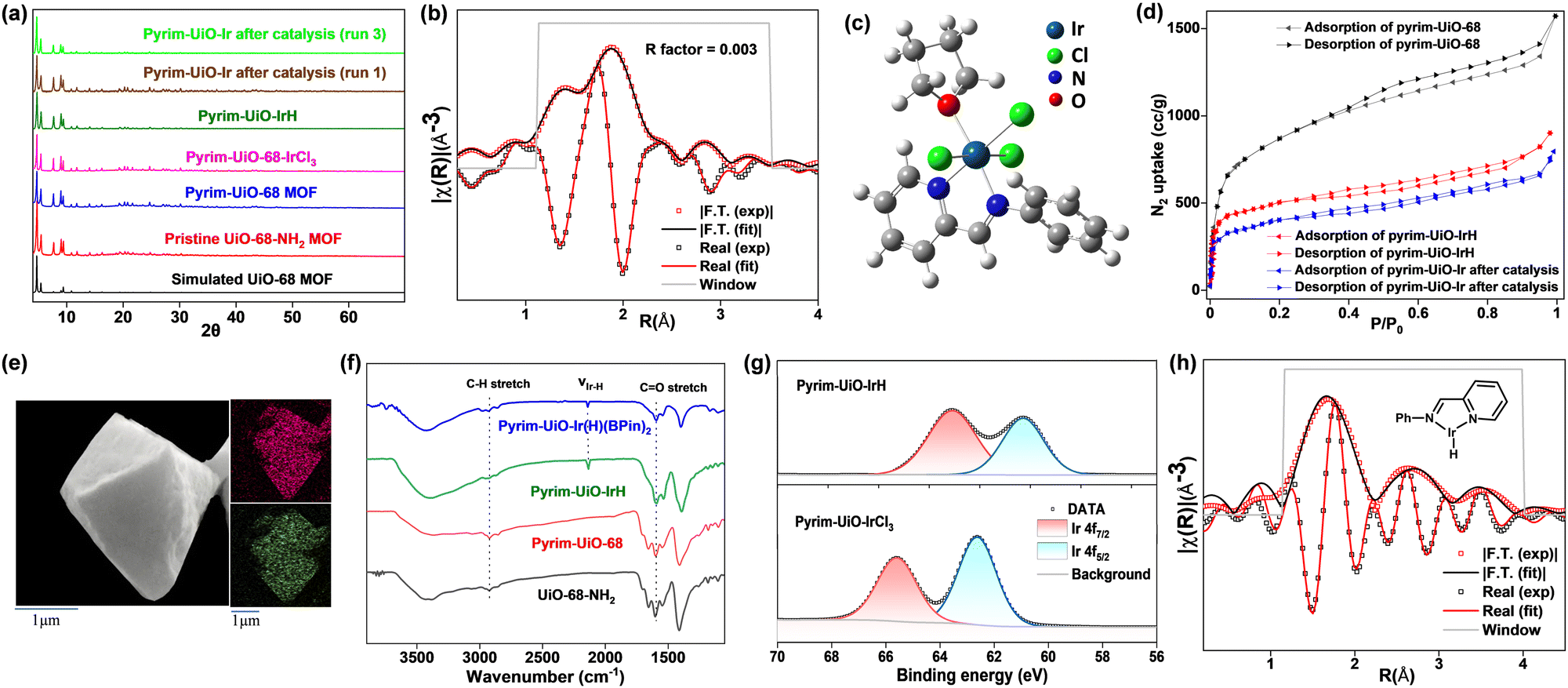 | ||
| Fig. 2 (a) PXRD patterns of simulated UiO-68 MOF (black), pristine UiO-68-NH2 MOF (red), pyrim-UiO-68 MOF (blue), pyrim-UiO-IrCl3 (magenta), pyrim-UiO-IrH (olive), pyrim-UiO-Ir after run 1 (brown) and pyrim-UiO-Ir after run 3 (green). (b) EXAFS spectra (red and black hollow squares) and fits (red and black solid lines) of pyrim-UiO-IrCl3 in the R space from 1.15–3.5 Å. (c) DFT optimised structure of (pyrim)IrCl3(THF) molecule. (d) BET nitrogen sorption isotherms of pyrim-UiO-68 (black), pyrim-UiO-IrH (red) and pyrim-UiO-Ir after catalysis (blue) measured at 77 K. (e) SEM image of a pyrim-UiO-IrH particle along with elemental mapping of Zr and Ir. (f) IR (KBr) spectrum of UiO-68-NH2 (black), pyrim-UiO-68 MOF (red), pyrim-UiO-IrH (green) and pyrim-UiO-IrH(BPin)2 (blue). (g) Ir 4f XPS spectra of pyrim-UiO-IrCl3 and pyrim-UiO-IrH. (h) EXAFS spectra (red and black hollow squares) and fits (red and black solid lines) of pyrim-UiO-IrH in the R space from 1.15–4.0 Å. | ||
Inductively coupled plasma-optical emission spectroscopy (ICP-OES) analysis of the digested pyrim-UiO-IrH revealed a Ir-loading of ∼32% with respect to the pyridylimine moiety, corresponding to the formula of Zr6(μ3-O)4(μ3-OH)4(C26H16N2O4Ir0.32H0.32)6. Pyrim-UiO-IrH has a BET surface area of 1245 m2 g−1, and a pore size of 0.8 nm, which is lower than that of pristine pyrim-UiO MOF, due to the incorporation of iridium-moiety within the MOF's pores (Fig. 2d). Scanning electron microscopy-energy dispersive X-ray analysis (SEM-EDX) mapping of pyrim-UiO-IrH displayed uniform distributions of Ir and Zr ions through the MOF particle (Fig. 2e). The IR spectrum (KBr) of pyrim-UiO-IrH showed a characteristic νIr–H stretching frequency at 2140 cm−1 (Fig. 2f). X-ray absorption near edge structure (XANES) showed the Ir L3-edge energy of pyrim-UiO-IrH at 11.216 KeV, which is 3 eV lower than that of pyrim-UiO-IrCl3 and 1.0 eV higher than Ir(0) foil, suggesting a formal +1 oxidation state for Ir ion in pyrim-UiO-IrH (Fig. S18, ESI†). The existence of IrI-species in pyrim-UiO-IrH was further confirmed by X-ray photoelectron spectroscopy (XPS), which displayed Ir 4f7/2 and 4f5/2 binding energy peaks at 60.3 eV and 63.1 eV, respectively, and ∼2.1–2.3 eV lower than those of pyrim-UiO-IrCl3 and IrCl3 (Fig. 2g). The EXAFS analysis of pyrim-UiO-IrH at the Ir L3-edge suggests the existence of mono pyridylimine-ligated iridium(I)-hydride moiety within the MOF (Fig. 2h).
