
Recent advances in Ru-based electrocatalysts for oxygen evolution reaction
Jie
Ying
a,
Jiang-Bo
Chen
b,
Yu-Xuan
Xiao
a,
Susana I.
Cordoba de Torresi
c,
Kenneth I.
Ozoemena
 *bd and
Xiao-Yu
Yang
*bd
*bd and
Xiao-Yu
Yang
*bd
aSchool of Chemical Engineering and Technology, Sun Yat-sen University, Zhuhai 519082, China
bState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Shenzhen Research Institute, Joint Laboratory for Marine Advanced Materials in Pilot National Laboratory for Marine Science and Technology (Qingdao), Wuhan University of Technology, Wuhan, 430070, China. E-mail: xyyang@whut.edu.cn
cInstituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes 748, 05508-000 São Paulo, Brazil
dMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Private Bag 3, Johannesburg 2050, South Africa. E-mail: kenneth.ozoemena@wits.ac.za
First published on 29th December 2022
Abstract
The oxygen evolution reaction (OER), as a four-electron transfer process with large overpotential loss that is also kinetically sluggish, is one of the most crucial electrochemical processes to convert renewable energy into chemical fuels. Ruthenium (Ru)-based electrocatalysts for the OER have several advantageous features including high activity, pH-universality and the lowest price in the noble metal family. However, the instability of Ru-based catalysts in the OER is a central hurdle limiting their widespread application. This review focuses on recent developments made in the field of Ru-based OER catalysts. In the coverage, first the characteristics of Ru-based OER catalysts such as the OER mechanism, intrinsic activity and stability issue are discussed. Then, recently reported Ru-related catalysts falling in the Ru metal and Ru compound subclasses are described with special emphasis being given to a discussion of activity/stability-enhancing strategies. Finally, several challenges and future perspectives in this field are summarized. It is anticipated that this review will promote a better understanding of Ru-based OER catalysts and lead to new strategies to design active and stable Ru-materials.
 Jie Ying | Jie Ying received his Ph.D. degree from Wuhan University of Technology in 2016. He then joined the group of Prof. Zhongwei Chen at University of Waterloo (2016) and Prof. Christoph Janiak at Heinrich-Heine-Universität Düsseldorf (2018) as a postdoctoral fellow. Currently, he is an associate professor in Sun Yat-sen University. His scientific interest includes design and synthesis of nanocomposite and their application in catalysis and electrocatalysis. |
 Kenneth I. Ozoemena | Kenneth I. Ozoemena is Professor of Materials for Energy & Electrochemistry at the University of the Witwatersrand (Wits), South Africa. Prior to joining Wits in 2017, he had worked at the South Africa's Council for Scientific and Industrial Research as Chief Research Scientist and Research Group Leader of the CSIR Electrochemical Energy Technologies (2009–2017). His current research interests are focused on energy storage and conversion systems and electrochemical sensors. |
 Xiao-Yu Yang | Xiao-Yu Yang received his Ph.D. from Jilin University (co-educated at FUNDP of Belgium). After a postdoctoral fellowship at the FUNDP, he worked as a “Chargé de Recherches” at the FNRS in Belgium. He is currently working as full professor at WUT, co-professor at SMSEGL, and visiting professor at HU. His research is aimed at hierarchical assembly techniques and hierarchical structured materials for the applications in energy, environment, catalysis, bioengineering, and ocean-engineering. |
1. Introduction
Global energy consumption is growing at a dramatic rate. Because over 80% of energy used in the world originates from unsustainable fossil fuels, this level will inevitably lead to exhaustion of supplies and a serious level of CO2 emission.1,2 Thus, clean and renewable energy sources, such as solar and wind, are attractive sustainable and environmentally clean alternatives. However, the inherent intermittency and localization of energy derived from solar and wind sources are impediments to the utilization of these alternatives.3–5 One very promising approach to circumvent these drawbacks is to transform the energy derived from these sources into storable and transportable fuels such as H2.6–8 Hydrogen gas produced by the hydrogen evolution reaction (HER) occurring at the cathode in electrolytic water splitting, can be stored and ultimately reacted with oxygen using fuel cell technology to generate clean, carbon free energy.9–11 However, a bottleneck exists in the H2 production process as a consequence of the sluggishness of the simultaneous anodic oxygen evolution reaction (OER), which occurs via a four electron–proton coupled pathway that requires high energy to overcome a kinetic barrier.12–15 Therefore, to carry out the HER in an efficient manner, low-cost, highly active and stable electrocatalysts for the OER are required.Ruthenium (Ru), the least expensive member of the Pt-related noble metal family, has a price that is only 20% that of Pt.16 Ru-based materials that have suitable binding energies for reactive intermediates and high thermostability have emerged as attractive catalysts for many reactions, including the OER,17–19 HER,20–22 oxygen reduction reaction,23–25 CO oxidation reaction,26–28 and so on. A large effort has been expended in the past decade to identify Ru-based materials, particularly RuO2, which can serve as efficient OER catalysts.29–31 To date, Ru-based materials are still one of the best OER catalysts owing to the most suitable adsorption/desorption effect towards oxygen, thus is close to the top of the volcano among frequently-used oxides (RuO2, IrO2, Co3O4, Mn3O4, etc.).32 Although numerous advances have been made in designing and fabricating highly active Ru-based catalysts, Ru-based materials normally suffer from serious stability issues especially under harsh water electrolysis conditions. For example, both metallic Ru and RuO2 display extreme instability as anode materials for the OER because they undergo oxidation at the high overpotentials required.33,34 Although the development of active catalyst is still the cornerstone, the focus in recent years is gradually shifting to the stability of developed catalysts with the rapid development of advanced characterization technique, which has enabled more detailed assessments to be made of the properties responsible for the activities and stabilities of Ru-based OER catalysts.
Since research in the field of Ru-based materials has very rapidly intensified in recent years and only a few reviews summarizing the latest advancement of Ru-based OER catalysts have been published,35,36 an overview of recent progress is needed to disseminate knowledge about strategies that have been developed for the design and synthesis of Ru-based catalysts for the OER. In this review, recently developed Ru-based catalysts with superior OER performance are summarized. Firstly, the OER mechanism, intrinsic activity and stability issue associated with the Ru-based catalysts are briefly introduced for the scientific understanding. Then, based on the recent advances in this research field, Ru-related materials are divided into two classifications: Ru metals and Ru-based compounds (Fig. 1). The former classification is divided into four subclassifications, i.e., structure/morphology engineering, single-atom design, alloy construction, heterostructure configuration and design of Ru–C composites. The latter subclassification includes Ru oxides, Ru chalcogenides and pyrochlore ruthenates. For each part, more attention has been focused on the discussion of activity/stability-enhancing strategies for catalyst design via salient examples. Finally, the remaining challenges and future perspectives in this flourishing field are proposed.
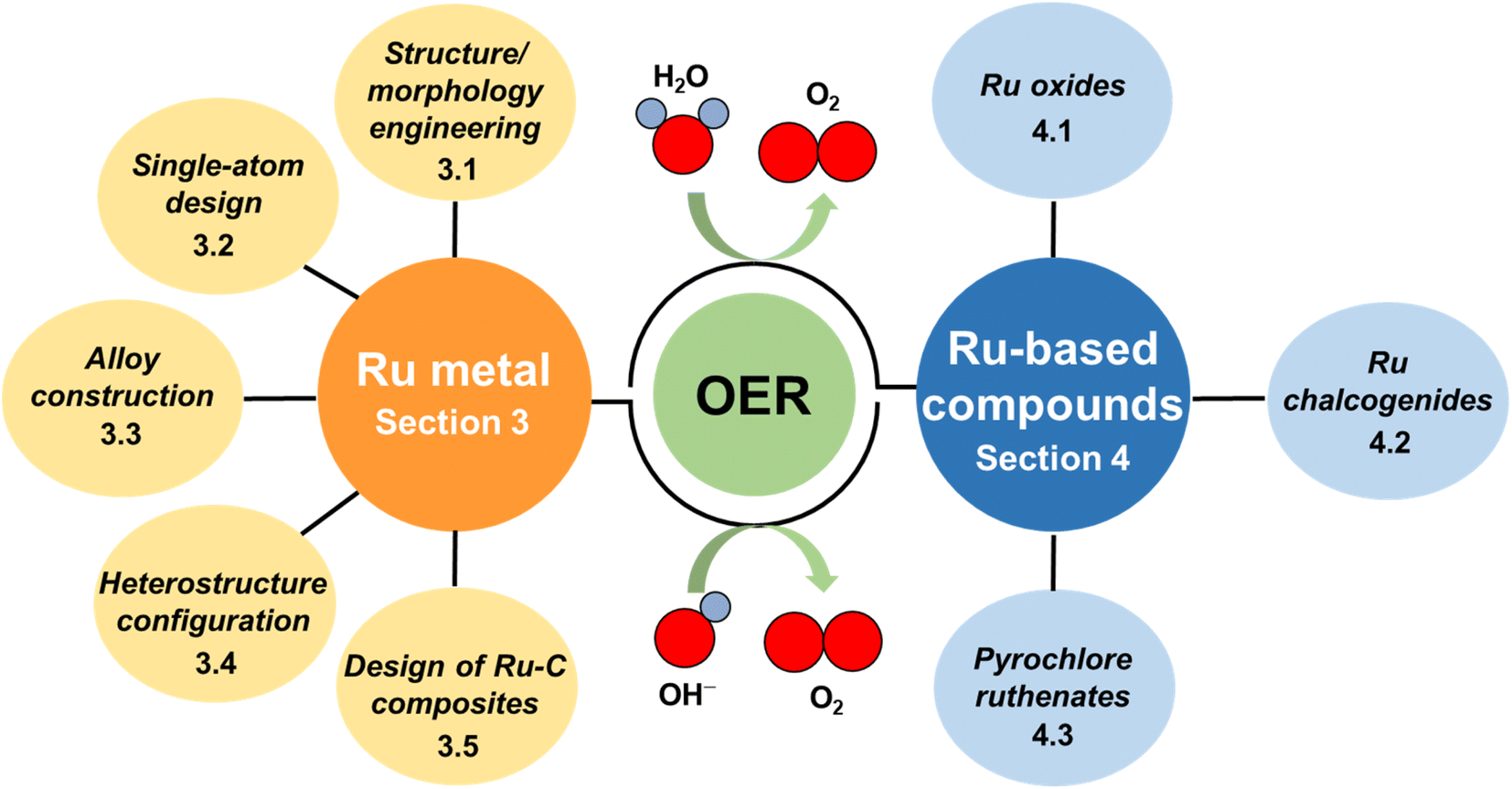 | ||
| Fig. 1 Diagram outlining the categories used to review of recent advances made in the development of Ru-based catalysts for the OER. | ||
2. General characteristics of OER
2.1 OER mechanism
During the OER, four electrons are transferred initially from water and then from several intermediates in stepwise manner to produce an oxygen molecule.37 It is proposed that the overall reaction occurs through multiple electron–proton oxidation processes in conjunction with absorption/desorption of various oxygen intermediates,38 which is called the absorbate evolution mechanism (AEM). Difficulties associated with detecting these intermediates has prevented formulation of a precise pathway for the OER in both acidic and alkaline/neutral media. Several possible routes have been suggested for each reaction,39 the most accepted of which involve the stepwise pathways displayed in Fig. 2A and B and eqn (1)–(8), where * represents active sites of the catalyst.40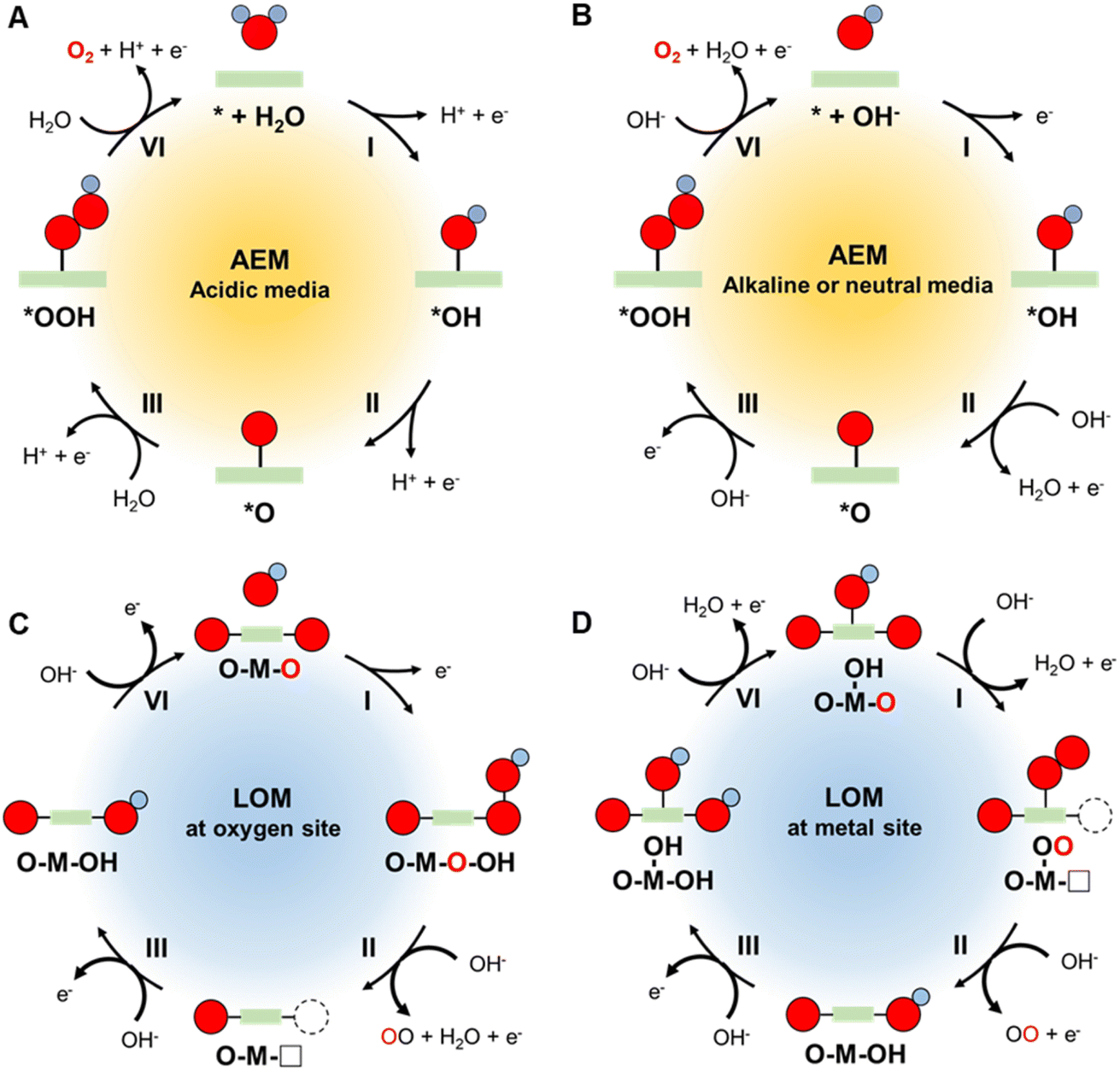 | ||
| Fig. 2 Schematic illustration of AEM pathways for the OER under (A) acidic and (B) alkaline/neutral conditions. Schematic illustration of LOM pathways for OER on (C) oxygen site and (D) metal site. | ||
In acidic media:
| * + H2O → *OH + H+ + e− | (1) |
| *OH → *O + H+ + e− | (2) |
| *O + H2O → *OOH + H+ + e− | (3) |
| *OOH → O2 + H+ + e− | (4) |
In alkaline or neutral media:
| * + OH− → *OH + e− | (5) |
| *OH + OH− → *O + H2O + e− | (6) |
| *O + OH− → *OOH + e− | (7) |
| *OOH + OH− → O2 + H2O + e− | (8) |
In these mechanisms, the adsorbed O-related species that become bound to the active sites of the catalyst include the three intermediates*OH, *O, *OOH. In the electrochemical oxidation process, oxidation of water/hydroxyl ions takes place to generate *OH, followed by deprotonation and further oxidation to produce *O and *OOH, and eventually O2. In acidic media, a water molecule undergoes initial loss of an electron, while in alkaline/neutral media, a hydroxide ion serves as the oxidation site.
