
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a flow system for decentralized electrochemical analysis of heavy metals using screen-printed electrodes: the importance of sensor stability
Serena
Laschi
 a,
Patrick Severin
Sfragano
a,
Francesco
Tadini-Buoninsegni
a,
Nathalie
Guigues
b and
Ilaria
Palchetti
*a
a,
Patrick Severin
Sfragano
a,
Francesco
Tadini-Buoninsegni
a,
Nathalie
Guigues
b and
Ilaria
Palchetti
*a
aDepartment of Chemistry “Ugo Schiff” (DICUS), Via della Lastruccia 3, 50019 Sesto Fiorentino, FI, Italy. E-mail: Ilaria.palchetti@unifi.it
bLaboratoire national de métrologie et d'essai (LNE), 1 rue Gaston Boissier, 75015 Paris, France
First published on 11th June 2024
Abstract
Year after year, the need for decentralized tools to tackle the monitoring of heavy metal levels in the environment gradually increases. In this context, suitable electrochemical methodologies are widely established and particularly attractive for the production of low-cost miniaturized field-deployable analytical platforms. This work focused on the development of an automatable portable system based on square-wave anodic stripping voltammetry (SWASV) for the on-line detection of heavy metals. The surface of the sensors is appropriately modified and coupled with a fluidic system equipped with an ad-hoc designed flow cell. A custom software tool was introduced to handle the remote-controlled potentiostat and automate the various steps of the procedure, including stirring operations, cleaning phases, SWASV measurements, and data collection. After studying technical and analytical challenges, the final system developed was applied to the simultaneous detection of Cd(II), Pb(II), and Cu(II) in solution, achieving sub-ppb detection limits. Additionally, the practical applicability of the method was successfully applied to river water samples collected from the Loire basin in France.
Introduction
Heavy metals are natural constituents of the marine and freshwater environment, but generally, they are found in very low concentrations.1 Rapid industrialization and urbanization are mainly responsible for the increase in heavy metal contamination, resulting in substantially accelerated rates of mobilization and movement in the environment.2–4 Indeed, municipal and industrial wastewaters, mine drainage, offshore oil and gas exploration, agricultural runoff, acid rain etc. have contributed to the increased metal load in natural waters being ultimately incorporated into aquatic sediments.2–4 Monitoring of heavy metals is indeed crucial to identify the sources and pathways in the aquatic environment. Consistent and smart monitoring contributes to the preservation and protection of aquatic ecosystems by avoiding negative effects on human health. Specifically, monitoring of heavy metal concentrations in waterbodies may involve direct measurements of water samples, or indirect monitoring of heavy metal load in sediments or organic tissues of local organisms. Nowadays, monitoring methodologies of water samples consist of discrete or cumulative, manual or automatic sampling, followed by laboratory analysis, using analytical techniques such as inductively coupled plasma-mass spectrometry (ICP-MS), atomic absorption spectroscopy (AAS), and inductively coupled plasma-atomic emission spectroscopy (ICP-AES).5On the other hand, on-site heavy metal detection in a waterbody, reducing the pre-treatment step to a minimum and maintaining the most original characteristics of heavy metals, has shown a number of advantages compared to the traditional approaches for heavy metals distribution assessment in waters involving manual sampling, off-site detection, and possible contamination.6 Electrochemical devices have been widely proposed for trace heavy metal detection since they require simple analytical procedures.7–12 They are low-cost and well-suited for miniaturization and automatic in situ measurements with minimal sample manipulation.10,11 Electrochemical systems also allow for fast analyses, resulting in valid experimental data obtained mostly in real-time or in a few minutes. By using electrochemical techniques, on-line monitoring of water samples becomes possible, providing dynamic data of relevance for biogeochemical survey.13 Nevertheless, specific developments are still required for such applications, particularly to improve sensitivity, limits of detection (LODs), and automation. Thus, there is the need for innovative tools equipped with a multi-sensor platform that can allow the electrochemical analysis of a broad spectrum of analytes with remote control and data processing.14
In this work, a stand-alone device for on-line electrochemical monitoring of heavy metals based on square-wave anodic stripping voltammetry (SWASV) was developed. The system consists of a self-standing setup equipped with an ad-hoc designed flow cell, a properly modified sensor, a pumping system for sample introduction, and a portable remote-controlled potentiostat. In addition, a custom software tool was optimized and exploited to automatically select and alternate the various analytical steps required for the procedure, including stirring operations, cleaning stages, and measurement phases. Different technical and analytical challenges were studied and reported. The developed procedure is able to perform the simultaneous detection of Cd(II), Pb(II), and Cu(II) below sub-ppb levels in less than 4 min. An experimentally optimized automated cleaning step introduced via software allows for the regeneration of the sensor surface and its reuse for multiple analyses over time. The entire fluidic setup is organized to limit the working volume and therefore the final waste. Preliminary experiments carried out on river samples collected from the Loire basin in France demonstrated the applicability of the proposed system to detect trace levels of heavy metals in real matrices.
Experimental
Reagents
Methocel® 90HG was purchased from Fluka (Milan, Italy). Mercury acetate, Suprapur® grade hydrochloric, nitric acid, and acetic acids were purchased from Merck (Milan, Italy). The water used for the preparation of solutions was from a Milli-Q System (Millipore S.p.A., Milan, Italy). Heavy metal stock solutions were prepared by diluting Cd(II), Pb(II), and Cu(II) standard solutions AAS grade (Merck, Milan, Italy).Sensors
Screen-printed carbon-based electrodes (SPCEs) were purchased from Ecobioservices and Researches SrL (Sesto Fiorentino, Italy). They consist of a graphite working electrode (diameter 3 mm), a graphite counter electrode, and a silver pseudo-reference electrode. In addition, the silver electrical contacts were covered by a graphite layer to prevent oxidation phenomena during storage.The surface of the screen-printed graphite working electrode was then modified using a specific coating mixture to make it suitable for the detection of certain heavy metals. This coating mixture was prepared according to Palchetti et al.,15 and, briefly, it was prepared by mixing 100 mg of mercury acetate with 100 μL of acetic acid in 10 mL of Milli-Q H2O. To 1.5 mL of this solution, 3.5 mL of H2O and 6.25 mg of Methocel® 90HG were added. Once homogeneous, 1 μL of this mixture was cast onto the surface of the working electrode and dried in the air at room temperature.
The external standard 3 M KCl Ag/AgCl electrode (diameter: 4 mm) was purchased from Merck (Milan, Italy).
Instrumentation
A handheld battery-powered Bluetooth-controlled potentiostat, namely EmStat4X assisted by PSTrace 5.10 (Palmsens, Netherlands), was used to perform the electrochemical measurements. Through a proprietary switchbox, the potentiostat was connected to a software-controllable magnetic mini-stirrer (topolino, IKA), to synergically activate mixing operations during the analyses. The SPCEs were connected to the EmStat4X potentiostat through a specially adapted electrical edge connector. Square-wave anodic stripping voltammetry (SWASV) conditions were: conditioning potential: −0.15 V for 60 s, deposition potential: −1.1 V for 120 s, equilibration time: 30 s, square-wave amplitude (Ep): 28 mV, step potential (Es): 3 mV, frequency (f): 15 Hz.Liquid circulation inside the fluidic system was established through positive displacement by using a miniaturized software-controllable stepping motor peristaltic pump (Goso, 0760-87997182, China). All software-assisted mechanisms were controlled through an in-house custom-made script developed under the software developer kit of PalmSens and run by a portable tablet computer on Windows 10 (Microsoft Corporation, USA).