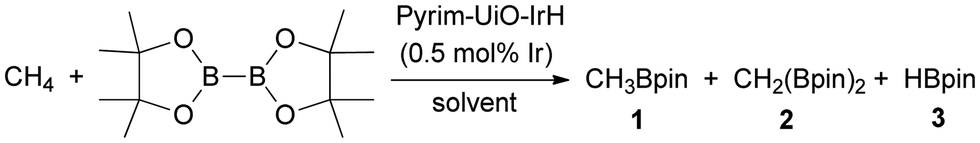
Pyrim-UiO-IrH catalyzed methane borylation reactions were conducted in a high-pressure batch reactor using B2pin2 as the borylating agent and the limiting reagent in a suitable solvent under a desired methane pressure. Initial trials using 0.2 mmol of B2pin2 and pyrim-UiO-IrH (0.5 mol% Ir) in several different solvents at 130 °C under 40 bar of CH4 for 24 h revealed that cyclohexane gave the highest yield of CH3Bpin (98%) with complete conversion of B2pin2 (entries 1–4, Table 1). CH4 borylation in other solvents such as THF, toluene, heptane and DMF gave lesser yields of CH3Bpin, due to the formation of various byproducts such as borylated solvents, CH2(Bpin)2, pinBOBpin, and HOBpin.20 Further screening of reaction temperature, CH4 pressure, catalyst loading, and reaction time showed that 0.5 mol% Ir- loading, 130 °C, 40 bar of CH4, and 24 h of reaction are optimal for selective mono-borylation of methane (entry 1, Table 1). The borylation reaction did not occur below 100 °C, however, the formation of a significant amount of HOBpin, pinBOBpin and C6H11Bpin was observed at 150 °C (entries 6 and 7, Table 1). The borylation reactions under 20 bar and 30 bar of CH4 afforded CH3Bpin in only 26% and 47% yields, respectively (entries 8 and 9, Table 1), presumably due to the lower solubility of CH4 at lower pressure. Pyrim-UiO-IrH catalyzed CH4 borylation under optimal reaction conditions afford CH3Bpin in 98% yield, giving rise to the highest TON of 196 (entry 1, Table 1). Gas chromatography (GC) analysis of the crude reaction mixture showed the exclusive formation of CH3Bpin with no detectable diborylated product CH2(Bpin)2.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | P (bar) | Time (h) | Conv. of B2pin2 | %Yield of 1b (selectivity)c | TON |
| a Reaction conditions: 1.9 mg of pyrim-UiO-IrH (1.02 μmol of Ir), 51 mg B2pin2 (0.2 mmol), 2 mL solvents. b Yield was determined by GC as GC peak area (CH3Bpin)/peak area (total boron species) × 100. c Selectivity was calculated as GC peak area (CH3Bpin)/peak area (total products) × 100. | |||||||
| 1 | C6H12 | 130 | 40 | 24 | 100 | 98 (98) | 196 |
| 2 | THF | 130 | 40 | 24 | 89 | 29 (33) | 58 |