Recently, an alternative mechanism named lattice oxygen-mediated mechanism (LOM) has been proposed, which involves the surface lattice oxygen redox.41–43 In the AEM, the surface of catalyst is regarded to be stable with only valence state change of the active site. In contrast, the catalyst surface is not stable in the LOM, in which the lattice oxygen is activated and participates in the oxygen evolution process. Two OER pathway via LOM with different active centers has been proposed. As depicted in Fig. 2C, activated lattice oxygen can readily act as an active site, which directly receives OH− by means of nucleophilic attack to produce the *OOH species.44 The release of O2 generates an oxygen vacancy site, which is adsorbed by OH− to form *OH intermediate. Other than this pathway, another LOM mechanism takes the metal site as the active center to adsorb OH− and then undergoes the deprotonation reaction (Fig. 2D).45 The surface reconstruction allows the conjunction of the *O species and activated lattice oxygen to generate *OOH species, which is regarded as a newly generated oxygen molecule to follow the subsequent O2 release. Therefore, the lattice oxygen of catalysts is activated and participates in both LOM pathways under OER process.
2.2 Intrinsic activity
Since all steps in the OER mechanism are thermodynamically uphill, the step with the largest positive ΔG is rate-determining.46 Previous studies have shown that differences between the binding free energies of *OH and *OOH (ΔG*O − ΔG*OH) on many metal oxides are large (3.2 ± 0.2 eV), which is highly relevant to the OER kinetics.47 As a result, a superior OER catalyst should not have too strong or too weak bonding energies for the O-related intermediates. Rossmeisl et al. showed that the theoretical OER activity can be expressed in terms of the negative change of the Gibbs free energy (−ΔG) as a function of the oxygen binding energy (Fig. 3A).48 Accordingly, catalyst that have *O binding energies of ca. 2.3 eV should have high OER activities corresponding to the dashed line in the free energy vs. binding energy plot. Moreover, the volcano section in the bottom part of Fig. 3A also demonstrates that OER activity is influenced by steps for formation of *O (red line) and *OOH (green line) for weak and strong *O surface binding energies, respectively. Based on first principle calculations, Man et al. showed that the relationship existing between the negative theoretical overpotential of metal oxides and their binding free energy difference (ΔG*O − ΔG*OH) correlates with their OER activities (Fig. 3B).49 Because it has the optimal binding interactions between metal–O species and the intermediates, RuO2 displays the highest activity among various metal oxides. The very weak bonding interactions between *OOH and the catalyst cause the generation of the *OOH intermediate (ΔG3) to be thermodynamically much more difficult than those of the other intermediates (*OH, *O) (Fig. 3C).50 Thus, these considerations suggest that the performance of OER catalysts can be improved by enhancing surface bonding interactions with *OOH and thus shifting their chemisorption free energy for *OOH to more negative values. It is important to note that the performance of OER catalysts not only correlates with the thermodynamics of intermediate binding interactions, but it is also influenced by other factors such as medium pH, and catalyst crystallinity, morphology/structure and particle size.51–54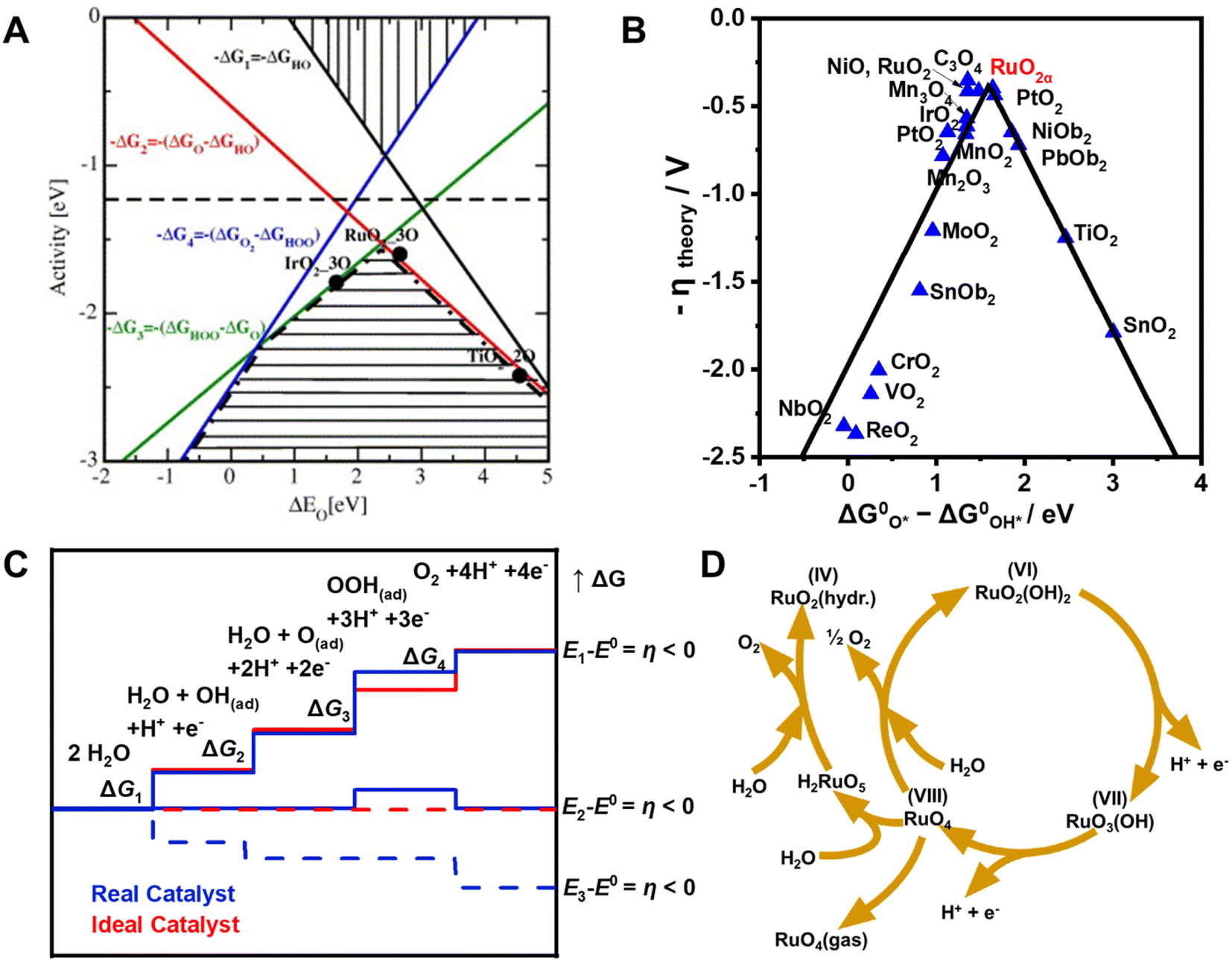 | ||
| Fig. 3 (A) Theoretical OER activity, defined as the negative change of Gibbs free energy (−ΔG), as a function of the oxygen binding energy. Reproduced with permission from ref. 48. Copyright 2007, Elsevier. (B) “Volcano”-shaped relationships between OER activity and binding energy of ΔG*O − ΔG*OH in some metal oxides. Reproduced with permission from ref. 49. Copyright 2011, Wiley-VCH. (C) Plot of Gibbs free energy of reactive species and intermediates (horizontal lines) of the OER versus the reaction coordinate. Blue lines and red lines indicate energetics of a real (typical) catalyst and an ideal catalyst, respectively, at three different electrode potentials. Dashed lines indicate energetics at the electrode potential where all thermochemical barriers disappear (“thermochemical overpotential”). Reproduced with permission from ref. 50. Copyright 2010, Wiley-VCH. (D) Model for the oxygen evolution and corrosion on Ru and RuO2 electrodes. Reproduced with permission from ref. 56. Copyright 1983, The Electrochemical Society. | ||
2.3 Stability issue
It is well-known that Ru-based catalysts are highly unstable under the harsh conditions present in the OER. The benchmark OER catalyst RuO2 undergoes dissolution at overpotentials above 1.4 V due to the extensive oxidation,55 and the dissolution rate of metallic Ru in the OER process is several orders of magnitude higher than that of its oxides.33 For example, Paoli et al. found that 90% of metallic Ru dissolves after 15 min of continuous electrolysis at 1.5 V, while only 30% of RuO2 is destroyed under the same operation conditions.55 Over the past decade, great progress has been made in gaining an understanding of the details involved in degradation and dissolution of Ru in the OER, and this has led to formulation of the degradation mechanism illustrated in Fig. 3D.56 The instability of Ru in this process is ascribed to the formation of unstable RuO4 species through involvement of lattice oxygen.57 In the pathway, the formed RuO4 intermediate releases from the electrode and diffuses into the solution, leading to a reduction of the electrode mass, which can be quantitatively determined using inductively coupled plasma mass spectrometry (ICP-MS).58–60 The generation of ruthenate(VIII+) ion in the electrolyte solution was also confirmed by utilizing in situ reflection spectroscopy rotating ring-disk electrode measurements.61 Moreover, it was found that the onsets of OER and dissolution of Ru occur simultaneously, indicating that dissolution is induced by the OER. Since parts of the dissolved RuO4 described in Fig. 3D would not be recovered by redeposition as the reaction progress, the amount of dissolved RuO4 largely hampers the catalyst stability.A wide range of studies have been conducted to enhance the stabilities of metallic and oxidized Ru-based catalysts for the OER, and these efforts have led to development of strategies to design and fabricate the high-performance Ru-based materials described in the following sections and summarized in Table 1.
