
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupported rhenium catalysts for the hydrogenation of levulinic acid derivatives: limits and potential†
Riccardo
Bacchiocchi
ab,
Jacopo
De Maron
ab,
Tommaso
Tabanelli
 *ab,
Daniele
Bianchi
c and
Fabrizio
Cavani
ab
*ab,
Daniele
Bianchi
c and
Fabrizio
Cavani
ab
aDipartimento di Chimica Industriale Toso Montanari, Università di Bologna, Viale Risorgimento 4, 40136 Bologna, Italy. E-mail: tommaso.tabanelli@unibo.it
bCenter for Chemical Catalysis-C3, Alma Mater Studiorum Università di Bologna, Viale Risorgimento 4, 40136 Bologna, Italy
cIstituto Eni Donegani, Via Fauser 4, 28100 Novara, Italy
First published on 10th January 2023
Abstract
Levulinic acid derivatives, such as alkyl levulinates, are suitable starting reactants for the production of fuel components, namely γ-valerolactone (GVL), alkyl valerates, pentanol, and pentylvalerate (PV). The reactions were performed in batch, without any additional solvents, by investigating the catalytic activity of several Re-based catalysts. In this way, we confirmed the crucial role of the support acidity in promoting the ring-opening of GVL and its consecutive reduction to valeric compounds. In the optimised conditions, bimetallic Re-Ru-O/HZSM-5 yielded methyl valerate (MV) and valeric acid (VA) with a productivity of 512 mmol gmetal−1 h−1, one of the highest reported in the literature to date. Rhenium can also foster the reduction of valeric acid/esters to PV through the formation of 1-pentanol and its efficient esterification/transesterification with the starting material. However, we also proved that Re-based catalysts undergo leaching of the active phase in the presence of carboxylic acids, especially by working in neat with VA, thus limiting the recyclability of the catalytic material. Furthermore, the over-reduction of rhenium affects catalytic performance, suggesting not only that a pre-reduction step is unnecessary, but also that it could be detrimental for the catalyst activity.
1. Introduction
Starting from the industrial revolution, human activities have affected the carbon cycle not only by adding more carbon dioxide to the atmosphere, but also by affecting the ability of forests, soils, and oceans to capture and store CO2 from the air.1 Lignocellulosic biomass (LCB) is one of the most abundant renewable feedstocks and may replace fossil raw materials in chemical and fuel production.2,3 For instance, the pyrolysis of LCB can yield a bio oil containing carboxylic acids that can be upgraded to gasoline by means of a ketonization/hydrotreating process.4 Alternatively, LCB can be used to produce several significant building blocks such as syngas (by means of gasification), methanol (from syngas), ethanol (by glucose fermentation), and ethylene (by ethanol dehydration). These molecules can be further combined to produce valuable bio-based chemicals, such as methyl methacrylate.5 Among the bioderived platform molecules obtainable from LCB, levulinic acid (LA) was selected by the U. S. Department of Energy as one of the top twelve most valuable molecules and with the greatest potential, with a market value of approximately 4000 Tonnes per year in 2020.6,7 In particular, the most investigated strategy for the exploitation of LA is its hydrogenation towards fuel additives, solvents, and other added-value bio-based chemicals and, in this context, heterogeneous and homogeneous catalysts are widely used.8 In literature, most works deal with the conversion of LA and its ester towards γ-valerolactone (GVL) because LA is the most easily obtainable compound, either by hydrogenation with H2 or by catalytic H-transfer hydrogenation with light, bio-based alcohols, in both liquid- and gas-phase continuous flow reactors.9–16The most recent advances in literature, however, have also led to different consecutive reduction products.17 For instance, 2-methyltetrahydrofuran (2-MTHF) was proposed as an efficient fuel additive for gasoline formulations and as an eco-friendly solvent alternative to THF.18–21 Another significant product is 1,4-pentadiol (1,4-PDO), thanks to its potential use as a renewable monomer for the production of polyesters.22,23 Lastly, the preparation of valeric acid and/or its esters (VA/VE) was reported over a variety of heterogeneous, bifunctional catalysts, in particular those combining a support displaying Brønsted acidity with an active (i.e. group VIII) hydrogenation metal.24 The most promising results on this topic are reported in Table 1.25–31
| Catalyst | Reaction condition | Conversion (%) | Yield (%) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| Solvent | T (°C) | P H2 (bar) | Time (h) | GVL | VA + VE | Other | |||
| a Only valeric acid. | |||||||||
| Ru/SBA-SO3H | Ethanol | 240 | 40 | 6 | 100 | 1 | 94 | 1 | 25 |
| Ru/HZSM-5 | 1,4-Dioxane | 200 | 40 | 10 | 100 | 8 | 91 | — | 26 |
| Pd/HZSM-5 | Ethanol | 240 | 40 | 8 | 100 | 8 | 90 | 2 | 27 |
| Co/HZSM-5 | Ethanol | 240 | 30 | 3 | 100 | 1 | 97 | — | 28 |
| Ru/HZSM-5 | 1,4-Dioxane | 220 | 30 | 10 | 100 | 1 | 86 | — | 29 |
| Nb–Cu/ZPS | Water | 150 | 30 | 4 | 100 | — | 99a | — | 30 |
| Pt/HMFI | — | 200 | 8 | 6 | 100 | — | 99a | — | 31 |
A high activity and selectivity towards VA/VE were reported for all catalysts, but in most cases both an additional organic solvent and a high molar excess of hydrogen were required, compared to LA. Furthermore, some authors reported leaching issues (e.g. dealumination or desulfurization of the support) which were often difficult to solve. As shown in Table 1, in the majority of works in literature dealing with LA hydrogenation, solvents were used (e.g. 1,4-dioxane, water, alcohols, long-chain hydrocarbons); some studies, however, showed promising results also with neat LA. For instance, Weckhuysen et al. obtained the complete conversion of LA yielding GVL by using Ru supported on TiO2 and H-β as co-catalyst, while Shimizu et al. showed that Pt/HMFI is a suitable catalyst for the one-pot hydrogenation of LA to VA.31,32
A much more challenging task is the selective reduction of the carboxylic group of LA and VA. In this context, bimetallic catalysts – in which one metal facilitates the heterolytic H2 cleavage for hydrogenation/hydrogenolysis steps (e.g. Ru, Pt, Pd, Rh, Ir, Co, Ni, Cu), while the second metal (e.g. a promoter such as Sn, Mo, Cr, Re) fosters the activation/adsorption of the carbonyl group of the acid/ester molecule – are the most investigated, in many cases showing a substantial synergy.33 Promoters are redox oxyphilic species which enhance the reducibility of carboxylic groups.34 For instance, Toba et al. reported good results in the hydrogenation of mono- and di-carboxylic acids towards the corresponding alcohols and diols using a Ru–Sn–Al2O3 catalyst in a batch reactor (i.e. VA but not LA).35 Li et al. hydrogenated acetic acid to ethanol at 100 °C over an Ir–MoOx/SiO2 catalyst on a tubular flow reactor.36 A catalyst consisting of copper chromite mixed with an alkali or alkaline-earth metal component and an inorganic matrix component has been patented for the vapour-phase hydrogenation of methyl esters to fatty alcohols under fixed-bed conditions.37 Davis et al. investigated the effect of ReOx on the reduction of propionic acid over Pd-supported catalysts; likewise, Liang et al. studied the role of Re in Re–Ru/C bimetallic catalysts for the hydrogenation of succinic acid.38,39
Several works suggest that the oxyphilic nature of rhenium is responsible for the interaction between active metal sites and carboxylic groups; however, Re may also activate H2 by promoting dehydrogenation reactions;40 hence, rhenium-based catalysts may selectively reduce carboxylic compounds. As an example, Toyao's group investigated the hydrogenation of aromatic carboxylic acids over a Re/TiO2 catalyst, which selectively yielded the corresponding alcohols.41 Through density functional theory (DFT) calculations, they also demonstrated that Re and ReOx showed a higher affinity towards carboxyl groups as well as a lower affinity towards benzene moiety in comparison to other metal surfaces, thus proving that Re has an oxyphilic nature.42,43
Compared to LA, levulinic esters (LE) can be obtained directly from cellulose by means of alcoholysis with good to excellent yields, without the need for a further esterification step, thus paving the way towards their direct exploitation as bio-based platform molecules.44–46 In particular, methyl, ethyl, and n-butyl levulinate are interesting thanks to their physicochemical properties.47 In fact, they might find applications not only as specialty chemicals, but also as additives in the fragrance and petrochemical industries.48–50 Moreover, LE are acid-free compounds and this makes them a promising alternative to levulinic acid as the starting material for the production of γ-valerolactone and its consecutive reduction products.51–54 In fact, the use of LA as reactant often causes the deactivation of the catalyst due to the leaching of the supported metal generated by acid media, and/or the fouling of the catalyst due to the oligomerization/polymerization of unsaturated intermediates.55 Therefore, the catalytic conversion of LE might be more attractive for industrial applications.
