
Mechanistic variances in NO release: ortho vs. meta isomers of nitrophenol and nitroaniline†
Prahlad Roy
Chowdhury
 ,
Monali
Kawade
and
G. Naresh
Patwari
*
,
Monali
Kawade
and
G. Naresh
Patwari
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: naresh@chem.iitb.ac.in
First published on 23rd April 2024
Abstract
The NO release following 266 nm photolysis of ortho and meta isomers of nitrophenol and nitroaniline shows a bimodal translational energy distribution, wherein the slow and fast components originate from dynamics in the S0 and T1 states, respectively. The translational energy distribution profiles for any NO product state show a higher slow-to-fast (s/f) branching ratio for the ortho isomer in comparison with the meta isomer. The observed variation in the s/f branching ratio vis-à-vis the ortho and meta isomers is attributed to the presence of intramolecular hydrogen bonding between the ortho substituent and NO2 moiety, which favours the roaming mechanism.
Nitroaromatic compounds produce nitric oxide (NO) upon irradiation, which modulates the oxidative capacity of the atmosphere.1,2 Measurements on the translational energy distribution of NO fragments following UV photolysis of nitroaromatic compounds show the presence of slow and fast components (a bimodal behaviour), which has been interpreted as the presence of two distinct pathways for NO release.3,4 The absence of a direct route to produce NO from nitroaromatic compounds suggests that both pathways must proceed via nitro-to-nitrite photoisomerization. The 266 nm excitation of nitrobenzene to high-lying electronic states (S4/S3) results in ultrafast relaxation to the T1 state with a timescale of about 200 fs, and thereafter the dynamics on the T1 surface is much slower with the lifetime of about 90 ps, wherein the nuclear dynamics will dominate.5 These observations are in accordance with the hypothesis that the topography of the T1 surface influences the dynamics of NO formation.3 Based on electronic structure calculations, the fast NO component is attributed to a direct elimination channel from the T1 state featuring an oxaziridine ring-type (tight) transition state.3,4,6 On the other hand, the slow translational energy component of the NO radical is attributed to nitro-to-nitrite photoisomerization in the ground state, either arising from an oxaziridine ring-type and/or a NO2 roaming transition state. The NO2 roaming mechanism originates from the frustrated dissociation of the C–N bond in nitrobenzene and the NO2 radical is trapped in a shallow potential well around the phenyl radical and “roams” to form phenyl nitrite (C6H5ONO), and the release of NO radical with low translational energy is attributed to the presence of an exit barrier for O–NO bond dissociation.4 Thus far, it has been inferred