| Classification | Catalyst | Electrolyte | Overpotential (mV) at 10 mA cm−2 | Tafel slope (mV dec−1) | Stability (V or mA cm−2 @ h) | Ref. |
|---|---|---|---|---|---|---|
| Ru metals | Ru nanoparticles | 0.5 M H2SO4 | 202 | ∼70 | 10 mA cm−2 @ 10 h | 62 |
| Ru branched nanoparticles | 0.1 M HClO4 | 180 | ∼52 | 4 mA cm−2 @ 10 h | 63 | |
| Ru/CoFe-LDHs | 1 M KOH | 198 | ∼39 | 200 mA cm−2 @ 24 h | 64 | |
| Ru1–Pt3Cu | 0.1 M HClO4 | 220 | Not given | 10 mA cm−2 @ 28 h | 29 | |
| Ru–N–C | 0.5 M H2SO4 | 267 | ∼53 | 1.5 V @ 30 h | 65 | |
| High-entropy Ru alloys | 0.5 M H2SO4 | 258 | ∼84 | 10 mA cm−2 @ 10 h | 66 | |
| RuCu nanosheets | 1 M KOH | 234 | Not given | 10 mA cm−2 @ 12 h | 67 | |
| RuCu nanosheets | 0.5 M H2SO4 | 236 | Not given | Not given | 67 | |
| Ru@IrOx | 0.05 M H2SO4 | 282 | ∼69 | 1.55 V @ 24 h | 68 | |
| Ru oxides | RuO2 nanowires | 0.5 M H2SO4 | 234 | Not given | 5 mA cm−2 @ 20 h | 69 |
| RuO2 nanowires | 1 M KOH | 224 | Not given | 5 mA cm−2 @ 20 h | 69 | |
| RuO2 nanosheets | 0.5 M H2SO4 | 199 | ∼38 | 10 mA cm−2 @ 6 h | 70 | |
| Co-doped RuO2 | 0.5 M H2SO4 | 169 | ∼47 | 10 mA cm−2 @ 50 h | 71 | |
| S–RuFeOx | 0.1 M HClO4 | 187 | ∼40 | 1 mA cm−2 @ 50 h | 72 | |
| Mn–RuO2 | 0.5 M H2SO4 | 158 | ∼43 | 10 mA cm−2 @ 10 h | 73 | |
| RuIrCaOx | 0.5 M KHCO3 | 250 | Not given | 10 mA cm−2 @ 200 h | 74 | |
| (Ru–Co)Ox | 1 M KOH | 171 | Not given | 10 mA cm−2 @ 10 h | 75 | |
| SrRuIrOx | 0.5 M H2SO4 | 190 | ∼39 | 10 mA cm−2 @ 1500 h | 76 | |
| Cr0.6Ru0.4O2 | 0.5 M H2SO4 | 178 | ∼58 | 10 mA cm−2 @ 10 h | 77 | |
| WErRuO2−δ | 0.5 M H2SO4 | 168 | ∼67 | 10 mA cm−2 @ 500 h | 78 | |
| Ru chalcogenides | RuTe2 | 1 M KOH | 275 | ∼53 | 10 mA cm−2 @ 12 h | 79 |
| Amorphous RuTe2 | 0.5 M H2SO4 | 245 | Not given | 10 mA cm−2 @ 24 h | 80 | |
| Amorphous RuTe2 | 1 M KOH | 285 | Not given | Not given | 80 | |
| RuS2 | 1 M KOH | 282 | ∼103 | 10 mA cm−2 @ 20 h | 81 | |
| Ru/RuS2 | 0.5 M H2SO4 | 201 | ∼47 | 10 mA cm−2 @ 24 h | 82 | |
| Pyrochlore ruthenates | Pb2Ru2O7−x | 0.1 M NaOH | Not given | ∼45 | 10 mA cm−2 @ 2 h | 83 |
| Y2Ru2O7−δ | 0.1 M HClO4 | 190 | ∼55 | 1 mA cm−2 @ 8 h | 84 | |
| Y1.8Cu0.2Ru2O7−δ | 1 M H2SO4 | Not given | ∼52 | 1 mA cm−2 @ 6 h | 85 | |
| Y1.7Sr0.3Ru2O7 | 0.5 M H2SO4 | 264 | ∼45 | 10 mA cm−2 @ 28 h | 86 | |
| Nd2Ru2O7 | 0.1 M HClO4 | 210 | ∼48 | 1 mA cm−2 @ 6 h | 87 | |
| Yb2Ru2O7 | 0.1 M HClO4 | 310 | ∼91 | Not given | 87 |
3. Ru metals
3.1 Structure/morphology engineering
Boosting the catalytic performance of Ru metals for the OER requires that the number and utilization of active sites be increased, and that corrosion resistance be enhanced.40 Structural/morphological engineering approaches that enhance specific surface areas and mass/electron transfer rates can be employed to alter the properties of Ru metals in favorable ways.88–90Highly branched Ru nanoparticles with controllable crystallinity and surface facets have been generated by Tilley et al. using rational adjustment of the ratios of dodecylamine (DDA) surfactant to Ru precursor in the preparative process.63 In this effort, polycrystalline Ru nanoparticles were formed using high ratios of DDA surfactant to Ru precursor, while low ratios lead to formation of Ru branched nanoparticles that have exposed low-index facets (Fig. 4A). Compared with polycrystalline Ru nanoparticles, faceted Ru branched analogs exhibit higher activity with an overpotential of 180 mV (Fig. 4B) (note: in this review, all potentials are versus the reversible hydrogen electrode and all overpotentials are obtained at a current density of 10 mA cm−2) and high stability reflected in only a 85 mV overpotential increase after 4 h. It was observed that Ru atoms on low-index facet Ru branches are more resistant to dissolution, while most of those in the polycrystalline Ru branches are readily dissolved because they are at low coordination sites (Fig. 4C). The finding that faceted Ru branches with large numbers of retained active surface atoms display greatly enhanced properties suggests that controlling the crystallinity and surface configuration of Ru-based catalysts is a potential strategy for designing new OER catalysts.
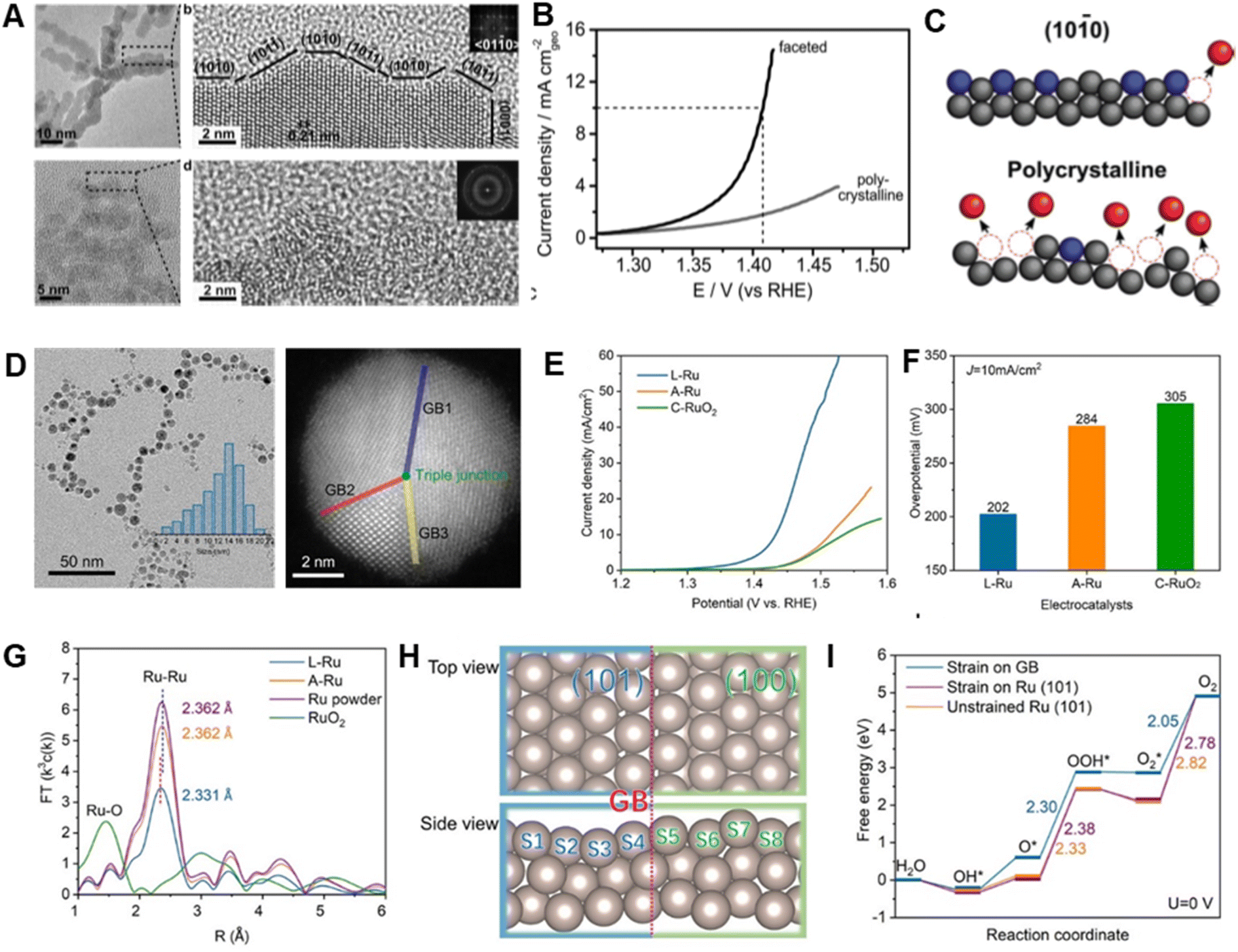 | ||
| Fig. 4 (A) TEM image of faceted Ru branched nanoparticles (top) and polycrystalline Ru nanoparticles (bottom). (B) Potentiodynamic curves of the OER performance of faceted Ru branched nanoparticles and polycrystalline Ru nanoparticles, obtained in 0.1 M HClO4 solution. (C) Schematic of the proposed structural changes affecting the OER activity and stability of faceted Ru branched nanoparticles (top) and polycrystalline Ru nanoparticles (bottom). Reproduced with permission from ref. 63. Copyright 2019, Wiley-VCH. (D) TEM and of HAADF-STEM images of Ru nanoparticles with grain boundaries. (E and F) OER performance of electrodes based on different Ru catalysts. (G) FT k3-weighted EXAFS spectra of different Ru catalysts. (H) Atomic model of Ru(101)–Ru(100) grain boundaries. (I) Free-energy landscape of near Ru(101)–Ru(100) grain boundaries, strain-free Ru(101) surface, and monocrystal Ru(101) surface. Reproduced with permission from ref. 62. Copyright 2020, The American Chemical Society. | ||
Strain engineering can be utilized to adjust electronic structures and optimize adsorption abilities of catalysts toward reaction intermediates by turning atomic distances on the surface.91,92 This approach has emerged as a powerful tool for improving the performances of various electrocatalysts.93–95 For example, Du et al. took the advantage of the laser-ablation-in-liquid technique to create strain by introducing grain boundaries into activity enhanced Ru OER catalysts.62 The prepared Ru catalyst with numerous grain boundaries operates at a low overpotential of 202 mV for the OER in acidic media, which is 100 mV lower than that of commercial RuO2 (305 mV) (Fig. 4D–F). The novel Ru catalyst also displays superior durability, by not undergoing significant degradation over a 10 h working period. Density functional theory (DFT) calculations demonstrate that compressive strain at grain boundaries increases the electron density on Ru atoms, lowers the d-band center (Fig. 4G) and weakens adsorption of OER intermediates (Fig. 4H and I), ultimately leading to enhanced catalytic performance.
3.2 Single-atom design
Considering that the particle size markedly affects the number of active sites, decreasing the size of catalysts form their bulk to ultrafine nanoparticles and clusters can provide ultrahigh atomic utilization and high catalytic performance. Due to the high surface energy, catalyst supports are important to avoid aggregation and/or overgrowth of the prepared nanoparticles/clusters with desired sizes.96,97 Recently, reducing the sizes of catalyst to singe atoms are becoming rising hotspots, which can make more efficient use of metal atoms. This design feature has aroused interest in exploring catalytically active components of single atoms and interfacial interactions that occur between active sites and supports.98–100 The construction of Ru single-atom catalysts not only enhances their OER activity/stability but also improves the economics of noble metal utilization. In addition, the catalytic performance of single atom catalysts can be influenced by the nature of supporting materials, since the electronic structures of stabilized single atoms are highly dependent on the local coordination environments of anchored sites on supports.Recently, many supports, such as layered double hydroxides (LDHs),101–103 metals,29,104 and carbons,105–107 have been developed to stabilize Ru single atoms and to create superior electrocatalytic performance. LDHs, having two-dimensional structures, are interesting platforms for stabilization of single atom catalysts because they contain two-dimensional flat facets, ultrathin thicknesses and high surface areas.108,109 For example, single atom Ru anchored on the surface of CoFe-LDHs was prepared by Sun et al. using a simple two-step procedure involving co-precipitation of CoFe-LDHs and reduction of Ru precursor (Fig. 5A and B).64 The synthesized CoFe-LDHs containing 0.45 wt% Ru exhibit a very low overpotential of only 198 mV and a negligible loss of activity after 1000 cycles of voltage sweeps between 1.35 and 1.5 V in alkaline solution. The enhanced OER activity and stability of this catalyst can be attributed to its respective optimal adsorption free energy for *OOH and improved resistance to formation of high oxidation states of Ru owing to strong synergetic electron coupling between single-atom Ru and LDHs (Fig. 5C and D).
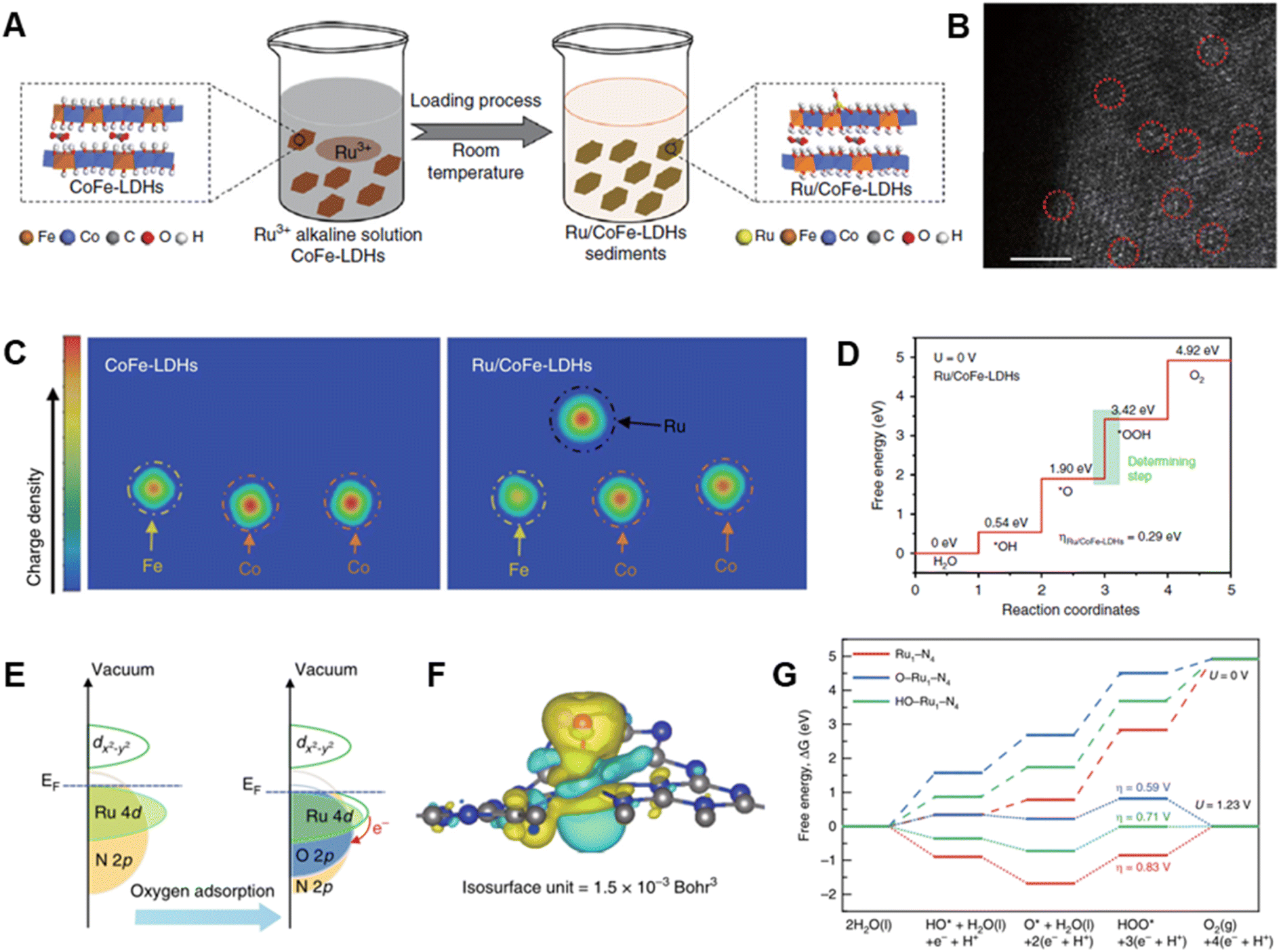 | ||
| Fig. 5 (A) Schematic illustration of the hydrolysis-deposition procedure to form Ru/CoFe-LDHs. (B) The Cs-corrected STEM image of Ru/CoFe-LDHs nanosheet. (C) The differential charge density of elements in CoFe-LDHs and Ru/CoFe-LDHs from a computational simulation. (D) Gibbs free-energy diagram for the four steps of the OER on Ru/CoFe-LDHs. Reproduced with permission from ref. 64. Copyright 2019, Springer Nature. (E) Schematic illustration of the effect of oxygen adsorption on the electronic structure of Ru–N–C. (F) Electron density difference plot of the O–Ru1–N4. Yellow and light green contours represent electron accumulation and deletion, respectively. (G) Free energy diagram for the OERs on Ru1–N4, O–Ru1–N4, and HO–Ru1–N4. Reproduced with permission from ref. 65. Copyright 2019, Springer Nature. | ||
In the OER promoted by Ru-based catalysts, oxygen evolution always occurs along with Ru dissolution.110,111 The latter phenomenon is linked to oxidative released by the lattice oxygen evolution reaction (LOER), which leads to peeling off of active oxygen-coordinated Ru moieties.55 A method to lessen the contribution of the LOER, and thus improve stability, involves supporting single-atom Ru on a matrix in the absence of oxygen coordination, such as on an acid-resistant N-doped carbon where strong Ru–N bonds exist. A single-atom Ru–N–C catalyst of this type was fabricated by Yao et al. by using incipient wetness impregnation of a Ru precursor into carbon support followed by pyrolysis.65 The as-prepared Ru–N–C catalyst has outstanding OER activity with a 267 mV overpotential and it does not undergo decomposition after 30 h operation in acidic media. The high OER activity and stability of this Ru–N–C catalyst is a consequence of its O–Ru1–N4 structure which displays greater charge transfer from Ru though dynamic pre-adsorption of a single oxygen atom (Fig. 5E–G).