To the best of our knowledge, there are no reports in literature on the direct hydrogenation of LA or LE to yield 1-pentanol (1-PAO). The properties of 1-PAO and its derivatives (i.e. dipentyl ether, pentyl valerate) make them suitable biofuels, thanks to their reduced particulate emissions and the consequent improvement in air quality.56–59 Pentyl valerate (PV) has appropriate polarities, better volatility, and higher ignition properties as a biofuel for gasoline and jet fuel applications.60 In particular, the hydrogenation of GVL to PV in the liquid phase with pentanol as reactant/solvent has been investigated using noble metals supported on SiO2–Al2O3- and Cu-based catalysts.61–65 The one-pot upgrading of GVL to PV was achieved with 60.6% of yield using a bifunctional 5% Pd/HY catalyst in a batch reactor system also without pentanol as reactant/solvent under relatively harsh conditions (260 °C, 80 bar of H2, 30 h, solvent n-octane).66
Herein, we report on the conversion of levulinic acid derivatives to 1-pentanol and/or pentyl valerate using a batch reactor in solvent-free conditions over Re-based catalysts. First, the hydrogenation of methyl levulinate (ML) was investigated in a one-pot approach aiming to directly obtain VA/VE, 1-PAO, and PV. Then Re-based catalysts were examined also in the hydrogenation of valeric acid/ester (VA/VE) to evaluate the catalytic performance for the selective hydrogenation of carboxylic groups, with a special focus on both catalyst stability and the possible leaching of active species.
2. Experimental
All catalysts were prepared by incipient wetness impregnation (IWI) to obtain a 5 wt% rhenium on supports. For the IWI procedure, an aqueous solution containing a proper amount of NH4ReO4 was loaded on the support powder (usually 1.9 g); then it was oven-dried at 120 °C for 4 h and calcined at 500 °C for 3 h (ramp 5 °C min−1) in static air to remove NH4 ions and H2O from the precursor and support. For the reduction step, the powder was exposed to an H2/N2 flow (100 ml min−1, 10 vol% of H2) at 350 °C for 1 h (ramp 10 °C min−1). For the preparation of bimetallic catalysts, the NH4ReO4 solution was loaded first (in order to obtain a 5 wt% of Re on the support) and the sample was oven-dried at 120 °C before loading a solution of RuCl3·H2O (to obtain 1 wt% of Ru on support). Silica and zeolite supports were delivered respectively by GRACE and Zeolyst (see ESI†).Catalytic tests, the analysis system of post-reaction solutions, and the calculations for conversion, yields, and molar balance are described in detail in the “Experimental” section of the ESI.† The catalysts were characterized by means of N2-physisorption at 77 K with the BET method, MP-AES, XRD, TPR, and ATR-IR. Instrument specifications and analysis procedures are described in detail in the “Catalyst's characterization” section of the ESI.†
In most research described in literature, the precursor used for the synthesis of Re-based catalysts is NH4ReO4, for which XPS and XANES analyses confirm that the oxidation state of rhenium remains +7 after thermal treatment.43,67 Since all calcinations in this study were obtained in static air, it has been assumed that the Re oxidation state was not affected and that – as a consequence of the thermal treatment in the air – all NH4ReO4 was decomposed into Re2O7. For the sake of simplicity, “Re2O7/support catalyst” has been shortened to “Re–O/support”.
3. Results and discussion
3.1 Supported Re–O catalysts
First, we investigated the effect of the support on the catalytic activity of Re-based materials for the hydrogenation of methyl levulinates (ML) to GVL and other consecutive reduction products. In particular, Table 2 shows the performances of Re–O/SiO2 and Re–O/HZSM-5 at 210 °C and 40 bar of hydrogen for 4 h. As mentioned in the Experimental section in the ESI,† by working with neat methyl levulinate the molar ratio H2/ML in the reaction system was equal to only 1.4; therefore, the reaction system needed hydrogen refilling to keep the overall pressure constant during tests and to make the occurrence of the desired consecutive reduction reactions possible. Interestingly, both catalysts were active for the target reaction, with Re–O/HZSM-5 showing a higher ML conversion than Re–O/SiO2 (88% and 72% respectively), whereas the GVL yield was around 66% in both cases. Remarkably, Re promoted the hydrogenation of the carboxylic group of GVL leading to 2-MTHF (obtained with an underestimated yield of around 1% due to its higher volatility during the depressurization of the autoclave after the reaction). On the other hand, the higher acidity of Re–O/HZSM-5 in respect to Re–O/SiO2 led to the formation of valeric acid and methyl valerate. These compounds may be obtained via two main pathways: (i) the hydrogenation of ML carbonyl group to 4-hydroxypentanoic acid/ester (4-HPA/E) followed by its further dehydration and the saturation of the resulting double bond, or (ii) the consecutive ring-opening and hydrolysis of GVL to 4-HPA/E, which may then follow the previous reaction pathway.Starting from these results, it was decided to investigate the influence of a pre-reduction treatment on the catalytic activity. In literature, a pre-reduction treatment of Re-based materials is usually carried out with H2 diluted in an inert gas (5–30 vol%) at 500 °C before testing the materials. It has been reported that bulk Re2O7, when heated in a hydrogen atmosphere, sublimes at 180 °C before its reduction.68 Conversely, when Re2O7 is mixed with Pt or Pd, a complete reduction to metallic Re occurs below 200 °C without any loss. Firstly, the stability of the supported Re-species upon thermal treatment was verified by means MP-AES of the calcined materials and results are reported in Table 1S.† The amount of rhenium measured this way was in good agreement with the expected value of 5 wt%, suggesting that the metal-support interaction stabilizes rhenium species. Then, a pre-reduction treatment was carried out at 350 °C under a flow of 10 vol% H2 in N2 on Re–O/HZSM-5 before catalytic testing. The reduced catalyst was labelled Re-R/HZSM-5. Surprisingly, the pre-reduction treatment caused a significant drop in both the catalytic activity and the selectivity: not only did ML conversion decrease from 88% to 40%, but also GVL yield dropped from 44% to 16%, while Others' yields increased from 7% to 11% compared to the non-reduced Re–O/HZSM-5 material (“Others” include 2-MTHF, 4-HPA/E, and unidentified products). The GC-FID analysis of the reaction mixture revealed the presence of heavier, high-retention-time compounds. Unfortunately, GC-MS analysis did not clarify their structure, but it is known that GVL can undergo decarboxylation to produce unsaturated compounds, such as butenes or pentenoic species, which may oligomerize to heavy products.55,69
All in all, the drop in conversion of ML and the different product distribution indicate that a catalyst's over-reduction is detrimental for catalytic performance, as observed also in other cases, i.e. in the hydrogenolysis of glycols to alcohols or alkanes (e.g. from glycerol to propanols).70–72 Therefore, the nature of the support seems to play a side role in catalytic activity, while ML conversion and GVL yield seem to be mainly influenced by the oxidation state of rhenium.
For this reason, the reducibility of catalysts was investigated by means of H2-TPR. Fig. S2† shows the H2-TPR profiles of Re–O/SiO2 and Re–O/HZSM-5, which are characterized by H2 consumption centred at 415 °C and 385 °C, respectively. The lower reduction in temperature displayed by Re–O/HZSM-5 may suggest either a better dispersion of Re-species, which results in a high active surface or, from another point of view, a weaker interaction in respect to the one between rhenium and SiO2, which decreases the temperature required for Re reduction. For both samples, the molar ratio between the hydrogen consumed and Re2O7 was around 5. Assuming an initial Re oxidation state of +7 (see Experimental), the observed H2 consumption means that the average oxidation state of rhenium species will be +2 in the reduced material. It should be noted that Re2+ oxides (namely ReO phases) are not stable and were only detected in particular conditions on rhenium-metal surfaces.73 Conversely, rhenium usually forms oxides with oxidation states equal to +4, +6, and especially +7. Therefore, it may be argued that after reduction Re is more likely to be present as a mixture of metallic Re(0) and Re(IV) (ReO2 phases), even though the presence of even higher oxidation states cannot be completely ruled out. The TPR characterization also showed that the reduction occurred in a single event and not in multiple steps, suggesting that the formation of reduced species such as Re(0) may generate a further reduction of the surrounding oxide, possibly by H2 spillover, so that the reduction of Re7+ all the way down to Re(0) occurred within a very narrow temperature range.