Fluidic system
A block scheme of the portable system used in this work is reported in Fig. 1a. As mentioned in the previous section, the sample is collected by a miniaturized peristaltic pump and introduced inside a fluidic system completed by a magnetic mini-stirrer. Once processed, the sample is conveyed inside a sealed container for its disposal under safety conditions. The handheld potentiostat controls the sensing apparatus and the magnetic stirrer, while being connected wirelessly to a computer for remote control. Two setups are available for the fluidic system, as depicted in Fig. 1b. A first setup (Setup 1) is equipped with an ad hoc designed flow cell, which enables the use of SPCEs for flow-through applications. Particularly, the cell was mechanically realized in poly(methyl methacrylate) and presents a hollow volume of 2.5 mL, a height of 3.4 cm, and lies on a round base with a diameter of 3 cm. A schematic drawing of the cell is reported in Fig. 1c. Notably, the overflow condition is exploited: the body of the cell is equipped with an inlet that allows it to be filled from the bottom; a watertight inspectable lid made in polytetrafluoroethylene seals the cell and forces the flow to exit from the outlet, placed on the top. The SPCE is inserted into a gasket-equipped slit of the lid and tightened by two screws. An additional O-ring gasket placed at the entrance of the cell allows for a watertight seal of the lid itself. The cell assures the continuous flow of the reagents around the working electrode.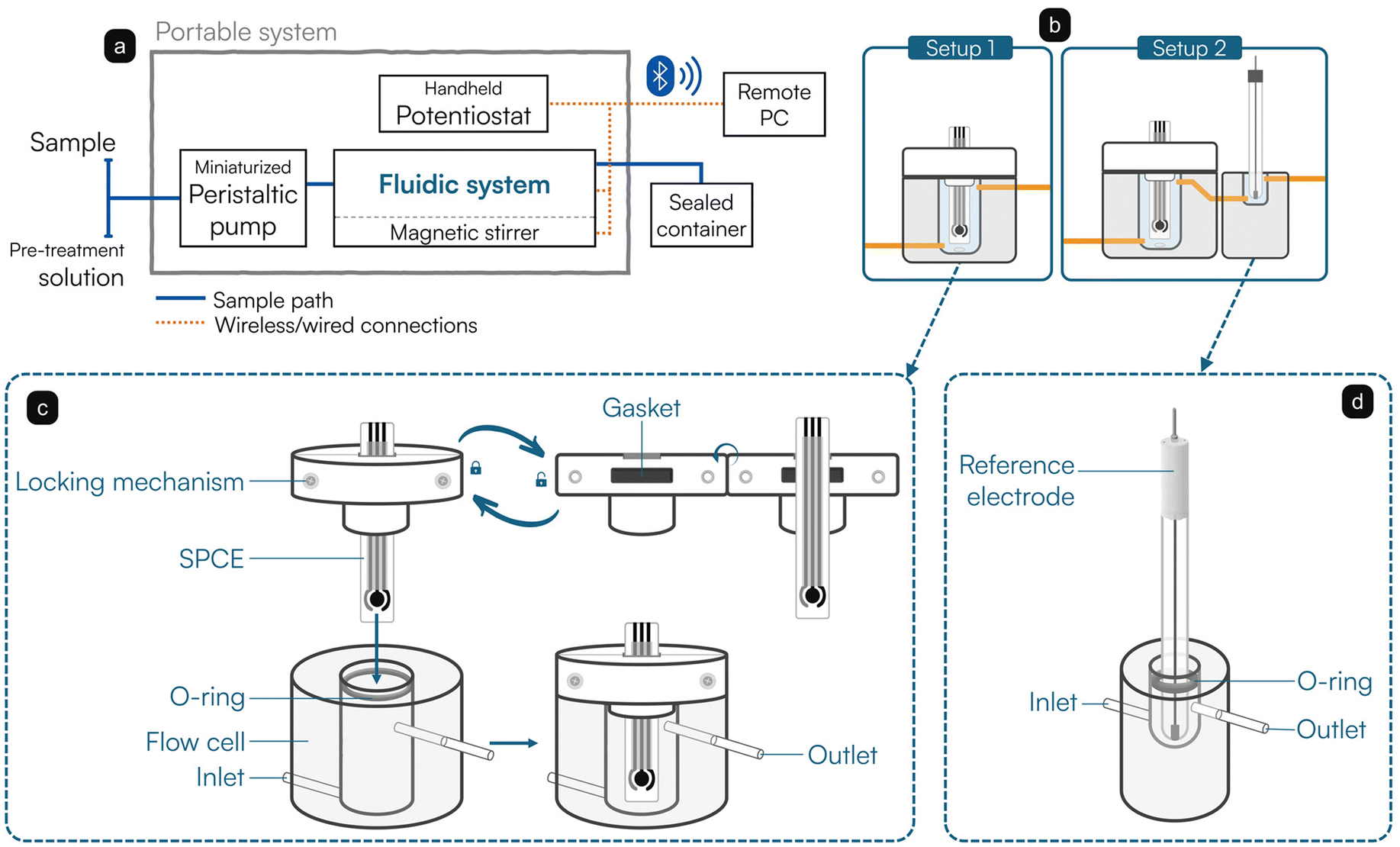 | ||
| Fig. 1 Flow system setup used. (a) Block diagram showing the main parts of the system. (b) Two setups for the fluidic system: a single flow cell for the SPCE (left) or in series with a second cell for the external reference electrode (right). (c) Schematic drawing of the ad hoc designed flow cell for the SPCE. The top part is represented by a watertight lid featuring a gasket-equipped slit for the introduction of the SPCE inside the cell. The lid can be opened along the horizontal axis and locked with two screws. Inside, the flow cell presents an O-ring to tightly seal it when the lid is introduced. Once sealed, the inner portion of the cell is interfaced to external elements through inlet and outlet. (d) Schematic drawing of the external reference electrode flow cell. | ||
In a second version (Setup 2), the outlet of the flow cell is connected in series with another ad hoc designed flow cell (Fig. 1d), hosting a volume of approximately 0.5 mL and housing an external Ag/AgCl reference electrode (3 M KCl). The continuity of the flow stream between the two cells guarantees the electrical connection, whereas the position of the reference electrode downstream of the flow avoids possible contaminations of the testing solution deriving from any leakage from the porous septum. Polyvinylchloride tubes (Elkay Lab, 116-0549-160, inner diameter: 2.29 mm) were used to connect the various elements of the fluidic system. The total volume of the complete fluidics was experimentally estimated to be ranging approximately between 3.0 (Setup 1) and 3.5 mL (Setup 2).
Measurement procedure
Initially, a new modified SPCE is positioned in its flow cell by fixing it in the watertight lid using the appropriate screws. Prior to starting with a measurement session, the SPCE-coated surface is activated to make it suitable for heavy metal deposition (“Activation”). This activation consists of applying −1.1 V for 300 s in 0.1 HCl under stirring to reduce Hg in situ at a metallic level and, thus, obtain a stable mercury film (Hg film) onto the graphite surface of the working electrode. Subsequently, 10 SWASV scans are carried out until low and stable background signals were obtained.15 Once this procedure is completed, a constant potential of −0.15 V is applied to the sensor until use, in order to maintain the stability of the modified surface.Sampling can be carried out in continuous or discontinuous flow. In a continuous flow setup, the sample is introduced into the fluidics, where a T-shaped fitting, connected to the same peristaltic pump, allows its in-line acidification before arriving at the flow cell. The discontinuous flow sampling is carried out as for the continuous procedure but by stopping the pump. In this case, the sample is acidified before its introduction into the fluidics. All measurements reported in this work were performed in discontinuous flow sampling. Thus, either the blank (0.1 M HCl/acetate buffer) or an acidified sample is introduced by the pump into the fluidics at a flow rate of 0.5 mL min−1. Once the circuit is completely filled (total running time: 7 min), the pump is stopped and the measurement can start, following the steps indicated in Table 1. After a conditioning phase (“1. Conditioning”) in which the sensor is subjected to a potential of −0.15 V under continuous stirring (to further enhance the cleaning of the sensor's surface), an accumulation potential is applied to deposit and preconcentrate the metals on the surface through amalgam formation16,17 (“2. Accumulation”). The method proceeds with an equilibration step, during which the potential is maintained at the same value as the previous phase, but without stirring (“3. Equilibration”). After 30 seconds, a SWASV scan is performed in quiescent conditions7,18,19 (“4. Measurement”). Once the measurement phase is terminated, the sensor is kept at the stand-by potential of −0.15 V until the next measurement (“5. Stand-by”). During the analysis, stirring, applied potential, and duration time are all controlled by the custom software tool that automatically performs the steps mentioned above. Additionally, after carrying out the measurement, the script saves and stores the SWASV scan collected. The total duration of the analysis amounts to 3 min and 50 s.
| Step name | Applied potential (V) | Duration time (s) | Stirring (on/off) |
|---|---|---|---|
| 1. Conditioning | −0.15 | 60 | On |
| 2. Accumulation | −1.10 | 120 | On |
| 3. Equilibration | −1.10 | 30 | Off |
| 4. Measurement | SWASV scan | 20 | Off |
| 5. Stand-by | −0.15 | Until the next measurement | On |
Real sample collection and analysis
River water samples were collected during a campaign conducted in the Loire river basin (France) in seven different points going from the Massif Central to Orléans. Water samples were filtered on site and within 2 h of sampling, through a 0.45 μm filter (Teflon filters, Durapore, Millipore, USA), then acidified (pH < 2) with ultrapure HNO3, and stored in polyethylene bottles (ELVETEC, France) until use. All bottles were previously washed in the laboratory with ultrapure 10% HNO3, followed by Milli-Q water and stored in Milli-Q water until use.Trace elements were measured without pre-concentration by ICP-MS (PQ3, VG from VG Elemental/Thermo)20 following the standard EN ISO 17924-2 for metals measurement in water.21 The detection limits for the different trace elements are in the range of 0.05–0.1 μg L−1, and the uncertainty associated, expressed as relative standard deviation (RSD), was 10%.