Metallic Pt has a dramatically lower dissolution rate than that of Ru because it undergoes relatively weaker bonding with oxygen species,60 a property which can be utilized to decrease the contribution of LOER. In this regard, Li et al. prepared a series of PtCux/Ptskin core–shell structures containing dispersed single-atom Ru (Ru1–PtxCuy) via acid or electrochemical etching of unstable Cu.29 A volcano relationship was observed to exist between the lattice constants of the PtCu alloys and their OER activities. The optimized catalyst, Ru1–Pt3Cu, displays a 90 mV lower overpotential and 10 times longer lifetime than those of commercial RuO2. These observations demonstrate that the compressive strain in the Pt skin effectively adjusts the electronic structure and redox ability of the anchored single-atom Ru, thereby providing optimized adsorption of oxygen intermediates and high oxidation/dissolution resistance by efficiently blocking over-transference of electrons from Ru to O-containing ligands.
3.3 Alloy construction
Alloying other metals is regarded as an effective strategy to tune the catalytic properties of the host metal because it generates heteroatom bonds and modifies lattice parameters and electronic structures.21,112–115 Consequently, the electrocatalytic performance of Ru-based alloys can be remarkably enhanced.Abruña et al. synthesized a series of Ru–M (M = Co, Ni, Fe) alloy catalysts using a simple impregnation method.116 Alloying Ru with metals creates catalysts that have significantly improved OER activities in alkaline media in comparison with that of commercial Ru catalyst. DFT calculations demonstrate that the enhancement of OER performances of Ru–M alloyed catalysts can be attributed to weakening of O binding with Ru caused by Co, Ni or Fe incorporation. Moreover, the electrocatalytic performance of Ru-based alloys can be further improved by integrating them into particular structures, such as a two-dimensional structure, which provide larger surface areas and greater exposure of atoms acting as active sites.117–119 Huang et al. reported the synthesis of channel rich RuCu bimetallic nanosheets, which consist of crystallized Ru and amorphous Cu.67 The RuCu nanosheets have much improved OER activity in comparison with those of RuCu nanoparticles and commercial Ir/C in both alkaline and acidic electrolytes. In addition, the nanosheets display a very small potential increase during 21.5 h chronoamperometry test at 10 mA cm−2. In this study, a high ratio of both Ru4+/Ru0 and Cu2+/Cu0/1+, together with the synergistic effect between Ru and Cu are pivotal factors leading to the enhanced OER activity/stability.
High-entropy alloys (HEAs) comprised of at least five uniformly distributed metals have attracted increasing interest from both a theoretical and experimental viewpoint.120–122 The high degree of synergy between each metal endows an HEA with lattice distortion, high entropy and the cocktail effect, which lead to a significant enhancement of catalytic performance.123–126 Recently, Fujita et al. described novel nanoporous HEAs with hierarchical porosity that were fabricated by dealloying.66 The HEAs, containing up to 14 elements including Al, Ag, Au, Co, Cu, Fe, Ir, Mo, Ni, Pd, Pt, Rh, Ru and Ti, have high catalytic activities and durabilities for the OER in acidic media that are superior to those of commercial Pt/graphene and IrO2 catalysts. Characterization studies on these materials show that their crystal structures, overall morphologies and element compositions are well retained throughout a durability test in the acidic media, except for small decreases taking place in the proportion of Al in the alloy and insignificant surface roughening possibly caused by the dissolution of surface Al in the acidic media. The superior OER performance of these Ru-based catalysts can be ascribed to their unique features, including hierarchical porous structures, high surface areas, and highly dispersed elements.
3.4 Heterostructure configuration
Normally, synergistic effects associated with incorporation of Ru and other materials, such as metals, oxides, sulfides, phosphides and carbons into heterostructured catalysts effectively improve electrocatalytic activities and stabilities.35,127–129Qiao et al. reported a study of heterostructured Ru@IrOx catalysts that undergo charge redistribution between highly strained and disordered Ru cores and partially oxidated Ir shells through the metal–metal oxide heterojunction (Fig. 6A).68 Because of the synergistic electronic and structural interactions associated with the elevated valence of the Ir shell and the decreased valence of the Ru core, heterostructured Ru@IrOx require low overpotentials of 282 mV and they display a high activity retention of 90% over a 24 h period in acidic media (Fig. 6B and C). The authors suggest that one factor responsible for the high stabilities of heterostructured Ru@IrOx is the presence of stable chemical states of both Ru and Ir species, which hinders generation of high-valence intermediate species that readily dissolve during OER process. In addition, the core–shell nanostructure of Ru@IrOx offers efficient surface protection of active Ru sites toward oxidation/dissolution.
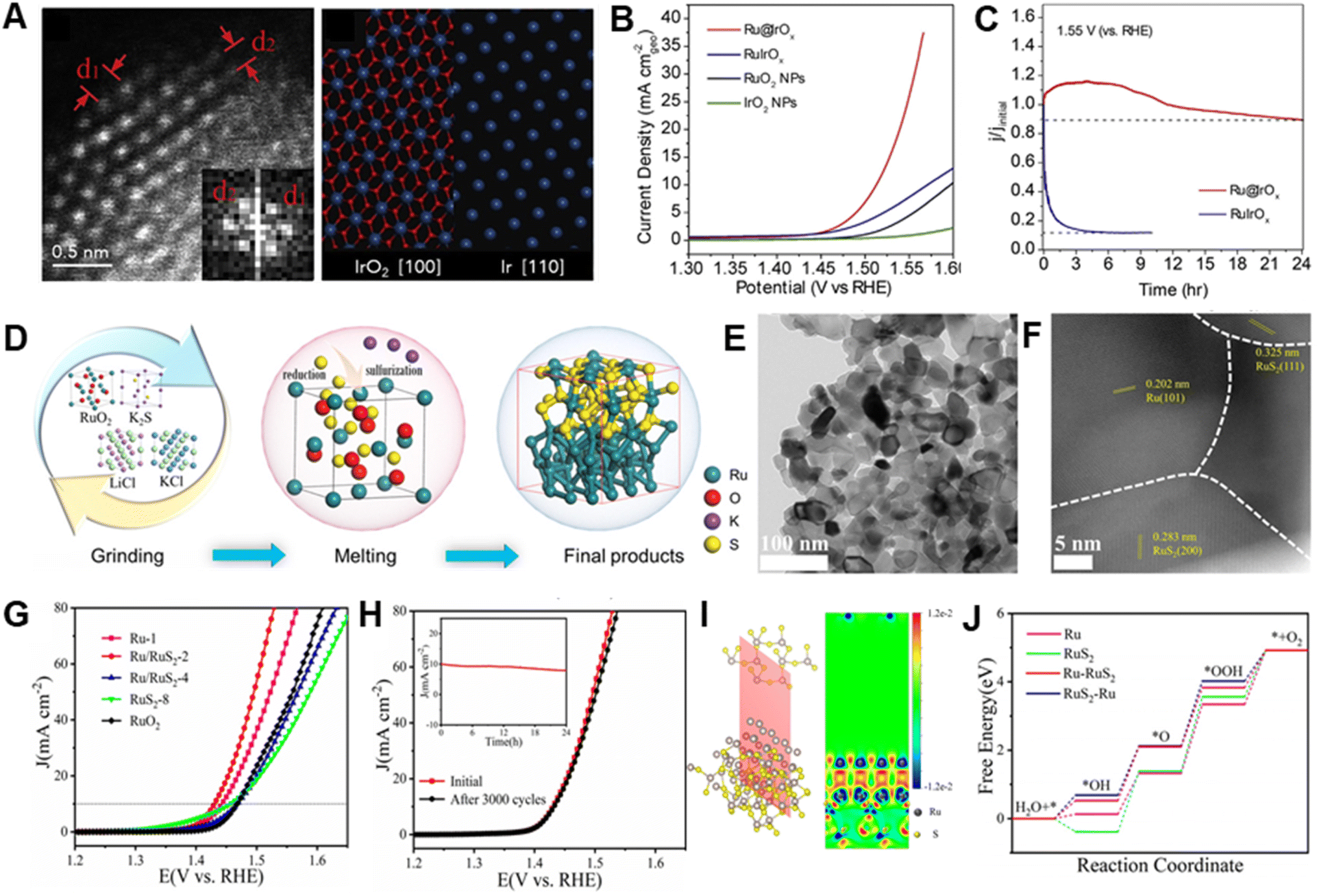 | ||
| Fig. 6 (A) HAADF-STEM image of a randomly chosen Ru@IrOx core–shell icosahedral nanocrystal along the [112] axis and projected structural models of tetragonal IrO2 and Ir along their [100] and [110] axes, respectively. (B) LSV curves normalized by using geometrical surfaces of various electrocatalysts in N2-saturated 0.05 M H2SO4 solutions. (C) Current–time chronoamperometric response of Ru@IrOx and RuIrOx electrocatalysts at 1.55 V. Reproduced with permission from ref. 68. Copyright 2019, Elsevier. (D) Preparation method of Ru/RuS2 heterostructure. (E and F) HRTEM image and STEM image of Ru/RuS2 heterostructure. (G) LSV curves with iR correction of various electrocatalysts. (H) LSV curves before and after 3000 cycles and inserted i–t chronoamperometric response of Ru/RuS2. (I) Charge density difference of two-dimensional slice on RuS2–Ru model. (J) Gibbs free energy diagrams of Ru, RuS2, Ru–RuS2 and RuS2–Ru models for OER. Reproduced with permission from ref. 82. Copyright 2021, Wiley-VCH. | ||
Diverse active materials might be beneficial to different stepwise reactions, while rational design of heterostructures enables these separate reactions to take place on diverse sites at close quarters, accordingly accelerating the rates of overall OER reaction.130 Mu et al. reported that a thin Ru/RuS2 nanosheet heterostructure can be generated using an isochronous reduction–sulphuration strategy (Fig. 6D–F).82 The as-prepared Ru/RuS2 heterostructure has outstanding catalytic performance for the OER in acidic solution, by exhibiting a very low overpotential (201 mV) and excellent long-term stability (no obvious activity decline after 3000 CV cycles) (Fig. 6G and H). DFT calculations indicate that charge redistribution over many interfaces improves the electronic state configuration of heterostructured Ru/RuS2 (Fig. 6I), leading to formation of surface electron-deficient Ru sites for optimized adsorption of oxygen intermediates (Fig. 6J) that causes a decrease in the thermodynamic energy barriers, thus enhancing OER performance.
3.5 Design of Ru–C composites
Ru catalysts can also be supported on various carbon substrates in the form of Ru–C composites, which have been widely reported to be highly active and stabile catalysts.131–136 The carbon supports used for this purpose, including graphene, carbon nanosheet and porous carbon, have many advantageous features such as super conductivity, high specific area and strong metal–carbon interactions. However, because carbon is extremely unstable during the OER, little effort has been devoted to designing carbon supported high performance Ru-based OER catalysts.Very recently, Zou et al. reported the synthesis of Ru nanoclusters with particle size of 2–3 nm supported on boron and nitrogen co-doped carbon nanotubes (Ru@B,N-CNTs) by a mild solution blending and post calcination method.137 The as-obtained Ru@B,N-CNTs diplays a low overpotential of 315 mV, outperforming the activities of B,N-CNTs (467 mV), Ru@B-CNTs (378 mV), Ru@N-CNT (409 mV) and commercial RuO2 (390 mV). Moreover, Ru@B,N-CNTs also exhibits a high durability by retaining its activity even after 36 h chronopotentiometry test at 10 mA cm−2 in base media, which is ascribed to stability enhancement caused by the carbon support through synergistic modulation of the electronic structure via the N and B doping.
