
Hydrothermal synthesis of stable lead-free Cs4MnBi2Cl12 perovskite single crystals for efficient photocatalytic degradation of organic pollutants†
Xiao-Feng
Qi
,
Fei
Zhang
,
Zhi-Peng
Chen
,
Xu
Chen
 ,
Mo-Chen
Jia
,
Hui-Fang
Ji
* and
Zhi-Feng
Shi
*
,
Mo-Chen
Jia
,
Hui-Fang
Ji
* and
Zhi-Feng
Shi
*
Key Laboratory of Materials Physics of Ministry of Education, School of Physics and Microelectronics, Zhengzhou University, Daxue Road 75, Zhengzhou, 450052, China. E-mail: shizf@zzu.edu.cn; jifang@zzu.edu.cn; Fax: +86-371-67766629; Tel: +86-150-9333-9165
First published on 23rd February 2023
Abstract
Recently, the photocatalytic degradation of organic pollutants using halide perovskite materials has increasingly attracted people's attention. However, the stability issues and the lead toxicity of such materials cast a shadow over their practical applications. In this study, we successfully synthesized non-toxic Cs4MnBi2Cl12 single crystals (SCs) using a hydrothermal method. Joint experiment–theory characterizations reveal the good stability of Cs4MnBi2Cl12. Furthermore, the application of Cs4MnBi2Cl12 as a photocatalyst in the degradation of organic pollutants was demonstrated for the first time. In detail, rhodamine B (RhB), methylene blue and Sudan Red III dissolved in ethanol can be degraded and decolorized by Cs4MnBi2Cl12 SCs under light irradiation. About 97% RhB can be degraded in 7 min, and the degradation efficiency is hardly decreased over ten cycles. Besides, the photocatalytic degradation mechanisms were studied, and the results show that the superoxide (˙O2−) plays a leading role in the photocatalytic process. The degradation pathways of organic pollutants were subsequently analyzed by high-performance liquid chromatography–mass spectrometry, involving three stages of N-deethylation, ring opening, and mineralization. Encouraged by the remarkable structural and optical stability of Cs4MnBi2Cl12 SCs, such lead-free perovskite materials may have broad application prospects in the field of photocatalysis.
Introduction
Recently, with the rapid population growth and the development of global industrialization, the pollution of organic dyes has become increasingly high, which has damaged the ecosystem seriously and threatened the sustainable development of human society.1 Under these circumstances, the development of advanced treatment technologies for organic pollutant degradation is an important pursuit and certainly a worthwhile subject. Solar energy is abundant, and has shown immense potential in tackling environmental problems and photocatalysis has proven to be effective in the degradation of organic dyes.2 The photocatalytic reaction uses semiconductor materials as catalysts to degrade, decolorize, and detoxify macromolecular organic pollutants under sunlight, and convert them into inorganic small molecular substances, such as CO2 and H2O.3 This treatment technology possesses the advantages of low energy consumption, simple operation, and mild reaction conditions.4 Currently, the experimentally explored photocatalyst materials include metal oxides (TiO2, ZnO, SnO2, and WO3),5–7 metal sulfides (CdS, PbS, and ZnS),8 nitrides (C3N4 and GaN),9 phosphides (InP and GaP),10,11 and so on. For example, Zhang synthesized a novel phosphorus (P)-doped fiber tubular graphitic carbon nitride (g-C3N4)/g-C3N4/titanium nitride (TiN) composite (FCN/CN/TiN).12 FCN/CN/TiN has been endowed with remarkably high photocatalytic performance with a degradation efficiency of 94%, and can still maintain the stability of the material and degradation efficiency after four cycles. Although the materials mentioned above have shown good performance in photocatalytic reactions, they still face some drawbacks including fast recombination rates of photo-generated carriers, low light utilization, complex preparation process, etc.13–15 Therefore, from the application point of view, it is imperative to search for alternative semiconductor materials with a low carrier recombination rate and cost-effectiveness as photocatalysts to address the above issues.In recent years, metal-halide perovskite materials have increasingly received attention due to their great capabilities in cost-effective and large-scale manufacturing in optoelectronic devices, including solar cells, photodetectors, lasers, and light-emitting diodes.16–22 Compared to the intensive efforts in optoelectronic applications, the investigation of perovskites as photocatalysts in pollutant degradation is relatively less intensive. In fact, the intrinsically large light absorption coefficient, long charge carrier lifetime, tunable bandgap, and solution-processed method provide metal-halide perovskites with broad prospects for high-performance photocatalysts, and much progress in this field has been witnessed in the past several years, including photocatalytic reduction of CO2,23 hydrogen production,24 NO removal,25 and organic pollutant degradation. For example, CsPbX3 (X = Cl, Br) perovskite quantum dots (QDs) can decompose methyl orange (MO) solution into a colorless solution within 100 min.26 The eosin-B dye can be degraded by CsPbCl3 QDs under visible light irradiation within 140 min.27 CsPbBr3 nanocrystals can degrade methylene blue (MB) solution within 60 min, and the photocatalytic degradation rate is 99.18%.28 Nevertheless, the inherent toxic element lead contained in lead-halide perovskites and their instability cast a shadow over their practical applications. Thanks to the rich combinatorial chemistry of perovskite materials, researchers have developed some stable and lead-free perovskite materials and applied them in organic pollutant degradation. For example, Bresolin et al. synthesized the (CH3NH3)3Bi2I9 powder, which can effectively degrade Rhodamine B (RhB) and MB solutions under light irradiation.29 Analogously, Akinbami et al. observed that Cs3Bi2Br9 nanoparticles can be used for photocatalytic degradation of RhB and MB solutions.30 Besides, Cs2AgInCl6 nanocrystals were used for photocatalytic degradation of Sudan Red III dyes, about 98.5% of Sudan Red III solution was degraded within 16 min.31 Moreover, Cs2AgBiBr6 also has photocatalytic properties and can be used to degrade RhB solution, and the photocatalytic efficiency can be significantly improved when precious metals such as Pt and Au are deposited on its surface32 while the above three lead-free perovskite materials are characterized by indirect bandgap nature, resulting in a low light utilization efficiency. Moreover, they all have relatively short carrier lifetimes, thus it is difficult to ensure that the photoexcited charge carriers have sufficient time to reach the active sites to complete the surface redox reaction.
In this study, we successfully synthesized the non-toxic and stable Cs4MnBi2Cl12 single crystals (SCs) using a hydrothermal method, which are characterized by direct bandgap and long carrier lifetime. The Cs4MnBi2Cl12 SCs can degrade and decolorize RhB, MB and Sudan Red III in ethanol-based photocatalytic systems, demonstrating a high degradation efficiency and stability. About 97% RhB can be degraded in 7 min, and the degradation efficiency does not decrease significantly after ten cycles. After the degradation process, the photocatalyst can be recovered by filtration and cleaning, showing the characteristics of convenient recovery, and high recovery and utilization rate. The photocatalytic degradation mechanisms show that the dye molecules have undergone the mineralization process, and the main active species are the superoxide radical (˙O2−), and the degradation pathways of organic pollutants involve three stages of N-deethylation, ring opening, and mineralization. The above results show that the lead-free Cs4MnBi2Cl12 SCs with a simple synthesis method and high degradation efficiency are safe and efficient photocatalytic materials.