Re–O/HZSM-5 was then selected for an investigation of the effect of temperature on the hydrogenation of ML, because this catalyst showed the highest activity toward the consecutive reduction products of GVL. The results of these tests are reported in Fig. 1. At 180 °C, ML conversion was below 40%, the main product was GVL, and no traces of valeric acid or its ester were detected. Increasing the temperature from 180 °C to 210 °C led to more than twice as much ML conversion and triple the GVL yield, while valerates (MV and VA) also started to form. Remarkably, a further increase in temperature up to 230 °C did not lead to substantial differences in product distribution. The molar balance was good in all the temperature ranges investigated (>86%), demonstrating that the formation of heavy compounds was limited even by working in neat conditions. A qualitative analysis of the gaseous phase in GC-MS (Fig. S3a†) revealed the presence of light products such as methane, CO2, and especially dimethyl ether (DME). Significantly, methyl pentyl ether (MPE), also, was detected in the gas phase using Re–O/HZSM-5. This is indirect evidence that Re, if combined with a support showing Brønsted acid sites, may catalyse the reduction of ML to the corresponding alcohol. In fact, it is well known that strong Brønsted acidic sites such as those shown by the HZSM-5 support catalyse the dehydration/condensation of alcohols.74 As previously mentioned, the initial molar ratio H2/ML in the reaction system is equal to only 1.4; therefore, the reaction system needed hydrogen refilling to (i) keep the total pressure constant at 40 bar during the tests, and (ii) make the desired consecutive reduction reactions possible. Nevertheless, the production of light, volatile compounds – mainly DME (bp −24 °C at atmospheric pressure), but also MPE – led to a further increase of the internal pressure, thus limiting or even hindering the addition of more molecular hydrogen, which is crucial for promoting further reductions. This was confirmed by the calculations (detailed in ESI†) of both hydrogen conversion (H2 conversion) and methanol balance (MeOH balance), whose trends as a function of the reaction temperature are reported in Fig. S4.† Interestingly, H2 conversion increased threefold when the temperature was increased from 180 °C up to 210 °C. However, it remained almost stable with a further increase up to 230 °C. At the same time, MeOH molar balance trends (related to liquid-phase products) showed a dramatic temperature-related decrease from 85% at 180 °C to 30% at 230 °C. MeOH missing from the balance was supposed to form the DME found among gaseous products by GC-MS. These results suggests that a significant partial pressure of DME in the headspace of the autoclave affected the hydrogen refilling by limiting the amount of H2 that could be added. This is likely to be the reason why the results obtained at 230 °C are comparable to what was observed at 210 °C.
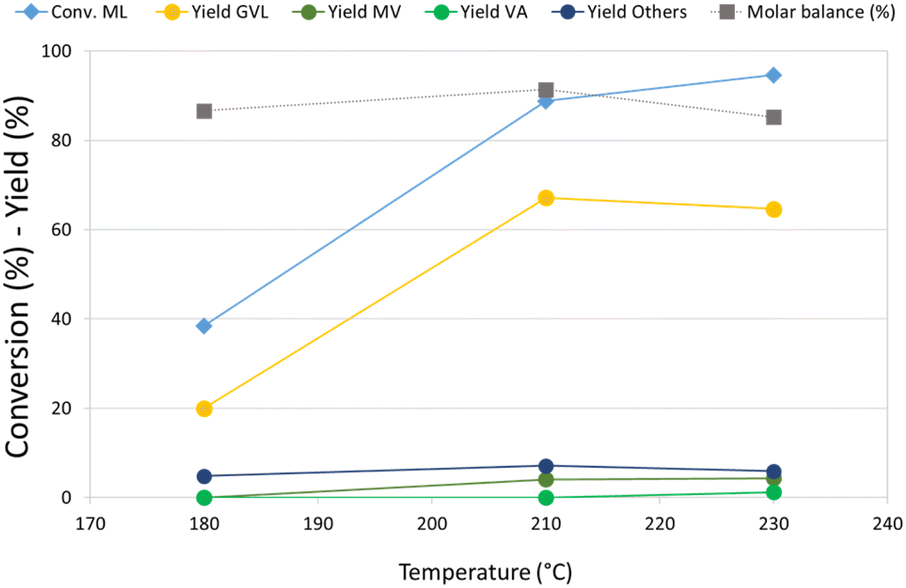 | ||
| Fig. 1 Effect of temperature on ML hydrogenation over Re–O/HZSM-5. Reaction conditions: solvent free (ML neat), T = variable, PH2 = 40 bar, t = 4 h, catalyst loading = 5 wt% [mcat/mML]. Others = 2-MTHF, 4-HPA/E and unidentified products. | ||
3.2 Supported Ru-Re-O catalysts
In the attempt to further improve catalyst performance, it was decided to combine a hydrogenation metal (Ru) with our main active metal (Re), by preparing a bimetallic system (Re-Ru-O/HZSM-5, 5 wt% Re and 1 wt% Ru), which was investigated in different reaction conditions for the hydrogenation of ML.The influence of H2 pressure has been investigated by means of experiments in the 10-to-40 bar pressure range: the results can be found in the “Effect of hydrogen pressure on catalytic activity” section of the ESI (Fig. S5†). Briefly, it was found that working at 40 bar was beneficial for the desired reaction and the all the following catalytic tests were carried out at this pressure.
Temperature (Fig. 2) was found to have a crucial influence on the reaction outcome, as shown in Fig. 2a. Thanks to its higher hydrogenating activity, at 180 °C Re-Ru-O/HZSM-5 showed results similar to those observed with Re–O/HZSM-5 at 230 °C. An increase in temperature up to 210 °C yielded up to 17% and 8% of MV and VA, while at the same time GVL yield decreased from 59% to 34%. A further increase in temperature boosted consecutive reductions and MV + VA were obtained with 65% yield, while GVL yield dropped to 5%. Notably, during tests over Re-Ru-O/HZSM-5 it was possible to refill hydrogen without interruption thanks to the enhanced hydrogenating activity of the catalyst, which competes kinetically with the acid-catalysed condensation of alcohols (particularly methanol) that was favoured at high temperature with Re–O/HZSM-5. The molar balance not exceeding 80% is probably due to the formation of over-reduction products such as light hydrocarbons. Indeed, a qualitative GC-MS analysis of the headspace detected light products such as methane, CO2, DME, but also products of ML reduction such as MPE, MV, n-butane, and n-pentane (Fig. S3b†). Finally, Others' yield (including unidentified heavy products, 2-MTHF, methyl 4-hydroxypentanoate, and traces of methyl pentyl ether (MPE) and pentyl valerate (PV)) remained constant with temperature at around 7%. Time-resolved experiments were carried out at 230 °C under 40 bar of H2 pressure (Fig. 2b) to assess the feasibility of promoting consecutive reduction reactions toward 1-pentanol and pentyl valerate. A ML conversion as high as 75% was achieved after only 1 h of reaction, and further increased (up to over 99%) after 2 hours already. Initially, GVL was the main product with a 22% yield after 1 hour, but it was replaced by valerates already after 2 hours (MV and VA). MV and VA were detected already after 1 h, and their yield increased over time, reaching a peak of 65% after 4 h, underlining the significant activity of the catalyst in the reduction of the so-formed hydroxy acid/ester to valerates. At the same time, it becomes evident that valerates may be further converted to consecutive products such as 1-pentanol, methyl pentyl ether, and pentyl valerate (grouped together under the term “1-pentanol derivatives = 1-PAO derivatives”), obtained with a yield of 6% after 24 h. Products of valerates reduction were observed in gas-phase qualitative GC-MS analysis as well (Fig. S3c†). This finding, together with the worsening of the molar balance between 4 h and 24 h of reaction, suggests that the formation of gaseous products causes a decrease in the total yield of liquid phase products over time, leading to the observed decrease of the molar balance. The comparison of the catalytic activity of Re–O/HZSM-5, Ru–O/HZSM-5 and Re-Ru-O/HZSM-5 at optimised conditions (230 °C, 4 h, 40 bar of H2) are shown in Fig. S9† again proving the beneficial effect of the co-presence of both Re and Ru in order to improve both MV + VA yields and molar balances.
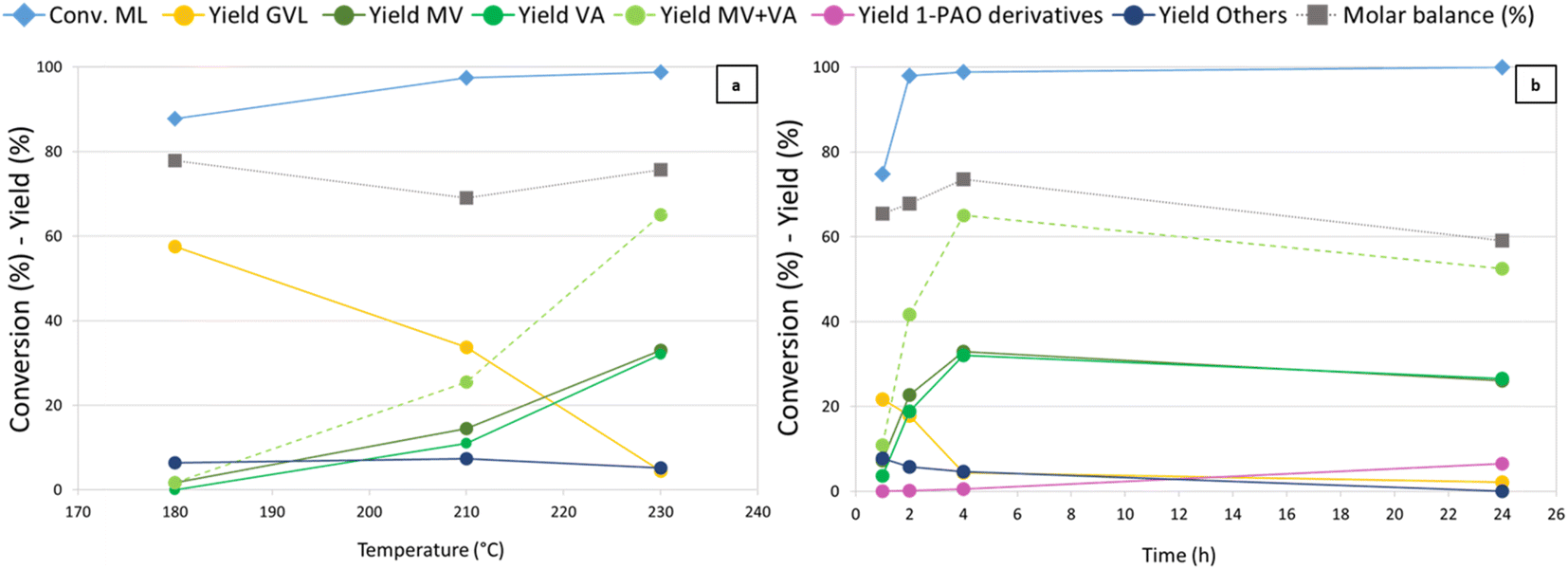 | ||
| Fig. 2 ML hydrogenation over Re-Ru-O/HZSM-5. Reaction conditions: solvent free (ML neat), PH2 = 40 bar, catalyst loading = 5 wt% [mcat/mML]. (a) Effect of temperature for a 4 h reaction time and (b) effect of reaction time at 230 °C. Others = 2-MTHF, 4-HPA/E and unidentified products. | ||
3.3 Characterisation of Re-Ru-O/HZSM-5 catalyst
The reducibility of Re-Ru-O/HZSM-5 before and after reaction was characterized by means of TPR Fig. S6†) and compared to the one of their monometallic analogues (Fig. S2†). The Re–O/HZSM-5 catalyst showed only one sharp peak at 380 °C, whereas the bimetallic Re-Ru-O/HZSM-5 showed one main reduction peak centred at 255 °C and a small one at 166 °C. These temperature correspond to the reduction of the Re and Ru oxide species respectively, as explained by Baranowska et al.75 In their study, these authors investigated bimetallic Ru–Re/Al2O3 with different Ru/Re ratios (i.e., 90/10, 75/25, 50/50) and found that Ru is reduced at lower temperature than Re; moreover, the presence of increasingly high amounts of Ru progressively reduces the temperature required by Re reduction. Hence, the sharp peak at 166 °C in the H2-TPR profile for the fresh Re-Ru-O/HZSM-5 can be attributed to the reduction of the Ru species, which subsequently foster rhenium reduction at a lower temperature (255 °C) than the one required by Re–O/HZSM-5 (380 °C) by means of H2 spillover. Similarly, the Re-Ru-O/HZSM-5 catalysts after ML hydrogenation under 40 bar of H2 at 230 °C for 4 h, showed a broad reduction peak at 260 °C and a sharp peak at 115 °C (Fig. S6b†). Again, the hydrogen consumption during the analysis was measured, and for the freshly calcined Re-Ru-O/HZSM-5 it was assumed that all Ru was present as RuO2 and that all Re was in a + 7 initial oxidation state. Under these assumptions, after the complete reduction of Ru(IV) to Ru(0), the molar ratio between the remaining consumed H2 and the supported Re2O7 was around 5, suggesting that – as it was for Re–O/HZSM-5 – Re is present as a mixture of Re0 and ReO2 after reduction. Conversely, during the reduction of the spent Re-Ru-O/HZSM-5, a much lower amount of hydrogen was consumed. In particular, the molar ratio H2/(Re + Ru) was 5 times lower than for the fresh catalyst, thus suggesting that the supported Re2O7 and RuO2 were partially reduced in situ to Ru(0) and Re(0) during the reaction with ML.This high degree of reduction adversely affects catalyst performances as demonstrated by a catalytic test carried out over Re-Ru-R/HZSM-5 (the pre-reduction treatment was carried out at 350 °C under a flow of 10 vol% H2 in N2 on Re-Ru-O/HZSM-5 before the catalytic test, Fig. S7†). The relevant results showed the same trend as that observed for monometallic Re supported on HZSM-5. In fact, the performance of the pre-reduced bimetallic catalysts was worse than the one of the same freshly calcined material: ML conversion was 83%, GVL yield was 69%, and only 7% of MV + VA yield was obtained under the same reaction conditions.
In consideration of the information collected so far, we calculated the valeric acid/esters productivity of our catalyst in the best conditions and compared it with the values reported by other authors in literature (Table 3). Among the studies which employed a solvent and focused on the hydrogenation of LA (Table 3, entry 1–6), 2 wt% Pd/HZSM-5 showed high yield (90%) in VA + VE at 240 °C for 8 h in ethanol, but also high productivity, around 495 mmolVA+VE gPd−1 h−1 (Table 3, entry 3).27 Indeed, to the best of our knowledge, the direct HDO of neat ML to valeric acid/esters has been reported for the first time in the present work, with a VA + VE yield of 65% achieved with a bimetallic catalyst containing 5 wt% Re and 1 wt% Ru supported over HZSM-5, after a 4 h reaction at 230 °C, corresponding to a productivity of 512 mmolVA+VE gRe+Ru−1 h−1 (Table 3, entry 9). This productivity is comparable to that of catalysts which work with solvent, and it is at least threefold higher than the highest VA + VE productivity of 169 mmolVA gPd−1 h−1 reported by Shimidzu et al. (Table 3, entry 7).31 Conversely, when only Ru is considered as the active metal for hydrogenation, the corresponding productivity increased up to 3073 mmolVA+VE gRu−1 h−1. As a matter of fact, only He et al. have recently reported a higher productivity, equal to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 514 mmolVA+VE gRu−1 h−1, by working at 220 °C with 40 bar of H2 using 1 wt% Ru/HZSM-5 over neat ethyl levulinate (EL), this being the highest productivity reported to date.76
514 mmolVA+VE gRu−1 h−1, by working at 220 °C with 40 bar of H2 using 1 wt% Ru/HZSM-5 over neat ethyl levulinate (EL), this being the highest productivity reported to date.76
| Entry | Catalyst | Reaction conditions | Productivity (mmolVA+VE gmetal−1 h−1) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Reactant | Solvent | T (°C) | P H2 (bar) | Time (h) | ||||
| a Calculated considering all metal loading. b Calculated considering only Ru loading. | ||||||||
| 1 | 5 wt% Ru/SBA-SO3H | LA | Ethanol | 240 | 40 | 6 | 63 | 25 |
| 2 | 1 wt% Ru/HZSM-5 | LA | 1,4-Dioxane | 200 | 40 | 10 | 391 | 26 |
| 3 | 2 wt% Pd/HZSM-5 | LA | Ethanol | 240 | 40 | 8 | 495 | 27 |
| 4 | 10 wt% Co/HZSM-5 | LA | Ethanol | 240 | 30 | 3 | 278 | 28 |
| 5 | 3 wt% Ru/HZSM-5 | LA | 1,4-Dioxane | 220 | 30 | 10 | 115 | 29 |
| 6 | 4–40 wt% Nb–Cu/ZPS | LA | Water | 150 | 30 | 4 | 39 | 30 |
| 7 | 5 wt% Pt/HMFI | LA | — | 200 | 8 | 6 | 169 | 31 |
| 8 | 5–1 wt% Re-Ru-O/HZSM-5 | ML | — | 230 | 40 | 4 | 512 | This worka |
| 9 | 5–1 wt% Re-Ru-O/HZSM-5 | ML | — | 230 | 40 | 4 | 3073 | This workb |
| 10 | 1 wt% Ru/La–Y (or HY) | EL | — | 220 | 40 | 0.5 | 11514 |
76 |
3.4 VA and MV hydrogenation
Considering the ability of Re-supported materials to effectively catalyse the formation of valerates starting from ML, we decided to focus our efforts on the hydrogenation of valerates (VA and MV) with the aim of promoting the formation of 1-pentanol (1-PAO) and/or pentyl valerate (PV) following a “two-step” strategy (namely from ML to VA/MV and then from VA/MV to 1-PAO derivatives).Table 4 reports the catalytic activity of bimetallic materials (e.g., Re-Ru-O/HZSM-5 and Re-Ru-O/SiO2), and monometallic materials (Re–O/SiO2 and Re–O/HZSM-5) for VA hydrogenation at 210 °C, 40 bar of hydrogen, for 4 h. VA conversion was generally low, being the one obtained over Re–O/SiO2 the highest (35%). Significantly, all materials displayed low selectivity to 1-PAO due to its consecutive fast esterification with excess VA that formed PV in its place (as also demonstrated later in this manuscript).
| Catalyst | Conv. VA (%) | Yield (%) | Leaching Re (%) | |
|---|---|---|---|---|
| 1-PAO | PV | |||
| Re-O/SiO2 | 35 | 1 | 27 | 26.7 |
| Re-Ru-O/SiO2 | 10 | <1 | 7 | 30.5 |
| Re-Ru-O/HZSM-5 | 17 | 0 | 7 | 42.9 |
| Re–O/HZSM-5 | 15 | <1 | 8 | 45.0 |
Notably, by carrying out the hydrogenation of neat VA, we were able to highlight a crucial issue which is often dramatically underrated in literature: rhenium leaching. Generally speaking, leaching of rhenium was higher for the HZSM-5-supported catalyst (≈45%) than for the SiO2-supported catalyst (27–30%). Previously, it was suggested that the lower reduction temperature displayed by Re–O/HZSM-5 in respect to Re–O/SiO2 may indicate either a better dispersion of Re-species on the zeolite support, which results in a higher active surface, or else that the rhenium–SiO2 interaction is stronger compared to Re-HZSM-5 (see Fig. S3, ESI†). In agreement with H2-TPR results, the lower leaching values for silica-supported catalysts may indicate that rhenium is more strongly bonded to SiO2 than to HZSM-5.
Fig. 3 shows the catalytic results obtained in MV hydrogenation at 210 °C, 40 bar of hydrogen, for 4 h with Re–O/SiO2 and Re–O/HZSM-5. Over Re–O/SiO2 the conversion of MV and product yields were low (X MV ≈ 15%, PV = 7% and 1-PAO = 1%), but the amount of Re lost due to leaching was very low as well (1.1%).
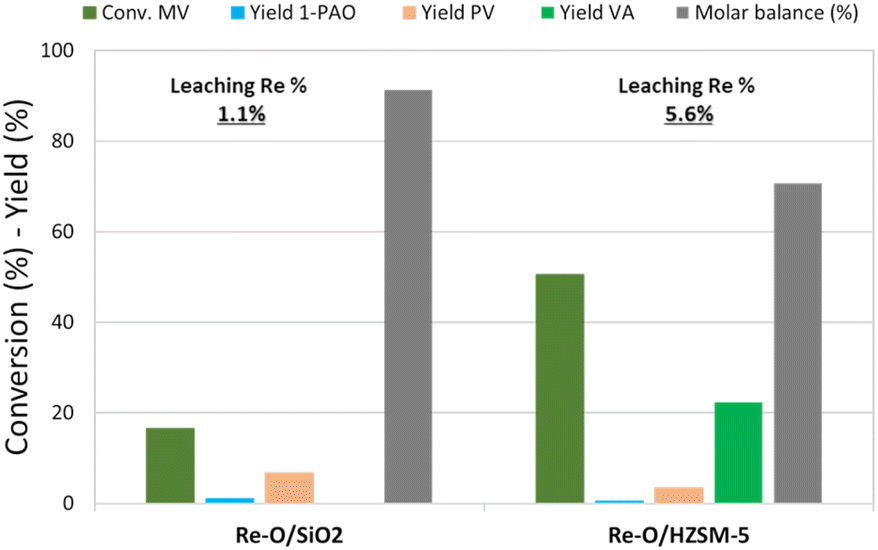 | ||
| Fig. 3 MV hydrogenation over monometallic catalysts. Reaction conditions: solvent free (neat MV), T = 210 °C, PH2 = 40 bar, t = 4 h, catalyst loading = 5 wt% [mcat/mMV] | ||
On the other hand, the performance of Re–O/HZSM-5 in the same conditions was quite different: in fact, MV conversion was as high as 50% and the main product was VA with a yield of 17%, while PV, 1-PAO, and methyl pentyl ether (MPE) were obtained in small amounts. As a consequence, the molar balance obtained was much lower than the one obtained with Re–O/SiO2. This evidence suggests that during the hydrogenation of MV different supports foster different reaction pathways: for instance, the strongly acidic HZSM-5 trigger mainly the hydrolysis of MV to VA and other side reactions that worsen the molar balance by forming light incondensable compounds and/or heavy carbonaceous residues. At the same time, Re loss due to leaching with Re–O/HZSM-5 (5.6%) was higher than with Re–O/SiO2.
Generally speaking, the use of MV as the reactant significantly reduces the leaching of Re for both Re–O/SiO2 and Re–O/HZSM-5 (see Table 4), strongly suggesting that VA is the main actor in the active-phase dissolution because of its acidic characteristics. In this context, the higher metal leaching suffered by the zeolite-supported catalyst can be explained by its high activity towards MV hydrolysis to VA.
MV hydrogenation was further investigated as a function of the reaction time over Re–O/SiO2 (Fig. 4).
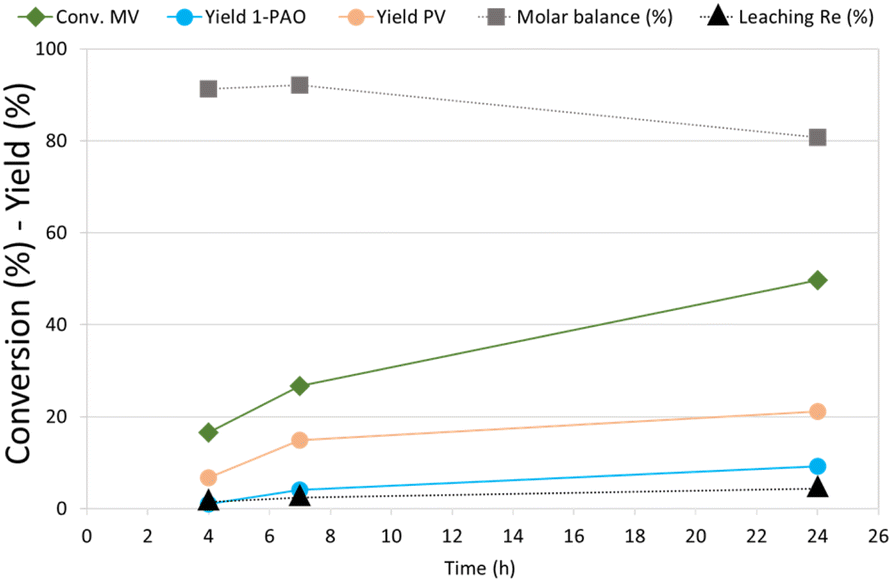 | ||
| Fig. 4 Effect of reaction time in MV hydrogenation over Re–O/SiO2. Reaction conditions: solvent free (neat MV), T = 210 °C, PH2 = 40 bar, t = 4 h, catalyst loading = 5 wt% [mcat/mMV] | ||
By increasing the reaction time from 4 to 7 h the conversion of MV increased from around 15% to 27%, while PV and 1-PAO yields, reached 15% and 4% respectively. Unfortunately, increasing further the reaction time to 24 h did not produce a proportional increase in the target products yield, (PV = 21% and 1-PAO = 10%) due to consecutive reactions that worsened the molar balance (81%). However, the leaching of rhenium (4.3%) was limited even after 24 h of reaction. As a matter of fact, VA was not detected among reaction products because the activity of Re–O/SiO2 for MV hydrolysis to VA is low: as a consequence, the loss of active phase was much lower than the one obtained during the hydrogenation of VA.
Since the progressive rhenium leaching may negatively impact the recyclability of the catalysts, Re–O/SiO2 was recovered by filtration, washed with acetone, dried and recycled for another run. Results obtained by working with both VA and MV after 4 h, at 210 °C and 40 bar of hydrogen are shown in Fig. S8.† Interestingly, when VA is used as reactant, the extensive leaching of Re (26.7%) results in a dramatic loss of activity of the recycled catalyst. On the other hand, when the hydrogenation is carried out on MV the leaching is strongly limited (1.1%) and the catalytic performance of the recycled is comparable to the one of the fresh catalyst. Moreover, in order to exclude the contribution of any homogenous rhenium species a dedicated Sheldon test was carried out during the hydrogenation of VA with Re–O/SiO2 and the outcome is reported in Fig. S9.† After 2 h of reaction the catalyst was removed by filtration from the reaction mixture and thereafter VA conversion remained constant during the following 2 h, suggesting that despite VA fosters extensive leaching of Re, the resulting soluble Re-species do not contribute to the overall catalytic activity.
3.5 The reaction network: reactivity of intermediates
The reaction mechanism of VA/VE over Re–O/SiO2 was investigated by loading intermediates and products under the previously investigated reaction conditions (210 °C, 4 h, 40 bar). Fig. 5 shows the results obtained by loading the autoclave with neat valeric acid, valeraldehyde, 1-pentanol and pentyl valerate, respectively. As previously seen, VA conversion yielded mainly PV with very low amounts of 1-PAO.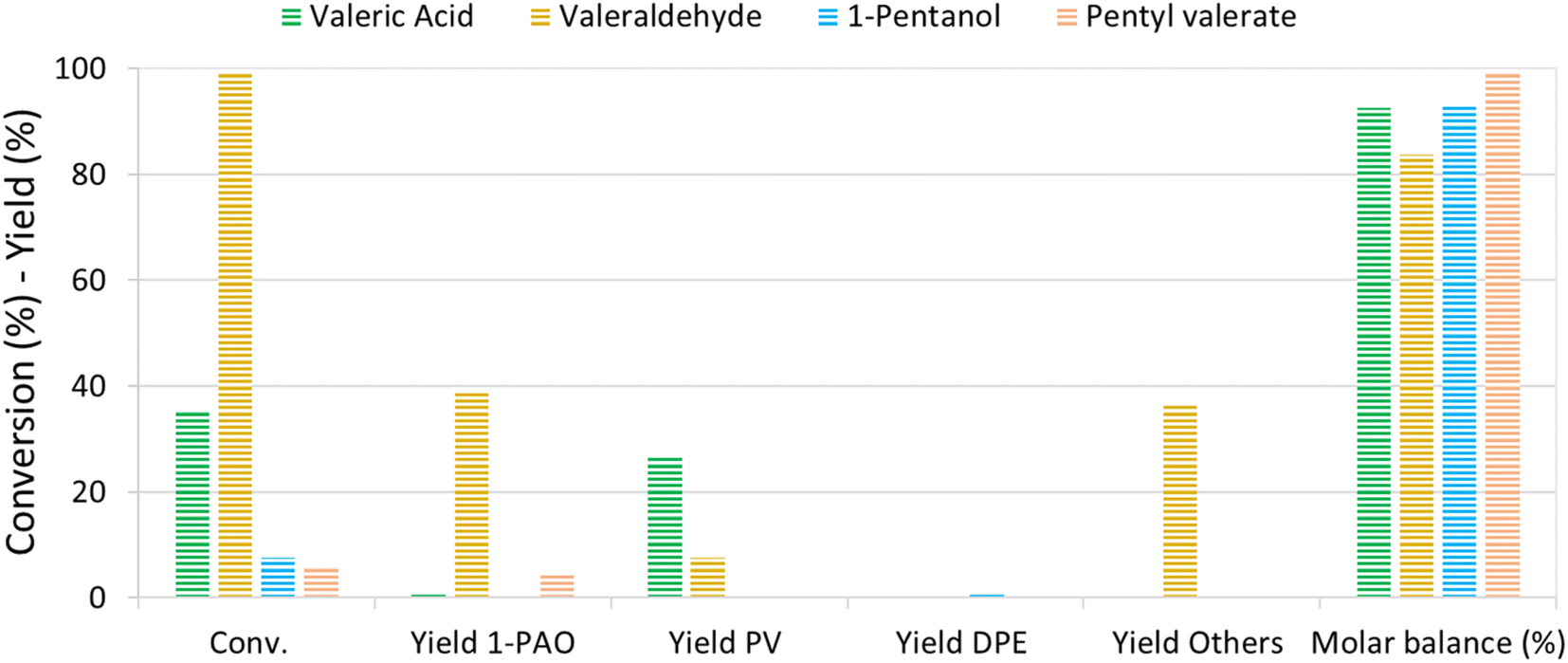 | ||
| Fig. 5 Investigation of the reaction pathway loading intermediates and products in the presence of Re–O/SiO2. Reaction conditions: solvent free, T = 210 °C, PH2 = 40 bar, t = 4 h, catalyst loading = 5 wt% [mcat/mreactant] | ||
Interestingly, valeraldehyde showed a conversion >99%; 1-PAO yield was around 40%, whereas Others' yield reached 37%. Among Others, the compounds with longer carbon chains have been identified as C10 aldehydes and alcohols formed via the aldolic condensations and hydrogenation of the obtained aldehydes; C10 alkenes and alkanes were found too. PV was detected with a yield of 8% and was probably formed via a Cannizzaro-like disproportionation and consecutive esterification or directly via a Tishchenko reaction.
Interestingly, when 1-PAO was charged as reactant, proved to be a quite stable compound in the absence of strong Brønsted acidic sites over the catalyst, showing a conversion of only 7%. Likewise, PV showed a low conversion of about 6% and only 1-PAO was detected as the product. For each test, the headspace of the autoclave after reaction was analysed qualitatively by means of GC-MS: n-butane and n-pentane were detected in every test.
Summarizing all the information collected so far, the overall reaction scheme reported in Scheme 1 may be proposed.
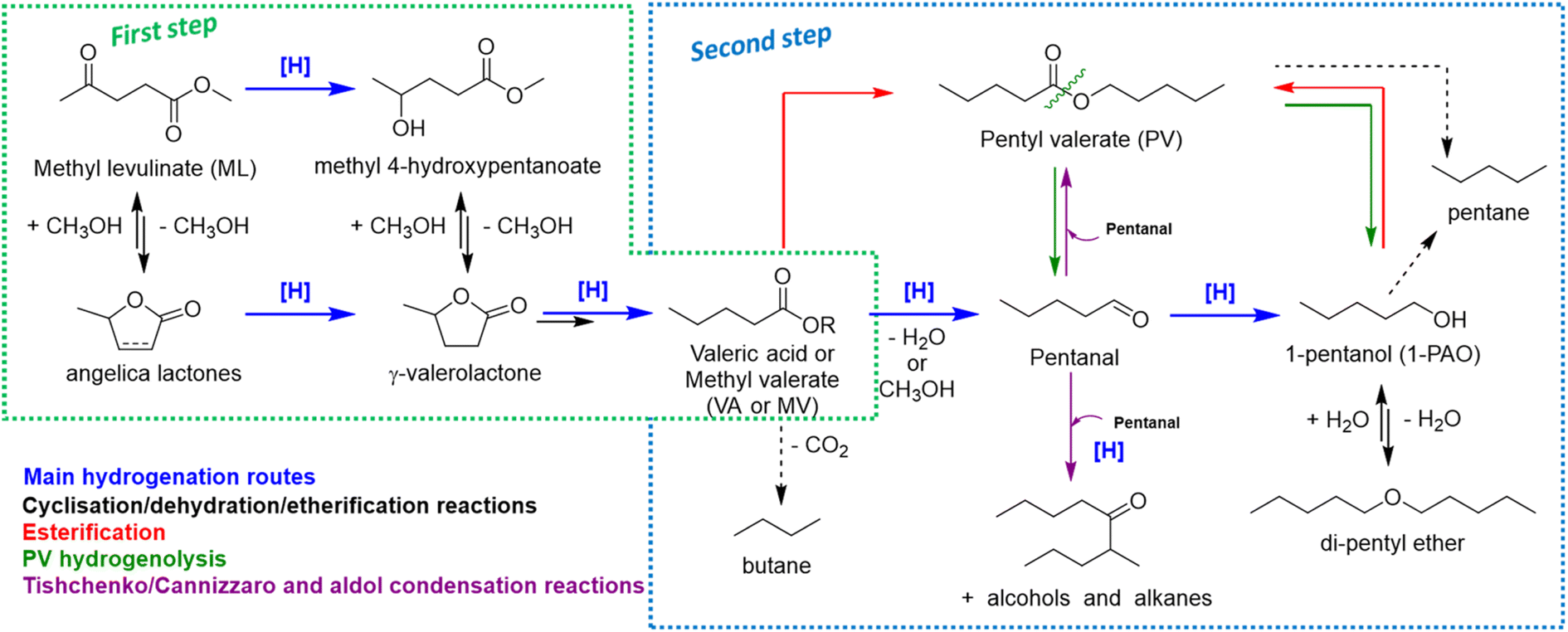 | ||
| Scheme 1 Proposed reaction scheme from ML to 1-PAO derivatives through the formation of VA/MV. | ||
During VA hydrogenation, valeric acid is partially reduced to highly reactive valeraldehyde (pentanal), even if it has not been detected, or partially decomposed through decarboxylation reaction yielding n-butane (Scheme 1, second step). Under standard reaction conditions, valeraldehyde is an unstable intermediate which might react in several ways. Hydrogenation to 1-PAO is the main reaction obtained by working with a hydrogen excess and/or limited concentration of the aldehyde; however, by working in neat conditions, aldol condensation to C10 aldehydes and alcohols and Tishchenko or Cannizzaro disproportion to PV were effectively induced. Indeed, when the aldehyde was formed as an intermediate (i.e., starting from VA), no traces of C10 were found in the liquid phase. PV may be formed through the esterification of VA with 1-PAO, but also via a Tishchenko or Cannizzaro reaction of valeraldehyde. With regard to 1-PAO, its stability lead consecutive hydrogenolysis reaction unfavourable, leading to only traces of n-pentane and di-pentyl ether (DPE). As a matter of fact, 1-PAO effectively reacted in the presence of VA to yield PV under these reaction conditions. Lastly, PV showed limited reactivity; the only detected product was 1-PAO. The absence of VA suggests that an important contribution of ester hydrolysis reaction back to VA and 1-PAO can be ruled out. Even in the case of MV, valeric acid was not detected in the reaction mixture, indicating that VA is not a crucial intermediate in alkyl valerate hydrogenation. These data suggest that alkyl valerate hydrogenolysis may occur by splitting into aldehyde and alcohol (green arrows). However, it cannot be ruled out that the splitting occurs between oxygen and the alkyl chain, yielding pentane and valeric acid; the latter can be converted as detailed above. Previous results of MV hydrogenation suggest that Re–O/SiO2 mainly privileges the first pathway, whereas Re–O/HZSM-5 privileges the second one.77–79
4. Conclusions
In summary, this work reports on the catalytic hydrogenation of levulinic acid derivatives, mainly methyl levulinate (ML), under H2 atmosphere over supported Re-based catalysts without the need for any additional solvents. The nature of the support may lead to different reaction pathways. It has been suggested that rhenium over-reduction is detrimental to catalytic performance, but the influence and role of its oxidation state has not yet been fully elucidated.With regard to the one-pot hydrogenation of ML to 1-PAO, bimetallic Re/Ru deposited on zeolite showed an interesting performance, leading to MV + VA yields of up to 65%; also, only traces of 1-PAO derivatives were found after 4 h at 230 °C. The calculated productivity of MV + VA in these conditions, at the best of our knowledge, was the second-highest ever reported in literature.
Considering that the increase of the reaction times only slightly increases the 1-PAO derivatives yield, a two-step approach for a more efficient production of these compounds has been proposed. Indeed, while in the first step MV/VA can be produced with an acceptable yield from ML, a second step for the further conversion of valeric compounds to 1-PAO, PV, or DPE has been investigated. We thus found that, starting from VA, a 27% PV yield could be achieved after 4 h at 210 °C. Nevertheless, an extensive rhenium leaching (of around 30–40%) has been observed with the use of neat VA. Interestingly, by working with MV, this issue can effectively be limited to below 4%, obtaining a PV yield of around 20% in the optimized condition over Re–O/SiO2.
In the near future, further study will be crucial for highlighting the reaction mechanism over the surface and identifying an effective strategy for stabilizing Re species, thus increasing catalyst stability and recyclability.
Author contributions
Riccardo Bacchiocchi: conceptualization, methodology, investigation, validation, visualization, and writing – original draft. Jacopo De Maron: conceptualization, methodology, investigation, discussion and suggestions. Tommaso Tabanelli: conceptualization, investigation, validation, visualization, and writing – review and editing. Daniele Bianchi: resources, discussion, funding acquisition, review and editing, and supervision. Fabrizio Cavani: conceptualization, resources, discussion, funding acquisition, review and editing, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Eni SpA is acknowledged for the PhD grant assigned to RB.Notes and references
- IPCC, Climate Change 2014 Synthesis Report, 2014 Search PubMed.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- J. De Maron, L. Bellotti, A. Baldelli, A. Fasolini, N. Schiaroli, C. Lucarelli, F. Cavani and T. Tabanelli, Sustainable Chem., 2022, 3, 58–75 CrossRef CAS.
- J. De Maron, M. Eberle, F. Cavani, F. Basile, N. Dimitratos, P. J. Maireles-Torres, E. Rodriguez-Castellón and T. Tabanelli, ACS Sustain. Chem. Eng., 2021, 9, 1790–1803 CrossRef CAS.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass, 2004, 1, 76 Search PubMed.
- V. Rajendaren, S. M. Saufi and M. A. K. M. Zahari, Biomass Convers. Biorefin., 2022 DOI:10.1007/s13399-022-03444-7.
- K. Yan, C. Jarvis, J. Gu and Y. Yan, Renew. Sustain. Energy Rev., 2015, 51, 986–997 CrossRef CAS.
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657–1667 CrossRef CAS PubMed.
- A. Hommes, A. J. ter Horst, M. Koeslag, H. J. Heeres and J. Yue, Chem. Eng. J., 2020, 399, 125750 CrossRef CAS.
- B. Mallesham, P. Sudarsanam, B. Venkata Shiva Reddy, B. Govinda Rao and B. M. Reddy, ACS Omega, 2018, 3, 16839–16849 CrossRef CAS PubMed.
- A. M. R. Galletti, C. Antonetti, V. De Luise and M. Martinelli, Green Chem., 2012, 14, 688–694 RSC.
- G. Grillo, M. Manzoli, F. Bucciol, S. Tabasso, T. Tabanelli, F. Cavani and G. Cravotto, Ind. Eng. Chem. Res., 2021, 60, 16756–16768 CrossRef CAS.
- P. Blair Vásquez, T. Tabanelli, E. Monti, S. Albonetti, D. Bonincontro, N. Dimitratos and F. Cavani, ACS Sustain. Chem. Eng., 2019, 7, 8317–8330 CrossRef.
- T. Tabanelli, Curr. Opin. Green Sustainable Chem., 2021, 29, 100449 CrossRef CAS.
- T. Tabanelli, E. Paone, P. Blair Vásquez, R. Pietropaolo, F. Cavani and F. Mauriello, ACS Sustain. Chem. Eng., 2019, 7, 9937–9947 CrossRef CAS.
- S. Dutta and N. S. Bhat, ChemCatChem, 2021, 13, 3202–3222 CrossRef CAS.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez De María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- I. Obregõn, I. Gandarias, N. Miletic, A. Ocio and P. L. Arias, ChemSusChem, 2015, 8, 3483–3488 CrossRef PubMed.
- I. Obregón, I. Gandarias, M. G. Al-Shaal, C. Mevissen, P. L. Arias and R. Palkovits, ChemSusChem, 2016, 9, 2488–2495 CrossRef PubMed.
- Y. B. Huang, A. F. Liu, Q. Zhang, K. M. Li, W. B. Porterfield, L. C. Li and F. Wang, ACS Sustain. Chem. Eng., 2020, 8, 11477–11490 CrossRef CAS.
- T. Mizugaki, Y. Nagatsu, K. Togo, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, Green Chem., 2015, 17, 5136–5139 RSC.
- M. Li, G. Li, N. Li, A. Wang, W. Dong, X. Wang and Y. Cong, Chem. Commun., 2014, 50, 1414–1416 RSC.
- Z. Yu, X. Lu, J. Xiong and N. Ji, ChemSusChem, 2019, 12, 3915–3930 CrossRef CAS PubMed.
- T. Pan, J. Deng, Q. Xu, Y. Xu, Q. X. Guo and Y. Fu, Green Chem., 2013, 15, 2967–2974 RSC.
- W. Luo, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2014, 320, 33–41 CrossRef CAS.
- M. Muñoz-Olasagasti, A. Sañudo-Mena, J. A. Cecilia, M. L. Granados, P. Maireles-Torres and R. Mariscal, Top. Catal., 2019, 62, 579–588 CrossRef.
- P. Sun, G. Gao, Z. Zhao, C. Xia and F. Li, ACS Catal., 2014, 4, 4136–4142 CrossRef CAS.
- Z. Yi, D. Hu, H. Xu, Z. Wu, M. Zhang and K. Yan, Fuel, 2020, 259, 3–6 CrossRef.
- N. Karanwal, D. Verma, P. Butolia, S. M. Kim and J. Kim, Green Chem., 2020, 22, 766–787 RSC.
- K. Kon, W. Onodera and K. I. Shimizu, Catal. Sci. Technol., 2014, 4, 3227–3234 RSC.
- W. Luo, U. Deka, A. M. Beale, E. R. H. Van Eck, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2013, 301, 175–186 CrossRef CAS.
- M. Tamura, Y. Nakagawa and K. Tomishige, Asian J. Org. Chem., 2020, 9, 126–143 CrossRef CAS.
- J. Pritchard, G. A. Filonenko, R. Van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- M. Toba, S. I. Tanaka, S. I. Niwa, F. Mizukami, Z. Koppány, L. Guczi, K. Y. Cheah and T. S. Tang, Appl. Catal. Gen., 1999, 189, 243–250 CrossRef CAS.
- Z. Wang, G. Li, X. Liu, Y. Huang, A. Wang, W. Chu, X. Wang and N. Li, Catal. Commun., 2014, 43, 38–41 CrossRef CAS.
- D. Tharuk and W. Carrick, Copper chromite hydrogenation catalysts for production of fatty alcohols, WO 2012/074841, 2012.
- X. Di, C. Li, B. Zhang, J. Qi, W. Li, D. Su and C. Liang, Ind. Eng. Chem. Res., 2017, 56, 4672–4683 CrossRef CAS.
- J. D. Kammert, A. Chemburkar, N. Miyake, M. Neurock and R. J. Davis, ACS Catal., 2021, 11, 1435–1455 CrossRef CAS.
- M. L. Gothe, K. L. C. Silva, A. L. Figueredo, J. L. Fiorio, J. Rozendo, B. Manduca, V. Simizu, R. S. Freire, M. A. S. Garcia and P. Vidinha, Eur. J. Inorg. Chem., 2021, 2021, 4043–4065 CrossRef CAS.
- T. Toyao, S. M. A. H. Siddiki, A. S. Touchy, W. Onodera, K. Kon, Y. Morita, T. Kamachi, K. Yoshizawa and K. I. Shimizu, Chem.–Eur. J., 2017, 23, 1001–1006 CrossRef CAS PubMed.
- T. Toyao, S. M. A. H. Siddiki, Y. Morita, T. Kamachi, A. S. Touchy, W. Onodera, K. Kon, S. Furukawa, H. Ariga, K. Asakura, K. Yoshizawa and K. I. Shimizu, Chem.–Eur. J., 2017, 23, 14848–14859 CrossRef CAS PubMed.
- T. Toyao, K. W. Ting, S. M. A. H. Siddiki, A. S. Touchy, W. Onodera, Z. Maeno, H. Ariga-Miwa, Y. Kanda, K. Asakura and K. ichi Shimizu, Catal. Sci. Technol., 2019, 9, 5413–5424 RSC.
- Y. B. Huang, T. Yang, Y. T. Lin, Y. Z. Zhu, L. C. Li and H. Pan, Green Chem., 2018, 20, 1323–1334 RSC.
- J. Zhang, S. Bin Wu, B. Li and H. D. Zhang, ChemCatChem, 2012, 4, 1230–1237 CrossRef CAS.
- A. M. Raspolli Galletti, C. Antonetti, S. Fulignati and D. Licursi, Catalysts, 2020, 10, 1–2 CrossRef.
- H. Ariba, Y. Wang, C. Devouge-Boyer, R. P. Stateva and S. Leveneur, J. Chem. Eng. Data, 2020, 65, 3008–3020 CrossRef CAS.
- A. Démolis, N. Essayem and F. Rataboul, ACS Sustain. Chem. Eng., 2014, 2, 1338–1352 CrossRef.
- H. Joshi, B. R. Moser, J. Toler, W. F. Smith and T. Walker, Biomass Bioenergy, 2011, 35, 3262–3266 CrossRef CAS.
- L. Negahdar, M. G. Al-Shaal, F. J. Holzhäuser and R. Palkovits, Chem. Eng. Sci., 2017, 158, 545–551 CrossRef CAS.
- H. Chen, Q. Xu, D. Zhang, W. Liu, X. Liu and D. Yin, Renewable Energy, 2021, 163, 1023–1032 CrossRef CAS.
- Z. Yang, Y. B. Huang, Q. X. Guo and Y. Fu, Chem. Commun., 2013, 49, 5328–5330 RSC.
- Y. Kuwahara, Y. Magatani and H. Yamashita, Catal. Today, 2015, 258, 262–269 CrossRef CAS.
- D. Ren, X. Wan, F. Jin, Z. Song, Y. Liu and Z. Huo, Green Chem., 2016, 18, 5999–6003 RSC.
- P. M. Ayoub and J.-P. Lange, Process for converting levulinic acid into pentanoic acid, WO 2008/142127, 2008.
- L. Wei, C. S. Cheung and Z. Huang, Energy, 2014, 70, 172–180 CrossRef CAS.
- N. Yilmaz and A. Atmanli, Fuel, 2017, 191, 190–197 CrossRef CAS.
- J. Campos-Fernandez, J. M. Arnal, J. Gomez, N. Lacalle and M. P. Dorado, Fuel, 2013, 107, 866–872 CrossRef CAS.
- M. Marchionna, R. Patrini, F. Giavazzi, M. Sposini and P. Garibaldi, World Pet. Congr. Proc., 2000, 3, 38–45 Search PubMed.
- J. P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479–4483 CrossRef CAS PubMed.
- K. G. Martínez Figueredo, E. M. Virgilio, D. J. Segobia and N. M. Bertero, Chempluschem, 2021, 86, 1342–1346 CrossRef PubMed.
- K. G. Martínez Figueredo, D. J. Segobia and N. M. Bertero, Energy Convers. Manag., 2022, 13, 0–11 Search PubMed.
- K. G. Martínez Figueredo, E. M. Virgilio, D. J. Segobia and N. M. Bertero, React. Chem. Eng., 2022, 7, 1997–2008 RSC.
- W. Li, Y. Li, G. Fan, L. Yang and F. Li, ACS Sustain. Chem. Eng., 2017, 5, 2282–2291 CrossRef CAS.
- C. E. Chan-Thaw, M. Marelli, R. Psaro, N. Ravasio and F. Zaccheria, RSC Adv., 2013, 3, 1302–1306 RSC.
- K. Yan, T. Lafleur, X. Wu, J. Chai, G. Wu and X. Xie, Chem. Commun., 2015, 51, 6984–6987 RSC.
- B. Rozmysłowicz, A. Kirilin, A. Aho, H. Manyar, C. Hardacre, J. Wärnå, T. Salmi and D. Y. Murzin, J. Catal., 2015, 328, 197–207 CrossRef.
- C. Bolivar, H. Charcosset, R. Frety, M. Primet, L. Tournayan, C. Betizeau, G. Leclercq and R. Maurel, J. Catal., 1975, 39, 249–259 CrossRef CAS.
- J. Q. Bond, D. Martin Alonso, R. M. West and J. A. Dumesic, Langmuir, 2010, 26, 16291–16298 CrossRef CAS PubMed.
- Y. Amada, Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal. B Environ., 2011, 105, 117–127 CrossRef CAS.
- M. Tamura, Y. Amada, S. Liu, Z. Yuan, Y. Nakagawa and K. Tomishige, J. Mol. Catal. Chem., 2014, 388–389, 177–187 CrossRef CAS.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 3213–3226 CrossRef CAS.
- M. T. Greiner, T. C. R. Rocha, B. Johnson, A. Klyushin, A. Knop-Gericke and R. Schlögl, Z. Phys. Chem., 2014, 228, 521–541 CrossRef CAS.
- C. Bandinelli, B. Lambiase, T. Tabanelli, J. De Maron, N. Dimitratos, F. Basile, P. Concepcion, J. M. L. Nieto and F. Cavani, Appl. Catal. Gen., 2019, 582, 117102 CrossRef CAS.
- K. Baranowska, J. Okal and N. Miniajluk, Catal. Lett., 2014, 144, 447–459 CrossRef CAS.
- J. He, Z. Wu, Q. Gu, Y. Liu, S. Chu, S. Chen, Y. Zhang, B. Yang, T. Chen, A. Wang, B. M. Weckhuysen and T. Zhang, Angew. Chem., Int. Ed., 2021, 60(44), 23713–23721 CrossRef CAS PubMed.
- L. Chen, J. Fu, L. Yang, Z. Chen, Z. Yuan and P. Lv, ChemCatChem, 2014, 6, 3482–3492 CrossRef CAS.
- M. W. Schreiber, D. Rodriguez-Niño, O. Y. Gutiérrez and J. A. Lercher, Catal. Sci. Technol., 2016, 6, 7976–7984 RSC.
- J. Chen and Q. Xu, Catal. Sci. Technol., 2016, 6, 7239–7251 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2se01583h |
| This journal is © The Royal Society of Chemistry 2023 |