| 3 | Toluene | 130 | 40 | 24 | 80 | 35 (44) | 70 |
| 4 | Heptane | 130 | 40 | 24 | 84 | 72 (86) | 144 |
| 5 | C6H12 | 130 | 40 | 18 | 87 | 85 (98) | 170 |
| 6 | C6H12 | 110 | 40 | 24 | 20 | 19 (95) | 38 |
| 7 | C6H12 | 150 | 40 | 24 | 100 | 78 (78) | 156 |
| 8 | C6H12 | 130 | 20 | 24 | 27 | 26 (96) | 52 |
| 9 | C6H12 | 130 | 30 | 24 | 48 | 47 (98) | 94 |
| 10 | C6H12 | 130 | 40 | 48 | 100 | 82 (82) | 164 |
Importantly, pyrim-UiO-IrH was at least 24 times more active than its homogeneous control ([Ph(pyrim)(PhCO2Me)2]IrH, which produced only 4% CH3Bpin under the identical reaction conditions and Ir-loading (0.5 mol%) (Fig. 3a). The much higher catalytic activity of pyrim-UiO-IrH than its homogeneous Ir analogue is likely due to the greater stability of the pyrim-IrH species via active-site isolation at the linkers that prevent intermolecular decomposition.
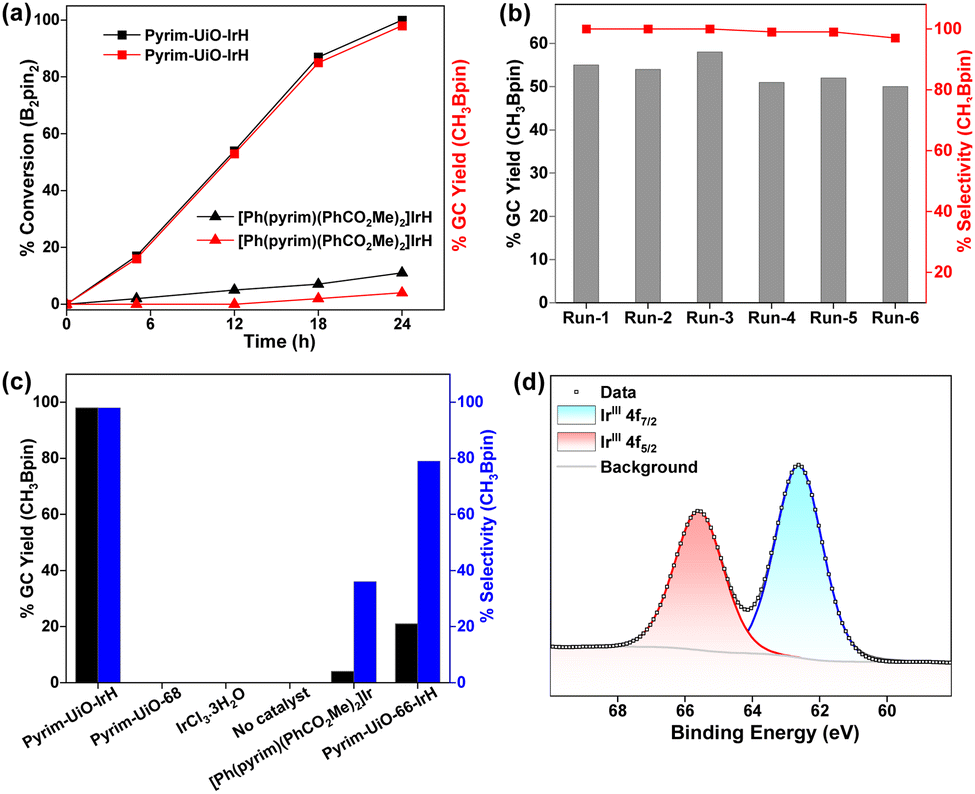 | ||
| Fig. 3 (a) Plot of %GC conversion and %GC yield of CH3Bpin vs time for CH4 borylation using pyrim-UiO-IrH (0.5 mol% Ir) and [Ph(pyrim)(PhCO2Me)2]IrH (0.5 mol% Ir) under identical conditions. (b) Plot for the %GC yield and %selectivity of CH3Bpin at various runs in the recycling of pyrim-UiO-IrH (0.5 mol% Ir). (c) Comparison of the catalytic activity for the conversion of CH4 to CH3Bpin over pyrim-UiO-IrH with other catalysts under identical reaction conditions. Conditions: 1.9 mg of pyrim-UiO-IrH (1.02 μmol of Ir) or equivalent Ir-loading for other catalysts, 2 mL C6H12, 130 °C, 40 bar CH4 and 24 h. (d) Ir 4f XPS spectrum of pyrim-UiO-IrH(Bpin)2. | ||
As a heterogeneous catalyst, pyrim-UiO-Ir could be recycled and reused at least 5 times with consistent activity without noticeable changes in structure or crystallinity, leading to the total TON up to 980 (Fig. 3b). No borylated product was detected when the methane borylation was carried out in the absence of MOF catalyst or with the pristine pyrim-UiO-68 or with IrCl3·3H2O (entries 15–17, Table S1, ESI†). The production of CH3Bpin ceased upon removing the solid MOF pyrim-UiO-IrH from the reaction mixture, suggesting that the catalytic moiety is embedded within the MOF (Fig. S6, ESI†). A catalytic reaction with 40 bar N2 instead of CH4 and 0.2 mmol B2Pin2 was conducted at 130 °C in cyclohexane for 24 h. In this reaction, no CH3Bpin product was formed, signifying CH4 as the only carbon