Results and discussion
In this work, the Cs4MnBi2Cl12 perovskite SCs were synthesized using a slow cooling crystallization method, as schematically illustrated in Fig. 1a. Firstly, the mixture of CsCl, MnCl2 and BiCl3 were thoroughly ground. Then, the mixture was dissolved in HCl solution. After a continuous sintering at 180 °C for 12 h, Cs4MnBi2Cl12 crystals can be precipitated by slowly cooling to room temperature at a cooling rate of 10 °C h−1. Fig. 1b illustrates the schematic crystal structure of Cs4MnBi2Cl12. One can see that Cs4MnBi2Cl12 with the triangular R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group belongs to layered double perovskite structure, in which Mn2+ and Bi3+ cations coordinate with six Cl− to form two octahedral blocking units, and the [MnCl6]4− octahedral layer is sandwiched between two [BiCl6]3− layers by angle sharing to form a two-dimensional perovskite structure, which endows it with excellent stability and tunable photoelectric properties.33,34 The triangular structural characteristics of the synthesized Cs4MnBi2Cl12 SCs were experimentally confirmed by X-ray diffraction (XRD). As shown in Fig. 1c, the Cs4MnBi2Cl12 SCs have only the diffraction patterns from {001} planes, belonging to a triangular Rm space group (cell parameters: a = b = 7.5426 Å, c = 36.9150 Å, α = β = 90°, γ = 120°), and the full-width at half-maximum (FWHM) of (0012) diffraction peak reaches 0.12°, suggesting a high crystallinity. Fig. 1d displays the X-ray photoelectron spectrometer (XPS) survey spectrum of Cs4MnBi2Cl12 SCs, where relevant signals of C, Cs, Mn, Bi, and Cl were confirmed, and the binding energy signals of Cs 3d, Mn 2p, Bi 4f, and Cl 2p were presented in Fig. S1 (ESI†) after calibrating with the C 1s peak at 284.57 eV. Fig. 1e and Fig. S2 (ESI†) show the typical scanning electron microscope (SEM) images of Cs4MnBi2Cl12 crystals, which have a regular rhombic octahedral structure similar to that of double perovskites, and their surfaces are flat without obvious voids, which further indicates that the as-prepared samples have high crystallinity. Energy dispersive spectroscopy (EDS) elemental mapping analysis in Fig. 1f shows that Cs, Mn, Bi, and Cl elements are uniformly distributed in the octahedral microcrystal. Quantitative analysis of the EDS spectrum shown in Fig. S3 (ESI†) gives a quantified atomic ratio (%) of 8.90
m space group belongs to layered double perovskite structure, in which Mn2+ and Bi3+ cations coordinate with six Cl− to form two octahedral blocking units, and the [MnCl6]4− octahedral layer is sandwiched between two [BiCl6]3− layers by angle sharing to form a two-dimensional perovskite structure, which endows it with excellent stability and tunable photoelectric properties.33,34 The triangular structural characteristics of the synthesized Cs4MnBi2Cl12 SCs were experimentally confirmed by X-ray diffraction (XRD). As shown in Fig. 1c, the Cs4MnBi2Cl12 SCs have only the diffraction patterns from {001} planes, belonging to a triangular Rm space group (cell parameters: a = b = 7.5426 Å, c = 36.9150 Å, α = β = 90°, γ = 120°), and the full-width at half-maximum (FWHM) of (0012) diffraction peak reaches 0.12°, suggesting a high crystallinity. Fig. 1d displays the X-ray photoelectron spectrometer (XPS) survey spectrum of Cs4MnBi2Cl12 SCs, where relevant signals of C, Cs, Mn, Bi, and Cl were confirmed, and the binding energy signals of Cs 3d, Mn 2p, Bi 4f, and Cl 2p were presented in Fig. S1 (ESI†) after calibrating with the C 1s peak at 284.57 eV. Fig. 1e and Fig. S2 (ESI†) show the typical scanning electron microscope (SEM) images of Cs4MnBi2Cl12 crystals, which have a regular rhombic octahedral structure similar to that of double perovskites, and their surfaces are flat without obvious voids, which further indicates that the as-prepared samples have high crystallinity. Energy dispersive spectroscopy (EDS) elemental mapping analysis in Fig. 1f shows that Cs, Mn, Bi, and Cl elements are uniformly distributed in the octahedral microcrystal. Quantitative analysis of the EDS spectrum shown in Fig. S3 (ESI†) gives a quantified atomic ratio (%) of 8.90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.95:3.97:23.13 (4.56:1:2.03:11.86) for Cs:Mn:Bi:Cl, which is close to the stoichiometry of the Cs4MnBi2Cl12 material. Fig. 1g shows the high-resolution transmission electron microscope (TEM) image of the Cs4MnBi2Cl12 crystal, and two sets of clear lattice fringes were identified with the interplanar spacings of 3.60 Å and 3.76 Å, corresponding to the (2
:1.95:3.97:23.13 (4.56:1:2.03:11.86) for Cs:Mn:Bi:Cl, which is close to the stoichiometry of the Cs4MnBi2Cl12 material. Fig. 1g shows the high-resolution transmission electron microscope (TEM) image of the Cs4MnBi2Cl12 crystal, and two sets of clear lattice fringes were identified with the interplanar spacings of 3.60 Å and 3.76 Å, corresponding to the (2![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 3 and (20 crystal planes of triangular Cs4MnBi2Cl12. The corresponding Fourier transformation (FFT) image displayed in upper-right pane verifies the single-crystalline characteristics of the Cs4MnBi2Cl12 crystal.
3 and (20 crystal planes of triangular Cs4MnBi2Cl12. The corresponding Fourier transformation (FFT) image displayed in upper-right pane verifies the single-crystalline characteristics of the Cs4MnBi2Cl12 crystal.
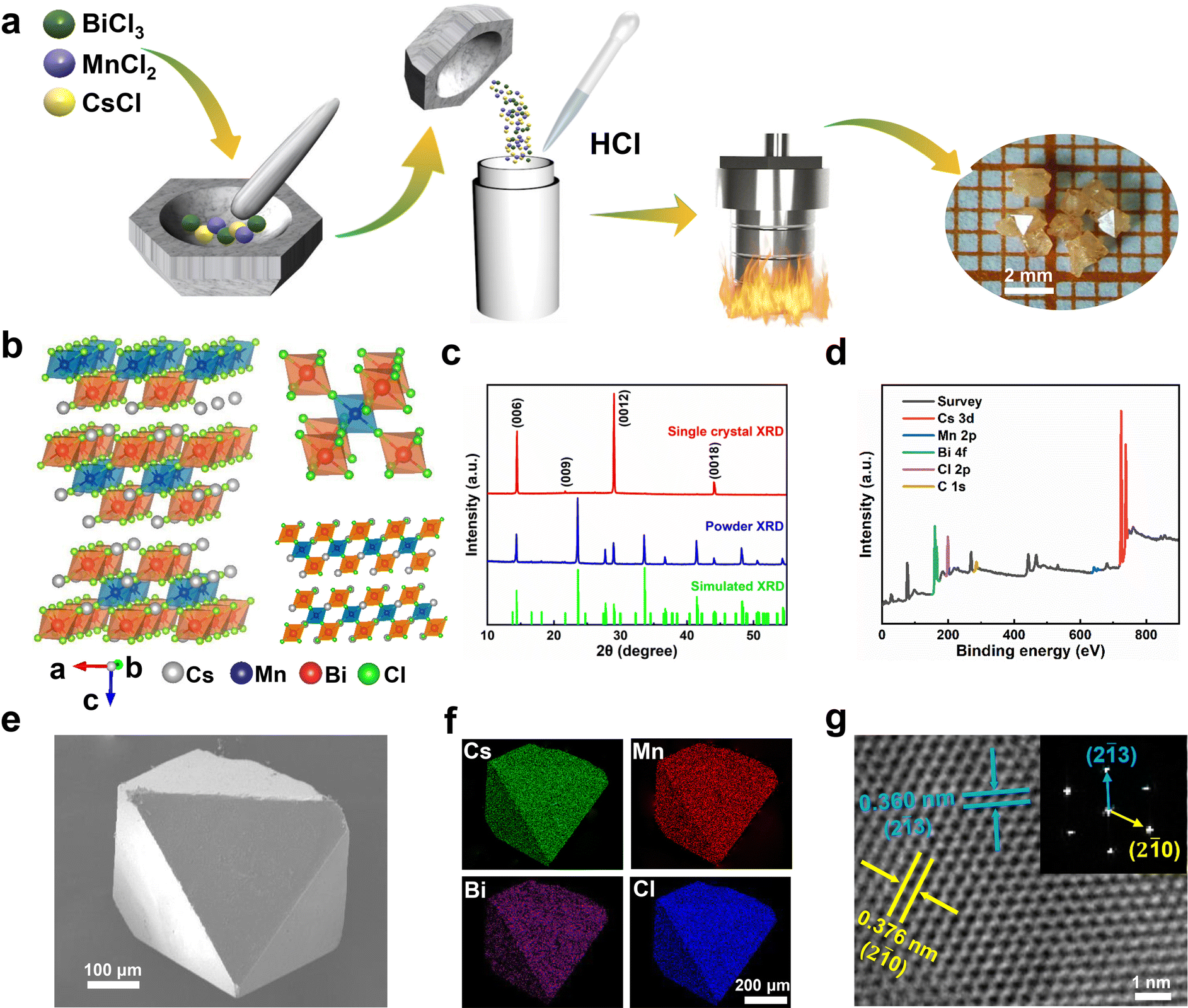 | ||
| Fig. 1 (a) Schematic diagram of the synthesis process of Cs4MnBi2Cl12 SCs. (b) Left: Schematic illustration of the crystal structure of Cs4MnBi2Cl12. Right: Isolated two types of octahedral blocking units (top) and the crystal lattice view along the [110] direction (bottom). (c) XRD patterns of Cs4MnBi2Cl12 SCs and powder. (d) XPS spectrum of Cs4MnBi2Cl12 SCs. (e) SEM image of an individual Cs4MnBi2Cl12 crystal, and (f) the corresponding EDS elemental distributions of Cs, Mn, Bi, and Cl, respectively. (g) High-resolution TEM images of Cs4MnBi2Cl12 SCs. The inset shows the corresponding FFT patterns. | ||
To understand the electronic structure of Cs4MnBi2Cl12, the electronic band structure and density of states (DOS) of Cs4MnBi2Cl12 were calculated by density functional theory (DFT).35 As shown in Fig. 2a, Cs4MnBi2Cl12 exhibits a direct bandgap at the F symmetry point with a bandgap energy of 3.22 eV. The corresponding DOS calculation in Fig. 2b shows that the conduction band minimum (CBM) of Cs4MnBi2Cl12 is mainly composed of Bi 6p and Cl 3p orbitals, and its valence band maximum (VBM) mostly consists of Mn 3d and Cl 3p orbitals. That is to say, the [MnCl6]4− and [BiCl6]3− polyhedra contribute the most electronic states in the energy band, and Cs contributes less to the CBM and the VBM. The isosurface plots of wave function |Ψ|2 of the CBM and the VBM are shown in Fig. 2c and d, and the charge density is concentrated in [MnCl6]4− and [BiCl6]3− groups, isolated by Cs+ ions.