In the case of electrochemical measurements, the following treatment was performed: 0.06 mL of acetic acid 100% Suprapur® were added to 9.94 mL of sample, in order to achieve 0.1 M acetate as the final concentration. Once treated, the sample was pumped for 15 min into the fluidics at 0.5 mL min−1 as flow rate, then stopped and submitted to SWASV analysis. A cleaning step was applied between subsequent analyses. Sample analysis was performed using the same modified SPCE. Quantification was performed either by the standard addition method or by interpolation with the calibration curve.
Results and discussion
From batch to flow anodic stripping voltammetry: overall characterization
Despite the environmental concern of Hg, mercury-based electrodes still play an important role in anodic stripping voltammetry thanks to the wide cathodic window, the excellent sensitivity in heavy metal determination, the wide range of metals that could be detected, and the simple surface renewal resulting in high reproducibility. Numerous efforts in the introduction of alternative less-toxic materials such as gold,22 bismuth,23 or antimony,24 are reported in the literature. Nevertheless, mercury-based electrodes remain the recommended approach when it comes to detecting simultaneously multiple heavy metals in traces, as recently highlighted by Dossi et al.14 and Xu et al.25 Therefore, when alternatives are not sufficient to provide the necessary performances, an interesting strategy could be represented by the minimization of the impact by reducing the amount of mercury used for the analysis.In a previous paper, our group15 proposed the in-batch use of mercury-coated screen-printed sensors, prepared beforehand in the lab and ready to use on-site. In this way, handling, transport, and disposal of toxic Hg(II) solutions upon performing decentralized measurements is avoided, given that the coating is pre-deposited on the electrode surface. The sensor previously described15 consists of (i) a miniaturized screen-printed carbon working electrode, modified with a coating mixture containing a small amount of mercury salt entrapped in a cellulose derivative, (ii) a silver pseudo-reference electrode, and (iii) a carbon counter electrode. The analytical procedure consisted of performing the analyses in a beaker and by manually substituting the heavy-metal-containing samples after processing them. This manual operation indeed entails high risks of environmental contamination and spilling, with potential threats to the safety of the operator. An interesting and safer advancement of this protocol could be represented by a miniaturized fluidic system operating in a closed circuit. Particularly, the sample (e.g., coming from water sources) could be collected by a peristaltic pump and introduced directly inside a flow cell where the sensor is located. Once the measurement is over, the contaminated sample can be directed through the outlet and inside a sealed container for its future disposal in safe conditions.
In this work, a fluidic system based on an ad hoc designed flow cell was thus developed. More in detail, in the presented setup, a compact peristaltic pump collects and directs the sample inside a sealed poly(methyl methacrylate) cell, where the modified SPCE is housed through a watertight lid and connected externally to a handheld potentiostat. The lid seals the cell and ensures a particular overflow mechanism, thus guaranteeing liquid circulation through the cell, while keeping it isolated and avoiding incoming and outgoing contaminations. Underneath the cell, a computerized mini-stirrer provides mixing operations when required. Once the sample is correctly introduced inside the flow cell, the software-assisted steps indicated in Fig. 2 are started, as previously reported in the Materials and Methods section. Briefly, after activation, a conditioning step is applied; then the software moves to the accumulation step, where the heavy metals potentially present inside the sample are accumulated and deposited onto the surface of the modified working electrode. Subsequently, an equilibration step is carried out, followed by the measurement through SWASV. Once completed, the system applies a stand-by potential. A cleaning step to remove the analyzed sample and wash the fluidics can be introduced, as discussed in the following sections. The cycle can start over from the conditioning step when a new sample is introduced.
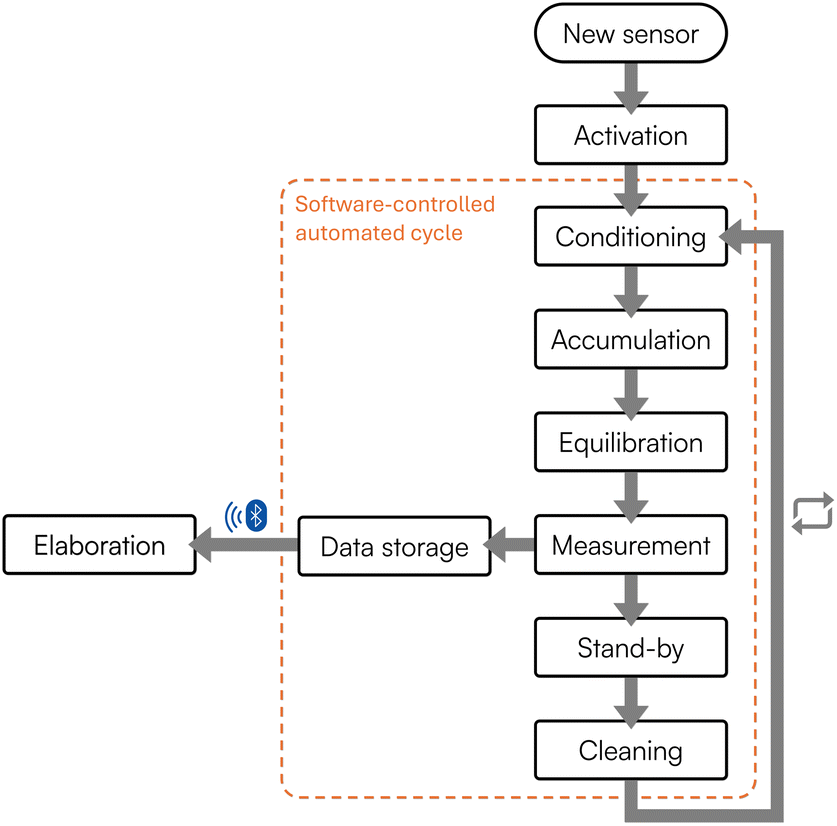 | ||
| Fig. 2 Schematic representation of the software tool developed to handle and automatize the procedure for the electrochemical analysis of heavy metals. | ||
Preliminary experiments were thus devoted to check if the sensor could be used for consecutive measurements in a flow-based stripping voltammetric analysis. The main parameters onto which the evaluation and optimization were focused were (i) the overall applicability of the setup, (ii) the study of a potential memory effect, (iii) the stability of the reference electrode, and (iv) the stability of the modified working electrode.
To assess the first point, namely the overall performance of the setup (Setup 1 of Fig. 1b), a calibration curve was performed. This allowed for the evaluation of the system's sensitivity using Pb(II) as a model analyte. For this purpose, after the conditioning step and a SWASV scan in 0.1 M HCl (blank solution), different solutions at increasing Pb(II) concentrations (in a range from 10 to 200 ppb) were tested. Each solution was analyzed in triplicate (i.e., three consecutive scans on the same solution); between two different solutions, a complete cleaning phase with 0.1 M HCl solution was performed.