source for CH3Bpin formation (Section S4.2, ESI†). Importantly, pyrim-UiO-68-IrH was almost four times more active and significantly chemoselective than pyrim-UiO-66-IrH MOF, having similar UiO-topology but smaller pore sizes in methane borylation due to the facile diffusion of the substrates and formation of smaller monoborylated products within the pores of UiO-68 MOFs via shape-selective catalysis (Fig. S10 and Section S4.5.2, ESI†).
The reaction between pyrim-UiO-IrIH and B2Pin2 at 130 °C for 30 min forms pyrim-UiO-IrIII(Bpin)2(H), which was characterized by XPS and IR spectroscopy (Fig. 2f and 3d). Pyrim-UiO-IrIII(Bpin)2(H) showed similar catalytic activity to pyrim-UiO-68-IrIH, suggesting IrIII(Bpin)2(H) species as the potential catalytic intermediate. We propose that pyrim-UiO-Ir catalyzed methane borylation proceeds via a IrIII–IrV–IrIII catalytic cycle,20,22 in which the oxidative addition of methane to pyrim-UiO-IrIII(Bpin)2(H) (INT-1) first form seven coordinated pyrim-UiO-IrV(CH3)(H)2(Bpin)2 (INT-2) followed by reductive elimination of CH3Bpin to generate pyrim-UiO-IrIII(Bpin)(H)2 (Fig. 4). Further oxidative addition of B2Pin2 followed by reductive elimination of HBpin regenerates pyrim-UiO-IrIII(Bpin)2(H). DFT calculation suggests that the boron-assisted oxidative addition of methane is the turn-over limiting step requiring a free activation energy of 34.9 kcal mol−1, similar to the reported 1,10-phenanthroline- and diphosphine-ligated Ir catalysts.20,22
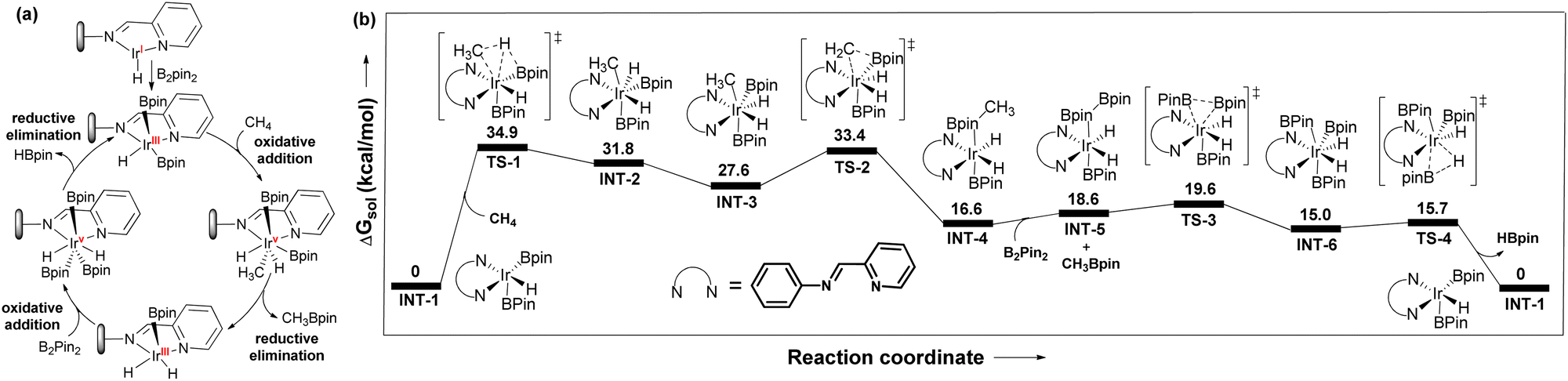 | ||
| Fig. 4 (a) Proposed catalytic cycle of pyrim-UiO-IrH catalyzed methane borylation. (b) DFT-calculated free energy profile at 403 K for pyrim-UiO-IrH catalyzed methane borylation reaction. | ||
However, the higher catalytic activity of pyrim-UiO-Ir in methane borylation is attributed to the facile oxidative addition of methane to the IrIII(H)(Bpin)2 species as opposed to the sterically encumbered IrIII(Bpin)3 species in other systems. In conclusion, we have developed a porous MOF-supported iridium catalyst for chemoselective monoborylation of methane, affording CH3Bpin in 98% yield, which is much higher than the previous reports. This work highlights the importance of MOFs in developing heterogeneous catalysts for chemoselective functionalization of methane and other hydrocarbons.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305–308 CrossRef CAS PubMed.
- T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056–3058 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 10096–10115 CrossRef CAS PubMed.
- W. Taifan and J. Baltrusaitis, Appl. Catal., B, 2016, 198, 525–547 CrossRef CAS.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS.
- A. K. Cook, S. D. Schimler, A. J. Matzger and M. S. Sanford, Science, 2016, 351, 1421–1424 CrossRef CAS.
- K. T. Smith, S. Berritt, M. González-Moreiras, S. Ahn, M. R. Smith, M.-H. Baik and D. J. Mindiola, Science, 2016, 351, 1424–1427 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- I. Senkovska, F. Hoffmann, M. Fröba, J. Getzschmann, W. Böhlmann and S. Kaskel, Microporous Mesoporous Mater., 2009, 122, 93–98 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- M. Rimoldi, A. J. Howarth, M. R. DeStefano, L. Lin, S. Goswami, P. Li, J. T. Hupp and O. K. Farha, ACS Catal., 2017, 7, 997–1014 CrossRef CAS.
- M. Kalaj and S. M. Cohen, ACS Cent. Sci., 2020, 6, 1046–1057 CrossRef CAS PubMed.
- B. Pramanik, R. Sahoo and M. C. Das, Coord. Chem. Rev., 2023, 493, 215301 CrossRef CAS.
- G. Cai, P. Yan, L. Zhang, H. C. Zhou and H. L. Jiang, Chem. Rev., 2021, 121, 12278–12326 CrossRef CAS PubMed.
- X. Zhang, Z. Huang, M. Ferrandon, D. Yang, L. Robison, P. Li, T. C. Wang, M. Delferro and O. K. Farha, Nat. Catal., 2018, 1, 356–362 CrossRef CAS.
- X. Feng, Y. Song, Z. Li, M. Kaufmann, Y. Pi, J. S. Chen, Z. Xu, Z. Li, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 141, 11196–11203 CrossRef CAS PubMed.
- S. Ahn, D. Sorsche, S. Berritt, M. R. Gau, D. J. Mindiola and M.-H. Baik, ACS Catal., 2018, 8, 10021–10031 CrossRef CAS.
- Q. Chen, A. Dong, D. Wang, L. Qiu, C. Ma, Y. Yuan, Y. Zhao, N. Jia, Z. Guo and N. Wang, Angew. Chem., 2019, 131, 10781–10786 CrossRef.
- O. Staples, M. S. Ferrandon, G. P. Laurent, U. Kanbur, A. J. Kropf, M. R. Gau, P. J. Carroll, K. McCullough, D. Sorsche, F. A. Perras, M. Delferro, D. M. Kaphan and D. J. Mindiola, J. Am. Chem. Soc., 2023, 145, 7992–8000 CrossRef CAS PubMed.
- H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- R. Newar, W. Begum, N. Antil, S. Shukla, A. Kumar, N. Akhtar, Balendra and K. Manna, Inorg. Chem., 2020, 59, 10473–10481 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01568a |
| This journal is © The Royal Society of Chemistry 2024 |