4. Ru-based compounds
4.1 Ru oxides
RuO2 have been extensively regarded as an effective catalyst for OER.138–140 However, both its oxidation at high potentials and release of lattice oxygen during the OER cause its deactivation or dissolution to occur readily.33 Therefore, rational designs of RuO2-based materials that have appropriate morphologies/compositions and electronic structures might enable circumvention of these problems.In one approach, Li et al. prepared defect-rich ultra-thin RuO2 nanosheets via a molten salt method involving directly mixing molten NaNO3 and RuCl3 at room temperature (Fig. 7A and B).70 As OER catalysts, the RuO2 nanosheets operate at a low overpotential of 199 mV and exhibit 14.9 and 80.6 times improved specific and mass activity, respectively in comparison with those of commercial RuO2. Ru vacancies in the RuO2 nanosheets have been shown to play a valuable role by markedly weakening the binding energy of O* with respect to that of OOH* (Fig. 7C), which results in a decrease in energy consumption in the conversion of O* to OOH*, and thereby improving the OER activity. Because of their high structural integrity, the RuO2 nanosheets display superior stability, undergoing only a slightly increase in overpotential at 10 mA cm−2 current density after a 6 h chronopotentiometry test.
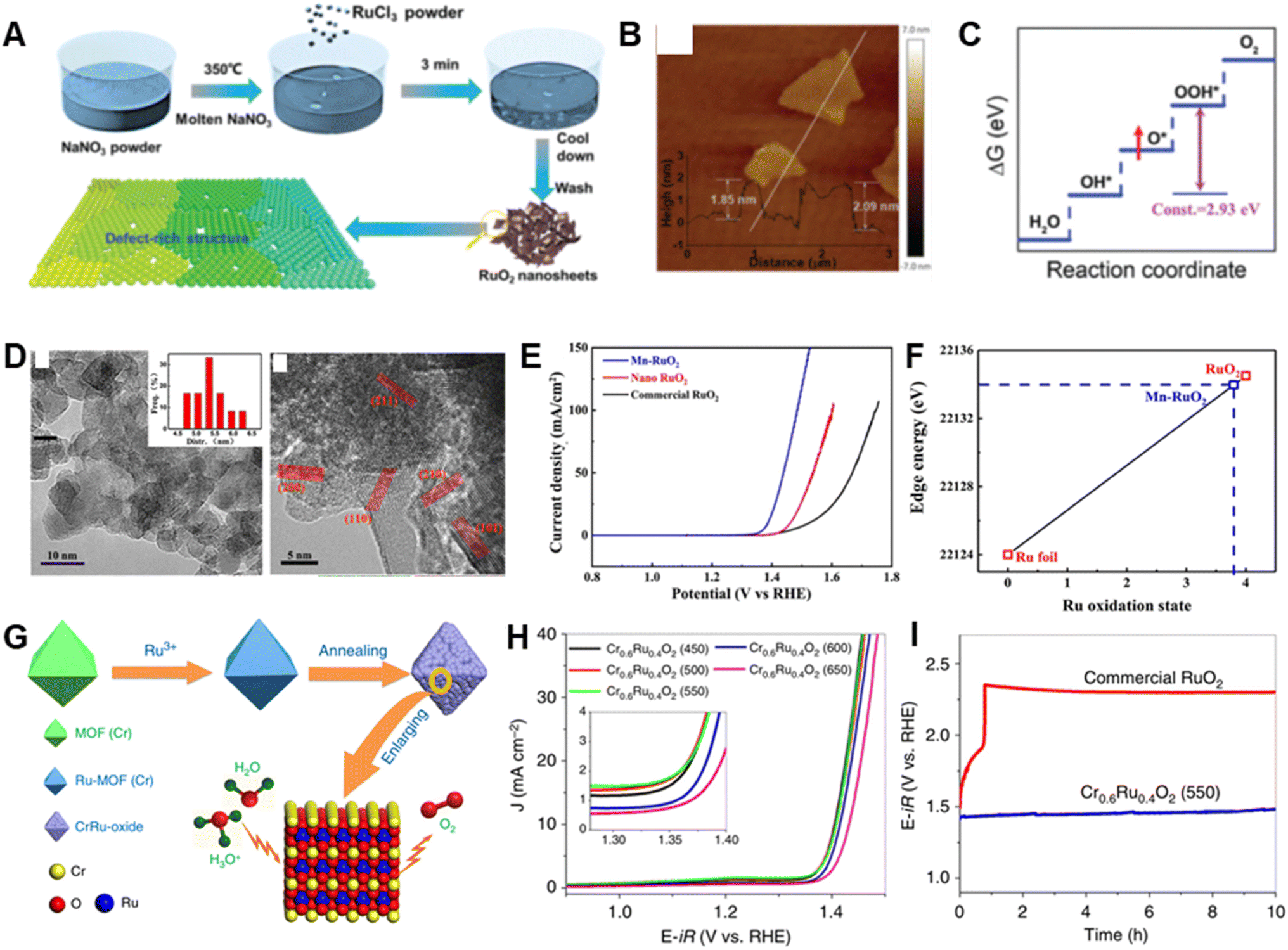 | ||
| Fig. 7 (A) Schematic illustration for the synthesis procedure of the RuO2 nanosheets. (B) AFM image of RuO2 nanosheets; the inset of (e) shows the thickness of RuO2 nanosheets. (C) Schematic showing the origin of OER activity improvement upon introducing a Ru vacancy. Reproduced with permission from ref. 70. Copyright 2020, Royal Society of Chemistry. (D) HRTEM images of Mn–RuO2 and nanocrystal size distribution (inset). (E) OER polarization curves of Mn–RuO2, nano RuO2 and commercial RuO2. (F) Plot of Ru oxidation state and absorption energy. Reproduced with permission from ref. 73. Copyright 2019, The American Chemical Society. (G) Schematic illustration of the preparation of Cr0.6Ru0.4O2 electrocatalysts for the OER in acid media. (H) Electrocatalytic OER activities of Cr0.6Ru0.4O2 nanoparticles annealed at temperature from 450 to 650 °C. (I) Chronopotentiometry performance under constant current density of 10 mA cm−2 up to 10 h. Reproduced with permission from ref. 77. Copyright 2019, Springer Nature. | ||
Doping with heteroatoms has proven to be an effective strategy to alter the intrinsic electrochemical performance of RuO2 through modification of surface-active species, the coordination environment and electronic structure.140–143 For example, Chen et al. showed that Mn-doped RuO2 nanoparticles have a 158 mV overpotential and display a negligible activity decline after long-time operation in an acid media (Fig. 7D and E).73 The Mn ion dopant in RuO2 causes a change in the d-band center of active sites on the RuO2(101) lattice and a decrease in the surface-adsorbate states, which results in a lower free energy change for the rate-determining step, and improved OER activity. This study identified two factors that are responsible for the high stability of this OER catalyst, one being protection against dissolution of Mn which has a protective effect on Ru. The other factor is the relatively lower valence of Ru (+3.8) created upon Mn incorporation (Fig. 7F), which counters the normal tendency of Ru-based oxides with ultrahigh valence states to easily dissolve during the OER process.
In addition, the electrocatalytic behavior of RuO2 can be readily regulated by generating mixed metal oxides.75,90,144 Chen et al. described a low Ru-content binary Cr–Ru oxide material derived from MIL-101 (Cr) (Fig. 7G), which shows a very low overpotential of 178 mV and maintains activity after the 10 h chronopotentiometry test at 10 mA cm−2 in acidic media (Fig. 7H and I).77 Ru is pivotal in inducing the generation of active rutile-structured CrO2 based on the observation that direct pyrolysis of MIL-101(Cr) precursor results in formation of only inactive Cr2O3. The higher activity Cr–Ru oxide is related to its modified electronic structure, which decreases the energy barrier for the generation of *OOH, and its improved stability results from the lower occupation of RuO2 at the Fermi level by incorporation of Cr.
4.2 Ru chalcogenides
Some ruthenium chalcogenides (RuSx or RuTex) have been explored as possible OER catalysts.82 For example, a low-temperature sulfuration method was used by Wang et al. to prepare pyrite-type RuS2 nanoparticles (Fig. 8A).81 The rational control of both disorder and defects in RuS2 was achieved by altering the sulfuration temperature (Fig. 8B–D). Low-crystalline RuS2, possessing optimized disorder and defects, is produced at low sulfuration temperature. This material exhibits higher OER activity with a 282 mV overpotential in comparison with those of various Ru-based and pyrite-type catalysts. It was found that more active sites are generated in conjunction with an increasing degree of disorder, while increasing the defects results in the improvement of intrinsic activity by upshifting the d-band center of Ru, thereby optimizing adsorption free energies toward oxygen intermediates. Moreover, during the OER process, a surface RuS2–RuOx core–shell structure with facilitated charge transfer is formed, which is more likely to be the actual active sites for the outstanding OER activity.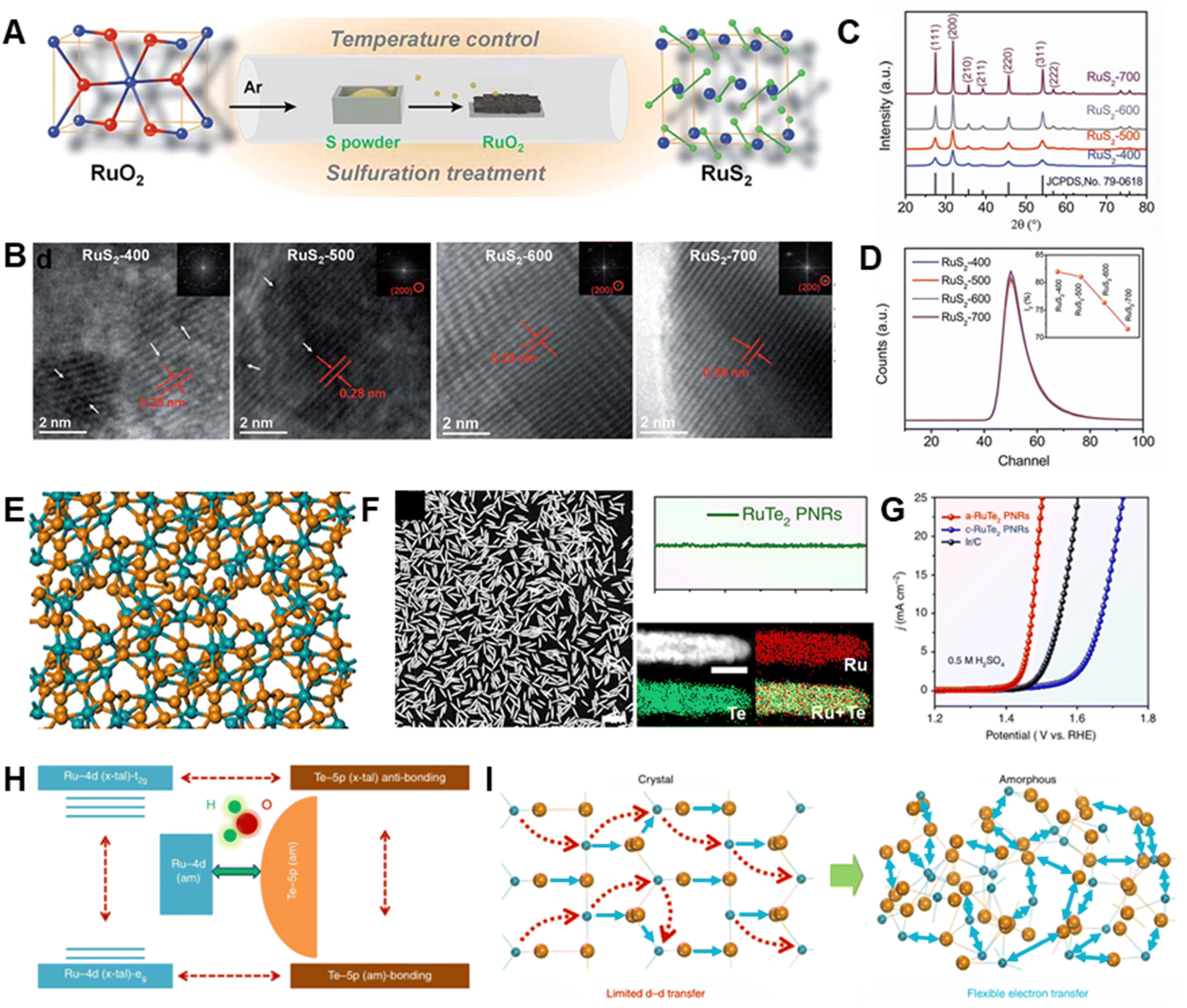 | ||
| Fig. 8 (A) Schematic illustration of the RuS2 nanoparticle synthesis. (B) HRTEM images, (C) XRD patterns and (D) of various RuS2 nanoparticles. Reproduced with permission from ref. 81. Copyright 2019, Royal Society of Chemistry. (E) Local atomic configurations of amorphous RuTe2. (F) Morphology and composition profile analysis of amorphous RuTe2. (G) OER polarization curves of amorphous RuTe2 porous nanorods, crystalline RuTe2 porous nanorods and commercial Ir/C in 0.5 M H2SO4. (H) Schematic diagram illustrating the amorphous RuTe2 enhancement of OER performance via highly efficient site-independent electron-transfer. (I) Schematic diagram of the electronic activity enhancement in amorphous structure. Reproduced with permission from ref. 80. Copyright 2019, Springer Nature. | ||
Recently, Huang et al. developed a robust electrocatalyst based on amorphous RuTe2 porous nanorods with bullet-like profile (a-RuTe2 PNRs; Fig. 8E and F), which has a low overpotential of 245 mV for the OER, which is far lower than those of crystalline RuTe2 porous nanorods and the benchmark electrocatalyst Ir/C (Fig. 8G).80 In this work, the local distortion-strain effect in a-RuTe2 PNRs adjusts Te–pπ coupling and increases intra- and inter-orbital electron-transfer between Ru active sites (Fig. 8H and I), thus promoting catalytic activity. A two-electrode system for acidic overall water splitting comprised of a-RuTe2 PNRs as both the cathode and anode material displays enhanced stability in comparison with that of commercial Ir/C‖Pt/C. During a 24 h chronopotentiometry test, Ir/C and Pt/C severely aggregate while the porous structure and bullet-like morphology of a-RuTe2 PNRs are largely maintained even in 5.0 M H2SO4 at 60 °C for 1 h because of their excellent chemical stability. It is noted that the presence of defects in amorphous structure can be filled with oxygen atoms to generate RuOxHy species during the OER process, which are beneficial to accelerate the reaction process. Although the examples described above demonstrate the huge potential of Ru chalcogenides in accelerating the OER, these materials are still rare and remain relatively unstudied.