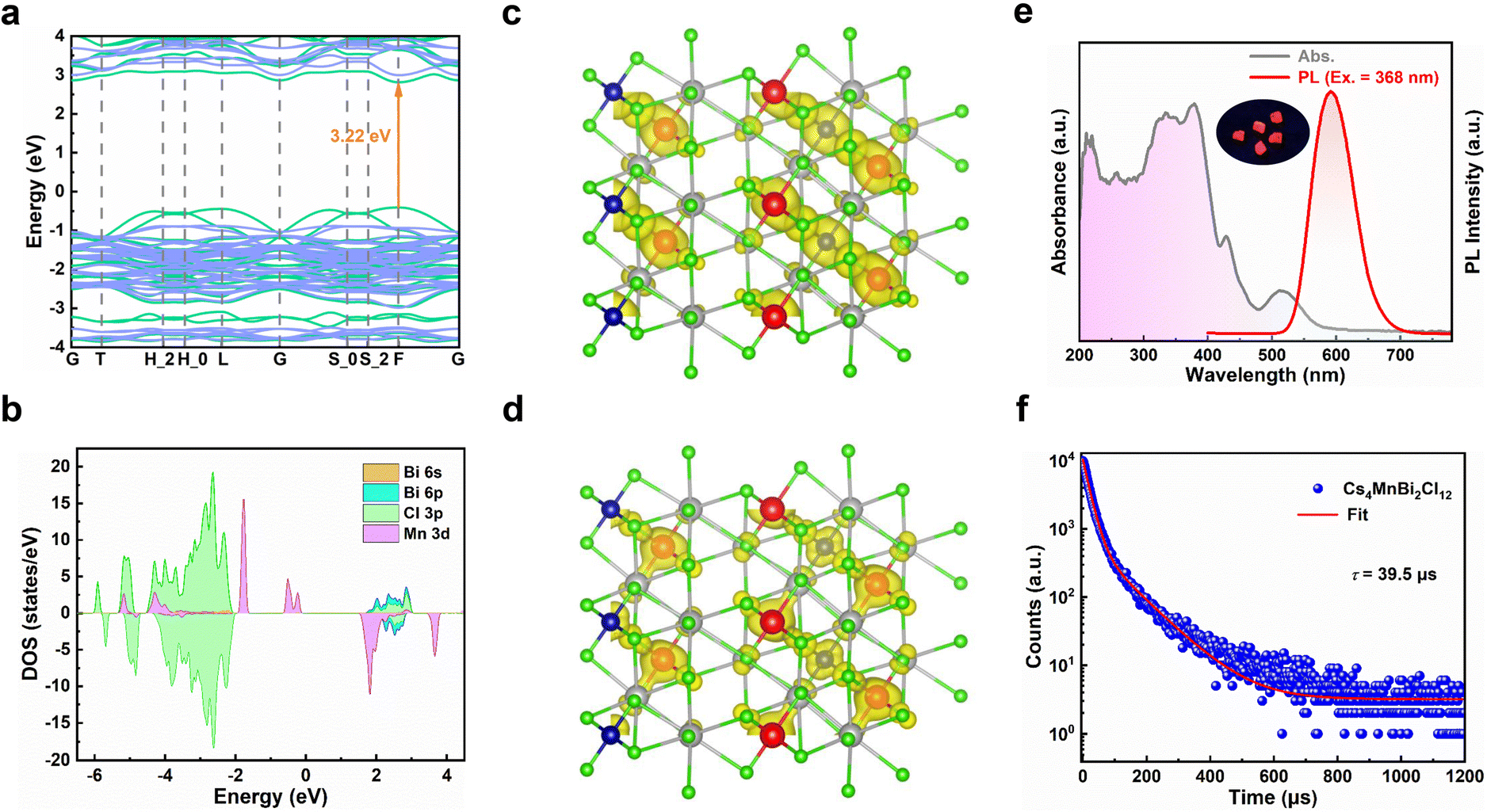 | ||
| Fig. 2 (a) Calculated band structure, and (b) the corresponding DOS diagram of Cs4MnBi2Cl12. The isosurface plots of the wave function |Ψ|2 of (c) CBM and (d) VBM. (e) ultraviolet-visible absorption and PL spectra of Cs4MnBi2Cl12 SCs. The inset shows the photograph of Cs4MnBi2Cl12 SCs under ultraviolet light irradiation (365 nm). (f) Time-resolved PL decay curve of Cs4MnBi2Cl12 SCs. | ||
The optical properties of Cs4MnBi2Cl12 SCs were studied by ultraviolet-visible absorption and photoluminescence (PL). As shown in Fig. 2e, the Cs4MnBi2Cl12 SCs have a broad absorption region (gray line), which start to absorb light at around 600 nm and have a strong absorption in the ultraviolet region.36 Such a wide absorption region is conducive to absorbing more energy that supports efficient photodegradation. It should be mentioned that the strong absorption in the ultraviolet region comes from the 6s2 → 6s1p1 transition of Bi3+ ions.37,38 A fully allowed 1S0 → 1P1 transition is observed near 210 nm, and the peak at 267 nm belongs to 1S0 → 3P2 forbidden transition, while the 1S0 → 3P1 transition is responsible for the strong peaks at 330 and 352 nm. This splitting is due to the dynamic Jahn–Teller distortion effect of the excited state caused by the coupling of lattice vibration and Bi 6p–Cl 3p antibonding orbital coupling.39 In addition, two weak absorption peaks at 430 and 515 nm were also detected in the visible region, corresponding to the spin-forbidden 6A1(S) → 4T1(G) and 6A1(S) → 4T2(G) d–d transitions of the Mn2+, respectively.40,41 From the Tauc plot analysis of the absorption spectra (Fig. S4, ESI†), the bandgap of Cs4MnBi2Cl12 SCs is determined to be 3.01 eV, similar to that of the reported data.36
Preliminary optical studies (Fig. 2e, red line) show that the Cs4MnBi2Cl12 SCs exhibit a strong and broad emission peak at 592 nm with a large FWHM of 75 nm, which is mainly caused by the spin-forbidden d–d transition (4T1 → 6A1) of paired Mn2+ ion octahedra with magnetic coupling.42 The inset of Fig. 2e displays a photograph of Cs4MnBi2Cl12 SCs under ultraviolet light irradiation (365 nm), showing an intense orange–red light. To further confirm the emission mechanisms of Cs4MnBi2Cl12, the PL spectra under different excitations and PL excitation (PLE) spectra under different emissions were measured. As shown in Fig. S5a (ESI†), the peak position and FWHM of the PL spectra show little dependence on the excitation wavelength ranging from 310 to 520 nm, suggesting that the broad red emission originates from the same radiative decay path rather than superposition of multiple defect recombination centers.43 Likewise, the nondependence of PLE spectra (Fig. S5b, ESI†) on the emission wavelength also confirms that the orange–red emission originates from the relaxation of the same excited state—d–d transition of Mn ions.36,44 In general, the PL intensity associated with the permanent defects in materials is characterized by a sublinear dependence on the excitation power, and an undesired emission saturation will occur when all defect states are filled.45 However, in the present case, a linear relationship of excitation power-dependent PL eliminates the possibility of permanent defect-related transition (Fig. S6, ESI†). Transient-state PL measurements were also performed to gain more insight into the carrier recombination of Cs4MnBi2Cl12 SCs. As shown in Fig. 2f, the PL decay curve of Cs4MnBi2Cl12 SCs excited by 368 nm monitoring at 592 nm can be fitted by three exponential decay functions, and the average PL lifetime is about 39 μs, which is much larger than the conventional lead-halide perovskites and other lead-free metal halides.46–50 Generally, the PL lifetime is inversely proportional to the density of trap states, where a longer lifetime is associated with less trap density. From the perspective of photocatalytic applications, a longer lifetime would reduce the possibility of carrier recombination, bringing about higher opportunity to degrade organic pollutants.31 Moreover, the longer PL lifetime implies a lower charge carrier recombination rate, thus the photoexcited charge carriers would have sufficient time to reach the active sites to complete the surface redox reaction.51
The stability of photocatalysts is an important index to evaluate whether it can have commercial application, so we carried out a detailed study on the stability of Cs4MnBi2Cl12 by combining theoretical and experimental analysis. First, the dynamic stability and thermodynamic stability of Cs4MnBi2Cl12 were studied by the first-principles calculations. As shown in Fig. 3a, no negative frequencies were observed in the calculated phonon band structure, which verifies the dynamic stability of Cs4MnBi2Cl12. Fig. 3b presents the first-principles molecular dynamics simulations at 300 K, and the small potential energy fluctuation implies a good thermodynamic stability of Cs4MnBi2Cl12. Furthermore, we extended the theoretical investigation into the decomposition enthalpy of Cs4MnBi2Cl12. As shown in Fig. 3c, the decomposition enthalpy of Cs4MnBi2Cl12 is much larger than that of CsPbBr3, CsPb2Br5, and other lead-free perovskite materials, indicating the better stability and greater advantages of Cs4MnBi2Cl12 for practical photocatalytic applications. Experimentally, the thermogravimetric analysis shows that Cs4MnBi2Cl12 SCs have a high thermal stability and exhibit no significant weight loss until 500 °C, as seen in Fig. 3d. Furthermore, we conducted the photostability test of the Cs4MnBi2Cl12 SCs by continuously illuminating the sample using a 365 nm ultraviolet lamp. As shown in Fig. 3e and Fig. S7 (ESI†), the absorption and PL spectra of Cs4MnBi2Cl12 SCs barely changed in terms of the spectral shape and peak intensity after 48 h of continuous irradiation, which suggests a good photostability of Cs4MnBi2Cl12 SCs. Considering the strong dependence of organic pollutant degradation on the solution environment, another stability test of Cs4MnBi2Cl12 SCs in different solvent environments was therefore conducted. Generally, water was deemed to be one of the fatal factors for conventional lead-halide perovskites, which are easily decomposed once exposed to water because of the hygroscopic halide salts.52 To evaluate the stability of Cs4MnBi2Cl12 SCs against water corrosion, the as-synthesized sample was therefore soaked in deionized water. As we expected, the Cs4MnBi2Cl12 SCs experienced a rapid fluorescence quenching in water environment and lost most of the brightness within 10 s of standing, as shown in the upper pane of Fig. 3f, which may be induced by the ionic nature of Cs4MnBi2Cl12. In sharp contrast, when we replaced the deionized water with alcohol, the bright orange–red luminescence typical to Cs4MnBi2Cl12 SCs can be stabilized even with a long-standing time for 60 h (bottom pane). Therefore, for stable and reproducible photocatalytic degradation by using Cs4MnBi2Cl12, alcohol is a very reliable solvent of choice. The above experimental results indicate the remarkable stability of the Cs4MnBi2Cl12 system, making it suitable as an environmentally friendly photocatalyst for the degradation of organic pollutants in ethanol.