As shown in Fig. 3a, upon performing SWASV measurements of the blank, namely 0.1 M HCl in the absence of metals, the behavior of the baseline can be ascribed to the cellulose derivative and the hydration of the cellulose-layer.15 This effect becomes evident only during the first scans performed with a new electrode and it is dramatically reduced during the subsequent scans.15 As reported in Fig. 3a and b, the sensor response shows a linear trend (r2 = 0.999) in the Pb(II) concentration range investigated, with good reproducibility (average RSD value around 8%). The flow system was then applied also to multielement analysis, specifically for the simultaneous detection of a solution containing 100 ppb each of Cd(II), Pb(II), and Cu(II). Fig. 3c displays the voltammograms obtained through SWASV in the absence (blank signal, dashed line) and the presence (solid line) of the three metals. The system shows to be sensitive to each of the metals tested. Moreover, it is interesting to note that the peak currents obtained for Pb(II) (17.3 ± 0.4 μA, n = 3) in this multielement analysis are indeed highly comparable to those obtained through the single analysis of Pb(II) alone (15.2 ± 0.5 μA, n = 3). This suggests that (i) the electrode surface maintains its efficiency, as it has no loss of sensitivity during the analysis of high concentrations of different metals, and more importantly (ii) that there is no need to carry out a further plating step in situ. Indeed, in anodic stripping voltammetry, it is usually advised to add a Hg salt also inside the solution under analysis to help regenerate in situ the Hg film potentially lost during the electrochemical scans.26–28 While it is reported that this practice can enhance even further the sensitivity of the method, it also dramatically augments the amount of Hg used and the risks of contamination. Nevertheless, herein, good sensitivity and low limits of detection were successfully achieved even without adding Hg in solution to regenerate the film, but by simply applying constantly a stand-by negative potential to the electrode (when not performing measurements) to keep the Hg film at a metallic level, hence considerably reducing the loss of the coating film.
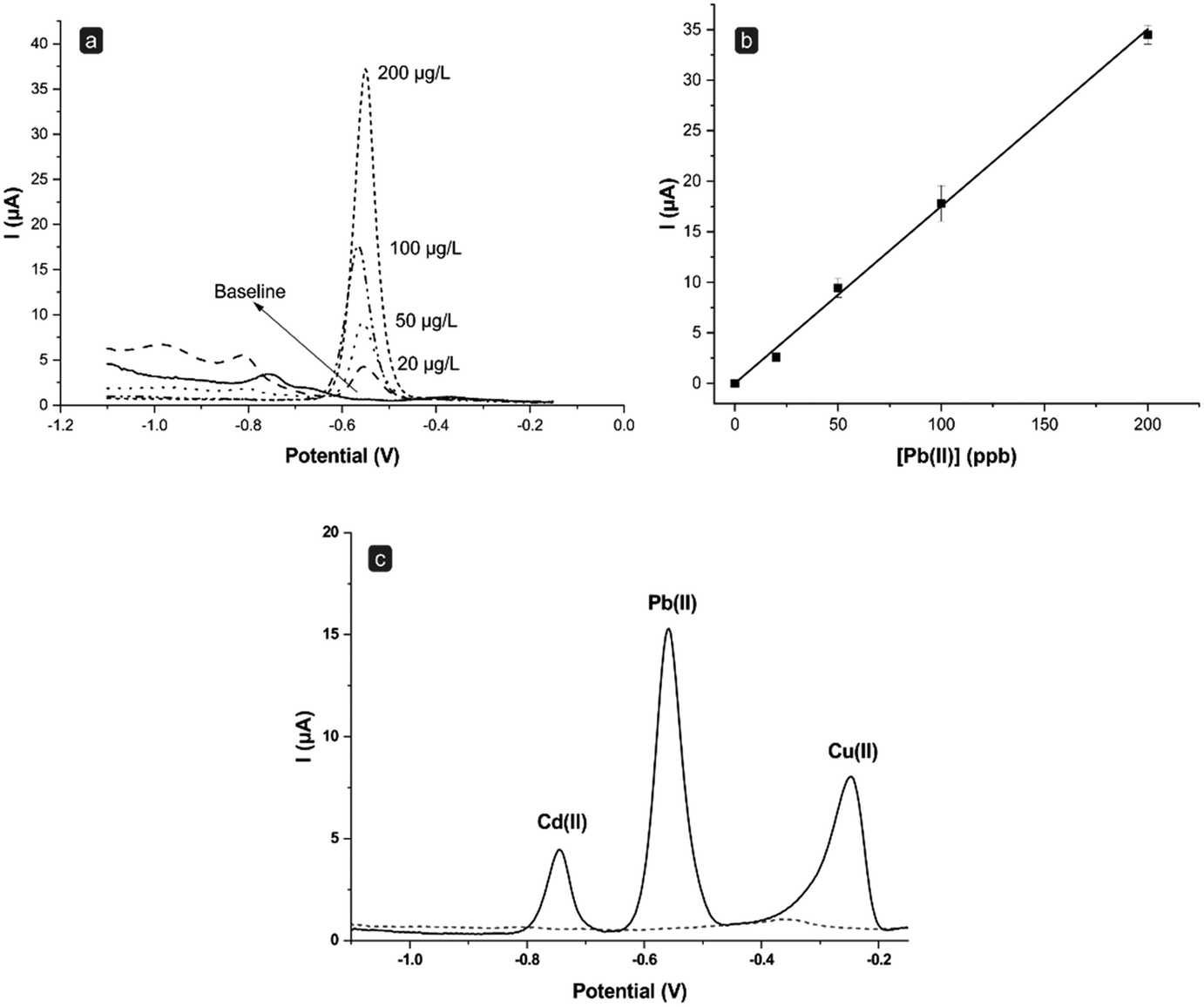 | ||
| Fig. 3 SWASV calibration curve of Pb(II) in 0.1 M HCl using the flow system, represented as (a) raw signals and (b) linear fitting obtained by interpolating the average peak current heights (n = 3). (c) SWASV raw signals obtained for a 0.1 M HCl solution containing 100 ppb each of Cd(II), Pb(II), and Cu(II) (solid line), compared to the signal of the blank (dashed line). | ||
Once the applicability of the method was assessed, the potential “memory effect” on the next measurement was studied. The memory effect has two components: one related to the fluidic system and one related to the incomplete oxidation of zero-valent metals during dissolution.15,29 The latter component is controlled by the application of a “conditioning step” (Table 1). The component related to the fluidic system can be considered negligible in the case of continuous flow measurements, in which the flow is never stopped, and fresh sample is continuously flowing inside the cell; nevertheless, it becomes more important when dealing with the discontinuous flow procedure, in which the pump is stopped and reactivated only for the collection of the next sample to be analyzed. In this case, it is necessary to be sure that the previously analyzed sample has been completely removed before performing the measurement.
Thus, a comparison between the blank signal obtained with an unused (new) sensor and that registered after performing a multianalyte calibration curve of Cd(II), Pb(II), and Cu(II) in a concentration range between 0 and 100 ppb was carried out (Fig. 4a). Notably, after the abovementioned multianalyte calibration, a washing step was applied by introducing a volume of 0.1 M HCl corresponding to a complete refilling of the fluidics. Indubitably, residual signals ascribable to the three metals previously analyzed are still present after the washing step; this means that a single refilling with 0.1 M HCl as a cleaning procedure is not sufficient to obtain a complete replacement of the solution into the fluidics and to restore a suitable blank signal.