that the slow translational energy component of the NO radical originates from the S0 state with both roaming and non-roaming (oxaziridine ring-type) mechanisms. Recent high-level electronic structure calculations have suggested that the topography of the T1/S0 crossing points influences the slow and fast translational energy components of NO, which could involve oxaziridine or roaming-type geometries, and the roaming pathway has a relatively higher (4.61 eV) barrier.7
The ability of T1 surface topography to direct the NO release via the oxaziridine or roaming-type mechanisms, and thereby influence the relative yields of the fast and slow translational energy components, was investigated in several ortho-substituted nitrobenzenes by probing a specific NO fragment (v = 1; J = 50.5) state.8 In these cases the appearance of the bimodal translation energy distribution profile of the NO radical was dependent on the nature of the substituent. The substituents that were capable of forming intramolecular hydrogen bonding with the NO2 group, such as OH and NH2 groups, favoured the slow component over the fast component. In other words, the slow-to-fast (s/f) branching ratio between the components of the total translation energy distribution profile of NO was greater than one (s/f > 1) in the case of o-nitrophenol and o-nitroaniline. On the other hand, a reverse trend with s/f < 1 was observed for o-nitroanisole and o-nitrotoluene, wherein the substituents do not hydrogen bond with NO2. The experimentally observed results were interpreted on the basis of two-dimensional potential energy surfaces (2D-PESs) in the T1 state and it was inferred that the minimum energy path favours the NO2 roaming mechanism for the substituents, in the ortho position, capable of hydrogen bonding (OH and NH2), while in the other cases (H, CH3, OCH3 and CF3 groups), the NO release via an oxaziridine ring-type mechanism is preferred.8 On the other hand, the variation in the substituents at the para position had only a marginal influence on the s/f branching ratio.9
A pertinent question that arises at this state is: does the intramolecular hydrogen bonding between the NO2 and the ortho substituent influence the NO release mechanism? To test this hypothesis 2D-PESs in the T1 state were calculated for two pairs of substituted nitrobenzenes in which the OH and NH2 groups were switched from the ortho position to the meta position and the corresponding 2D-PESs in the T1 state are shown in Fig. 1 and Fig. S2 (ESI†). These 2D-PESs clearly illustrate that the minimum energy path switches from the NO2 roaming mechanism to the oxaziridine ring-type mechanism for both OH and NH2 substituents when shifted from the ortho to meta position, respectively. The topographical aspects of the 2D-PESs remained unaltered with two distinct DFT functionals viz., B3LYP and M06-2X (compare Fig. 1 and Fig. S2, ESI†). Since the 2D-PES is the projection of the multidimensional potential on two nascent internal coordinates leading to NO release, it can be therefore hypothesised based on the minimum energy pathway in the 2D-PESs that the s/f ratio will be higher in the ortho isomer in comparison with the meta isomer.8