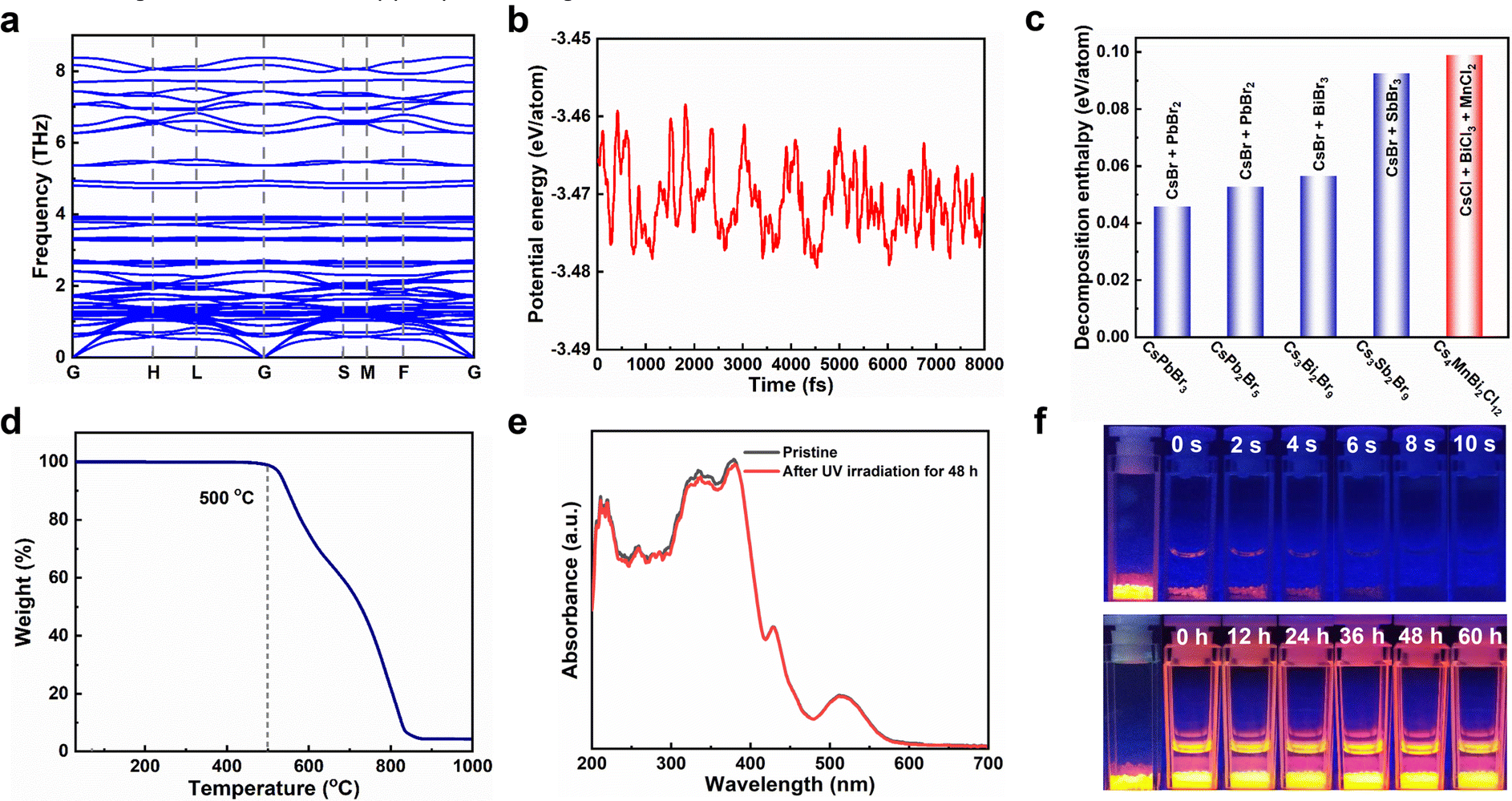 | ||
| Fig. 3 (a) Phonon band structures of the Cs4MnBi2Cl12. (b) Potential energy fluctuations of Cs4MnBi2Cl12 as a function of the molecular dynamic simulation step at 300 K. (c) Comparison of the decomposition enthalpy of Cs4MnBi2Cl12 with other halide perovskites. (d) Thermogravimetry analysis of the Cs4MnBi2Cl12 SCs. (e) Absorption spectra of the Cs4MnBi2Cl12 SCs before and after continuous ultraviolet light irradiation for 48 h. (f) Photographs of the Cs4MnBi2Cl12 SCs soaked in water (upper) and alcohol (bottom) for different times. | ||
The photocatalytic activity of Cs4MnBi2Cl12 SCs was experimentally evaluated by removing three organic dyes (RhB, MB, and Sudan III) in ethanol solution under a 300 W mercury lamp irradiation. Fig. 4a–c present a series of ultraviolet-visible absorption spectra of RhB, MB, and Sudan Red III at different irradiation times in the presence of Cs4MnBi2Cl12, and regular spectral evolution was revealed. Specifically, the absorption peak intensity of RhB decreases dramatically with increasing the irradiation time (Fig. 4a). Within a short period of 7 min, the characteristic absorption peak of RhB almost disappears, with a degradation rate as high as 97%, which suggests that the RhB solution was almost completely degraded. The inset of Fig. 4a presents the change of RhB solution color with different irradiation times, showing a regular change from dark purple to transparent. To verify the positive role of Cs4MnBi2Cl12 in the RhB degradation process, the control experiments were conducted. As shown in Fig. S8 (ESI†), RhB is hardly degraded under a mercury lamp irradiation without the Cs4MnBi2Cl12 catalyst. Also, RhB is difficult to be degraded with catalyst under dark conditions. These results indicate that Cs4MnBi2Cl12 is the real factor to enable degradation of RhB. For the other organic pollutants, the effect of photodegradation is also significant. As shown in Fig. 4b and c, MB and Sudan Red III dyes with the same concentration as RhB were employed for the identical experiments, and the regular color changes from blue cyan (MB) and orange (Sudan Red III) to transparent lend support to the view of almost complete degradation. In detail, the degradation rates of MB and Sudan III were 98.3% and 97.4%, respectively, within 16 min. The above results demonstrate that Cs4MnBi2Cl12 SCs show high photocatalytic activity for different dyes, among which the degradation performance of RhB is the best (Fig. 4d), so we will focus on RhB as the main research object in the following research. Subsequently, the degradation rates of RhB with different addition amounts of Cs4MnBi2Cl12 catalysts were investigated to describe the kinetics of the photocatalytic reaction. As shown in Fig. 4e, with the increase of catalyst content, the degradation rate of RhB first increases and then decreases, and the degradation rate reaches its maximum when the catalyst content is 5 mg. The Langmuir Hinshelwood (L–H) model can be used to describe the degradation rate under different catalyst contents more intuitively, in which the first-order model is expressed as –ln (Ct/Co) = k1t + b, where C0 is the initial concentration of dye, Ct is the concentration of dye under different degradation reaction times t, k1 is the first-order rate constant and b is a constant. By fitting the data, it can be found that the calculated k1 increases from 0.3914 to 0.4737 min−1 when the addition amount of Cs4MnBi2Cl12 increases from 3 to 5 mg (Fig. 4f), implying that the degradation rate increases with the increase of catalyst content, which is mainly because of the number of active sites available for adsorption that are proportional to the catalyst content. Theoretically, more catalyst addition means larger exposure area to light, and thus more reactive species (free radicals) can be produced.53,54 However, further increasing the catalyst content to 10 mg and above, light penetration through the solution would be inhibited due to an enhanced light scattering.55,56 As a result, the degradation rate of RhB decreases rather than increases. Table S1 (ESI†) summarizes the photocatalytic properties of Cs4MnBi2Cl12 and other perovskite materials. By comparison, the Cs4MnBi2Cl12 has a more superior photocatalytic activity.