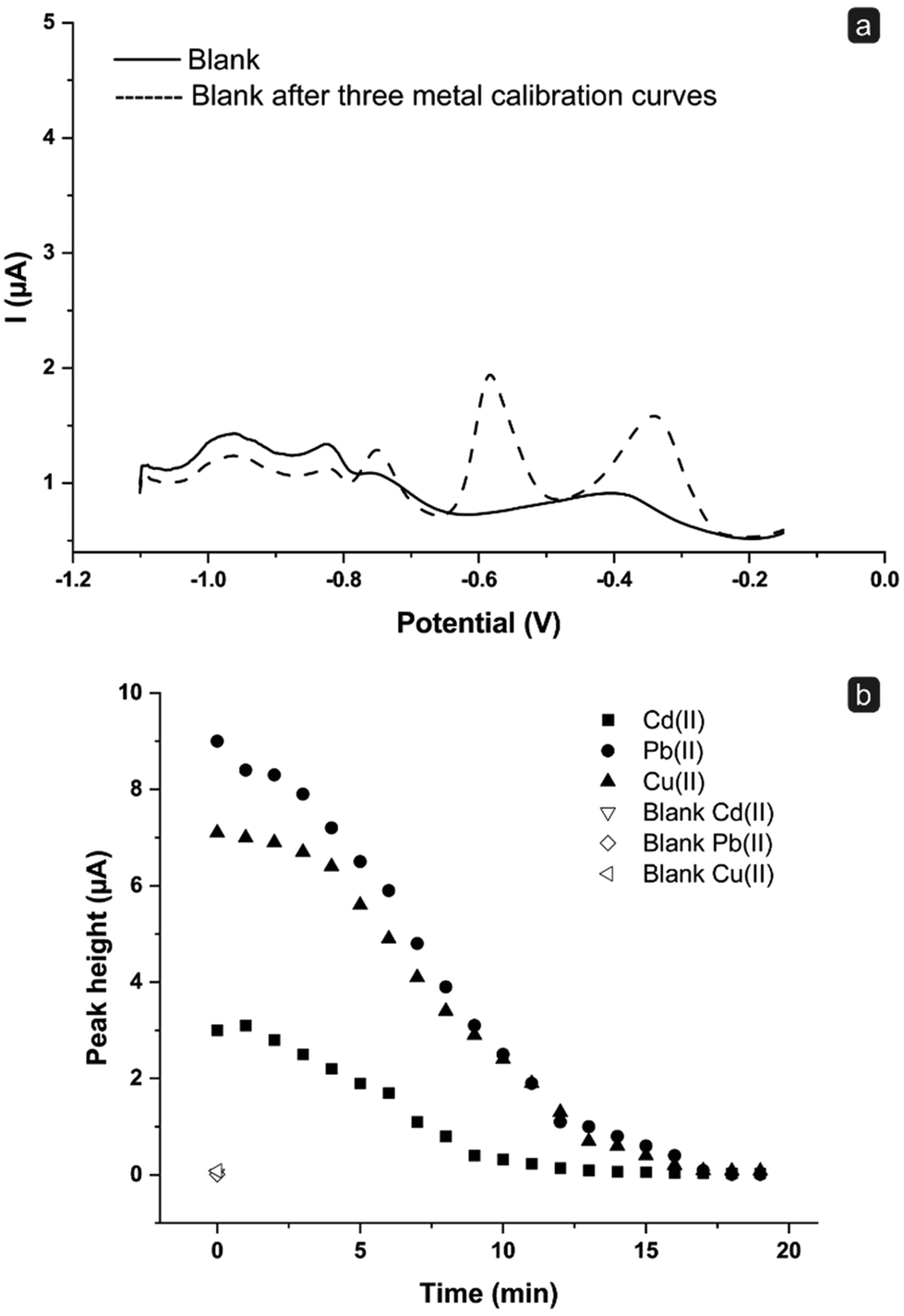 | ||
| Fig. 4 (a) SWASV signals for 0.1 M HCl (blank) recorded for a sensor as unused (new) (solid line) and after performing three complete multianalyte calibration curves of Cd(II), Pb(II), and Cu(II) in the concentration range 0–100 ppb. (b) Effect of the cleaning treatment: signal depletion of a 0.1 M HCl solution containing 100 ppb each of Cd(II), Pb(II), and Cu(II) during continuous washing of the fluidics using a 0.1 M HCl solution at a flow rate of 0.5 mL min−1; SWASV measurements were performed every 1 min; the empty symbols represent the blank signals recorded for each of the three metals before the multianalyte addition. | ||
Following this result, a series of experiments was performed to verify the volume of 0.1 M HCl solution to flush through the fluidic system in order to restore a clean blank signal. To this end, after a SWASV measurement in the presence of 100 ppb each of Cd(II), Pb(II), and Cu(II) (t = 0 min), portions of a 0.1 M HCl solution were introduced inside the cell at a flow rate of 0.5 mL min−1, with an interval time between the injections of 1 min. At the end of each interval time, the flow was stopped and a SWASV scan was registered. Fig. 4b shows the peak heights obtained for Cd(II), Pb(II), and Cu(II) at t = 0 min and then every 1 min. As noticeable, there is a decrease in the height of the current peaks, demonstrating a time-dependent removal process of the metals from the fluidics due to the replacement of the sample present in the cell by the incoming 0.1 M HCl fresh solution.
However, it has also to be noted that after 7 minutes (corresponding to the time required for a complete change of solution inside the fluidics) the blank signal is almost restored for Cd(II) (30% of residual signal), while it continues to be non-negligible for Pb(II) and Cu(II) (around 50% of signal still present). This confirms that a single filling cycle may not be sufficient to obtain an adequate cleaning. This condition is reached after 15 min, where a residual signal of around 3% is obtained for all the tree metals; this value also corresponds to the time required to totally refill and empty twice the flow system. Hence, this time was used for cleaning processes in further analyses. Notably, this cleaning time can be modulated and reduced by adjusting the flow rate.
The stability over time of the electrochemical setup was then tested. Two effects were taken into consideration, namely workout time and the number of measurements performed.
With this in mind, an experimental protocol was developed as follows: after the first activation phase of a new sensor, a 100 ppb Pb(II) solution was flushed into the system. SWASV scans were performed every hour in triplicate for two days, except for a night break of 10 hours, between day 1 and day 2, during which the electrode was kept at the standby potential of −0.15 V in 0.1 M HCl. The experiment collected a total number of 42 scans. The heights of the obtained current peaks were plotted as a function of the measurement time, obtaining the plot reported in Fig. 5. These results pointed out that, despite showing a stable current peak height during day one, a significant signal decrease was observed on day two. By studying the voltammograms obtained, it can also be noticed that the decrease in the peak height is associated with a peak-splitting phenomenon appearing from the second set of measurements of day 2 (Fig. 5). This peak splitting was already observed in other works14,30 and seems to be typical for long-term uses of Hg film-based sensors. Particularly, it can be ascribed to a deterioration of the mercury film during measurements, or of the screen-printed pseudo-reference electrode. Since the counter electrode used is composed of a graphite-based material, its potential degradation was not evaluated as it can be considered an inert material. The effect of replacing the pseudo-reference electrode with a classical reference electrode was first investigated. The fluidic system was thus adjusted to accommodate an external standard Ag/AgCl reference electrode (Setup 2 in Fig. 1b). Twelve measurements were carried out on day 1, after which the electrode was kept at the standby potential overnight to then continue the measuring cycle on days 2 and 3. As reported in Fig. 6a, the voltammogram obtained through SWASV on day 3 shows a deep degeneration of the peak shape over time, even if only 10 measurement cycles had been performed. This demonstrates that the signal's shape and intensity do not strictly depend on the number of cycles performed.