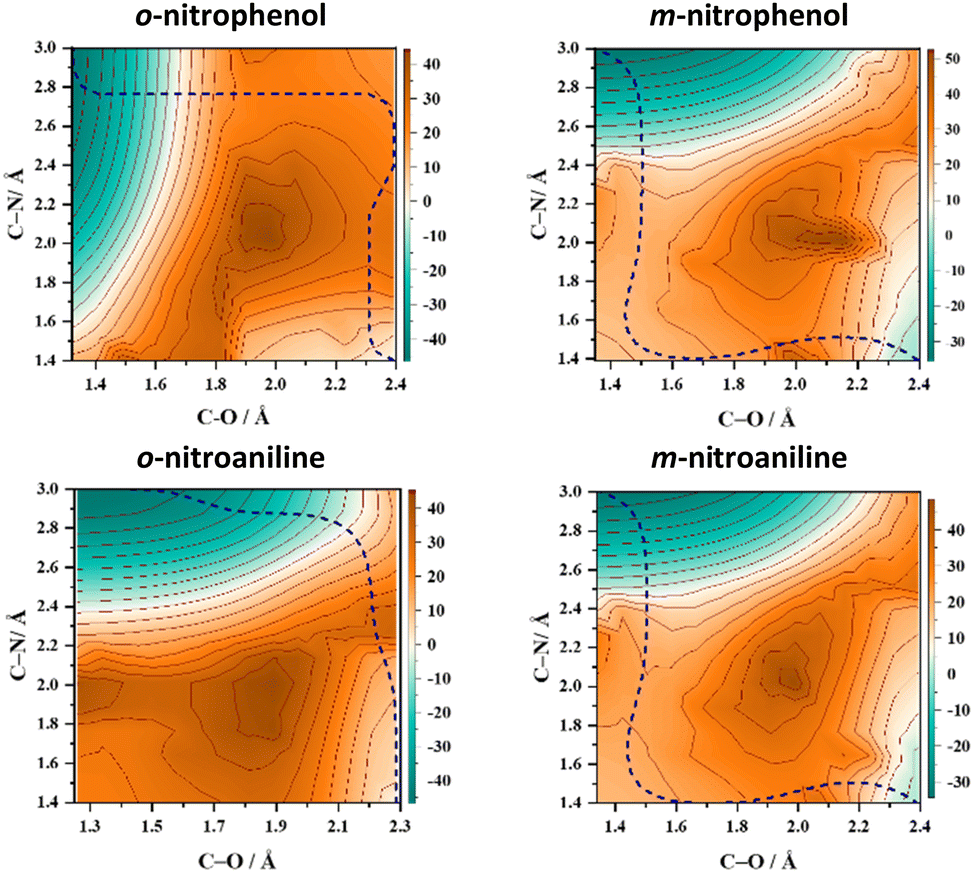 | ||
| Fig. 1 Two-dimensional potential energy surface (2D-PES) plots of o-nitrophenol, m-nitrophenol, o-nitroaniline, and m-nitroaniline calculated using the B3LYP/6-311++G(d,p) level of theory. The blue dashed lines represent the minimum energy paths (MEPs) connecting the starting structure (bottom right corner) with the formation of NO and the co-fragment phenoxy radical (top left corner) in the T1 state. The energy scale is in kcal mol−1 relative to the starting geometry on the T1 surface. The one-dimensional cut of the 2D-PES along the minimum energy path resulting in NO release is shown in Fig. S1 (ESI†) along with the selected geometries. | ||
The experimental realization of this conjecture based on the 2D-PES calculation was carried out by measuring the total translational energy distribution profiles of NO produced from o-nitrophenol, m-nitrophenol, o-nitroaniline, and m-nitroaniline using the velocity map imaging (VMI) technique10 and the results are presented in Fig. 2. In all the four NO product states that were probed, the translational energy distribution profiles show bimodal behaviour (see Fig. S3 and S4, ESI†) with the s/f branching ratio for the ortho isomer higher than the corresponding meta isomer (see Table 1). Additionally, the s/f ratio of o-nitrophenol is always higher than the o-nitroaniline, which is in accordance with the fact that the OH group forms stronger hydrogen bonds than the NH2 group. These observations clearly reinforce the role of intramolecular hydrogen bonding in favouring the slow component. In general, the contribution from the fast component increases with rotational quantum number J, in agreement with the earlier reports;4 however, this effect appears to be more prominent with an increase in vibrational quantum number v. It must be noted that the 2D-PES is a projection of the multidimensional potential in two-dimensions and hence a simplified model. The trends in the s/f branching ratio, therefore, are an indicator of the influence of the substituent to form intramolecular hydrogen bonding with the NO2 group resulting in the enhancement of the roaming mechanism, thereby validating the initial hypothesis.
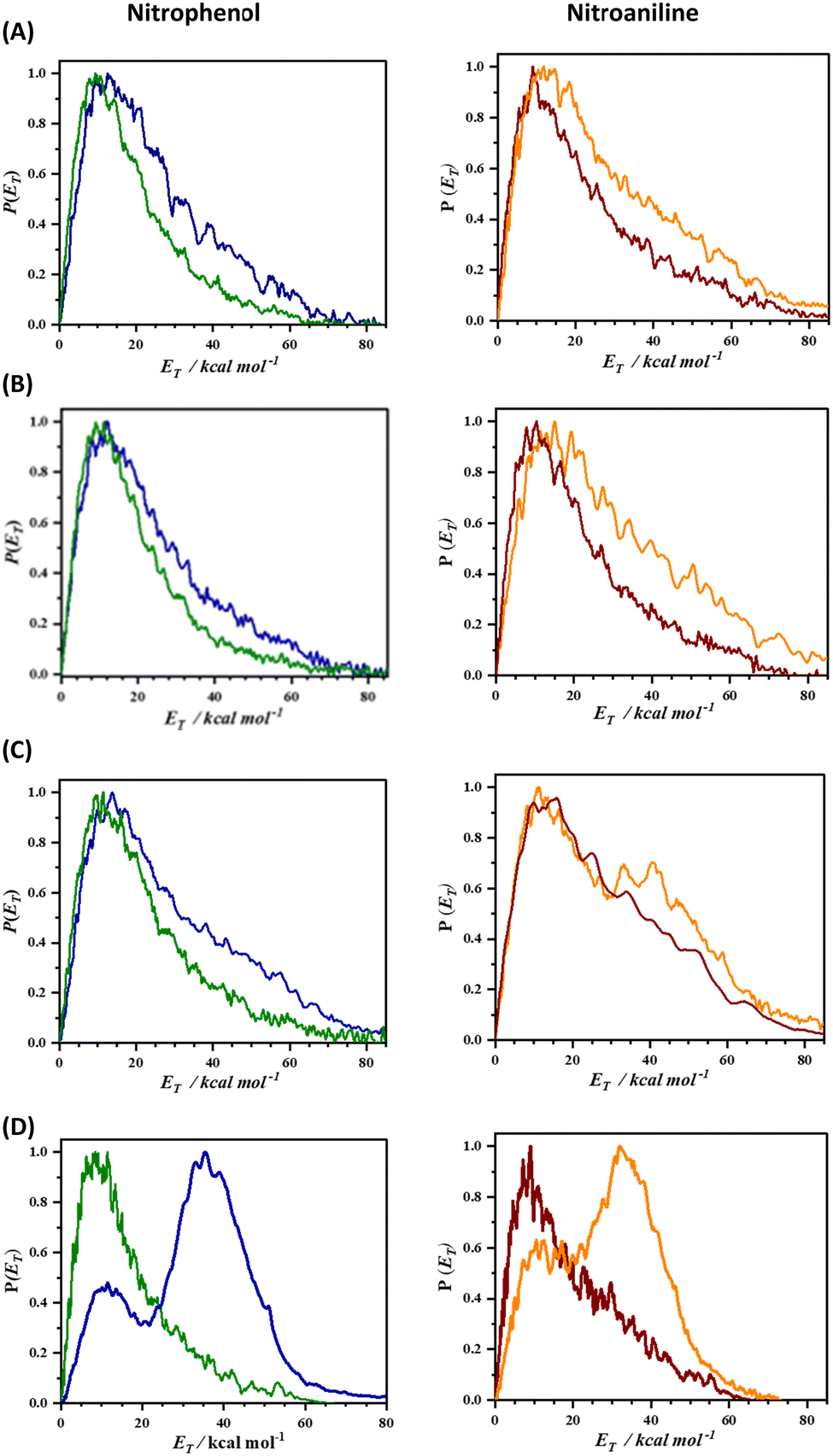 | ||
| Fig. 2 The total translational energy distribution profiles [P(ET)] of NO, produced from nitrophenol (left panel) and nitroaniline (right panel), probed for various product states of NO (A) v = 0; J = 21.5, (B) v = 0; J = 29.5, (C) v = 0; J = 33.5 and (D) v = 1; J = 50.5. The [P(ET)] profiles of the ortho and meta nitrophenols are depicted by solid green and blue curves, respectively, while ortho and meta nitroanilines are represented by solid brown and orange curves, respectively. It is noteworthy that the fast component in the [P(ET)] profiles of meta isomers is consistently higher than that of the ortho isomer across all the product states. | ||
| NO product state | Nitrophenol | Nitroaniline | ||
|---|---|---|---|---|
| ortho | meta | ortho | meta | |
| v = 0; J = 21.5 | 4.92 | 3.17 | 3.23 | 2.28 |
| v = 0; J = 29.5 | 5.40 | 3.78 | 3.67 | 2.38 |
| v = 0; J = 33.5 | 4.31 | 2.31 | 2.05 | 1.21 |
| v = 1; J = 50.5 | 3.85 | 0.33 | 1.92 | 0.45 |
The topology of the T1 surface regulates the preferential mechanism; however, it has been well established that the nature of the T1/S0 crossing points and the exit barrier on the S0 surface dictate the translational energy of the NO products.4,5,11–14 In order to assess the role of the S0 surface on the translational energy distribution of the NO products, 2D-PESs in the S0 state were also computed, which are depicted in Fig. 3 (and Fig. S5, ESI†). In the case of o-nitrophenol, an early (closer to the reactant) T1/S0 crossing point along the minimum energy path on the T1 surface is a loose transition state, reflecting the NO2 roaming mechanism. The NO2 roaming and the oxaziridine mechanism are distinguished by evaluating the difference between the two N–O bonds, which is depicted in Fig. S6 (ESI†). The roaming path is characterized by a sudden and late change in the difference between two N–O bonds, whereas the oxaziridine pathway is more gradual. On the S0 2D-PES, the pathway for NO release is corrugated with an exit barrier of about 14 kcal mol−1 (see Fig. 3) and the presence of several phenyl nitrite like intermediates, which ultimately result in slow NO release. Conversely, in the case of m-nitrophenol, a late (closer to the products) T1/S0 crossing point was observed along the minimum energy path on the T1 surface, which leads to the fast translational energy component. The slow translational energy component of the NO release is generally attributed to the dynamics on the S0 surface, which once again originates from the roaming and non-roaming mechanisms. Therefore, based on the present set of results, it can be inferred that intramolecular hydrogen bonding between the NO2 and the substituent in the ortho position plays a pivotal role in enhancing the roaming mechanism, thereby resulting in a higher s/f ratio for the ortho isomer relative to the meta isomer as well as higher s/f ratio for the o-nitrophenol in comparison with o-nitroaniline.
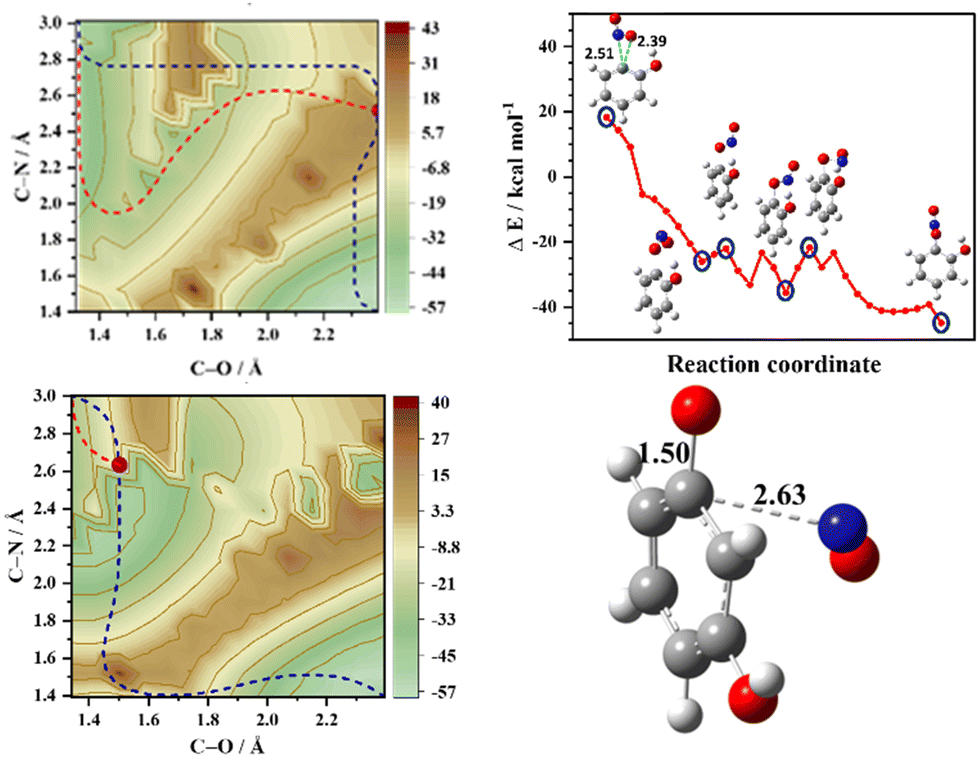 | ||
| Fig. 3 Ground state (S0) 2D-PES plots of o-nitrophenol (top left) and m-nitrophenol (bottom left) with the projected minimum energy path (MEP) that connects the reactant (bottom right corner) to the NO and phenoxy radical as products (top left corner) in the T1 state (blue dashed line). The purple dot represents the T1/S0 crossing point along the T1 MEP and the red dashed lines represent the MEPs in the S0 state. Note that the T1/S0 crossing for o-nitrophenol and m-nitrophenol is early and late relative to the reactant, respectively. The S0-MEP and selected structures after the T1/S0 crossing point for o-nitrophenol are shown (top right) along with the T1/S0 crossing point of m-nitrophenol (bottom right). The distances are shown in Å and the energy scale is in kcal mol−1 relative to the starting geometry in the T1 state. | ||
Summarising, the branching ratio of the slow-to-fast (s/f) translational energy components, for various NO product states, shows a higher value for the ortho isomer in comparison to the meta isomer. Furthermore, consistently higher s/f ratio for the o-nitrophenol over o-nitroaniline is attributed to the OH group's propensity to form stronger hydrogen bonds compared to the NH2 group. Therefore, it is inferred that the ortho isomer preferentially releases the slower NO fragments, which is attributed to the minimum energy roaming path. On the other hand, in the meta isomers, the faster component mediated via the oxaziridine pathway is enhanced. Thus, it has been illustrated that the hydrogen bonding ability of the substituent in the ortho position to the NO2 group favours the roaming mechanism.
A skimmed molecular beam of helium doped with the heated (330–350 K) reagents (o-nitrophenol, m-nitrophenol, o-nitroaniline, and m-nitroaniline; Sigma Aldrich) was intersected by a counter-propagating pump (266 nm) and probe lasers15 and the ensuing NO cations were detected via the A2Π − (X2Π) [(0,0); Q1 (J = 21.5; 29.5; 33.5)] and A2Π − (X2Π) [(1,0); P1J = 50.5]16,17 and imaged. Each image was collected for 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 shots, which were symmetrized using ImageJ software.18 Abel inversion was carried out by the Basis Set Expansion method (BASEX) to extract the translational energy distribution.19 The plane of polarization of both lasers is kept parallel to the plane of the detector and the laser flux was optimized such that the signal intensity for the NO fragment was observed only in the presence of both the pump and probe lasers. The total translational energy profiles [P(ET)], black traces in Fig. S3 and S4 (ESI†), were fitted to a bimodal empirical function,8,20,21
000 shots, which were symmetrized using ImageJ software.18 Abel inversion was carried out by the Basis Set Expansion method (BASEX) to extract the translational energy distribution.19 The plane of polarization of both lasers is kept parallel to the plane of the detector and the laser flux was optimized such that the signal intensity for the NO fragment was observed only in the presence of both the pump and probe lasers. The total translational energy profiles [P(ET)], black traces in Fig. S3 and S4 (ESI†), were fitted to a bimodal empirical function,8,20,21
| P(ET) = C·[(ET)a1·(EmaxT − ET)b1 + (ET)a2·(EmaxT − ET)b2] |
The experimental results were interpreted by calculating the two-dimensional potential energy surfaces (2D-PES) in the ground (S0) and first excited triplet (T1) states with the B3LYP/6-311++G(d,p) and M06-2X/6-311++G(d,p) levels of theory using the Gaussian 09 suite of programs.22 The 2D-PESs were mapped along two internal coordinates; the C–N bond (“C” referring to the carbon atom of the aromatic ring to which the nitrogen atom “N” of the nitro group is attached) and C–O bond (“O” referring to the oxygen atom of the nitro group that is away from the ortho substitution, which later forms a bond with the “C” atom of the aromatic ring leading to NO release). The distances R(C–N) and R(C–O) are varied in the range 1.4 to 3.0 Å and 1.25 to 2.3 Å, respectively, in 14 equidistant steps on each coordinate resulting in 196 data points. The minimum energy path on the 2D-PES for the release of NO from each of the substituted nitrobenzenes was evaluated using the Minimum Energy Path Surface Analysis (MEPSA) tool.23
PRC thanks PMRF for the research fellowship and MK is supported by the Women Scientists Scheme of the Department of Science and Technology (Grant no. SR/WOS-A/CS-18/2019). Financial support from the Science and Engineering Research Board of the Department of Science and Technology (Grant no. CRG/2022/005470) and the Board of Research in Nuclear Sciences (BRNS Grant no. 58/14/18/2020) is gratefully acknowledged. The authors wish to thank Ms Shaivi Kesari and Ms Bhawana for their help in carrying out some of the experiments. GNP wishes to thank Kaleidoscope-2023 for stimulating discussions.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- E. G. Alvarez, D. Amedro, C. Afif, S. Gligorovski, C. Schoemacker, C. Fittschen, J. F. Doussin and H. Wortham, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13294–13299 CrossRef CAS.
- G. Lammel and J. N. Cape, Chem. Soc. Rev., 1996, 25, 361–369 RSC.
- M. F. Lin, Y. T. Lee, C. K. Ni, S. Xu and M. C. Lin, J. Chem. Phys., 2007, 126, 064310 CrossRef PubMed.
- M. L. Hause, N. Herath, R. Zhu, M. C. Lin and A. G. Suits, Nat. Chem., 2011, 3, 932–937 CrossRef CAS PubMed.
- L. Saalbach, N. Kotsina, S. W. Crane, M. J. Paterson and D. Townsend, J. Phys. Chem. A, 2021, 125, 7174–7184 CrossRef CAS PubMed.
- Y. He, A. Gahlmann, J. S. Feenstra, S. T. Park and A. H. Zewail, Chem. – Asian J., 2006, 1, 56–63 CrossRef CAS PubMed.
- A. Giussani and G. A. Worth, J. Phys. Chem. Lett., 2024, 15, 2216–2221 CrossRef CAS PubMed.
- N. B. Bejoy, P. Roy Chowdhury and G. N. Patwari, J. Phys. Chem. Lett., 2023, 14, 2816–2822 CrossRef CAS.
- N. B. Bejoy and G. N. Patwari, J. Phys. Chem. A, 2023, 127, 7168–7169 CrossRef CAS PubMed.
- S. Mishra, N. B. Bejoy, M. Kawade, H. P. Upadhyaya and G. N. Patwari, J. Chem. Sci., 2021, 133, 128 CrossRef CAS.
- A. Giussani and G. A. Worth, J. Chem. Theory Comput., 2017, 13, 2777–2788 CrossRef CAS PubMed.
- J. Mewes, V. Jovanovic, C. M. Marian and A. Dreuw, Phys. Chem. Chem. Phys., 2014, 12393–12406 RSC.
- H. A. Ernst, T. J. A. Wolf, O. Schalk, N. González-García, A. E. Boguslavskiy, A. Stolow, M. Olzmann and A. N. Unterreiner, J. Phys. Chem. A, 2015, 119, 9225–9235 CrossRef CAS PubMed.
- A. Giussani and G. A. Worth, Phys. Chem. Chem. Phys., 2020, 22, 15945–15952 RSC.
- S. Singh, M. Kawade, P. R. Chowdhury and G. N. Patwari, J. Chem. Sci., 2023, 135, 1–10 Search PubMed.
- R. P. E. Engleman Jr, H. M. Peek and V. D. Baiamonte, 1970 DOI:10.2172/4128104.
- M. Sumida, Y. Kohge, K. Yamasaki and H. Kohguchi, J. Chem. Phys., 2016, 144, 64304–64312 CrossRef.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- S. M. Poullain, D. V. Chicharro, L. Rubio-Lago, A. García-Vela and L. Bañares, Philos. Trans. R. Soc., A, 2017, 375, 20160205 CrossRef PubMed.
- C. T. Matthaei, D. P. Mukhopadhyay and I. Fischer, J. Phys. Chem. A, 2021, 125, 2816–2825 CrossRef CAS PubMed.
- H. J. Deyerl, I. Fischer and P. Chen, J. Chem. Phys., 1999, 111, 3441–3448 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- I. Marcos-Alcalde, J. Setoain, J. I. Mendieta-Moreno, J. Mendieta and P. Gómez-Puertas, Bioinformatics, 2015, 31, 3853–3855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methodology, 2D-PES at the M0-62X/6-311++G (d,p) level, one dimensional cut of the T1-2D-PES along the MEP of NO release, VMI images and fitting of the [P(ET)] profiles, and T1/S0 crossing in nitroaniline. See DOI: https://doi.org/10.1039/d4cc01497a |
| This journal is © The Royal Society of Chemistry 2024 |