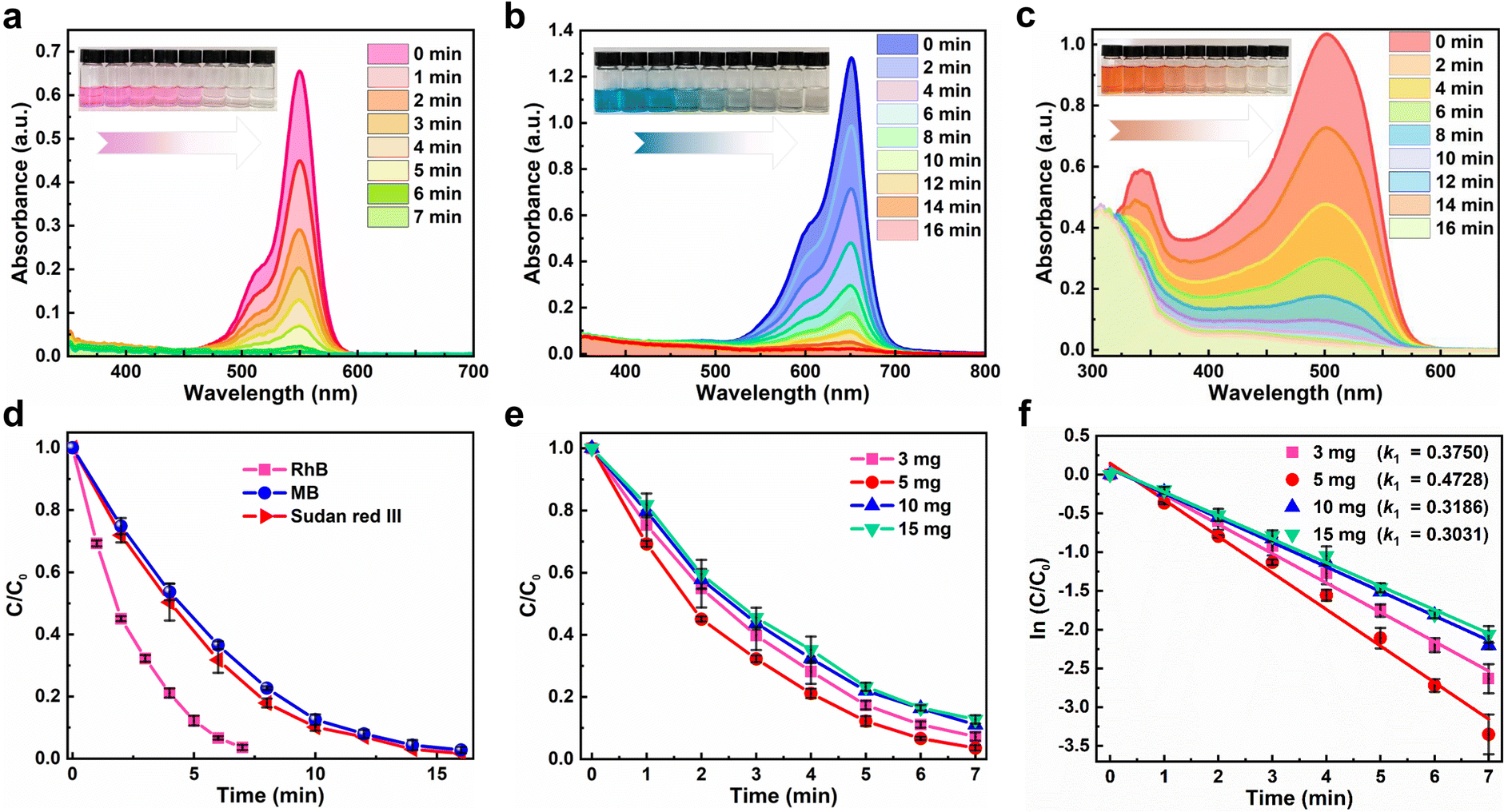 | ||
| Fig. 4 Ultraviolet-visible absorption spectra of (a) RhB, (b) MB, and (c) Sudan Red III at different irradiation times in the presence of Cs4MnBi2Cl12. The insets show the change of solution color with time. (d) Plots of C/C0versus the irradiation time for the degradation of RhB, MB, and Sudan Red III dyes. (e) Plots of C/C0versus the irradiation time for the degradation of RhB with different catalyst addition amount. (f) Corresponding reaction kinetic curves of the RhB degradation fitted using the first-order model. | ||
It is well known that the sustainability of photocatalysts plays a key role in their practical applications. In the present case, the sustainability of the Cs4MnBi2Cl12 photocatalyst was examined by performing continuous ten cycles of RhB degradation under the same conditions (air ambient, 30 °C, 50% humidity). As shown in Fig. 5a, the photocatalytic degradation time of Cs4MnBi2Cl12 SCs can be well maintained for 7 min over a ten cycle test; ten plotted curves repeat well and exhibit a reproducible response with time, indicating that the Cs4MnBi2Cl12 photocatalyst has a good cycling stability. From the application point of view, a robust cycling stability of catalysts can effectively reduce the cost of photocatalysis, opening opportunities for future practical applications. It is worth mentioning that such good sustainability is greatly superior to the lead-halide perovskite catalysts (CsPbBr3) reported by Fan et al.,57 where the photocatalytic efficiency of CsPbBr3 decays significantly over four cycles and the crystallinity of the recycled photocatalyst deteriorates seriously. Excitingly, this is not the case in our experiment. By comparing the XRD patterns of Cs4MnBi2Cl12 SCs before and after photocatalysis (Fig. 5b), one can observe that, after ten successive degradation cycles, the structural integrity of Cs4MnBi2Cl12 SCs was preserved well without the appearance of other additional diffractions, indicating that the photocatalyst is not decomposed even after ten photocatalytic cycles. Not only that, the Cs4MnBi2Cl12 photocatalyst, without any encapsulation and protection, also demonstrates a robust stability against oxygen and moisture degradation. As shown in Fig. 5c, after long-term storage for three months in ambient air, the Cs4MnBi2Cl12 SCs are able to maintain a good phase purity without the appearance of an impurity phase. Even if the Cs4MnBi2Cl12 SCs were re-used for photocatalytic experiment after being stored in air for three months, the degradation rate of RhB is not much different from before, as seen in Fig. 5d. Therefore, in terms of material stability, the Cs4MnBi2Cl12 possesses admirable and obvious advantages compared with other perovskite counterparts. This is no doubt the crucially important reason that ensuring the excellent sustainability of the photocatalytic degradation process. Furthermore, we investigated the effect of the size of Cs4MnBi2Cl12 on the degradation rate. As shown in Fig. S9a (ESI†), the photocatalytic experiments were carried out with Cs4MnBi2Cl12 powder and SCs, respectively. When the same mass of Cs4MnBi2Cl12 powder and SCs were used to degrade 10 mg L−1 of RhB dye, it can be found that the degradation rate of the powder is faster than that of the SCs, and the degradation can be completed in 6 min. In addition, photocatalytic cycling experiments were carried out with Cs4MnBi2Cl12 powder, as shown in Fig. S9b (ESI†), RhB could be completely degraded within 6 min of the first two cycles, but the time for complete degradation of RhB was gradually prolonged as the number of cycles continued to increase, which may be caused by the decomposition of Cs4MnBi2Cl12 powder after multiple degradation cycles. To confirm our suspicions, we monitored the XRD of Cs4MnBi2Cl12 powder under different cycles, as shown in Fig. S9c (ESI†), a small amount of the BiCl3 impurity phase appeared in the XRD patterns after the third cycle of catalytic testing, and the impurity peak of BiCl3 after the fifth cycle increased, indicating that the Cs4MnBi2Cl12 powder is easily decomposed into BiCl3 after multiple cycles, which is also the reason for the decrease in the degradation rate after multiple cycles. In contrast, although the degradation rate of Cs4MnBi2Cl12 SCs is slightly smaller than that of Cs4MnBi2Cl12 powder, Cs4MnBi2Cl12 SCs show better cycle stability, which is more suitable for practical applications in photocatalytic degradation. Therefore, we use Cs4MnBi2Cl12 SCs for photocatalytic experiments. The above results and discussions fully reflect the great application prospects of stable Cs4MnBi2Cl12 SCs in photocatalysis fields. The above results and discussions fully reflect the great application prospects of stable Cs4MnBi2Cl12 in photocatalysis fields.