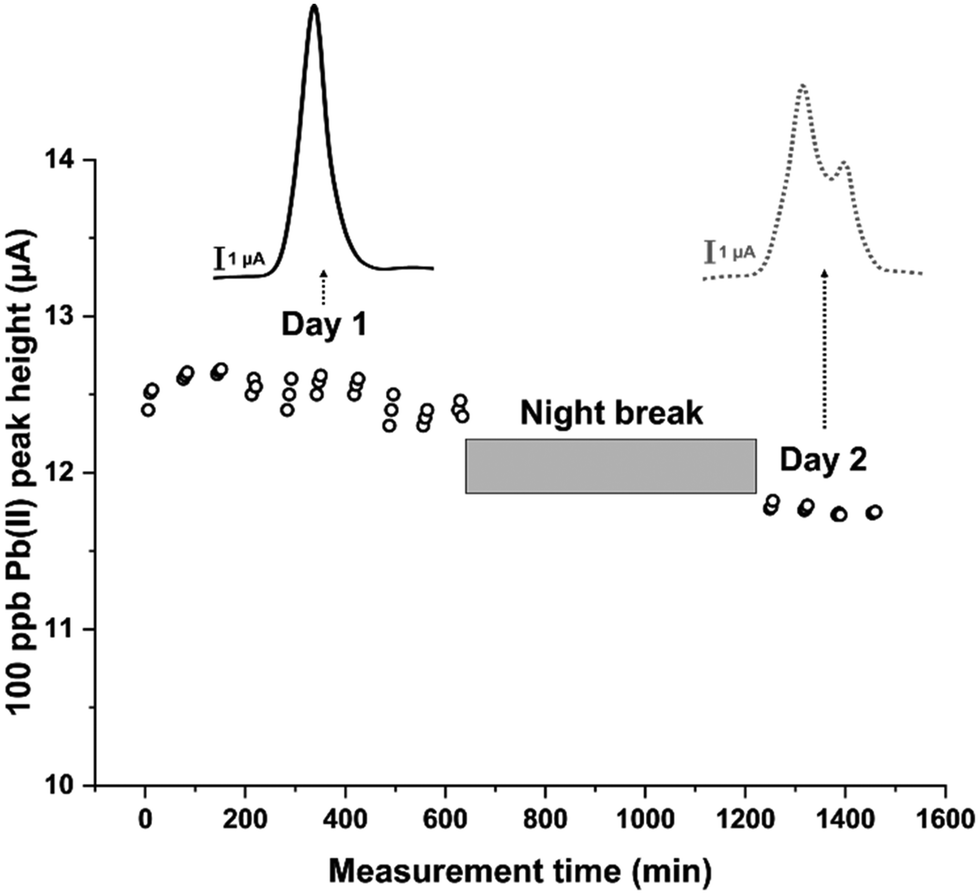 | ||
| Fig. 5 Stability of the modified sensors during a two days measurement session for the detection of 100 ppb Pb(II) in 0.1 M HCl; measurements were performed every hour in triplicate (except for a 10 hour night break) for a total of 42 scans. The scans above show the peak splitting phenomenon: comparison between the SWASV signal obtained on the first scan of Day 1 and that on Day 2. | ||
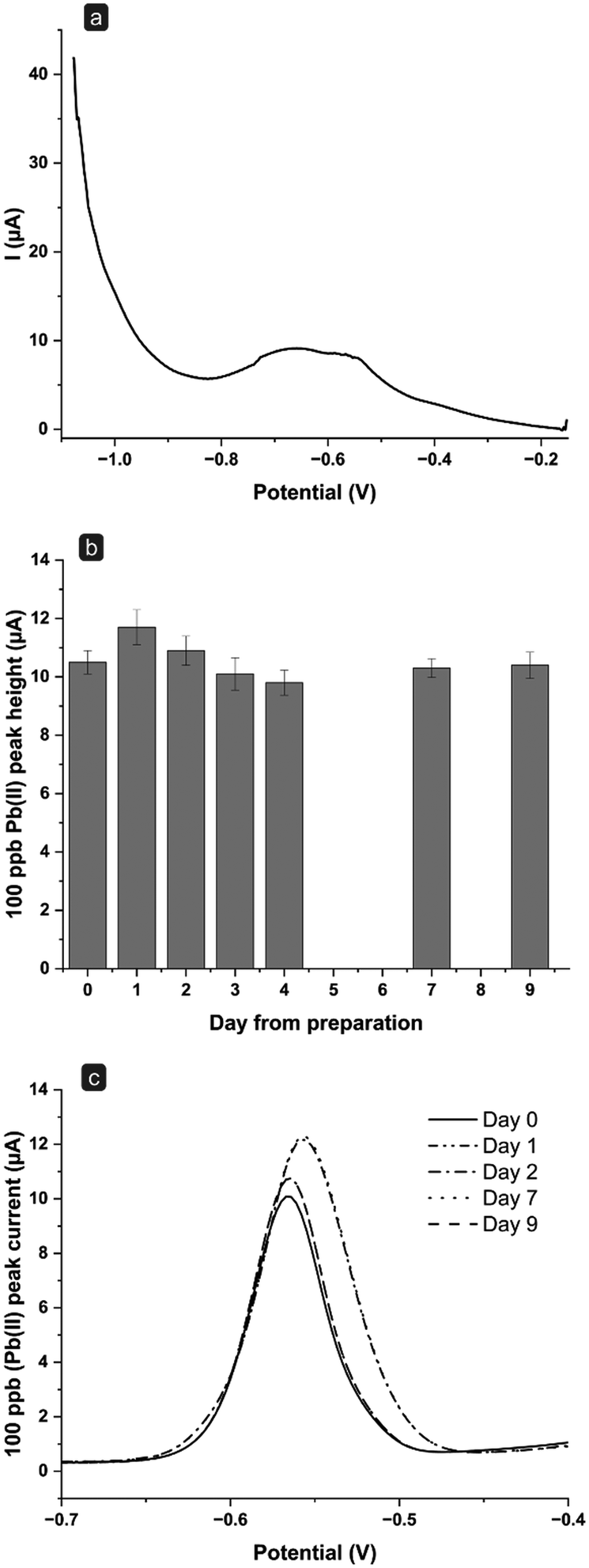 | ||
| Fig. 6 (a) SWASV signal of a 0.1 M HCl solution containing 100 ppb Pb(II) on Day 3 using an external Ag/AgCl reference electrode. (b) Peak heights and (c) voltammograms obtained for 100 ppb Pb(II) using 0.1 M acetate buffer (pH 4.75) as medium to study the stability of the flow system during consecutive days; the bars represent average peak currents resulting from SWASV (n = 6 per day, total number of measurements: 54); the potentials are referred to the external Ag/AgCl reference electrode. | ||
Nevertheless, it was concluded that the use of an external reference electrode did not appear to improve the stability of the system. Considering these results, the next step was to focus on the stability of the Hg-modified working electrode.
Indeed, the splitting of the peak could be related to the homogeneity of mercury film distribution after long-term use. Heavy metals could be deposited in areas with a different distribution of mercury film, causing the peak splitting.30 Problems related to the mercury-film homogeneity could be accelerated by the acidic medium (0.1 M HCl, pH = 1), and by the deposition potential (−1.1 V), which could lead to hydrogen evolution.27,31 Furthermore, the presence of chloride anions up to 1 M,32,33 could also affect the stability of the sensors causing the formation of mercury(I) chloride, even though a −0.15 V standby potential is applied after each measurement cycle. Hence, experiments were carried out testing a different and less acidic medium that did not contain Cl− ions. As for the previous experiments, the setup including the external reference electrode was maintained, as the lack of chlorides in the measurement solution could affect the stability of the pseudo-reference electrode. Fig. 6b reports the results over consecutive days obtained for 100 ppb Pb(II) solutions prepared with 0.1 M acetate buffer, pH 4.75. Acetate buffer has been extensively used in anodic stripping voltammetric analyses for the detection of heavy metals thanks to the enhanced stability of the Hg film in this medium, as reported in the literature.34–39
Using this setup, the Hg film remains stable for up to 9 days and more than 50 measurements (6 measurements were performed for each day). Notably, the peak obtained after 9 days was still well defined, with no evident splitting or shoulders (Fig. 6c). These results seem to confirm that the pH value of the medium and the presence of Cl− ions affect the sensor stability over time. However, it should be considered that, in the application on real sample analysis, a complete absence of Cl− ions in the tested sample will not be foreseeable. Nevertheless, reducing the contact time of the sensor with Cl− ions to the measurement step only (while maintaining it in a chloride-free medium, i.e., acetic buffer, during the standby step), could help in preserving its long-term stability.
After all the abovementioned optimizations, the new conditions were used to establish a multianalyte calibration curve for a solution containing simultaneously Cd(II), Pb(II), and Cu(II). To this end, a software script was introduced to control and automate the procedure. As previously mentioned, this tool allows to (i) recall and perform the different measurement steps, (ii) automate the saving of the single scans, and (iii) apply the stand-by potential for sensor stability. Fig. 7 shows the SWASV scans obtained for Cd(II), Pb(II), and Cu(II) in the concentration range of 0–100 ppb. The linear regression analysis, obtained by plotting the height of the peaks obtained for each concentration, gave the following equations: for Cd(II), y = (0.040 ± 0.001)x; for Pb(II), y = (0.152 ± 0.001)x; for Cu(II), y = (0.077 ± 0.002)x (the standard deviation was calculated for n = 3 different sensors). In all cases, linearity ranging from 1 to 100 ppb was obtained with a correlation factor of 0.9988 for Cd(II), 0.9991 for Pb(II), and 0.9979 for Cu(II). The limits of detection (LOD) for each metal were estimated as the concentration corresponding to three times the standard deviation of the blank divided by the slope of the calibration curve, and were 0.08, 0.02, and 0.08 ppb for Cd(II), Pb(II), and Cu(II), respectively. Likewise, the limit of quantification (LOQ) was evaluated considering ten times the standard deviation of the blank divided by the slope of the calibration function, resulting in values of 0.25, 0.07, and 0.26 ppb for Cd(II), Pb(II), and Cu(II), respectively. It is interesting to note that these limits of detection are lower than those obtained with the in-batch manually operated protocol previously published from the same research group.15 Moreover, optimizations of the sensitivity can be introduced by modulating the accumulation time of the metals over the surface of the working electrode, thus promising good versatility of the performance of the sensor according to the analytical needs. This adds up to the advantage of having the results starting from less than 4 min following a semi-automated software-assisted procedure.