4.3 Pyrochlore ruthenates
Because of their low Ru content and superior performance in acidic OER, pyrochlore ruthenates with the general formula A2Ru2O7 have recently become promising alternatives to well-known RuOx and IrOx catalysts.145–147 Moreover, sectional substitution of the A site in these materials by other elements offers many possibilities to adjust their catalytic performance.Specifically, Y2Ru2O7 is the most studied pyrochlore ruthenate for the OER, particularly in the acidic media.84,148,149 For example, Yang et al. reported a high-performance Y2Ru2O7−δ electrocatalyst that exhibits a low overpotential of 190 mV and superior stability in 0.1 M perchloric acid solution.84 Compared with commercial RuO2, this pyrochlore has a lower valence state and a lower band center energy for overlap between Ru 4d and O 2p orbitals. These properties are responsible for the high activity of Y2Ru2O7 that has an overpotential of ca. 190 mV and high stability with negligible change of OER activity after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 voltage sweep cycles. The presence of Y cations plays a beneficial role in enhancing the stability of Y2Ru2O7−δ under acidic conditions, by stabilizing the unstable ruthenium oxide through formation of a pyrochlore structure.
000 voltage sweep cycles. The presence of Y cations plays a beneficial role in enhancing the stability of Y2Ru2O7−δ under acidic conditions, by stabilizing the unstable ruthenium oxide through formation of a pyrochlore structure.
Furthermore, Müller et al. demonstrated that substitution of the A site in Y1.8M0.2Ru2O7−δ (M = Fe and Cu) alters the surface oxygen vacancy concentration by changing the O 2p band center, thereby tuning OER activity (Fig. 9A and B).85 Compared with commercial RuO2, Y1.8M0.2Ru2O7−δ has a much higher OER activity, which can be ascribed to the existence of weaker M–O bond along with coupling between the M d states and O 2p states. Y1.8M0.2Ru2O7−δ also has excellent stability under galvanostatic OER conditions at 1 mA cm−2 for 6 h. In spite of some losses of the A-site cation from the oxide, the pyrochlore crystal structure of Y1.8M0.2Ru2O7−δ remains unchanged during a stability test, suggesting that cation loss only occurs at or near the surface layers. In this respect, an in-depth study is needed to assess the possibility that reconstruction of the surface structure of Y1.8M0.2Ru2O7−δ alters catalytic ability.
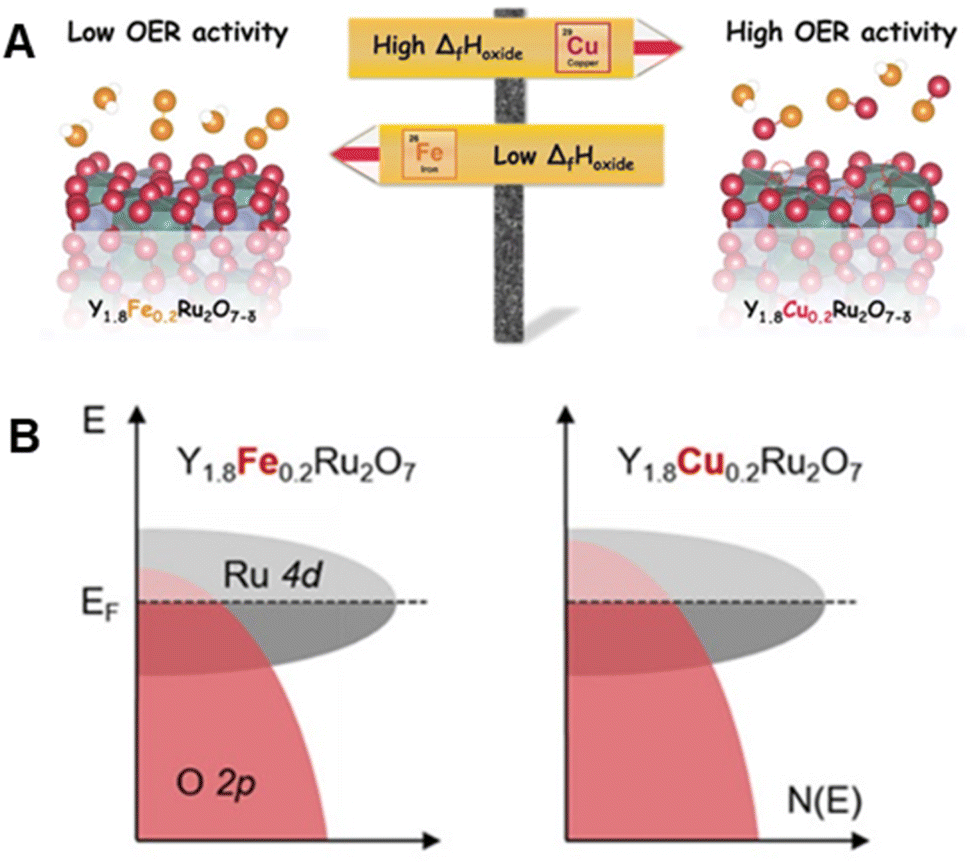 | ||
| Fig. 9 (A) Schematic illustration of the composition–performance relationship between Y1.8Fe0.2Ru2O7−δ and Y1.8Cu0.2Ru2O7−δ. (B) Schematic rigid band diagram for Y1.8Fe0.2Ru2O7−δ and Y1.8Cu0.2Ru2O7−δ illustrating an upshift of the O 2p band center. Reproduced with permission from ref. 85. Copyright 2020, The American Chemical Society. | ||
A possibility exists that interactions of non-completely filled orbitals of rare earth (RE) elements with filled orbitals on Ru that could modulate the electronic structure of Ru sites and promote altered electrocatalytic activities.150,151 This idea led to a study by Sun et al. of pyrochlore rare-earth ruthenates (RE2Ru2O7) containing a variety of rare-earth elements (Nd, Sm, Gd, Er, and Yb).87 Because of its weakening effect on Ru–O bonding, an increase in the radius of RE elements leads to an increase in the content of defective oxygens from 29.5% to 49.7% in the RE2Ru2O7 group. An increase in the defective oxygen content increases the gap between the 4d band center and the Fermi level of Ru, which weakens adsorption of oxygen intermediates and thereby enhances the OER performance. Among the RE2Ru2O7 catalysts investigated by Sun et al., Nd2Ru2O7 exhibits the lowest OER overpotential of 210 mV, a value that is 30 times higher than that of the commercial RuO2 catalyst. The results of a stability test, using 1 mA cm−2 in 0.1 M HClO4, showed that Nd2Ru2O7 maintains its performance over a 8 h period while the activity of RuO2 decreases in less than 1 h, demonstrating that RE atoms stabilize Ru active sites by preventing oxidation to form higher valence states that undergo ready dissolution.
5. Conclusion and perspective
Extensive studies of Ru-based materials have led to the development of highly active and stable catalysts for the OER, which should play a vital role in designing commercially viable water splitting. This review provides an overview of the latest developments made in studies of these electrocatalysts. The discussion, divided into two broad families of Ru-based materials including Ru metals and Ru compounds, gives special emphasis to the design of activity/stability-enhancing strategies by focusing on a limited number of recently reported systems. Notably, most of the strategies included in the review have the potential of being applicable to the development of other high-performance OER catalysts.Despite the significant progress that has been made in the field of Ru-based OER catalysts, several unresolved issues remain, including insufficient categories of compositions/morphologies/structures, the lack of in situ monitoring of the catalyst surface variations, and unsatisfied long-term stability in harsh conditions for industrial applications. To address these issues, the following research directions are recommended.
(1) Exploring new synthetic methodologies that can be employed to create Ru-based materials with greatly enriched numbers of active sites found in other noble metal containing materials. For instance, Pt- and Pd-based nanocrystals with high-indexed facets, hierarchically porous structures and chemically stable intermetallic compounds can be readily generated,152–155 while related Ru-based materials are difficult to prepare. The limited types of morphologies/structures/compositions that have been created thus far have greatly retarded the development of more efficient OER catalysts. Moreover, considering Ru is a noble metal, the development of efficient strategies for synthesis of Ru-based catalysts with low Ru loading to save the use of expensive noble metals is necessary. For example, construction of non-noble metal@Ru core–shell nanostructures with a thin Ru shell layer and doping of Ru atoms into transition metal oxides can improve the utilization rate of Ru, enhanced OER performance as well as display a higher cost-performance.156–158
(2) Developing theoretical studies to enable predictions of optimized structures/compositions of Ru catalysts with greatly enhanced activities/stabilities. Current theoretical studies generally use simplified models that questionably mimic the complexity of actual systems and processes. Thus, results arising from these calculations provide limited information to guide the design of new Ru-based catalysts. To investigate the physicochemical characteristics of OER catalysts in detail, models of reconstructed structures rather than pristine structures should be created. More attention should be paid to the partially reconstructed catalysts, which consist of the reconstructed species and pristine materials. Moreover, regarding the determination of free energy changes for different steps in the OER, the influence of other factors such as pH, surface thickness and external electric field should be considered.
(3) Conducting in situ/operando characterization techniques for investigating the true nature of catalytic sites and understanding the structure/morphology/composition evolution during the OER. Although remarkable improvements have occurred in understanding the degradation and dissolution of state-of-art OER catalysts, particularly in fundamental studies on model electrodes, the recently developed operando/in situ systems display qualitatively different results on dissolution mechanisms compared to those arising from experimental studies. Moreover, the current techniques cannot provide sufficient information to realize the real-world dynamic reconstruction of catalysts. Accordingly, to fully understand changes taking place in the physicochemical properties of catalysts that relate to activity and durability, advanced in situ/operando characterization techniques with high temporal, spatial and energy resolution are needed.
(4) Evaluating OER performance in real water electrolyzer systems instead of conventional three-electrode cells in order to determine bottlenecks that arise in commercial scale water splitting systems. The complexity of real water electrolyzers hampers full elucidation of the cause of catalyst deactivation. Thus, a knowledge bridge should be built between systems employed for fundamental research and practical applications. In addition, more research should be carried out to provide information needed to design high-performance electrocatalysts in commercial water electrolyzers. Moreover, compared with other kinds of catalysts, such as transition-metal-based materials,159,160 there are few studies dealing with the Ru-based OER catalysts in seawater, with results which are still far from being satisfactory. The OER process in seawater is mainly constricted by the parallel chlorine evolution reaction (ClER) and electrode blocking and corrosion by Cl−.161,162 Therefore, the development of highly OER selective, active and stable Ru-based catalysts for seawater splitting is highly required.
There is little doubt that investigations of high-performance Ru-based OER catalysts for electrochemical water splitting will continue to move forward and that commercial level systems will become available in the not too distant.
Conflicts of interest
There are no declared conflicts.Acknowledgements
Preparation of this review was supported financially by National Key Research and Development Program of China (2022YFB3805600, 2022YFB3805604), National Natural Science Foundation of China (52201286, 51861135313), Sino-German Center COVID-19 Related Bilateral Collaborative project (C-0046), FRFCU (2021qntd13), National 111 project (B20002), Program for Changjiang Scholars and Innovative Research Team in University (IRT_15R52), Guangdong Basic and Applied Basic Research Foundation (2019A1515110436, 2021A1515111131, 2022A1515011905, 2022A1515010137), Guangzhou Science and Technology Project (202102020463), Guangdong Province International Scientific and Technological Cooperation Projects (2020A0505100036), Shenzhen Science and Technology Program (JCYJ20210324142010029, GJHZ20210705143204014), Brazilian agencies CNPq (302173/2016-1), São State Research Foundation FAPESP (2015/26308-7) and South African Research Chair (DSI/NRF/Wits SARChI Chair; UID Number: 132739).References
- D. Welsby, J. Price, S. Pye and P. Ekins, Nature, 2021, 597, 230–234 CrossRef CAS PubMed.
- L. Shen, J. Ying, K. I. Ozoemena, C. Janiak and X.-Y. Yang, Adv. Funct. Mater., 2022, 32, 2110851 CrossRef CAS.
- X. Shi, Y. Qian and S. Yang, ACS Sustainable Chem. Eng., 2020, 8, 7097–7110 CrossRef CAS.
- M. R. Shaner, S. J. Davis, N. S. Lewis and K. Caldeira, Energy Environ. Sci., 2018, 11, 914–925 RSC.
- J. Ying, J. Li, G. Jiang, Z. P. Cano, Z. Ma, C. Zhong, D. Su and Z. Chen, Appl. Catal., B, 2018, 225, 496–503 CrossRef CAS.
- Y. Zhu, H. A. Tahini, Z. Hu, J. Dai, Y. Chen, H. Sun, W. Zhou, M. Liu, S. C. Smith, H. Wang and Z. Shao, Nat. Commun., 2019, 10, 1494 CrossRef PubMed.
- H.-Z. Yu, Y. Wang, J. Ying, S.-M. Wu, Y. Lu, J. Hu, J.-S. Hu, L. Shen, Y.-X. Xiao, W. Geng, G.-G. Chang, C. Janiak, W.-H. Li and X.-Y. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 27641–27647 CrossRef CAS PubMed.
- K. J. Warren, J. T. Tran and A. W. Weimer, Energy Environ. Sci., 2022, 15, 806–821 RSC.
- C. Vogt, M. Monai, G. J. Kramer and B. M. Weckhuysen, Nat. Catal., 2019, 2, 188–197 CrossRef CAS.
- Y.-X. Xiao, J. Ying, G. Tian, Y. Tao, H. Wei, S.-Y. Fan, Z.-H. Sun, W.-J. Zou, J. Hu, G.-G. Chang, W. Li, X.-Y. Yang and C. Janiak, Appl. Catal., B, 2019, 259, 118080 CrossRef CAS.
- Y.-X. Xiao, J. Ying, J.-B. Chen, Y. Dong, X. Yang, G. Tian, J. Wu, C. Janiak, K. I. Ozoemena and X.-Y. Yang, Chem. Mater., 2022, 34, 3705–3714 CrossRef CAS.
- L. Tian, X. Zhai, X. Wang, J. Li and Z. Li, J. Mater. Chem. A, 2020, 8, 14400–14414 RSC.
- Y. Yan, H. Cheng, Z. Qu, R. Yu, F. Liu, Q. Ma, S. Zhao, H. Hu, Y. Cheng, C. Yang, Z. Li, X. Wang, S. Hao, Y. Chen and M. Liu, J. Mater. Chem. A, 2021, 9, 19489–19507 RSC.
- F.-Y. Chen, Z.-Y. Wu, Z. Adler and H. Wang, Joule, 2021, 5, 1704–1731 CrossRef CAS.
- H. Wang, J. Ying, Y.-X. Xiao, J.-B. Chen, J.-H. Li, Z.-Z. He, H.-J. Yang and X.-Y. Yang, Electrochem. Commun., 2022, 134, 107177 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed.
- R. R. Rao, M. J. Kolb, N. B. Halck, A. F. Pedersen, A. Mehta, H. You, K. A. Stoerzinger, Z. Feng, H. A. Hansen, H. Zhou, L. Giordano, J. Rossmeisl, T. Vegge, I. Chorkendorff, I. E. L. Stephens and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2626–2637 RSC.
- Y. Hu, G. Luo, L. Wang, X. Liu, Y. Qu, Y. Zhou, F. Zhou, Z. Li, Y. Li, T. Yao, C. Xiong, B. Yang, Z. Yu and Y. Wu, Adv. Energy Mater., 2021, 11, 2002816 CrossRef CAS.
- S.-C. Sun, H. Jiang, Z.-Y. Chen, Q. Chen, M.-Y. Ma, L. Zhen, B. Song and C.-Y. Xu, Angew. Chem., Int. Ed., 2022, 61, e202202519 CAS.
- K. Li, Y. Li, Y. Wang, J. Ge, C. Liu and W. Xing, Energy Environ. Sci., 2018, 11, 1232–1239 RSC.
- S.-Y. Bae, J. Mahmood, I.-Y. Jeon and J.-B. Baek, Nanoscale Horiz., 2020, 5, 43–56 RSC.
- D. Li, Y. Liu, Z. Liu, J. Yang, C. Hu and L. Feng, J. Mater. Chem. A, 2021, 9, 15019–15026 RSC.
- J. Zhang, Y. Zhao, C. Chen, Y. C. Huang, C. L. Dong, C. J. Chen, R. S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed.
- L. Bai, Z. Duan, X. Wen, R. Si, Q. Zhang and J. Guan, ACS Catal., 2019, 9, 9897–9904 CrossRef CAS.
- Z. Yang, M. Xiang, Y. Zhu, J. Hui, Y. Jiang, S. Dong, C. Yu, J. Ou and H. Qin, Chem. Eng. J., 2021, 426, 131347 CrossRef CAS.
- B. K. Miller and P. A. Crozier, ACS Catal., 2021, 11, 1456–1463 CrossRef CAS.
- C. Wang, S. Liu, D. Wang and Q. Chen, J. Mater. Chem. A, 2018, 6, 11037–11043 RSC.
- O. Khalid, T. Weber, G. Drazic, I. Djerdj and H. Over, J. Phys. Chem. C, 2020, 124, 18670–18683 CrossRef CAS.
- Y. Yao, S. Hu, W. Chen, Z.-Q. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C.-R. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W.-X. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef CAS.
- L. Zhang, H. Jang, H. Liu, M. G. Kim, D. Yang, S. Liu, X. Liu and J. Cho, Angew. Chem., Int. Ed., 2021, 60, 18821–18829 CrossRef CAS PubMed.
- T. Kwon, H. Yang, M. Jun, T. Kim, J. Joo, J. Kim, H. Baik, J. Y. Kim and K. Lee, J. Mater. Chem. A, 2021, 9, 14352–14362 RSC.
- Y. Li, Y. Sun, Y. Qin, W. Zhang, L. Wang, M. Luo, H. Yang and S. Guo, Adv. Energy Mater., 2020, 10, 1903120 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- H. Over, ACS Catal., 2021, 11, 8848–8871 CrossRef CAS.
- J. Yu, Q. He, G. Yang, W. Zhou, Z. Shao and M. Ni, ACS Catal., 2019, 9, 9973–10011 CrossRef CAS.
- H. Sun and W. Jung, J. Mater. Chem. A, 2021, 9, 15506–15521 RSC.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- Y. Guo, T. Park, J. W. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Adv. Mater., 2019, 31, e1807134 CrossRef PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Y. Pi, Q. Shao, P. Wang, J. Guo and X. Huang, Adv. Funct. Mater., 2017, 27, 1700886 CrossRef.
- C. Huang, J. Zhou, D. Duan, Q. Zhou, J. Wang, B. Peng, L. Yu and Y. Yu, Chin. J. Catal., 2022, 43, 2091–2110 CrossRef CAS.
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647–4671 RSC.
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen, J. P. M. Veder, D. Guan, R. O`Hayre, M. Li, G. Wang, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 2002 CrossRef CAS PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- C. F. Dickens, C. Kirk and J. K. Nørskov, J. Phys. Chem. C, 2019, 123, 18960–18977 CrossRef CAS.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- C. Rong, X. Shen, Y. Wang, L. Thomsen, T. Zhao, Y. Li, X. Lu, R. Amal and C. Zhao, Adv. Mater., 2022, 34, 2110103 CrossRef CAS PubMed.
- L. Yan and B. Zhang, J. Mater. Chem. A, 2021, 9, 20758–20765 RSC.
- D. Ren, J. Ying, M. Xiao, Y.-P. Deng, J. Ou, J. Zhu, G. Liu, Y. Pei, S. Li, A. M. Jauhar, H. Jin, S. Wang, D. Su, A. Yu and Z. Chen, Adv. Funct. Mater., 2020, 30, 1908167 CrossRef CAS.
- J. Chen, J. Ying, Y. Xiao, Y. Dong, K. I. Ozoemena, S. Lenaerts and X. Yang, Sci. China Mater., 2022, 65, 2685–2693 CrossRef CAS PubMed.
- E. A. Paoli, F. Masini, R. Frydendal, D. Deiana, C. Schlaup, M. Malizia, T. W. Hansen, S. Horch, I. E. L. Stephens and I. Chorkendorff, Chem. Sci., 2015, 6, 190–196 RSC.
- R. Kötz, H. J. Lewerenz and S. Stucki, J. Electrochem. Soc., 1983, 130, 825–829 CrossRef.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- N. Hodnik, P. Jovanovič, A. Pavlišič, B. Jozinović, M. Zorko, M. Bele, V. S. Šelih, M. Šala, S. Hočevar and M. Gaberšček, J. Phys. Chem. C, 2015, 119, 10140–10147 CrossRef CAS.
- O. Kasian, S. Geiger, P. Stock, G. Polymeros, B. Breitbach, A. Savan, A. Ludwig, S. Cherevko and K. J. J. Mayrhofer, J. Electrochem. Soc., 2016, 163, F3099–F3104 CrossRef CAS.
- S. Cherevko, A. R. Zeradjanin, A. A. Topalov, N. Kulyk, I. Katsounaros and K. J. J. Mayrhofer, ChemCatChem, 2014, 6, 2219–2223 CrossRef CAS.
- R. Kötz, S. Stucki, D. Scherson and D. M. Kolb, J. Electroanal. Chem. Interfacial Electrochem., 1984, 172, 211–219 CrossRef.
- J.-Q. Wang, C. Xi, M. Wang, L. Shang, J. Mao, C.-K. Dong, H. Liu, S. A. Kulinich and X.-W. Du, ACS Catal., 2020, 10, 12575–12581 CrossRef CAS.
- A. R. Poerwoprajitno, L. Gloag, T. M. Benedetti, S. Cheong, J. Watt, D. L. Huber, J. J. Gooding and R. D. Tilley, Small, 2019, 15, e1804577 CrossRef PubMed.
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhang, Y. Kuang, Y. Li, Q. Ma, Z. Feng, W. Liu and X. Sun, Nat. Commun., 2019, 10, 1711 CrossRef PubMed.
- L. Cao, Q. Luo, J. Chen, L. Wang, Y. Lin, H. Wang, X. Liu, X. Shen, W. Zhang, W. Liu, Z. Qi, Z. Jiang, J. Yang and T. Yao, Nat. Commun., 2019, 10, 4849 CrossRef PubMed.
- Z. X. Cai, H. Goou, Y. Ito, T. Tokunaga, M. Miyauchi, H. Abe and T. Fujita, Chem. Sci., 2021, 12, 11306–11315 RSC.
- Q. Yao, B. Huang, N. Zhang, M. Sun, Q. Shao and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 13983–13988 CrossRef CAS PubMed.
- J. Shan, C. Guo, Y. Zhu, S. Chen, L. Song, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Chem, 2019, 5, 445–459 CAS.
- J. Yang, Y. Ji, Q. Shao, N. Zhang, Y. Li and X. Huang, Adv. Funct. Mater., 2018, 28, 1803722 CrossRef.
- Z. L. Zhao, Q. Wang, X. Huang, Q. Feng, S. Gu, Z. Zhang, H. Xu, L. Zeng, M. Gu and H. Li, Energy Environ. Sci., 2020, 13, 5143–5151 RSC.
- Y. Tian, S. Wang, E. Velasco, Y. Yang, L. Cao, L. Zhang, X. Li, Y. Lin, Q. Zhang and L. Chen, iScience, 2020, 23, 100756 CrossRef CAS PubMed.
- Y. Xue, J. Fang, X. Wang, Z. Xu, Y. Zhang, Q. Lv, M. Liu, W. Zhu and Z. Zhuang, Adv. Funct. Mater., 2021, 31, 2101405 CrossRef CAS.
- S. Chen, H. Huang, P. Jiang, K. Yang, J. Diao, S. Gong, S. Liu, M. Huang, H. Wang and Q. Chen, ACS Catal., 2019, 10, 1152–1160 CrossRef.
- L. Zhang, L. Wang, Y. Wen, F. Ni, B. Zhang and H. Peng, Adv. Mater., 2020, 32, 2002297 CrossRef CAS PubMed.
- C. Wang and L. Qi, Angew. Chem., Int. Ed., 2020, 59, 17219–17224 CrossRef CAS PubMed.
- Y. Wen, P. Chen, L. Wang, S. Li, Z. Wang, J. Abed, X. Mao, Y. Min, C. T. Dinh, P. Luna, R. Huang, L. Zhang, L. Wang, L. Wang, R. J. Nielsen, H. Li, T. Zhuang, C. Ke, O. Voznyy, Y. Hu, Y. Li, W. A. Goddard III, B. Zhang, H. Peng and E. H. Sargent, J. Am. Chem. Soc., 2021, 143, 6482–6490 CrossRef CAS PubMed.
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Nat. Commun., 2019, 10, 162 CrossRef PubMed.
- S. Hao, M. Liu, J. Pan, X. Liu, X. Tan, N. Xu, Y. He, L. Lei and X. Zhang, Nat. Commun., 2020, 11, 5368 CrossRef CAS PubMed.
- B. Tang, X. Yang, Z. Kang and L. Feng, Appl. Catal., B, 2020, 278, 119281 CrossRef CAS.
- J. Wang, L. Han, B. Huang, Q. Shao, H. L. Xin and X. Huang, Nat. Commun., 2019, 10, 5692 CrossRef CAS PubMed.
- Y. Zhu, H. A. Tahini, Y. Wang, Q. Lin, Y. Liang, C. M. Doherty, Y. Liu, X. Li, J. Lu, S. C. Smith, C. Selomulya, X. Zhang, Z. Shao and H. Wang, J. Mater. Chem. A, 2019, 7, 14222–14232 RSC.
- J. Zhu, Y. Guo, F. Liu, H. Xu, L. Gong, W. Shi, D. Chen, P. Wang, Y. Yang, C. Zhang, J. Wu, J. Luo and S. Mu, Angew. Chem., Int. Ed., 2021, 60, 12328–12334 CrossRef CAS PubMed.
- P. Gayen, S. Saha and V. Ramani, ACS Appl. Energy Mater., 2020, 3, 3978–3983 CrossRef CAS.
- J. Kim, P. C. Shih, K. C. Tsao, Y. T. Pan, X. Yin, C. J. Sun and H. Yang, J. Am. Chem. Soc., 2017, 139, 12076–12083 CrossRef CAS PubMed.
- D. A. Kuznetsov, M. A. Naeem, P. V. Kumar, P. M. Abdala, A. Fedorov and C. R. Müller, J. Am. Chem. Soc., 2020, 142, 7883–7888 CrossRef CAS PubMed.
- N. Zhang, C. Wang, J. Chen, C. Hu, J. Ma, X. Deng, B. Qiu, L. Cai, Y. Xiong and Y. Chai, ACS Nano, 2021, 15, 8537–8548 CrossRef CAS PubMed.
- H. Liu, Z. Wang, M. Li, X. Zhao, X. Duan, S. Wang, G. Tan, Y. Kuang and X. Sun, Sci. China Mater., 2021, 64, 1653–1661 CrossRef CAS.
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC.
- Q. Yao, B. Huang, Y. Xu, L. Li, Q. Shao and X. Huang, Nano Energy, 2021, 84, 105909 CrossRef CAS.
- Y. Tuo, W. Liu, C. Chen, Q. Lu, Y. Zhou and J. Zhang, Chem.–Asian J., 2021, 16, 2511–2519 CrossRef CAS PubMed.
- Z. Xia and S. Guo, Chem. Soc. Rev., 2019, 48, 3265–3278 RSC.
- X. Yang, Y. Wang, X. Tong and N. Yang, Adv. Energy Mater., 2022, 12, 2102261 CrossRef CAS.
- X. Wang, D. Luo, J. Wang, Z. Sun, G. Cui, Y. Chen, T. Wang, L. Zheng, Y. Zhao, L. Shui, G. Zhou, K. Kempa, Y. Zhang and Z. Chen, Angew. Chem., Int. Ed., 2021, 60, 2371–2378 CrossRef CAS PubMed.
- X. Liu, T. Liu, W. Xiao, W. Wang, Y. Zhang, G. Wang, Z. Luo and J.-C. Liu, Inorg. Chem. Front., 2022, 9, 4272–4280 RSC.
- Z. Hou, Z. Sun, C. Cui, D. Zhu, Y. Yang and T. Zhang, Adv. Funct. Mater., 2022, 32, 2110572 CrossRef CAS.
- Y.-J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS PubMed.
- Y. Dong, J. B. Chen, J. Ying, Y. X. Xiao, G. Tian, M. D. Symes and X. Y. Yang, Chem. Mater., 2022, 34, 8271–8279 CrossRef CAS.
- Y. Ren, Y. Tang, L. Zhang, X. Liu, L. Li, S. Miao, D. Sheng Su, A. Wang, J. Li and T. Zhang, Nat. Commun., 2019, 10, 4500 CrossRef PubMed.
- N. Zhang, X. Zhang, L. Tao, P. Jiang, C. Ye, R. Lin, Z. Huang, A. Li, D. Pang, H. Yan, Y. Wang, P. Xu, S. An, Q. Zhang, L. Liu, S. Du, X. Han, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 6170–6176 CrossRef CAS PubMed.
- X. Ye, C. Yang, X. Pan, J. Ma, Y. Zhang, Y. Ren, X. Liu, L. Li and Y. Huang, J. Am. Chem. Soc., 2020, 142, 19001–19005 CrossRef CAS PubMed.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 4587 CrossRef CAS PubMed.
- J. Jin, X. Han, Y. Fang, Z. Zhang, Y. Li, T. Zhang, A. Han and J. Liu, Adv. Funct. Mater., 2022, 32, 2109218 CrossRef CAS.
- Y. Hu, G. Luo, L. Wang, X. Liu, Y. Qu, Y. Zhou, F. Zhou, Z. Li, Y. Li, T. Yao, C. Xiong, B. Yang, Z. Yu and Y. Wu, Adv. Energy Mater., 2021, 11, 2002816 CrossRef CAS.
- Y. Yao, L. Zhao, J. Dai, J. Wang, C. Fang, G. Zhan, Q. Zheng, W. Hou and L. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208215 CAS.
- S. Wei, A. Li, J. C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W. C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed.
- L. Cao, Q. Luo, J. Chen, L. Wang, Y. Lin, H. Wang, X. Liu, X. Shen, W. Zhang, W. Liu, Z. Qi, Z. Jiang, J. Yang and T. Yao, Nat. Commun., 2019, 10, 4849 CrossRef PubMed.
- H. Yang, L. Shang, Q. Zhang, R. Shi, G. I. N. Waterhouse, L. Gu and T. Zhang, Nat. Commun., 2019, 10, 4585 CrossRef PubMed.
- C. Roy, B. Sebok, S. B. Scott, E. M. Fiordaliso, J. E. Sørensen, A. Bodin, D. B. Trimarco, C. D. Damsgaard, P. C. K. Vesborg, O. Hansen, I. E. L. Stephens, J. Kibsgaard and I. Chorkendorff, Nat. Catal., 2018, 1, 820–829 CrossRef CAS.
- J. Feng, Y. He, Y. Liu, Y. Du and D. Li, Chem. Soc. Rev., 2015, 44, 5291–5319 RSC.
- S. H. Chang, N. Danilovic, K.-C. Chang, R. Subbaraman, A. P. Paulikas, D. D. Fong, M. J. Highland, P. M. Baldo, V. R. Stamenkovic, J. W. Freeland, J. A. Eastman and N. M. Markovic, Nat. Commun., 2014, 5, 4191 CrossRef CAS PubMed.
- C. Roy, R. R. Rao, K. A. Stoerzinger, J. Hwang, J. Rossmeisl, I. Chorkendorff, Y. Shao-Horn and I. E. L. Stephens, ACS Energy Lett., 2018, 3, 2045–2051 CrossRef CAS.
- Y.-X. Xiao, J. Ying, G. Tian, X. Yang, Y.-X. Zhang, J.-B. Chen, Y. Wang, M. D. Symes, K. I. Ozoemena, J. Wu and X.-Y. Yang, Nano Lett., 2021, 21, 7870–7878 CrossRef CAS PubMed.
- Y.-X. Xiao, J. Ying, G. Tian, X.-Q. Zhang, C. Janiak, K. I. Ozoemena and X.-Y. Yang, Chem. Commun., 2021, 57, 986–989 RSC.
- J. Ying, G. Jiang, Z. P. Cano, Z. Ma and Z. Chen, Appl. Catal., B, 2018, 236, 359–367 CrossRef CAS.
- J. Ying, Z.-Y. Hu, X.-Y. Yang, H. Wei, Y.-X. Xiao, C. Janiak, S.-C. Mu, G. Tian, M. Pan, G. Van Tendeloo and B.-L. Su, Chem. Commun., 2016, 52, 8219–8222 RSC.
- H. Wang, Y. Yang, F. J. DiSalvo and H. D. Abruña, ACS Catal., 2020, 10, 4608–4616 CrossRef CAS.
- L. Jin, H. Xu, C. Chen, H. Shang, Y. Wang, C. Wang and Y. Du, ACS Appl. Mater. Interfaces, 2019, 11, 42123–42130 CrossRef CAS PubMed.
- Y. Xing, K. Wang, N. Li, D. Su, W.-T. Wong, B. Huang and S. Guo, Matter, 2020, 2, 1494–1508 CrossRef.
- J. Yang, Q. Shao, B. Huang, M. Sun and X. Huang, iScience, 2019, 11, 492–504 CrossRef CAS PubMed.
- E. P. George, D. Raabe and R. O. Ritchie, Nat. Rev. Mater., 2019, 4, 515–534 CrossRef CAS.
- Y. F. Ye, Q. Wang, J. Lu, C. T. Liu and Y. Yang, Mater. Today, 2016, 19, 349–362 CrossRef CAS.
- Q. Ding, Y. Zhang, X. Chen, X. Fu, D. Chen, S. Chen, L. Gu, F. Wei, H. Bei, Y. Gao, M. Wen, J. Li, Z. Zhang, T. Zhu, R. O. Ritchie and Q. Yu, Nature, 2019, 574, 223–227 CrossRef CAS PubMed.
- G. M. Tomboc, T. Kwon, J. Joo and K. Lee, J. Mater. Chem. A, 2020, 8, 14844–14862 RSC.
- T. Löffler, A. Ludwig, J. Rossmeisl and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 26894–26903 CrossRef PubMed.
- Y. Zhang, D. Wang and S. Wang, Small, 2022, 18, 2104339 CrossRef CAS PubMed.
- H. Li, J. Lai, Z. Li and L. Wang, Adv. Funct. Mater., 2021, 31, 2106715 CrossRef CAS.
- F. Zhang, X. Yu, J. Hu, L. Lei, Y. He and X. Zhang, Chem. Eng. J., 2021, 423, 130231 CrossRef CAS.
- S. Liu, X. Mu, P. Ji, Y. Lv, L. Wang, Q. Zhou, C. Chen and S. Mu, ChemCatChem, 2020, 12, 5149–5155 CrossRef CAS.
- Y. Zhao, X. Zhang, Y. Gao, Z. Chen, Z. Li, T. Ma, Z. Wu, L. Wang and S. Feng, Small, 2022, 18, 2105168 CrossRef CAS PubMed.
- Z.-J. Chen, G.-X. Cao, L.-Y. Gan, H. Dai, N. Xu, M.-J. Zang, H.-B. Dai, H. Wu and P. Wang, ACS Catal., 2018, 8, 8866–8872 CrossRef CAS.
- Q. Wang, M. Ming, S. Niu, Y. Zhang, G. Fan and J.-S. Hu, Adv. Energy Mater., 2018, 8, 1870135 CrossRef.
- J. Wang, Z. Wei, S. Mao, H. Li and Y. Wang, Energy Environ. Sci., 2018, 11, 800–806 RSC.
- Y. Li, F. Chu, Y. Liu, Y. Kong, Y. Tao, Y. Li and Y. Qin, Chem. Commun., 2018, 54, 13076–13079 RSC.
- J. Zhang, P. Liu, G. Wang, P. P. Zhang, X. D. Zhuang, M. W. Chen, I. M. Weidinger and X. L. Feng, J. Mater. Chem. A, 2017, 5, 25314–25318 RSC.
- Z.-L. Wang, K. Sun, J. Henzie, X. Hao, C. Li, T. Takei, Y.-M. Kang and Y. Yamauchi, Angew. Chem., Int. Ed., 2018, 57, 5848–5852 CrossRef CAS PubMed.
- Z. Peng, H. Wang, L. Zhou, Y. Wang, J. Gao, G. Liu, S. A. T. Redfern, X. Feng, S. Lu, B. Li and Z. Liu, J. Mater. Chem. A, 2019, 7, 6676–6685 RSC.
- M. Fan, X. Chen, M. Zhang, L. Cui, X. He and X. Zou, Inorg. Chem. Front., 2022, 9, 968–976 RSC.
- R. Liu, Y. Wang, D. Liu, Y. Zou and S. Wang, Adv. Mater., 2017, 29, 1701546 CrossRef PubMed.
- C. Wang, L. Jin, H. Shang, H. Xu, Y. Shiraishi and Y. Du, Chin. Chem. Lett., 2021, 32, 2108–2116 CrossRef CAS.
- L. Shi, A. Xu and T. Zhao, J. Phys. Chem. C, 2016, 120, 6356–6362 CrossRef CAS.
- J. Wang, H. Yang, F. Li, L. Li, J. Wu, S. Liu, T. Cheng, Y. Xu, Q. Shao and X. Huang, Sci. Adv., 2022, 8, eabl9271 CrossRef CAS PubMed.
- R. Madhu, A. Karmakar, S. Kumaravel, S. S. Sankar, K. Bera, S. Nagappan, H. N. Dhandapani and S. Kundu, ACS Appl. Mater. Interfaces, 2022, 14, 1077–1091 CrossRef CAS PubMed.
- K. Kishor, S. Saha, A. Parashtekar and R. G. S. Pala, J. Electrochem. Soc., 2018, 165, J3276–J3280 CrossRef CAS.
- V. A. Saveleva, L. Wang, W. Luo, S. Zafeiratos, C. Ulhaq-Bouillet, A. S. Gago, K. A. Friedrich and E. R. Savinova, J. Phys. Chem. Lett., 2016, 7, 3240–3245 CrossRef CAS PubMed.
- H. Liu, Z. Zhang, M. Li, Z. Wang, X. Zhang, T. Li, Y. Li, S. Tian, Y. Kuang and X. Sun, Small, 2022, 18, 2202513 CrossRef CAS PubMed.
- M. Kim, H. Ju and J. Kim, Chem. Eng. J., 2019, 358, 11–19 CrossRef CAS.
- J. Park, M. Shirai, G. Y. Jung, S. O. Park, M. Park, J. Ryu, S. K. Kwak and J. Cho, ACS Catal., 2018, 8, 9647–9655 CrossRef CAS.
- K. Sardar, E. Petrucco, C. I. Hiley, J. D. Sharman, P. P. Wells, A. E. Russell, R. J. Kashtiban, J. Sloan and R. I. Walton, Angew. Chem., Int. Ed., 2014, 53, 10960–10964 CrossRef CAS PubMed.
- P. Gayen, S. Saha and V. Ramani, Acc. Chem. Res., 2022, 55, 2191–2200 CrossRef CAS PubMed.
- S. Ji, Y. Qu, T. Wang, Y. Chen, G. Wang, X. Li, J. Dong, Q. Chen, W. Zhang, Z. Zhang, S. Liang, R. Yu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 10651–10657 CrossRef CAS PubMed.
- H. Xu, W. Yu, K. Pan, G. Wang and P. Zhu, Nano Res., 2021, 14, 720–729 CrossRef CAS.
- J. Ying, S. Lenaerts, M. D. Symes and X.-Y. Yang, Adv. Sci., 2022, 9, 2106117 CrossRef PubMed.
- J. Ying, X.-Y. Yang, G. Tian, C. Janiak and B.-L. Su, Nanoscale, 2014, 6, 13370–13382 RSC.
- Y. Dong, J. Ying, Y.-X. Xiao, J.-B. Chen and X.-Y. Yang, Chem.–Asian J., 2021, 16, 1878–1881 CrossRef CAS PubMed.
- L. Shen, J. Ying, G. Tian, M. Jia and X.-Y. Yang, Chem.–Asian J., 2021, 16, 1130–1137 CrossRef CAS PubMed.
- H. Hwang, T. Kwon, H. Y. Kim, J. Park, A. Oh, B. Kim, H. Baik, S. H. Joo and K. Lee, Small, 2018, 14, 1702353 CrossRef PubMed.
- D. Chen, Z. Pu, R. Lu, P. Ji, P. Wang, J. Zhu, C. Lin, H. W. Li, X. Zhou, Z. Hu, F. Xia, J. Wu and S. Mu, Adv. Energy Mater., 2020, 10, 2000814 CrossRef CAS.
- M. Qu, Y. Jiang, M. Yang, S. Liu, Q. Guo, W. Shen, M. Li and R. He, Appl. Catal., B, 2020, 263, 118324 CrossRef CAS.
- F. Zhang, L. Yu, L. Wu, D. Luo and Z. Ren, Trends Chem., 2021, 3, 485–498 CrossRef CAS.
- A. R. Jadhav, A. Kumar, J. Lee, T. Yang, S. Na, J. Lee, Y. Luo, X. Liu, Y. Hwang, Y. Liu and H. Lee, J. Mater. Chem. A, 2020, 8, 24501–24514 RSC.
- S. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- W. Zheng, L. Y. S. Lee and K. Y. Wong, Nanoscale, 2021, 13, 15177–15187 RSC.
| This journal is © The Royal Society of Chemistry 2023 |