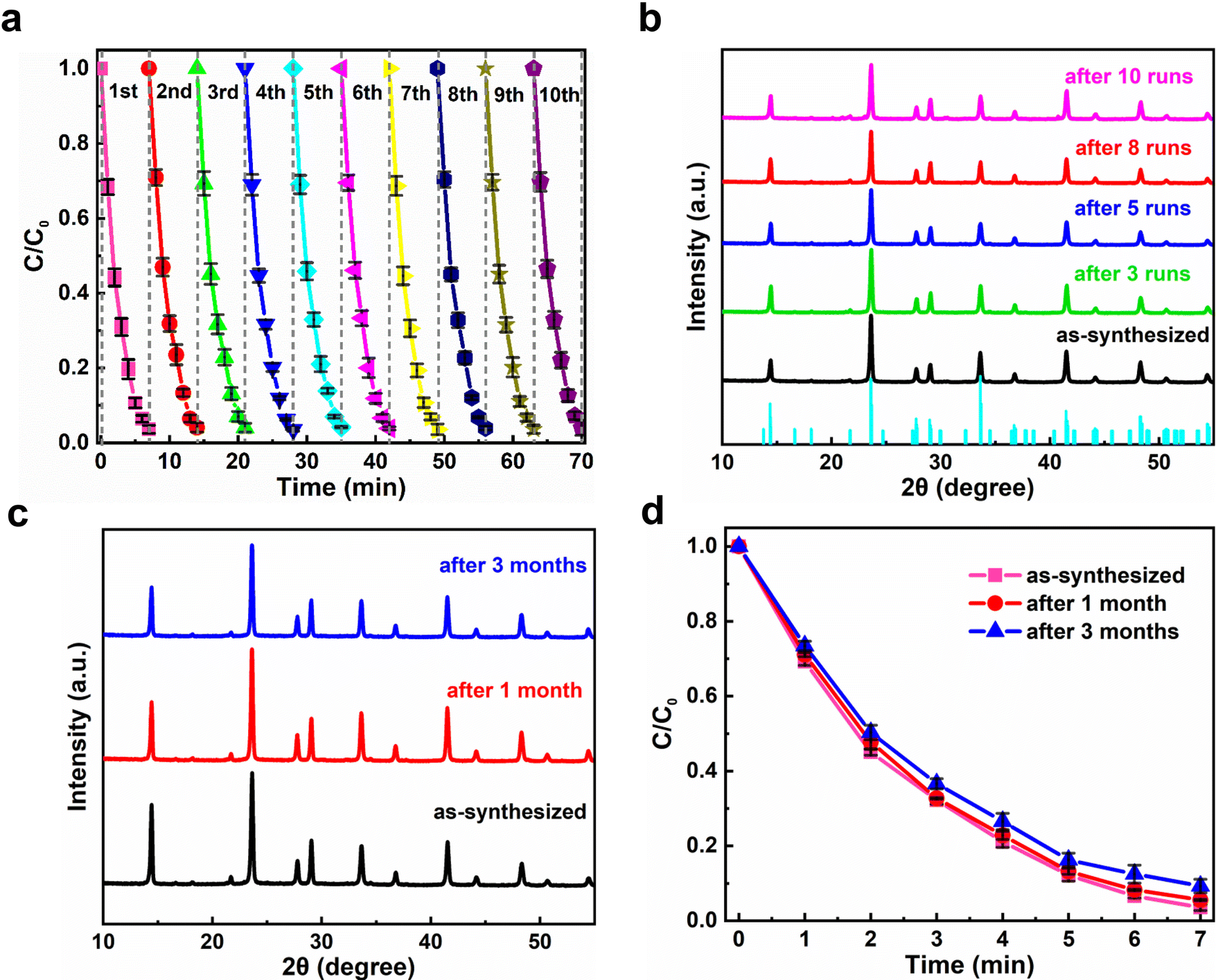 | ||
| Fig. 5 (a) Ten successive degradation cycle test of Cs4MnBi2Cl12 SCs under light irradiation in photocatalytic degradation of RhB. (b) XRD patterns of Cs4MnBi2Cl12 SCs before and after degradation cycle tests. (c) XRD patterns of Cs4MnBi2Cl12 after storage in air for one month and three months. (d) Plots of C/C0versus the irradiation time for the degradation of RhB with Cs4MnBi2Cl12 SCs after storage in air for one month and three months. | ||
Generally, during the photocatalytic reaction, electrons (e−), holes (h+), hydroxyl radical (˙OH) and superoxide (˙O2−) may exist as the active species.58 To examine the photocatalytic mechanisms, we therefore conducted the active radical capture experiments to identify the active species that re account for the degradation process. Fig. 6a plots the C/C0versus the irradiation time for the degradation of RhB with the addition of the free radical scavenger 2,2′,6,6′-tetramethylpiperidine-1-oxyl (TEMPO). One can observe that the photocatalytic degradation of dyes can be almost completely blocked after the TEMPO was added, suggesting that the photocatalytic degradation of dyes is a free radical-based process.59 That is to say, three active radicals including hydroxyl radical (˙OH), superoxide (˙O2−) and holes (h+) may involve in this photocatalytic degradation.60 Since ethanol can capture h+ active free radicals in the photocatalytic reaction, the most likely free radicals involved should be ˙OH and ˙O2−.61 To further make certain which free radical works, we add p-benzoquinone (p-BQ, ˙O2− scavenger) and isopropanol (IPA, ˙OH scavenger) to the ethanol solution of RhB for photocatalytic experiments, and their effects on the photocatalytic degradation efficiency of RhB were measured and compared. As shown in Fig. 6b, the IPA addition has little effect on the photocatalytic degradation of RhB, indicating that ˙OH does not participate in the degradation process. On the contrary, when p-BQ was added, the photocatalytic degradation was completely blocked, which suggests that ˙O2− plays an important role in the degradation process. Moreover, we conducted the electron paramagnetic resonance (EPR) spectra test at room temperature to further verify the existence of ˙O2− in the photocatalytic system, in which 5,5′-dimethyl-1-pyroline-N-oxide (DMPO) was used as the spin trapper. As shown in Fig. 6c, no signal of ˙O2− was observed under dark conditions, but the characteristic peaks of ˙O2− began to appear after the light irradiation for 2 min. With the increase of illumination time, the intensity of characteristic peaks also increases, which suggests that more ˙O2− is produced under added light irradiation. The above observations show that ˙O2− plays a leading role in the photocatalytic process, which is consistent with the results of active radical capture experiments discussed above.
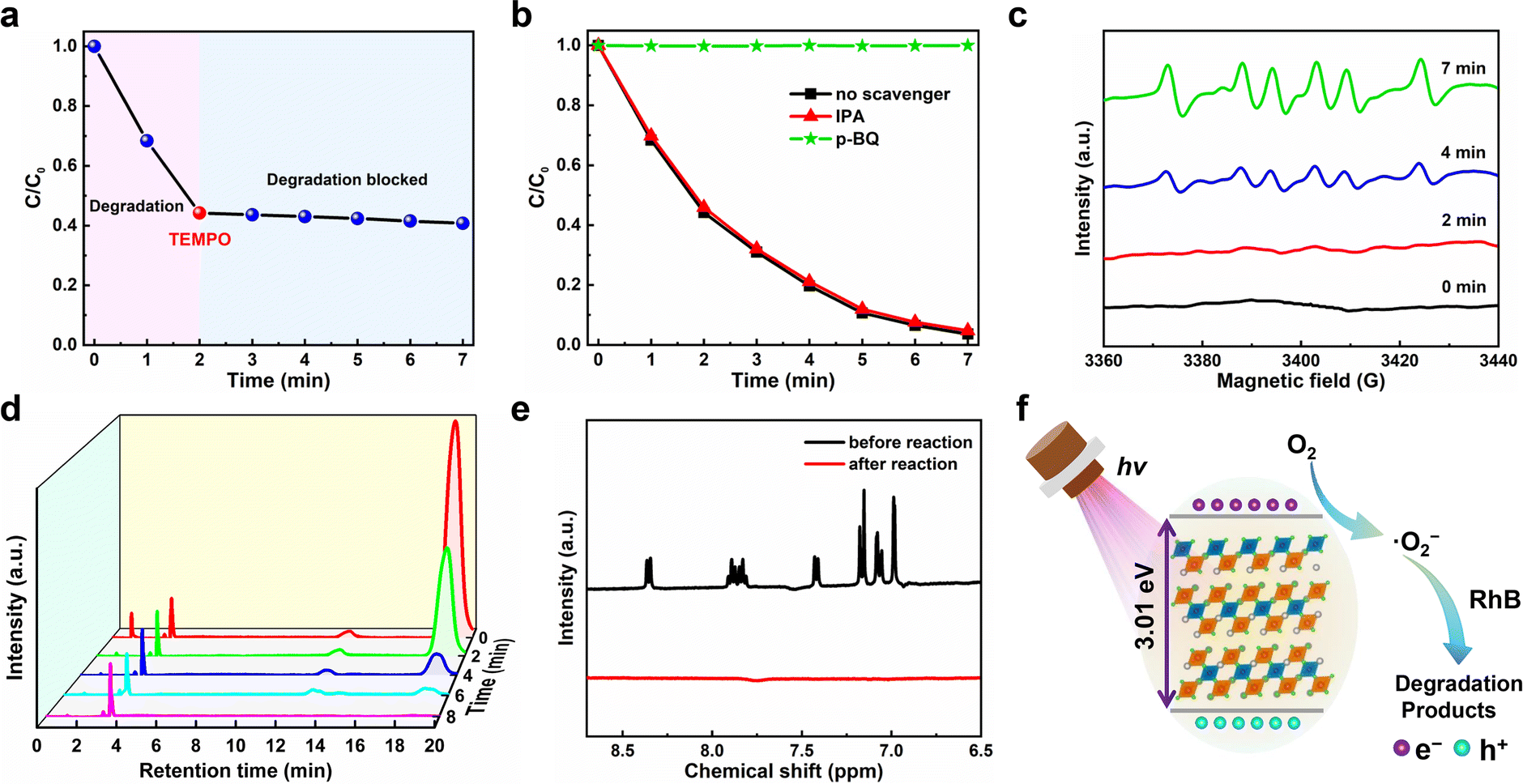 | ||
| Fig. 6 Plots of C/C0versus the irradiation time for the degradation of RhB (a) with the addition of TEMPO during the illumination, and (b) in the presence of different scavengers. (c) The DMPO spin-trapping EPR spectra of Cs4MnBi2Cl12 for ˙O2− under different light irradiation times. (d) HPLC spectra of RhB monitored at 550 nm versus the retention time. (e) 1H NMR analysis of RhB before and after photocatalysis. (f) Schematic of mechanisms for the photocatalytic reaction of RhB under light irradiation. | ||
The degradation process of RhB was further studied by high-performance liquid chromatography (HPLC). As shown in Fig. 6d, with the increase of radiation time, the characteristic HPLC peak of RhB at tR = 19.0 min gradually decreased and finally disappeared and no obvious new HPLC peaks appeared, which is consistent with the evolution trend of the ultraviolet-visible absorption spectra over the degradation process of RhB. Besides, the products after photocatalysis were examined by 1H nuclear magnetic resonance (1H NMR). As shown in Fig. 6e, compared with the RhB dye before photocatalysis, it was found that the characteristic peaks of H in the benzene rings of RhB dye molecules between 6.5 ppm and 9 ppm completely disappeared after photocatalysis, which proved the decomposition of the benzene ring in RhB; in other words, the complete degradation of RhB. Based on the analysis of the above experimental results, we propose the photocatalytic mechanisms of superoxide radical (Fig. 6f), and the main reaction processes for RhB degradation are described as follows:
| Cs4MnBi2Cl12 + RhB → Cs4MnBi2Cl12 − RhB (RhB*) | (1) |
| Cs4MnBi2Cl12 + hν → h+ + e− | (2) |
| e− + O2 → ˙O2− | (3) |
| ˙O2− + RhB* → degradation products | (4) |
In order to further determine the degradation pathway of RhB, the reaction solution was analyzed by HPLC–MS. Fig. S10 (ESI†) presents a series of MS spectra of the intermediates during RhB degradation, and their corresponding molecular formula are shown in Table S2 (ESI†). Based on the detected intermediates, we illustrated the possible degradation pathway of RhB. As shown in Fig. 7, in the photocatalytic degradation of RhB, N-deethylation was the main step leading to the production of series of intermediates.62,63 Since the C–N bond in the whole RhB molecule is the most unstable,64 it is most likely to be oxidized, broken and removed. Considering that there are two positions of C–N substituents in RhB molecule, so two ways of breaking exist. Firstly, RhB molecule was attacked by the photogenerated active radical (˙O2−), which showed the breaking of C–N bond and N-deethylation to obtain the product with m/z = 415. It continues to show the fracture of C–N bond, N-deethylation to obtain the products with m/z values = 387, 359 and 331. Note that the above degradation path shows a continuous process of N-deethylation. The further oxidative degradation of product with m/z = 331 showed an oxidative ring opening process,65 and thus the products with m/z = 139 and 154 were obtained. Finally, these compounds were further degraded into small molecular compounds and eventually mineralized into CO2, H2O, NO3− and NH4+. To sum up, the photocatalytic degradation pathway of RhB dye by Cs4MnBi2Cl12 SCs mainly includes three stages: N-deethylation, ring opening, and mineralization.