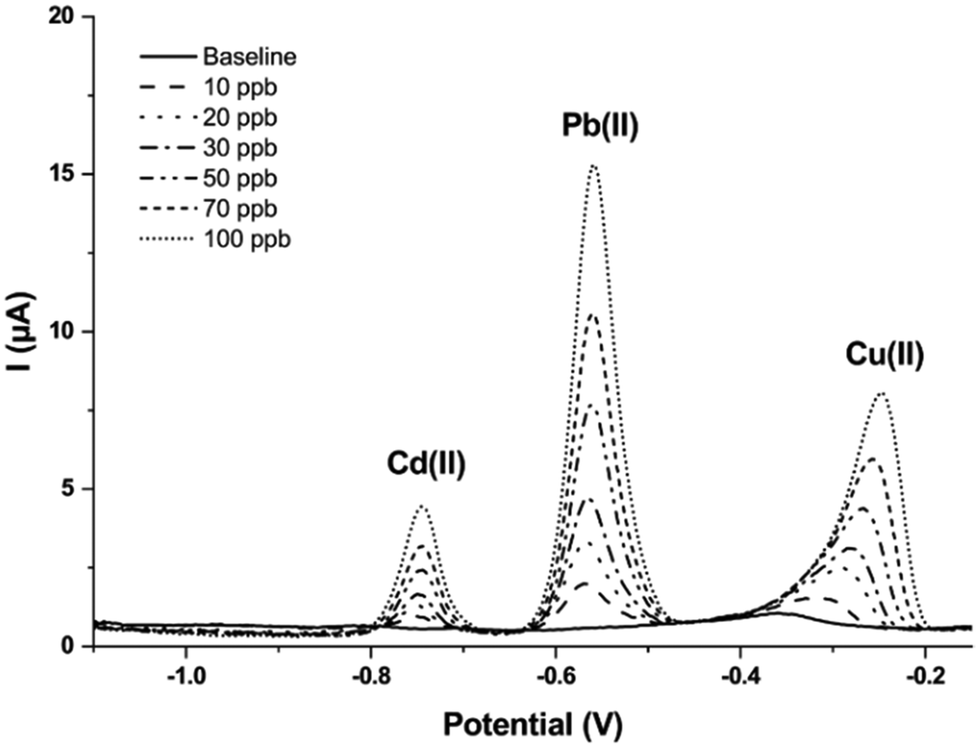 | ||
| Fig. 7 SWASV scans obtained for Cd(II), Pb(II), and Cu(II) in the range 0–100 ppb under optimized conditions; the measurements were performed using 0.1 M acetate buffer (pH 4.75) as medium. | ||
Real sample analysis
Once the analytical performances in standard solutions were established, real sample analysis was considered to evaluate the applicability of the system in waterbodies, and especially in river waters. Particularly, the system was tested on seven river water samples collected from the Loire River (France) and its upper tributaries in the Massif Central from a monitoring campaign for river water quality assessment. The samples were tested to evaluate the presence of Cd(II), Pb(II), and Cu(II), whose concentrations were determined through both the standard addition method and data interpolation using the calibration curve on standard solutions. This last procedure was carried out to evaluate the possibility of overcoming the use of the standard addition method, which is not a practical experimental approach when developing a system for continuous analysis. These results were then compared to those obtained independently, on a different aliquot of the same sample, by ICP-MS. The average results (n = 3) registered with all three methods are reported in Table 2. Notably, the values obtained with the two electrochemical-based methods are in good agreement. This result is extremely promising as it demonstrates that, for certain matrices, the standard addition method can be replaced by the use of the calibration curve fitting.| Sample | Cd(II) (ppb) | Pb(II) (ppb) | Cu(II) (ppb) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| SWASV standard addition method | SWASV calibration curve | ICP-MS | SWASV standard addition method | SWASV calibration curve | ICP-MS | SWASV standard addition method | SWASV calibration curve | ICP-MS | |
| 1 | ≤0.08 | ≤0.08 | ≤0.1 | ≤0.02 | ≤0.02 | ≤0.1 | 1.6 ± 0.3 | 0.8 ± 0.4 | 1.6 ± 0.1 |
| 2 | 1.2 ± 0.3 | 0.9 ± 0.2 | 4.6 ± 0.2 | ≤0.02 | ≤0.02 | ≤0.1 | 5.5 ± 0.4 | 4.3 ± 0.2 | 7.4 ± 0.1 |
| 3 | 1.4 ± 0.2 | 1.3 ± 0.2 | 2.5 ± 0.2 | ≤0.02 | ≤0.02 | ≤0.1 | 3.7 ± 0.4 | 3.4 ± 0.3 | 5.1 ± 0.1 |
| 4 | ≤0.08 | ≤0.08 | 0.9 ± 0.1 | 1.1 ± 0.3 | 0.8 ± 0.3 | 3.8 ± 0.1 | 2.9 ± 0.2 | 1.8 ± 0.3 | 3.8 ± 0.1 |
| 5 | ≤0.08 | ≤0.08 | 1.7 ± 0.3 | ≤0.02 | ≤0.02 | ≤0.1 | 1.0 ± 0.2 | 0.5 ± 0.2 | 1.6 ± 0.1 |
| 6 | ≤0.08 | ≤0.08 | ≤0.1 | 24.3 ± 0.6 | 24.1 ± 0.7 | 30.7 ± 0.2 | 23.0 ± 1.9 | 22.8 ± 0.9 | 24.1 ± 0.1 |
| 7 | ≤0.08 | ≤0.08 | ≤0.1 | ≤0.02 | ≤0.02 | ≤0.1 | 8.8 ± 0.9 | 6.7 ± 0.4 | 11.6 ± 0.1 |
However, a slight underestimation of the concentrations for all three metals in the case of SWASV analysis, when compared to ICP-MS data, is noticeable. This could be expected, since the electrochemical method does not foresee the matrix destruction, thus only the fraction of the metals available in solution is determined.40,41 Sample analysis also confirms the good sensitivity of the method, as already observed for the investigation of standard solutions.
Indeed, the electrochemical approach was able to quantify concentration values very close to the detection limit of the ICP-MS “gold standard” method. More importantly, these sensitivity levels were achieved after a very short analysis time for each sample (less than 4 min) and with a non-negligible advantage in terms of operating costs: only one sensor was used for the analysis of all samples (no replacement was needed).
Finally, considering the reduced waste produced (i.e., approximately 10 mL of waste per analysis), this portable flow system coupled with SWASV-based detection can be associated with a limited environmental impact. This makes the developed approach particularly interesting as a decentralized system for trace analysis, as it fulfils all the advantages of a transportable miniaturized system.
Conclusions
To tackle continuous decentralized heavy metal monitoring in waterbodies, an automatable and portable fluidic system was herein developed. It consisted of an ad-hoc designed flow cell housing a modified screen-printed electrode able to accumulate heavy metals on its surface. A custom software script was exploited to control wirelessly a handheld potentiostat, semi-automating the different steps of the procedure, including cleaning phases, stirring conditions, SWASV scans, and data collection. A study of the analytical challenges was reported, resulting in optimized conditions that were further implemented for the simultaneous detection of Cd(II), Pb(II), and Cu(II) in solution in less than 4 minutes. Good working electrode stability was demonstrated, allowing for the consecutive re-use of the sensor over multiple days (up to nine). Detection limits far below ppb levels (<0.1 μg L−1) were achieved, but versatile variations can be introduced by modulating the accumulation time of the metals. The applicability of the method was further successfully assessed by analyzing metal levels in river water samples collected from the Loire basin in France; indeed, a good agreement with the gold standard ICP-MS technology using both standard addition method and interpolation with the calibration curve approaches was found.Although imperative to perform multiplexed electrochemical analysis of trace metals, the use of Hg could raise concerns. Nevertheless, the amounts involved with this approach are indeed limited. Good sensitivities were achieved despite avoiding the common practice of adding Hg in solution to regenerate the Hg film, thus dramatically reducing the volumes required of such metal. Moreover, considering the preparation procedure, each sensor is covered by approximately 1 μg of mercury. Since each sensor has an autonomy of at least 100 measurements, which corresponds to around 1000 mL of produced waste, it can be noted that, in the worst-case scenario (i.e., the unlikely total release of the metal from the working electrode surface), the waste produced will contain a very small concentration of Hg (close to 1 μg L−1). The World Health Organization (WHO) established as 6 μg L−1 the limit of inorganic mercury in drinking water, as well as European Regulatory limit established this value as 1 μg L−1: this is the same concentration obtainable in the worst-case scenario. This sums up to the low volumes of waste collected in a safe sealed container. Lastly, substituting manual handling of the solutions with an automatable pump considerably lowers the risks of spilling and cross-contamination (while also reducing the possibility of damaging the Hg film since the sensor does not require to be transferred between various solutions). Overall, these advantages make this approach remarkably interesting where it is necessary to use a system for the remote analysis of trace metals while minimizing the environmental impact.