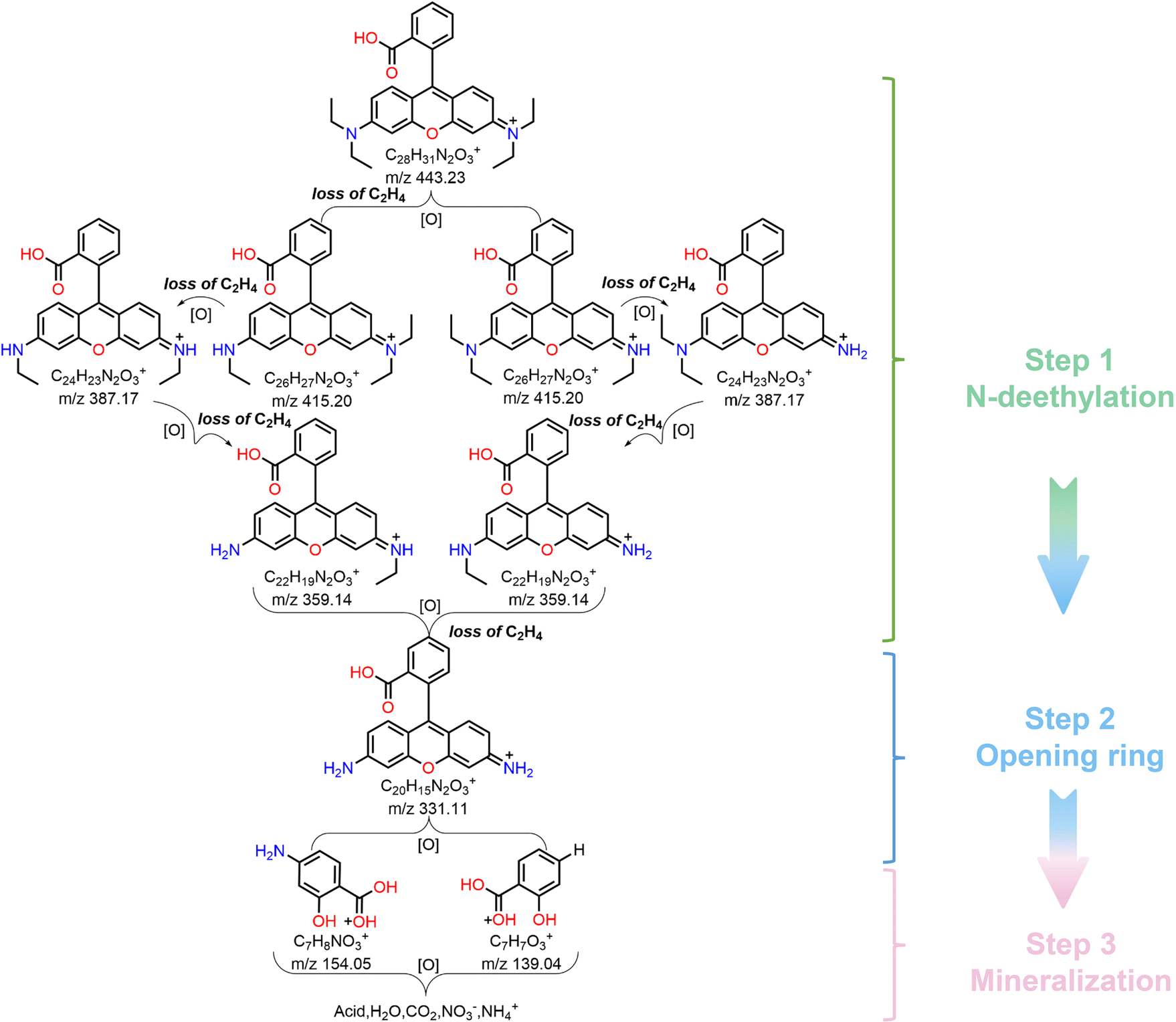 | ||
| Fig. 7 Photocatalytic degradation pathways of RhB dye under light irradiation. | ||
Conclusions
In conclusion, an alcohol based photocatalytic system was successfully developed based on non-toxic Cs4MnBi2Cl12 for the first time. The as-prepared Cs4MnBi2Cl12 SCs have the octahedral shape with excellent crystallization. Joint experiment–theory characterizations confirm the good thermal and light stability of Cs4MnBi2Cl12. Under light irradiation, the Cs4MnBi2Cl12 SCs exhibit an excellent photocatalytic activity, which could degrade ∼97% organic dye RhB within only 7 min, and the degradation efficiency has hardly decreased over ten successive cycles. Even after the Cs4MnBi2Cl12 catalysts were stored in ambient air for three months, they could function properly and the degradation rate can be almost maintained. Moreover, we investigated the photocatalytic mechanism and degradation pathway of RhB in the present system. The results reveal that the ˙O2− active radical plays a leading role in the photocatalytic process and the degradation pathway involves three stages of N-deethylation, ring opening, and mineralization. The nontoxicity, high photocatalytic efficiency, and robust stability of such Cs4MnBi2Cl12 crystals make their future application in photocatalysis field a real possibility.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (No. 12074347, 12004346, 61935009 and 12204426), the Science Foundation for Distinguished Young Scholars of Henan Province (No. 212300410019), the Support Program for Scientific and Technological Innovation Teams of Higher Education in Henan Province (No. 23IRTSTHN011), and the Open Fund of the State Key Laboratory of Integrated Optoelectronics (IOSKL2020KF04).Notes and references
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Y. Tachibana, L. Vayssieres and J. Durrant, Nat. Photonics, 2012, 6, 511–588 CrossRef CAS.
- W. Zhao, M. Adeel, P. Zhang, P. Zhou, L. Huang, Y. Zhao, M. A. Ahmad, N. Shakoor, B. Lou, Y. Jiang, I. Lynch and Y. Rui, Environ. Sci.: Nano, 2022, 9, 61–68 RSC.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
- Y. Liu, M. Zhang, C. H. Tung and Y. Wang, ACS Catal., 2016, 6, 8389–8394 CrossRef CAS.
- C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536–551 CrossRef CAS.
- Y. He, L. Zhang, M. Fan, X. Wang, M. L. Walbridge, Q. Nong, Y. Wu and L. Zhao, Sol. Energy Mater. Sol. Cells, 2015, 137, 175–184 CrossRef CAS.
- J. Yang, H. Yan, X. Wang, F. Wen, Z. Wang, D. Fan, J. Shi and C. Li, J. Catal., 2012, 290, 151–157 CrossRef CAS.
- B. AlOtaibi, S. Fan, D. Wang, J. Ye and Z. Mi, ACS Catal., 2015, 5, 5342–5348 CrossRef CAS.
- S. Yu, X.-B. Fan, X. Wang, J. Li, Q. Zhang, A. Xia, S. Wei, L.-Z. Wu, Y. Zhou and G. R. Patzke, Nat. Commun., 2018, 9, 4009 CrossRef PubMed.
- D. B. Grotjahn, V. M. Soto, E. J. Kragulj, D. A. Lev, G. Erdogan, X. Zeng and A. L. Cooksy, J. Am. Chem. Soc., 2008, 130, 6342–6344 CrossRef PubMed.
- M. J. Zhang, L. Tang, A. Duan, Y. Zhang, F. J. Xiao, Y. Zhu, J. J. Wang, C. Y. Fang and N. Yin, Chem. Eng. J., 2023, 452, 139068 CrossRef CAS.
- J. Qiu, X. Zhang, Y. Feng, X. Zhang, H. Wang and J. Yao, Appl. Catal., B, 2018, 231, 317–342 CrossRef CAS.
- M. Ge, Q. Li, C. Cao, J. Huang, S. Li, S. Zhang, Z. Chen, K. Zhang, S. S. Al-Deyab and Y. Lai, Adv. Sci., 2017, 4, 1600152 CrossRef PubMed.
- M. Sharma, T. Jain, S. Singh and O. P. Pandey, Sol. Energy, 2012, 86, 626–633 CrossRef CAS.
- H. Cho, S. H. Jeong, M. H. Park, Y. H. Kim, C. Wolf, C. L. Lee, J. H. Heo, A. Sadhanala, N. Myoung, S. Yoo, S. H. Im, R. H. Friend and T. W. Lee, Science, 2015, 350, 1222–1225 CrossRef CAS PubMed.
- Y. H. Kim, H. Cho and T. W. Lee, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11694–11702 CrossRef CAS PubMed.
- J. Y. Kim, J. W. Lee, H. S. Jung, H. Shin and N. G. Park, Chem. Rev., 2020, 120, 7867–7918 CrossRef CAS PubMed.
- Y. H. Kim, H. Cho, J. H. Heo, T. S. Kim, N. Myoung, C. L. Lee, S. H. Im and T. W. Lee, Adv. Mater., 2015, 27, 1248–1254 CrossRef CAS PubMed.
- F. Zhang, B. Yang, Y. Li, W. Deng and R. He, J. Mater. Chem. C, 2017, 5, 8431–8435 RSC.
- X. Yang, Y. Fu, R. Su, Y. Zheng, Y. Zhang and W. Yang, Adv. Mater., 2020, 32, 2002585 CrossRef CAS PubMed.
- C. Lin, S. Li, W. Zhang, C. Shao and Z. Yang, ACS Appl. Energy Mater., 2018, 1, 1374–1380 CrossRef CAS.
- S. Kumar, M. Regue, M. A. Isaacs, E. Freeman and S. Eslava, ACS Appl. Energy Mater., 2020, 3, 4509–4522 CrossRef CAS.
- Z. He, Q. Tang, X. Liu, X. Yan, K. Li and D. Yue, Energy Fuels, 2021, 35, 15005–15009 CrossRef CAS.
- B. Huo, J. Yang, Y. Bian, D. Wu, J. Feng, J. Zhou, Q. Huang, F. Dong and X. Tang, Chem. Eng. J., 2021, 406, 126740 CrossRef CAS.
- S. Das, T. Paul, S. Maiti and K. K. Chattopadhyay, Mater. Lett., 2020, 267, 127501 CrossRef CAS.
- Q. Zhang, X. Deng, C. Tan, Y. Zhou, X. Chen, X. Bai, J. Li, B. Tang, S. Li and H. Lin, J. Chem. Phys., 2020, 153, 024703 CrossRef CAS PubMed.
- Y. Zhao, H. Shi, X. Hu, E. Liu and J. Fan, Chem. Eng. J., 2020, 381, 122692 CrossRef CAS.
- B. M. Bresolin, S. B. Hammouda and M. Sillanpää, J. Photochem. Photobiol., A, 2019, 376, 116–126 CrossRef CAS.
- O. Akinbami, R. Moepya, G. N. Ngubeni, P. Tetyana, K. P. Mubiayi, M. J. Moloto and N. Moloto, J. Photochem. Photobiol., A, 2021, 419, 113460 CrossRef CAS.
- K. Li, S. Li, W. Zhang, Z. Shi, D. Wu, X. Chen, P. Lin, Y. Tian and X. Li, J. Colloid Interface Sci., 2021, 596, 376–383 CrossRef CAS PubMed.
- Z. Zhang, Y. Liang, H. Huang, X. Liu, Q. Li, L. Chen and D. Xu, Angew. Chem., Int. Ed., 2019, 58, 7263–7267 CrossRef CAS PubMed.
- B. Vargas, R. Torres-Cadena, D. T. Reyes-Castillo, J. Rodríguez-Hernández, M. Gembicky, E. Menéndez-Proupin and D. Solis-Ibarra, Chem. Mater., 2019, 32, 424–429 CrossRef.
- B. Vargas, R. Torres-Cadena, J. Rodríguez-Hernández, M. Gembicky, H. Xie, J. Jiménez-Mier, Y.-S. Liu, E. Menéndez-Proupin, K. R. Dunbar, N. Lopez, P. Olalde-Velasco and D. Solis-Ibarra, Chem. Mater., 2018, 30, 5315–5321 CrossRef CAS.
- F. Zhang, Z. Ma, Z. Shi, X. Chen, D. Wu, X. Li and C. Shan, Energy Mater. Adv., 2021, 5198145 CAS.
- J. H. Wei, J. F. Liao, X. D. Wang, L. Zhou, Y. Jiang and D. B. Kuang, Matter, 2020, 3, 892–903 CrossRef.
- A. Schmitz, L. L. Schaberg, S. Sirotinskaya, M. Pantaler, D. C. Lupascu, N. Benson and G. Bacher, ACS Energy Lett., 2020, 5, 559–565 CrossRef CAS.
- Y. El Ajjouri, V. S. Chirvony, N. Vassilyeva, M. Sessolo, F. Palazon and H. J. Bolink, J. Mater. Chem. C, 2019, 7, 6236–6240 RSC.
- S. Wu, W. Li, J. Hu and P. Gao, J. Mater. Chem. C, 2020, 8, 13603–13611 RSC.
- S. Fang, T. Wang, S. He, T. Han, M. Cai, B. Liu, V. I. Korepanov and T. Lang, Phys. Chem. Chem. Phys., 2022, 24, 9866 RSC.
- H. Zhang, J. Yao, Y. Yang and H. Fu, Chem. Mater., 2021, 33, 2847–2854 CrossRef CAS.
- X. Wang, N. Ali, G. Bi, Y. Wang, Q. Shen, A. Rahimi-Iman and H. Wu, Inorg. Chem., 2020, 59, 15289–15294 CrossRef CAS PubMed.
- N. P. Holzapfel, J. D. Majher, T. A. Strom, C. E. Moore and P. M. Woodward, Chem. Mater., 2020, 32, 3510–3516 CrossRef CAS.
- E. Sartori, M. Campolucci, D. Baranov, M. Zeng, S. Toso, G. Divitini, M. Ferretti, Z. Hens, L. Manna and F. Locardi, Nanoscale, 2022, 14, 305–311 RSC.
- R. Zhang, Z. Wang, X. Xu, X. Mao, J. Xiong, Y. Yang and K. Han, Adv. Opt. Mater., 2021, 9, 2100689 CrossRef CAS.
- C. Zhang, M. Zhang, W. Zheng, J. Wei, S. Wang, P. Huang, X. Cheng, T. Dai, Z. Chen and X. Chen, Nano Res., 2021, 15, 179–185 CrossRef.
- Y. Gao, Y. Cui, J. Li, Y. Xu, J. Hu and T. He, J. Phys. Chem. C, 2022, 126, 6694–6699 CrossRef CAS.
- Z. Ma, Z. Shi, C. Qin, M. Cui, D. Yang, X. Wang, L. Wang, X. Ji, X. Chen, J. Sun, D. Wu, Y. Zhang, X. J. Li, L. Zhang and C. Shan, ACS Nano, 2020, 14, 4475–4486 CrossRef CAS PubMed.
- F. Zhang, D. Yang, Z. Shi, C. Qin, M. Cui, Z. Ma, L. Wang, M. Wang, X. Ji, X. Chen, D. Wu, X. Li, L. Zhang and C. Shan, Nano Today, 2021, 38, 101153 CrossRef CAS.
- L. Zhou, J. F. Liao, Y. Qin, X. D. Wang, J. H. Wei, M. Li, D. B. Kuang and R. He, Adv. Funct. Mater., 2021, 31, 2102654 CrossRef CAS.
- J. G. Hou, S. Y. Cao, Y. Z. Wu, Z. M. Gao, F. Liang, Y. Q. Sun, Z. S. Lin and L. C. Sun, Chem. – Eur. J., 2017, 23, 9481–9485 CrossRef CAS PubMed.
- Z. C. Kong, J. F. Liao, Y. J. Dong, Y. F. Xu, H. Y. Chen, D. B. Kuang and C. Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- W. C. Oh, M. Chen, K. Cho, C. Kim, Z. Meng and L. Zhu, Chin. J. Catal., 2011, 32, 1577–1583 CrossRef CAS.
- T. G. Asere and G. D. Laing, Environ. Technol. Innovation, 2020, 20, 101126 CrossRef CAS.
- S. Ahmed, M. G. Rasul, R. Brown and M. A. Hashib, J. Environ. Manage., 2011, 92, 311–330 CrossRef CAS PubMed.
- C. C. Wang, C. K. Lee, M. D. Lyu and L. C. Juang, Dyes Pigm., 2008, 76, 817–824 CrossRef CAS.
- X. Qian, Z. Chen, X. Yang, W. Zhao, C. Liu, T. Sun, D. Zhou, Q. Yang, G. Wei and M. Fan, J. Cleaner Prod., 2020, 249, 119335 CrossRef CAS.
- K. A. Sultana, M. T. Islam, J. A. Silva, R. S. Turley, J. A. Hernandez-Viezcas, J. L. Gardea-Torresdey and J. C. Noveron, J. Mol. Liq., 2020, 307, 112931 CrossRef CAS.
- X. Lang and J. Zhao, Chem. – Asian J., 2018, 13, 599–613 CrossRef CAS PubMed.
- B. Tian, R. Dong, J. Zhang, S. Bao, F. Yang and J. Zhang, Appl. Catal., B, 2014, 158, 76–84 CrossRef.
- K. Katsiev, G. Harrison, H. Alghamdi, Y. Alsalik, A. Wilson, G. Thornton and H. Idriss, J. Phys. Chem. C, 2017, 121, 2940–2950 CrossRef CAS.
- T. S. Natarajan, M. Thomas, K. Natarajan, H. C. Bajaj and R. J. Tayade, Chem. Eng. J., 2011, 169, 126–134 CrossRef CAS.
- K. Yu, S. G. Yang, H. He, C. Sun, C. G. Gu and Y. M. Ju, J. Phys. Chem. A, 2009, 113, 10024–10032 CrossRef CAS PubMed.
- J. Chen, X. Xiao, Y. Wang and Z. Ye, Appl. Surf. Sci., 2019, 467, 1000–1010 Search PubMed.
- Z. He, C. Sun, S. Yang, Y. Ding, H. He and Z. Wang, J. Hazard. Mater., 2009, 162, 1477–1486 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc00185g |
| This journal is © The Royal Society of Chemistry 2023 |