Data availability
Data for this article are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the European Union (European Climate, Infrastucture and Environment Executive Agency), HORIZON-MISS-2022-OCEAN-01, project iMERMAID, grant number 101112824, is acknowledged.References
- V. Kumar, R. D. Parihar, A. Sharma, P. Bakshi, G. P. Singh Sidhu, A. S. Bali, I. Karaouzas, R. Bhardwaj, A. K. Thukral, Y. Gyasi-Agyei and J. Rodrigo-Comino, Chemosphere, 2019, 236, 124364 CrossRef CAS PubMed.
- S. A. Razzak, M. O. Faruque, Z. Alsheikh, L. Alsheikhmohamad, D. Alkuroud, A. Alfayez, S. M. Z. Hossain and M. M. Hossain, Environ. Adv., 2022, 7, 100168 CrossRef CAS.
- K. H. Hama Aziz, F. S. Mustafa, K. M. Omer, S. Hama, R. F. Hamarawf and K. O. Rahman, RSC Adv., 2023, 13, 17595–17610 RSC.
- N. A. A. Qasem, R. H. Mohammed and D. U. Lawal, npj Clean Water, 2021, 4, 36 CrossRef CAS.
- M. Jin, H. Yuan, B. Liu, J. Peng, L. Xu and D. Yang, Anal. Methods, 2020, 12, 5747–5766 RSC.
- J. Holmes, P. Pathirathna and P. Hashemi, TrAC, Trends Anal. Chem., 2019, 111, 206–219 CrossRef CAS.
- A. J. Borrill, N. E. Reily and J. V. Macpherson, Analyst, 2019, 144, 6834–6849 RSC.
- I. Palchetti, C. Upjohn, A. P. F. Turner and M. Mascini, Anal. Lett., 2000, 33, 1231–1246 CrossRef CAS.
- L. Quadrini, E. Salvadori, S. Laschi, M. Verrucchi, A. Gnerucci, A. Cagnini and I. Palchetti, Microchem. J., 2024, 200, 110330 CrossRef CAS.
- G. Zhao, T.-T. Tran, S. Modha, M. Sedki, N. V. Myung, D. Jassby and A. Mulchandani, Front. Chem., 2022, 10, 1–13 Search PubMed.
- I. Saar and H. Evard, Micromachines, 2023, 14, 1369 CrossRef PubMed.
- V. Pifferi, A. Testolin, C. Ingrosso, M. L. Curri, I. Palchetti and L. Falciola, Chemosensors, 2022, 10, 67 CrossRef CAS.
- J. Barton, M. B. G. García, D. H. Santos, P. Fanjul-Bolado, A. Ribotti, M. McCaul, D. Diamond and P. Magni, Microchim. Acta, 2016, 183, 503–517 CrossRef CAS.
- C. Dossi, D. Monticelli, A. Pozzi and S. Recchia, Chemosensors, 2018, 6, 37 CrossRef.
- I. Palchetti, S. Laschi and M. Mascini, Anal. Chim. Acta, 2005, 530, 61–67 CrossRef CAS.
- B. R. Wygant and T. N. Lambert, Front. Chem., 2021, 9, 809535 CrossRef CAS PubMed.
- Ø. Mikkelsen and K. H. Schrøder, Electroanalysis, 2003, 15, 679–687 CrossRef.
- C. Ariño, C. E. Banks, A. Bobrowski, R. D. Crapnell, A. Economou, A. Królicka, C. Pérez-Ràfols, D. Soulis and J. Wang, Nat. Rev. Methods Primers, 2022, 2, 62 CrossRef.
- G. Aragay, A. Puig-Font, S. Laschi, M. Mascini, L. Sanfilippo and A. Merkoçi, Proc. iEMSs 4th Bienn. Meet. - Int. Congr. Environ. Model. Softw. Integr. Sci. Inf. Technol. Environ. Assess. Decis. Making, iEMSs 2008, 2008, 3, 1414–1419.
- M. Baalousha, S. Stoll, M. Motelica-Heino, N. Guigues, G. Braibant, F. Huneau and P. Le Coustumer, Environ. Sci. Pollut. Res., 2019, 26, 5251–5266 CrossRef CAS PubMed.
- ISO, ISO 17294-2:2023(E). Water quality — Application of inductively coupled plasma mass spectrometry (ICP-MS) — Part 2: Determination of selected elements including uranium isotopes, 3rd edn, 2023 Search PubMed.
- S. Laschi, I. Palchetti and M. Mascini, Sens. Actuators, B, 2006, 114, 460–465 CrossRef CAS.
- Z. Zou, A. Jang, E. T. MacKnight, P.-M. Wu, J. Do, J. S. Shim, P. L. Bishop and C. H. Ahn, IEEE Sens. J., 2009, 9, 586–594 CAS.
- E. Tesarova, L. Baldrianova, S. B. Hocevar, I. Svancara, K. Vytras and B. Ogorevc, Electrochim. Acta, 2009, 54, 1506–1510 CrossRef CAS.
- K. Xu, C. Pérez-Ràfols, A. Marchoud, M. Cuartero and G. A. Crespo, Chemosensors, 2021, 9, 107 CrossRef CAS.
- S. Daniele, C. Bragato and M. A. Baldo, Anal. Chim. Acta, 1997, 346, 145–156 CrossRef CAS.
- V. Rehacek, I. Hotovy and M. Vojs, J. Phys.: Conf. Ser., 2007, 61, 982–986 CrossRef CAS.
- E. Fischer and C. M. van den Berg, Anal. Chim. Acta, 1999, 385, 273–280 CrossRef CAS.
- J. Zou, J. Liu, Y. Xie, G. Peng, L. Duan, D. Hu, S. Chen, F. Qu and L. Lu, Chem. Eng. J., 2023, 468, 143849 CrossRef CAS.
- G. Aragay, A. Puig-Font, M. Cadevall and A. Merkoçi, J. Phys. Chem. C, 2010, 114, 9049–9055 CrossRef CAS.
- V. Rehacek, K. Shtereva, I. Novotny, V. Tvarozek, V. Breternitz, L. Spiess and C. Knedlik, Vacuum, 2005, 80, 132–136 CrossRef CAS.
- M. A. Nolan and S. P. Kounaves, Anal. Chem., 1999, 71, 1176–1182 CrossRef CAS.
- H. P. Wu, Anal. Chem., 1994, 66, 3151–3157 CrossRef CAS.
- M. Wojciechowski and J. Balcerzak, Anal. Chim. Acta, 1991, 249, 433–445 CrossRef CAS.
- F.-M. Matysik, S. Matysik, A. M. O. Brett and C. M. A. Brett, Anal. Chem., 1997, 69, 1651–1656 CrossRef CAS.
- J. J. Triviño, C. Núñez, I. M.-S. Martín, M. Zúñiga and V. Arancibia, Int. J. Electrochem. Sci., 2022, 17, 220762 CrossRef.
- E. Nagles, V. Arancibia, C. Rojas and R. Segura, Talanta, 2012, 99, 119–124 CrossRef CAS PubMed.
- P. C. do Nascimento, M. da S. Marques, M. Hilgemann, L. M. de Carvalho, D. Bohrer, S. G. Pomblum and S. Schirmer, Anal. Lett., 2006, 39, 777–790 CrossRef CAS.
- J. Kruusma, C. E. Banks, L. Nei and R. G. Compton, Anal. Chim. Acta, 2004, 510, 85–90 CrossRef CAS.
- J. G. Rodríguez, I. Amouroux, M. J. Belzunce-Segarra, P. Bersuder, T. Bolam, M. Caetano, I. Carvalho, M. M. Correia dos Santos, G. R. Fones, J.-L. Gonzalez, S. Guesdon, J. Larreta, B. Marras, B. McHugh, F. Menet-Nédélec, I. Menchaca, V. Millán Gabet, N. Montero, M. Nolan, F. Regan, C. D. Robinson, N. Rosa, M. Rodrigo Sanz, M. Schintu, B. White and H. Zhang, Sci. Total Environ., 2021, 783, 147001 CrossRef PubMed.
- M. Pesavento, G. Alberti and R. Biesuz, Anal. Chim. Acta, 2009, 631, 129–141 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |