
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAptamers 101: aptamer discovery and in vitro applications in biosensors and separations
Lucy F.
Yang
,
Melissa
Ling
,
Nataly
Kacherovsky
and
Suzie H.
Pun
 *
*
Department of Bioengineering and Molecular Engineering and Sciences Institute, University of Washington, Seattle, Washington, USA. E-mail: spun@uw.edu
First published on 2nd May 2023
Abstract
Aptamers are single-stranded nucleic acids that bind and recognize targets much like antibodies. Recently, aptamers have garnered increased interest due to their unique properties, including inexpensive production, simple chemical modification, and long-term stability. At the same time, aptamers possess similar binding affinity and specificity as their protein counterpart. In this review, we discuss the aptamer discovery process as well as aptamer applications to biosensors and separations. In the discovery section, we describe the major steps of the library selection process for aptamers, called systematic evolution of ligands by exponential enrichment (SELEX). We highlight common approaches and emerging strategies in SELEX, from starting library selection to aptamer-target binding characterization. In the applications section, we first evaluate recently developed aptamer biosensors for SARS-CoV-2 virus detection, including electrochemical aptamer-based sensors and lateral flow assays. Then we discuss aptamer-based separations for partitioning different molecules or cell types, especially for purifying T cell subsets for therapeutic applications. Overall, aptamers are promising biomolecular tools and the aptamer field is primed for expansion in biosensing and cell separation.
 Lucy F. Yang | Lucy F. Yang completed her PhD at the University of Washington under the supervision of Prof. Suzie H. Pun. Her thesis work investigated SARS-CoV-2 spike protein-binding aptamers for diagnostics and therapeutics. She received her B.S. from the Massachusetts Institute of Technology. Her other research areas include polymers for drug delivery and microfluidics for cancer applications. |
 Melissa Ling | Melissa Ling graduated with a Bachelor's degree in Biomedical Engineering from Penn State and is currently a doctoral graduate student studying Molecular Engineering at the University of Washington. She has research and internship experience in chromatography and cell-based assays. Currently, she is interested in aptamer applications for cell purification and diagnostic devices. |
 Nataly Kacherovsky | Nataly Kacherovsky is a research scientist at Bioengineering department of the University of Washington, Seattle. She is leading the aptamer discovery group in Prof. Suzie Pun's lab. Nataly was born in Moscow, former USSR, and received her M.S. from Lomonosov Moscow State University in Molecular Genetics. She continued her studies under the guidance of Prof. Sergey Mirkin in the Institute of the Molecular Genetics trying to solve the mysteries of DNA H-form. After the collapse of the Soviet Union, she immigrated to Seattle, WA and joined the laboratory of Prof. Ted Young at the Biochemistry department at UW, studying the regulation of ADH2 transcription in yeast for 15 years. For a year she tried making living as an artist doing jewelry and pottery, but when the opportunity to join Prof. Pun came along, she didn't hesitate to jump on a new project. Nataly is passionate about aptamers, art, and Boston terriers. |
 Suzie H. Pun | Suzie H. Pun is the Washington Research Foundation Professor of Bioengineering and incoming Director for the Molecular Engineering and Sciences Institute. She is a fellow of the U.S. National Academy of Inventors (NAI) and American Institute of Medical and Biological Engineering (AIMBE). Suzie Pun received her B.S. from Stanford University and her PhD in Chemical Engineering from the California Institute of Technology. Her current work focuses on aptamer and polymer development for therapeutic and diagnostic applications in cancer, trauma, and infectious disease. |
Introduction
Aptamers are short, single-stranded nucleic acid molecular recognition agents that fold into 3D conformations and specifically bind to targets like proteins, peptides, small molecules, and metal ions. Most aptamers are discovered through a library selection process called systematic evolution of ligands by exponential enrichment (SELEX), which uses a desired target to enrich for binders among many random sequences. Utilized in research fields from biosensing to therapeutics, aptamers have versatile applications but have yet to reach their full potential.Aptamers are similar in function to antibodies, which are currently the most widely used molecular recognition agent. Aptamers can have very high affinity (as low as ∼10 picomolar1) and specificity (e.g. with the ability to distinguish between single amino acid differences in proteins2) for their targets. However, unlike antibodies, aptamers are chemically synthesized and thus are less expensive and faster to produce, more homogenous with less batch-to-batch variation, and more amenable to controlled chemical modifications.3–6 Moreover, DNA aptamers are more stable at a range of ionic conditions, pH, temperatures, and other storage conditions compared to protein antibodies7,8 (Table 1). Despite these advantages, aptamers currently lag behind in diversity of discovered sequences and commercial development compared to antibodies. Some potential causes include the labor-intensive nature of aptamer discovery and limitations due to early patent protection of aptamers.9 Today, however, advances in aptamer discovery and expiration of key patents around 2010 have allowed the field to flourish.
| Aptamer | Antibody | ||
|---|---|---|---|
| a Cost for DNA with no or simple chemical modifications (ex. thiol, amide, biotin) | |||
| Intrinsic qualities | Affinity | 10 pM to10 μM | 10 pM to10 μM |
| Specificity | Highly specific | Highly specific | |
| Molecular weight | 10–20 kDa | 150 kDa (IgG) | |
| Synthesis method | Chemical | Biological | |
| Stability | pH 5 to 9, –80 to 100 °C, liquid or dry | pH 5 to 8, sequence specific, empirically determined, −80 to 4 °C | |
| Application-related qualities | Selection speed | Days to weeks in vitro | Weeks in vivo |
| Variability | Very uniform | Batch to batch | |
| Commercial cost of 1 mg | ∼$50a | ∼$2000–5000 | |
| Chemical modifications | 5′ end, 3′ end, internal; controlled | Primary amine, carboxylic acid, thiol chemistries; difficult to control | |
RNA enzymes, the stepping stones for aptamers, are nucleic acids that can fold into complex structures and catalyze reactions, garnering research interest in the 1980s.10–13 In 1990, three research groups independently described iterative in vitro selection methods for RNA enzymes,14–16 which Tuerk and Gold coined SELEX.15 Ellington and Szostak expanded on the previous work and named the nucleic acids that were output of selections “aptamers”: ‘apta’ from aptus meaning ‘fitting’ in Latin.17 Many other major milestones in aptamer history occurred in quick succession. In 1992, the first DNA aptamers, thrombin-binding aptamer and specific dye-binding aptamers, were discovered.18,19 The first aptamers isolated by cell SELEX were reported in 1999 and bound to the parasite Trypanosoma brucei.20 In 2004, the first aptamer therapeutic drug, Macugen, was approved by the FDA to treat macular degeneration.21 Over time, technical advancements in DNA manipulation and sequencing, as well as new developments in selection strategies, have streamlined the selection progress and lowered the barrier to entry. However, the core idea behind SELEX remains unchanged for over 30 years. In that time, aptamer literature has grown from about 20 articles published in 1990 to over 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 articles published in 2020.
000 articles published in 2020.
In this review, we will describe aptamer selection and optimization strategies and then examine biomedical applications of aptamers in in vitro settings such as biosensing and cell separation. Overall, this review will prepare readers for entering the world of aptamers as it stands today, especially in light of the COVID-19 pandemic. For further reading, we recommend many other excellent general aptamer reviews22–25 as well as more focused reviews.26–30
Panning for gold: aptamer sequence discovery & optimization
As the primary method for discovering new aptamers, SELEX is the heart of aptamer research (Fig. 1). In SELEX, a starting library is incubated with desired molecular targets. In each selection round, bound sequences are recovered and PCR-amplified to create a new pool of aptamer sequences, which are used in the subsequent selection round. Typically, between five to 20 rounds are necessary to enrich the aptamer pool to find target-binding aptamers. In this section, we will cover basic concepts of and considerations for SELEX.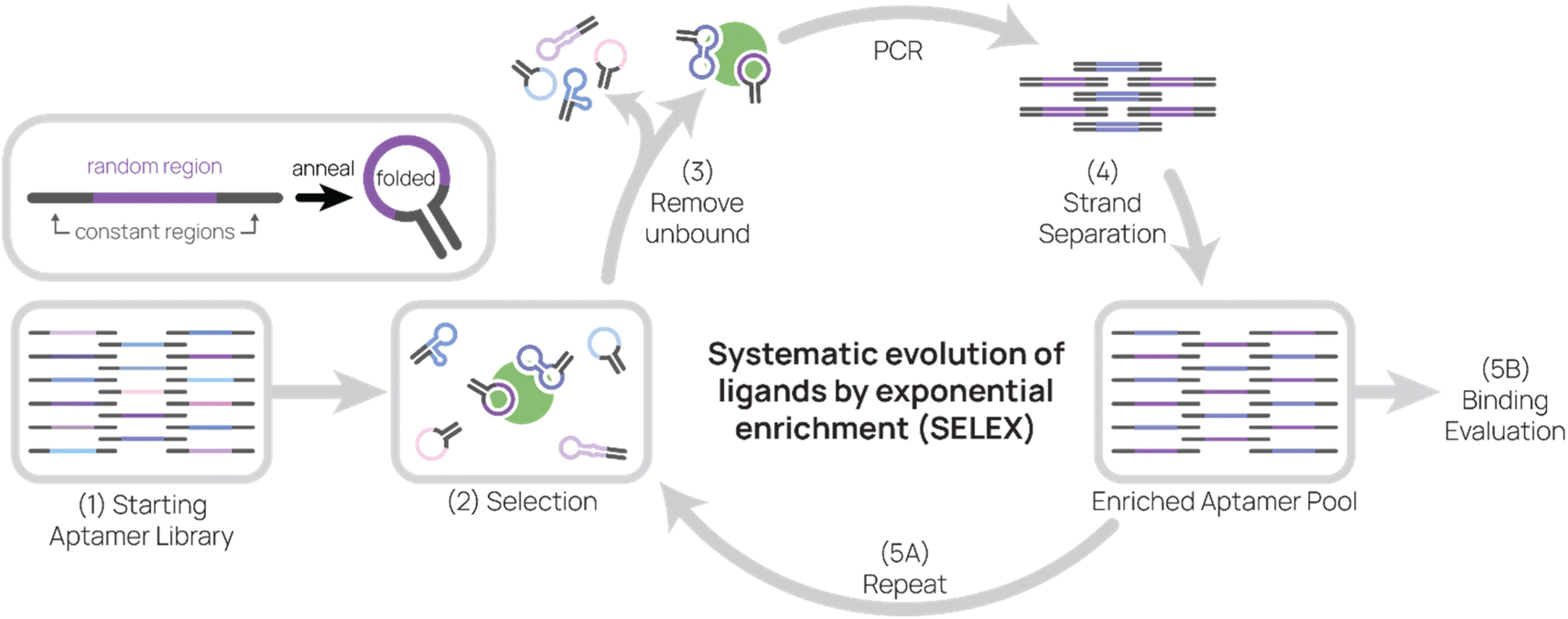 | ||
| Fig. 1 Systematic evolution of ligands by exponential enrichment (SELEX). Aptamers are composed of a random region and constant regions. After annealing, aptamers fold and adopt secondary structures that may allow them to bind to targets. (1) The starting aptamer library contains 1014–1015 unique sequences. (2) Then the aptamer library is incubated with target during selection. (3) Unbound aptamers are removed, and target-binding aptamers are eluted. (4) After conducting PCR on aptamers, sense and anti-sense strands are separated. (5) The enriched aptamer pool is used in the next round of selection and evaluated for binding. | ||
Starting library selection
The properties of the aptamer library are important to optimize for successful selection. In this section, we will focus on (A) the constant region, (B) the random region, (C) the library length, (D) pre-structured libraries, and (E) the type of nucleic acid.Aptamers typically consist of a random region flanked by two constant regions, which allow PCR amplification via complementary primers (Fig. 1, top left inset). The random region is theoretically unique for each aptamer in a starting library, and the longer the random region, the more possible candidate sequences there are. For example, a complete library containing a 40-nt random region would have 440 or ∼1024 unique sequences, which would weigh ∼50 kg in its entirety. Practically speaking, selection libraries are smaller and usually contain 1014–1015 unique sequences.31–33
The choice of aptamer length and random region length may influence SELEX success. Aptamers have been discovered in libraries with random regions as large as 220-nt and as short as 22-nt.31 One study compared the effect of six different random region lengths (16, 22, 26, 50, 70, 90) on target-binding sequence abundance and found that the target motif was most abundant in the 50-nt and 70-nt selections.34 In recent publications, a random region of 36- to 52-nt in length have been the most common, which is about 70- to 90-nt in total aptamer length. DNA synthesis costs also play a factor, as longer syntheses become more expensive and less efficient. For example, if the efficiency of adding a nucleotide base is 99.5%, an 80-mer aptamer is produced with 69% yield but a 160-mer aptamer is produced at 45% yield.
Pre-structured libraries are one strategy for increasing the probability of successful selection. One pre-structured library stabilizes the aptamer structure by incorporating a double-stranded stem formed by the constant regions. This predictable final structure also simplifies aptamer truncation designs.35 Another library strategy is guanine (G)-rich sequences in the random region to increase the probability of G quadruplexes,36 which are stable secondary structures formed by stacks of four guanidine units. Aside from their unique structure, G quadruplexes are of interest because they have important functions in DNA replication and repair, epigenetics, and other pivotal cellular processes.37 G quadruplexes are also often found in high-affinity aptamers,38,39 therefore increasing their frequency in the starting library may lead to higher chances of success.
Although early aptamer research focused on RNA aptamers, the majority of recently discovered and applied aptamers are DNA-based. Both DNA and RNA can form secondary and tertiary structures, though RNA backbone is more flexible. Compared to RNA, DNA is more stable and does not require reverse transcription step during amplification, thus making selection less tedious.24,29
Non-natural or chemically-modified nucleic acids also increase aptamer library diversity by increasing access to available binding epitopes on proteins.40 In one recent work, McCloskey et al. used alpha-L-threofuranosyl nucleic acid (TNA) to make backbone- and base-altered aptamers capable of undergoing in vitro selection and binding target proteins.41 These TNA aptamers (“threomers”) contain traditional bases as well as modified bases containing phenylalanine and tryptophan, which are planar aromatic amino acids often found at the antibody–antigen interface. From TNA libraries with ∼1014 unique instances of 40-nt random region, McCloskey et al. enriched for threomers that bind SARS-CoV-2 S1 protein or TNFα, and many threomers possessed nanomolar to sub-nanomolar affinities after optimization. However, some drawbacks of using threomers included the need for lengthy chemical synthesis (14 to 29 steps per TNA nucleoside triphosphate) and optimization of TNA polymerases. Similarly, the “click-SELEX” strategy uses alkyne-modified uridine (EdU) that are functioned by azide-modified groups.42 Unlike TNAs, EdU is commercially available and therefore DNA amplicons can be generated to include EdU in place of dT using conventional DNA polymerases and PCR methods. Click-SELEX can be employed with an azide that contains a group also found on the target's natural binders in order to improve binding probability.
Another non-natural nucleic acid strategy is to use mirror image aptamers made from L-RNA or L-DNA, called spiegelmers.43 Unrecognized by nucleases and DNA processing enzymes, spiegelmers are more stable in vivo but also are not easy to PCR amplify in selection. To circumvent the amplification problem, Williams et al. conducted SELEX with the enantiomer of the target peptide and D-RNA then synthesized the spiegelmers based on the discovered D-RNA sequence.44 However, challenges in generating target enantiomers limit the discovery of spiegelmers. The need for non-natural enantiomers is bypassed if the target is a symmetric molecule, such as biphenol A (BPA). In one recent work, Ren et al. developed BPA-binding spiegelmer for gold nanoparticle-based colorimetric detection of BPA.45
Selection targets
Incipient aptamer research identified binders of dyes on agarose beads,19 but today, aptamer binding targets have expanded to include metal ions, small molecules, peptides, and even molecules (usually receptor proteins or glycans) expressed on the surface of bacteria, virus, or human cells.A plethora of cell types have been used as selection targets,46 including cancer cells,39,47,48 T cells,33 and bacteria.49 The protocol for cell-SELEX is straightforward to perform and does not require many specialized tools.50 Cells are easily partitioned by centrifugation, leaving unbound aptamers in suspension. The cell-bound aptamers are eluted by incubating at high temperatures then separating by centrifugation. Aside from its simple protocol, cell-SELEX is advantageous because of its authentic target presentation on the cell surface and variety of targets.46 However, some challenges of cell-SELEX include binding reaction temperature and receptor identification. In our process, we conduct selection at 4 °C to avoid aptamer internalization, which would remove candidates from the pool. Consequentially, identified aptamers perform well at 4 °C, but may require optimization for binding at room temperature or 37 °C. However, cell-SELEX can also be performed at 37 °C because both internalized and surface-bound aptamers can be recovered upon cell lysis. Chemical extraction methods, such as TRIzol, can recover internalized RNA aptamers.51 The other major hurdle is identifying the aptamer's target, usually a membrane protein, which can be identified by pull-down and mass spectrometry.52 However, target identification is not straightforward because of non-specific pulldown and multiple potential targets. A newer method, stable-isotope labeling by amino acids in cell culture (SILAC) measures aptamer-specific pulldown by comparing pulldown in cells cultured with heavy lysine and arginine to that of “light” unlabeled cells.53 After optimization of binding, cross-linking, and lysis conditions for each aptamer, the SILAC-based workflow was used to identify two targets of aptamers with previously unknown receptors. Overall, there still remains many cell-targeting aptamers with unidentified targets and new strategies, and new approaches for receptor identification are needed.
Another prevalent selection target is recombinant protein. Unlike cell-based selection, protein-SELEX can be done at any temperature and does not require a receptor identification step. In fact, further studies, such as protein binding studies and cryo-EM,35 can precisely identify the binding epitope. However, the presentation of the protein may not be faithful to its native conformation, which may lead to aptamers that perform well in protein binding studies but not in its application. To circumvent this, hybrid cell- and protein-SELEX has been a successful approach. Pioneering this strategy, Hicke et al. used tumor cells then recombinant protein for selecting aptamers against tenascin-C, a protein implicated in tumor growth.47 Many aptamers were also identified by combined SELEX strategies. C42-aptamer, which targets human programmed cell death protein 1, was found after 13 rounds of protein then three rounds of cell SELEX.54 Wilner et al. discovered transferrin receptor-targeting RNA aptamers after selection against recombinant transferrin receptor protein for four rounds and Jurkat cells for the fifth round.55 SARS-CoV-2 spike protein-binding aptamer SNAP4 was found after six rounds of selection with HEK293 stably expressing spike protein and four rounds of protein SELEX.56 Using multiple protein targets within the same SELEX can also yield high affinity aptamers. Zhang et al. completed 13 selection rounds with wild-type SARS-CoV-2 S1, but then conducted five separate selection rounds using different variants of SARS-CoV-2 S protein.57
Another issue with protein-SELEX is that typically embedded or inward-facing portions of membrane proteins can be exposed during selection, potentially allowing aptamers to bind to an inaccessible location in the native state. One strategy to avoid this issue is to conduct selection with enveloped viruses. In “viro-SELEX”, the target membrane proteins are expressed on the viral envelope of a surrogate baculovirus, allowing native conformation expression.58 Similarly, Peinetti et al. conducted SELEX with target viruses to identify pathogen-binding aptamers.59 Lastly, protein-SELEX also struggles with enriching aptamers that bind to protein tags or solid supports. Many recombinant proteins are produced with a tag, such as His-tag or biotin, so the protein can be purified. In addition, many selection strategies utilize the tag to bind the protein to solid supports, which aid in partitioning. However, aptamers may bind to tags and solid supports instead of the target during SELEX, inadvertently enriching non-target binding aptamers.
For both cell- and protein-SELEX, negative selection or counter-selection is a common strategy used to reduce enrichment of off-target binders. During a round of selection, the aptamer pool is incubated with an undesired target, and those binders are depleted from the pool. For example, in some of our successful selections, the aptamer library is incubated with His-tagged non-target protein (negative selection), then after partitioning, the remaining sequences are incubated with His-tagged target protein (positive selection.)35,60
Partitioning bound and unbound aptamers
Separating binding and non-binding aptamers during selection is a key component of SELEX. With cell-SELEX, the partitioning is conveniently performed by centrifugation. For protein-SELEX, libraries are typically partitioned by two major methods. One method is capillary electrophoresis SELEX (CE-SELEX), which separates free aptamer, protein, and aptamer–protein complexes from each other due to varying electrophoretic mobility constants. CE-SELEX was first developed in 2004,61 and many variations exist today for improving certain aspects of the process: single step CE-SELEX (ssCE-SELEX) streamlines the selection in a single run to save time and materials;62 fraction collection CE-SELEX (FCE-SELEX) reduces contamination by loading PCR reagents directly into oil-sealed mixtures;63 and ideal-filter capillary electrophoresis (IFCE) tunes the ionic strength of the running buffer to migrate non-binding nucleotides in the opposite direction of nucleoprotein complexes, dramatically increasing the efficiency of separation by eliminating carry-over of non-binding aptamers.64 Overall, CE-SELEX offers tunable, quantifiable partitioning, but is limited by several factors. CE-SELEX can only process a small volume, which limits the library size and diversity sampled. CE-SELEX also requires size and charge differences between individual components and aptamer-target complex in order to be separated, and this is challenging for small molecule and membrane protein targets.Another approach for protein-SELEX is immobilization of protein onto magnetic spheres for magnetic separation. This technique was first used in 2005 as a less wasteful alternative to immobilization on columns,65 and today many SELEX strategies use magnetic spheres. Polyhistidine tags (His-Tag) on proteins are immobilized onto nickel-nitriloacetic acid (Ni-NTA) or cobalt-based magnetic spheres.32,35,66 Since this method only requires cheap, commercially available components like magnetic spheres and magnetic rack, protein SELEX with magnetic spheres is very accessible. Some drawbacks of magnetic selection include labor-intensive manual pipetting and dead volume during washing that may carry over non-binding aptamers. Other less frequently used methods also exist, such as repurposing surface plasmon resonance (SPR) chips67,68 and biolayer interferometry (BLI) biosensors69,70 for selection.
Recently, more efficient and accurate separation methods have emerged to improve the affinity and specificity of final aptamer candidates.71 One of these strategies is particle display developed by the Soh group, where emulsion PCR allows amplification and immobilization of each unique aptamer sequence to a particle, which is easily analyzed and collected by fluorescence-activated cell sorting.72 This technology also enables simultaneous screening for both affinity and specificity by labeling target and non-target, respectively, with different fluorescent colors.
Altogether, aptamer researchers today can choose from a diverse cast of SELEX targets and partitioning methods, not to mention combinations of multiple approaches and emerging techniques.
Secondary library preparation (PCR & strand separation)
After collecting aptamers that bind the target, it's necessary to amplify the pool and isolate the sense strand for further selection or binding analysis. The most common method for strand separation is PCR using a biotin-labeled reverse strand for pulldown. The PCR products are captured on streptavidin agarose and the desired strand is eluted by gentle denaturation followed by buffer exchange.50 The biotinylated PCR product pulldown method is high yield, commercially available, and quick, making it the most commonly used strand separation strategy. Another method employs lambda nuclease, which preferentially digests DNA with 5′ phosphorylation.73 A 5′ phosphorylated reverse primer is used in PCR, allowing specific digestion of the anti-sense strand. A third method for library preparation is asymmetric PCR.74,75 Forward and reverse primers are used in unequal molar ratios in order to preferentially amplify the sense strand. Then, the PCR product is separated by gel electrophoresis and ssDNA is eluted from the gel in a labor-intensive, low-yield process.Improving and evaluating library binding
Once a cycle of SELEX is completed, the resulting library can be used in another round of SELEX to further hone the library toward higher percentages of target-binding sequences.For each selection iteration, “knobs” can be adjusted to tune the stringency and desired results. For example, the stringency can be increased through: a decrease of aptamer library concentration; a decrease in incubation time or concentration of target; addition or increased concentration of competitors, such as bovine serum albumin (BSA), yeast tRNA, salmon sperm DNA, and dextran sulfate; addition or increased concentration of surfactants, such as Tween-20; an increase in wash steps; addition or increased concentration of counterselection target; a change in target, for instance to a smaller portion of the target protein or alternative cell line. Generally, these selection conditions are empirically determined. If conditions are too stringent, the amplification of the aptamer pool could be unviable or take a large number of PCR cycles. If conditions were too relaxed, the number of PCR cycles may be very small and successive pools may not increase binding signal. Because the first round of selection contains only unique aptamers, it is better to start with gentle conditions to avoid loss of potential binding candidates early in the selection process.
The aptamer library can be evaluated for target binding to monitor selection progress. Before modern methods, radioactive isotopes were used to label aptamers, but now, many SELEX protocols use fluorescently labeled primers. Apparent KD binding can be measured by methods like cell flow cytometry,56,66 dot blot assay,57 and direct ELISAs.56,76 Techniques that directly measure binding kinetics include surface plasmon resonance (SPR)32,77 and biolayer interferometry (BLI).35,56
Binding assessment between rounds is important for adjusting the SELEX strategy. For example, if no binding is observed, the strategy needs major reworking and SELEX can be restarted from an earlier round or restarted entirely from the beginning. When the binding signal is saturated in latter rounds, SELEX can conclude and the libraries can be sequenced. However, amplification of aptamer library to conduct binding studies requires significant time and materials investment, which can be more effectively spent on performing more SELEX rounds.
Lastly, the aptamer round libraries are sequenced and analyzed (Fig. 2). Next generation sequencing (NGS) is the primary tool for sequencing because it is commercially available, requires very little aptamer library relative to binding studies, and accurately outputs sequences. In our process, we typically read 100000 to 500000 complete aptamer sequences per library pool. To analyze the primary sequence, several software and web tools are commonly used. FASTAptamer ranks aptamer sequences by frequency and fold enrichment,78 Multiple Em for Motif Elicitation (MEME) Suite groups aptamers by motifs,79 G-quadruplex prediction software “Quadruplex forming G-Rich Sequences” (QGRS) predicts the presence of G-quadruplexes,80 FigTree visualizes sequences in a phylogenetic tree,81 and DNAMAN is an all-in-one molecular biology tool. Tools for secondary structure prediction include NUPACK, which predicts nucleic acid structure and aids in design,82 and Mfold, which predicts folding and hybridization based on free energy.83
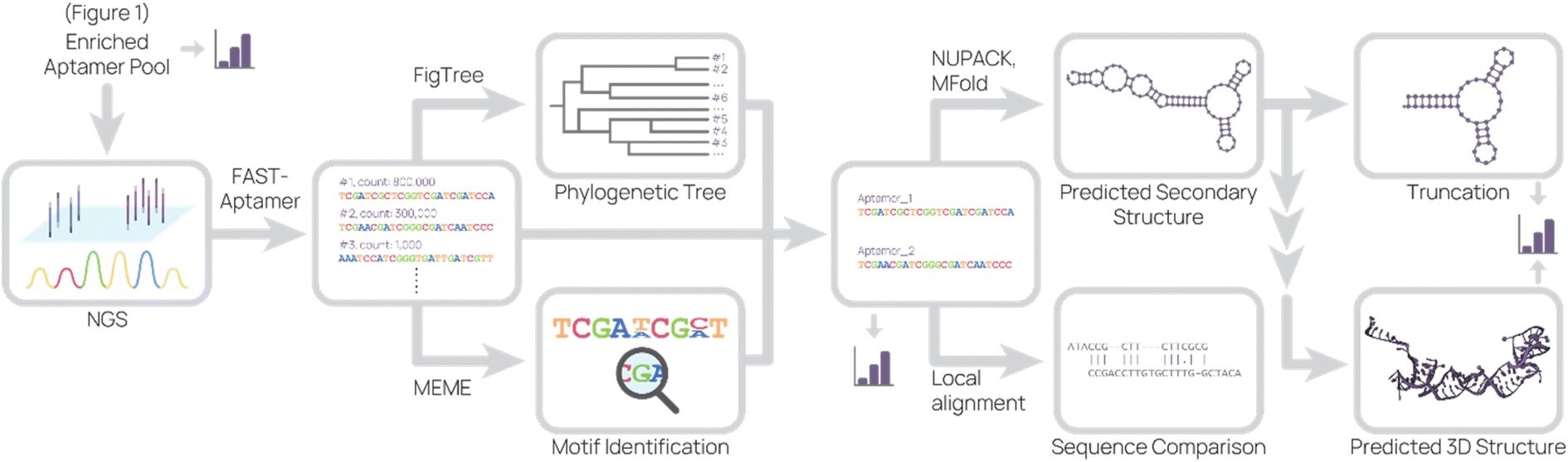 | ||
| Fig. 2 Representative sequence analysis pipeline. The enriched aptamer pool(s) from SELEX were sequenced, and the data set was analyzed to produce a list of aptamer sequences and their relative abundance. This list was used to generate a phylogenetic tree and to search for motifs. Combining information from these analyses, candidate aptamers were chosen for further investigation. The predicted 2D structure and sequence similarity to other aptamers were generated based on the candidate aptamer sequence. From the predicted 2D structure, truncations were designed and 3D structure were predicted. At several points (bar graph icon), binding studies were conducted. | ||
Based on these data, sequences can be grouped into families and consensus sequences picked to synthesize and further characterize. In an ideal selection, the binding sequences are ones with the highest frequency, the most common motif, increased frequency in later rounds, or greatest fold enrichment in one round. In some selections, however, the best-binding sequences may be less frequent or contain a less common motif.60 For example, it's possible that some binding sequences are less favored during amplification due to secondary structure.84 It is therefore prudent to pick sequences based off a minimal cut-off, such as sequences containing a unique motif and at least 1000 reads per million. Parasitic sequences can also appear in high frequency despite not being target binders, occluding the best-binding sequences.56 We hypothesize that parasitic sequences originate from PCR bias or contaminating oligos in reagents or equipment, but further investigation is needed.
In our experience, we have found that simple primary and secondary structure predictions are sufficient to continue aptamer characterization and applications. However, visualizing the aptamer binding site is important for understanding the binding mechanism. To that end, more computationally intensive methods are needed for tertiary structure prediction, which results in predicted 3D aptamer structures for in silico docking studies. There are several methods for tertiary structure prediction. In general, the aptamer sequence is converted to RNA for secondary and tertiary structure prediction, converted back to DNA, then refined by structural energy minimization.85 Molecular docking-based and machine learning-based methods are best reviewed in the cited articles due to the complex nature of these analyses.85,86 Overall, in silico experiments are useful to generate hypotheses, but further experiments are still necessary to define predicted aptamer–target interactions.
After picking aptamer sequences to test, the previously mentioned techniques for assessing aptamer library binding are also used to evaluate individual sequence binding. In our experience, if little enrichment was observed (i.e. later aptamer pools remained diverse) or no candidate sequences bind, the selection strategy needs revision. For example, the conditions could have been too harsh, removing potential binders, or the intended target was not presented properly (i.e. a recombinant cell membrane protein presented in a non-natural orientation or context.)
Other considerations
Typically, SELEX is conducted by manual pipetting, but microfluidic and computational approaches are under investigation for automating the process. Initially prototyped in 2006,87 microfluidic SELEX platforms can precisely control fluids and temperature in order to mix, incubate, and partition SELEX reagents from aptamer library introduction to secondary library generation. Several microfluidic systems that automate part of the SELEX process have been proposed,88 including systems that utilize magnetic particles or a class of molecularly imprinted polymers (MIPs) called sol–gel. Recently, Sinha et al. developed a microfluidic system capable of automating most of the process on-chip (negative selection, positive selection, PCR, competitive assay).89 Despite these advances, microfluidic SELEX requires substantial technical know-how and investment to set up, such as master molds for pouring polydimethylsiloxane (PDMS) and controllers for air valves and temperature regulation. In contrast, traditional SELEX uses tools readily available in most molecular biology labs (thermocycler, magnetic rack, etc.) therefore, additional investigation is needed to commercialize and disseminate the technology.Computational approaches to SELEX have also been investigated. In silico binding design can save time and reagents by performing docking simulations instead of tedious benchwork. This concept has been readily applied to de novo protein switches and biosenors.90,91 In addition, in silico SELEX can circumvent some inherent flaws in the process, such as avoiding the amplification of sequences that are preferentially PCR amplified or transcribed.92 Computationally-aided93 and combined computation and high-throughput array SELEX schematics72 have recently been conducted. However, in silico methods are hampered by the difficulties in accurately predicting aptamer structure, especially 3D DNA structures. A more accurate method for modeling is cryogenic electron microscopy (cryo-EM), which is difficult due to the flexibility and small size of short single-stranded DNA. For example, we recently developed a cryo-EM density-guided model of the DNA aptamer tJBA8.1 binding to its target transferrin receptor 1.94 However, our attempt to model the 3D structure of tJBA8.1 from its 2D prediction failed to match the cryo-EM map, indicating that in silico tools need further improvement to accurately predict structures and binding.
In vitro applications
Aptamers are being applied to biomedical problems in a multitude of use cases, from sensitive drug detection to being a drug itself. In this review, we focus on recent in vitro applications in two main sections: biosensing (with an emphasis on SARS-CoV-2 applications) and separations.Biosensing
Biosensors are systems that recognize biological inputs and output electrical, electrochemical, or optical signals indicating presence or absence of an analyte. Biosensors use biological recognition agents to detect the analyte and transducers to read signal.95 Biosensors typically produce results quickly (on the order of minutes to hours), in real-time, and sometimes continuously. Many biosensors are also easy to operate and inexpensive to produce, increasing the accessibility of the device. These key properties make biosensors suitable for applications such as point-of-care rapid diagnostics of diseases and monitoring of biomolecules in the environment.Not only are aptamers capable of molecular recognition like antibodies, but also they are smaller in size, easier to chemically modify, robust across various sample conditions, and more affordable to produce. These properties uniquely position aptamers as a recognition agent of choice in biosensing. The aptamer's compact size (∼101 kDa) is essential to decrease the distance between an electrode and the binding event, allowing for sensitive detection of electrochemical signal changes. Addition of functional groups and molecules onto nucleic acids is straightforward and controlled, and therefore less polydisperse and more consistent than chemistries conducted on antibodies.3–6 Lastly, aptamers can withstand harsh cleaning treatments and reuse unlike antibodies,96 allowing for repeated measurements on the same device.
Due to the aforementioned advantages, aptamers have been used in biosensors across many fields of study. In particular, the COVID-19 pandemic has shined a spotlight on biosensors for disease detection.97–100 Although PCR testing has been the gold standard, the need for rapid, point-of-care results led to the dominance of lateral flow assays and investigation into other rapid testing technologies, including aptamer-based detection methods. In this section, we will focus on biosensors organized by detection method through the lens of SARS-CoV-2 antigen detection.
Electrochemical biosensors
Electrochemical aptamer-based (E-AB) sensors are one type of label-free biosensor that employs conformational change of the aptamer upon binding to produce an electrical signal change (Fig. 3A). Typically, E-ABs require three electrodes, one of which is the aptamer-modified gold working electrode. A region of the aptamer containing redox reactive molecules, such as methylene blue or ferrocene, moves relative to the electrode after target binding. A “signal-on” E-AB produces the signal after binding, while a “signal-off” E-AB interrupts the signal after binding. The electrical signal change can be a change in potential, current, or impedance depending on the setup.101,102 Potential for high sensitivity, label- and sample processing-free prep, real-time detection of signal, and reusability give E-ABs distinct advantages over other diagnostic platforms.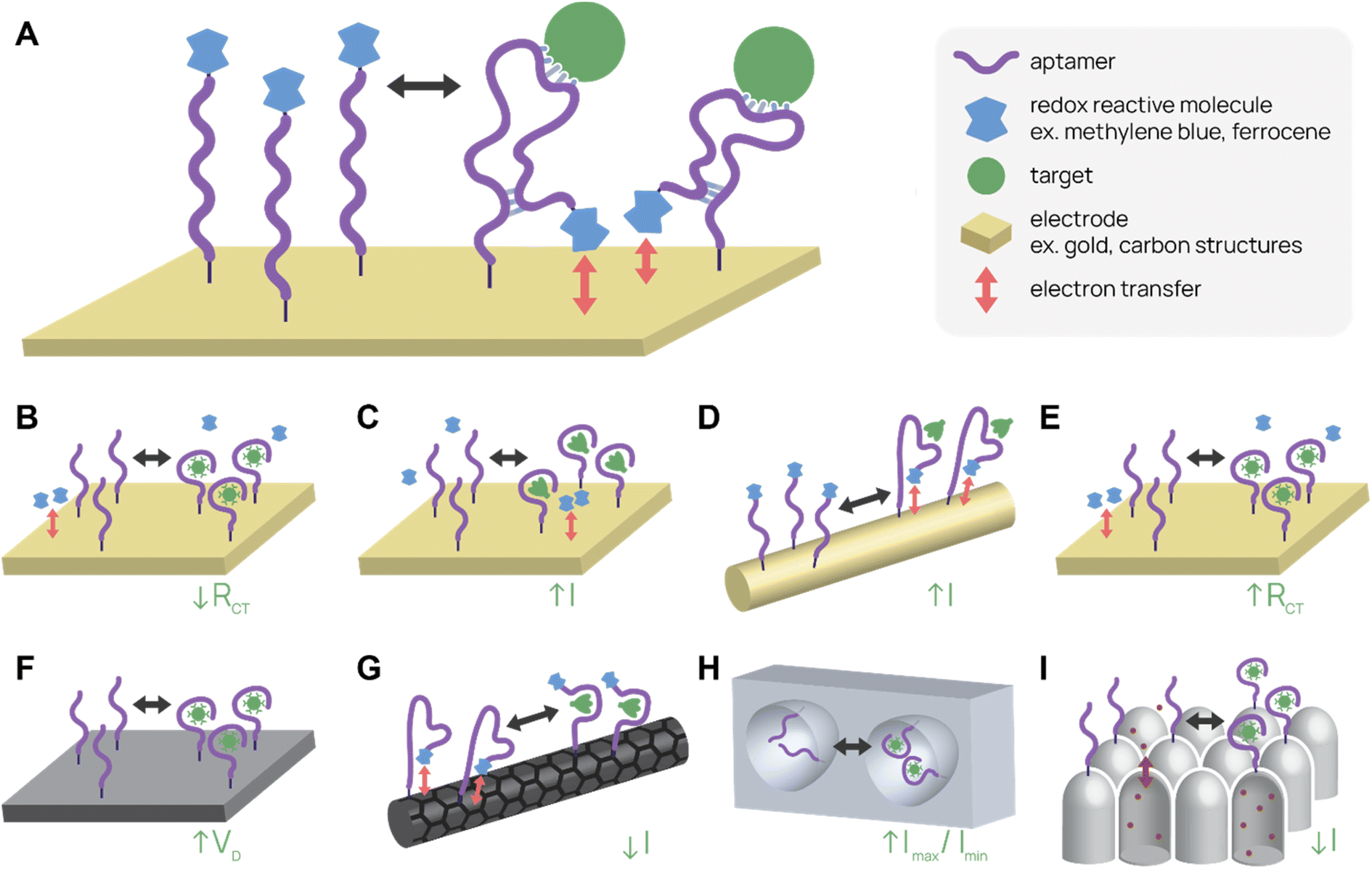 | ||
| Fig. 3 Electrochemical aptamer-based (E-AB) biosensor diagrams. (A) In a typical “signal-on” E-AB, aptamers change conformation after target binding, resulting in an increase in electrical signal. (B–I) SARS-CoV-2 detecting E-ABs. (B) Virus detection from Lasserre et al.110 (C) Spike protein detection from Martínez-Roque et al.111 (D) Spike protein detection from Idili et al.112 (E) Virus detection by “Cov-eChip” from Zhang et al.113 (F) Virus detection from Ban et al.114 (G) Spike protein detection from Curti et al.115 (H) Virus detection from Peinetti et al.59 (I) Virus detection from Shi et al.116 | ||
One of the most-cited E-AB platforms is a thrombin-detecting “signal-off” E-AB from the Plaxco group.103 A thrombin-binding aptamer was functionalized with methylene blue, a redox agent, and conjugated to a gold working electrode. When the aptamer bound thrombin, its new conformation shifted the methylene blue away from the electrode, reducing electron transfer and signal. This thrombin E-AB demonstrated rapid detection, nanomolar sensitivity, and reusability, igniting further research into E-ABs. Today, research groups have developed different electrode set ups and detection schemes, as well as expanded the menu of analytes, which now includes small molecule drugs (cocaine,104 ampicilin105), proteins (CRP,106 insulin107), and viruses (zika virus,108 avian flu,109 SARS-CoV-2 (ref. 59 and 110–116)).
Several recent publications describe aptamer-functionalized gold electrode E-ABs for detection of SARS-CoV-2 proteins and virus. Lasserre et al. used thin film gold electrodes (used in blood glucose test strips) functionalized with SARS-CoV-2 spike protein-binding aptamers to detect target by electrochemical impedance spectroscopy (Fig. 3B).110 The system detected 80 ng mL−1 (∼1 nM) of SARS-CoV-2 spike subunit 1 (S1) protein and distinguished between SARS-CoV-2 positive and negative clinical samples. In a different report, Martínez-Roque et al. described an aptamer-functionalized gold electrode E-AB to detect SARS-CoV-2 spike protein with sub-femtomolar sensitivity (0.007 to 700 fM dynamic range) (Fig. 3C).111 Idili et al. achieved picomolar precision (10−11 to 10−9 M dynamic range) detection of SARS-CoV-2 spike protein or receptor binding domain (RBD) with an aptamer-functionalized gold wire electrode (Fig. 3D).112 Lastly, Zhang et al. functionalized dimeric DNA aptamers to gold electrodes for SARS-CoV-2 protein and virus detection, named Cov-eChip (Fig. 3E).113 Notably, Cov-eChip detected SARS-CoV-2 with 80.5% sensitivity and 100% specificity in 73 unprocessed patient saliva samples, which outperformed all commercial and published rapid tests available.
There are also non-gold electrode E-ABs with similar detection strategies. Ban et al. recently reported a DNA aptamer-conjugated graphene field-effect transistor (GFET) platform for detecting SARS-CoV-2 spike protein and intact inactivated virus at <2 PFU mL−1 (Fig. 3F).114 This system was able to accurately distinguish between 10 positive and negative wild-type SARS-CoV-2 patient samples with 100% sensitivity and specificity. Curti et al. developed DNA aptamer-functionalized single-walled carbon nanotube screen-printed electrodes (SWCNT-SPEs) for detecting SARS-CoV-2 spike protein (Fig. 3G).115
In addition to utilizing electrical properties, E-ABs can use shape to specifically amplify detection signals. Peinetti et al. use aptamer-functionalized nanopores to confine and detect pseudotyped SARS-CoV-2 particles at as low as 104 copies per mL, even in undiluted human saliva (Fig. 3H).59 Remarkably, the platform can distinguish between UV-inactivated and intact SARS-CoV-2 pseudovirus because the pore entrance is narrow, increasing the variation in current signal upon virus binding. Shi et al. uses aptamer-functionalized nanochannels to detect as low as 1 fM of SARS-CoV-2 S1 protein and SARS-CoV-2 in clinical pharyngeal swabs (Fig. 3I).116 The analyte obstructs ion transport across the nanochannel and increases the zeta potential of nanochannel surface, resulting in a change in current.
Colorimetric biosensors
Colorimetric biosensors rely on color change to indicate detection. The most well-known example is the lateral flow assay (LFA), which were first pioneered in urine pregnancy tests117 and recently in COVID-19 rapid antigen tests. LFAs are portable, single-use paper devices that rely on molecular recognition agents, such as antibodies and aptamers, to capture and detect antigens. Typically, sample is applied to an absorbent pad and travels across the test strip by capillary action. When the target antigen is present, a test band appears because of antigen-specific accumulation of nanoparticles (Fig. 4A). LFAs are low-cost and easy to use, making them an effective tool for point-of-care diagnostics.118,119 Using aptamers in place of antibodies can further improve LFAs. Aptamers are easier to chemically modify, cost less to synthesize, are more consistent batch-to-batch, and possess longer shelf life.119 For example, aptamers can be commercially ordered with a thiol end group, which conjugates to gold nanoparticles with simple chemistry and consistent orientation. In contrast, antibodies need to be functionalized at existing chemical groups for covalent conjugation, which requires more complex chemistry and inconsistency in antibody orientation and number of functionalized sites.4,56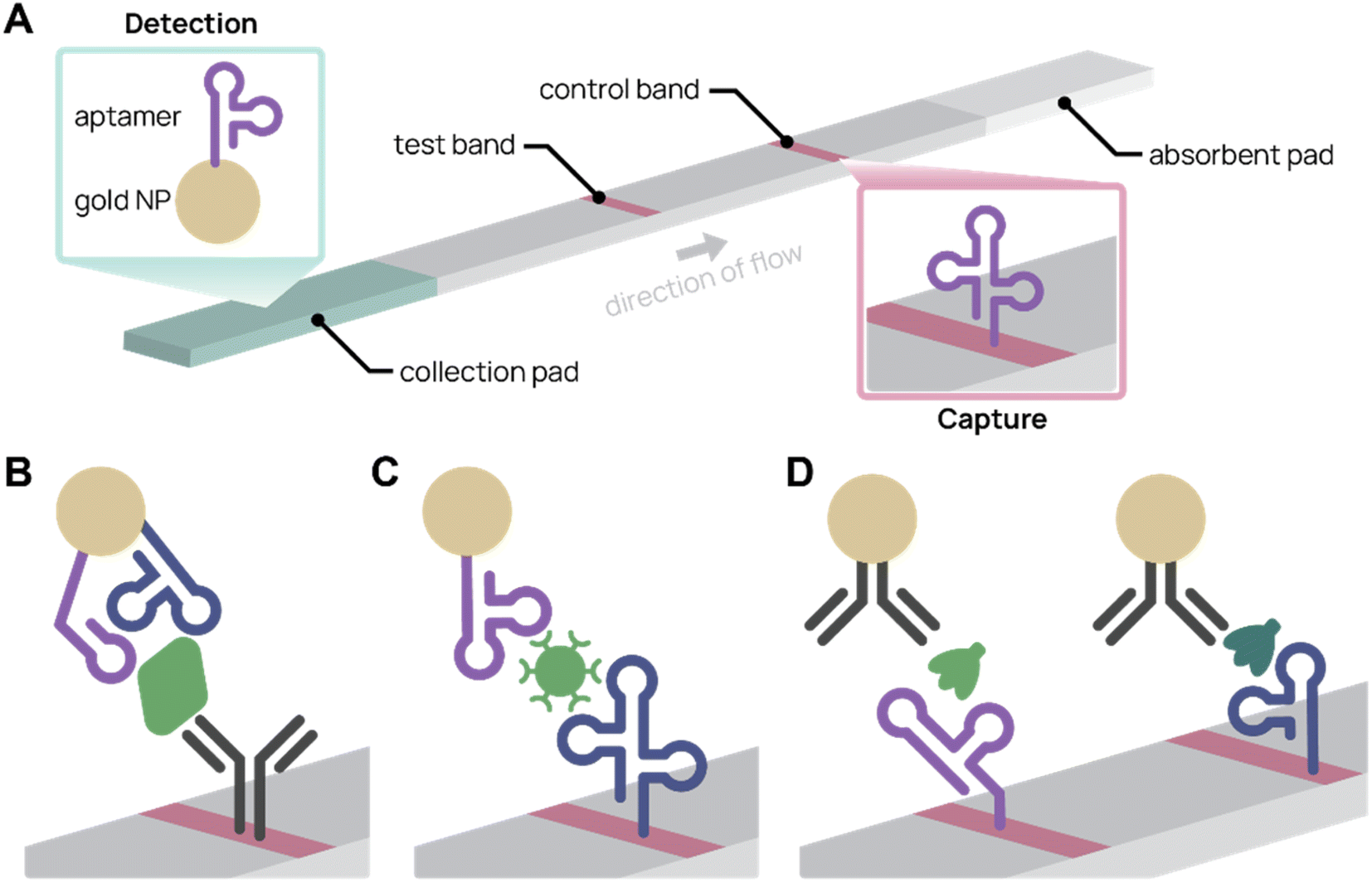 | ||
| Fig. 4 Aptamer lateral flow assay (LFA) diagrams. (A) In a typical aptamer sandwich LFA, target molecules within a sample bind both the aptamer-conjugated gold nanoparticle (NP) detection agent (green) and the aptamer capture agent (pink), turning the test band dark. (B)–(D) Schematics showing only the test bands during detection. (B) Aptamer-antibody LFA detecting SARS-CoV-2 nucleocapsid protein (green) with two different aptamer sequences on the gold NP. Adapted from Zhang et al.77 (C) Antibody-free aptamer LFA (“AptaFlow”) detecting SARS-CoV-2 virus (green) with one aptamer for capture and another for detection. Adapted from Yang et al.56 (D) Multiplex LFA for detecting two variants of SARS-CoV-2 spike protein (green and teal) using two test bands with unique aptamers. Adapted from Yang et al.60 | ||
Several aptamer LFAs have been developed for sensitive SARS-CoV-2 detection. Zhang et al. produced an LFA that uses an antibody to capture and two different nanoparticle-conjugated aptamers to detect as low as 20 pM SARS-CoV-2 nucleocapsid protein (Fig. 4B).77 We (the Pun group) developed several aptamer-based LFAs for SARS-CoV-2 detection by the spike protein.35,56,60 Notably, the antibody-free aptamer LFA (AptaFlow) uses two different SARS-CoV-2 spike protein binding aptamers for detection and capture, and can detect as low as 106 copies per mL of intact SARS-CoV-2 virus (Fig. 4C).56 In addition, we developed a multiplexed LFA that distinguishes between two variants of SARS-CoV-2 via variant-specific spike protein-binding aptamers (Fig. 4D).60
Other colorimetric biosensors have also been developed with aptamers in place of antibodies. Enzyme-linked immunosorbent assays (ELISAs) and enzyme-linked apta-sorbent assays (ELASAs) use antibodies or aptamers, respectively, to capture antigens. The molecular recognition agent is conjugated to an enzyme, such as horseradish peroxidase, to amplify signal by color-changing substrate. ELASAs are used to screen and characterize aptamers against SARS-CoV-2,120 but have limited diagnostic applications because they are not rapid and require lab processing.
Lastly, gold nanoparticle aggregation colorimetric biosensors rely on color change when aptamer-conjugated gold nanoparticles aggregate in the presence of antigen.121 Aithal et al. developed a gold nanoparticle agglomeration test using SARS-CoV-2 spike protein-binding aptamer-conjugated gold nanoparticles. The platform detected inactivated SARS-CoV-2 virus at concentrations as low as 3.54 × 106 copies per mL.122 Although rapid, gold nanoparticle aggregation biosensors require a absorbance reader to observe results near its limit of detection, which is not practical for point-of-care diagnostics.
Fluorescence biosensors
Fluorescence-based detection is also a key investigation area for aptamer biosensors. Although aptamers can be simply substituted in for antibodies in fluorescence read-out assays similar to ELASAs,111 another strategy is using a fluorophore and quencher to deliver signal. In one mode similar to “signal-on” E-ABs, the aptamer is designed such that the fluorophore is separated from the quencher when the aptamer binds to its target and changes conformation. Conversely, the aptamer can be designed to bring the fluorophore and quencher close together upon binding, inhibiting signal.123 Fluorophore-quencher aptamer biosensors have been designed for small molecule drugs and proteins.124,125 Some strategies leverage aptamer's special properties, such as taking advantage of the aptamer's DNA composition in order to specifically amplify signal. However, like ELASAs, fluorescence biosensors require an instrument to read results, limiting their appeal in COVID-19 diagnostics, which emphasizes point-of-care accessibility and speed.Chauhan et al. developed a net-shaped DNA probe (“DNA Net”) to detect whole SARS-CoV-2 virus by fluorescence.126 DNA Net was designed with three precisely spaced aptamers to bind to three sites on the trimeric spike protein. Upon aptamer binding, quencher-conjugated complementary DNA is displaced, releasing the reporter signal. DNA Net detected as low as 1000 copies per mL of SARS-CoV-2 virus, a sensitivity equivalent to standard PCR testing.
In a different strategy, Liu et al. designed an aptamer-assisted proximity ligation assay (Apt-PLA) that fluoresces during qPCR amplification.127 When two aptamer probes bind to the target protein, the ligation region of both probes are brought together, allowing ligase to connect the two probes. This completed stretch of DNA is amplified during real-time PCR, resulting in signal via a fluorescent reporter molecule. Apt-PLA detected SARS-CoV-2 nucleocapsid protein with a limit of detection of 37.5 pg mL−1, which is on par with commercial ELISA tests.
Gravimetric biosensors
Mass-sensitive detection is another label-free application avenue for aptamers. Microcantilevers are regularly used in microfluidic platforms for real-time, label-free mass measurements. When conjugated to the surfaces of microcantilevers, aptamers can immobilize their targets and cause a signal change in the microcantilever. In stress mode, the additional mass on the cantilever surface causes the microcantilever to bend, which can be measured by a laser and detector.128 Aptamer-functionalized microcantilevers have recently been applied to detect liver toxin microcystin-leucine–arginine (MC-LR),129 epithelial tumor marker Mucin 1,130 and tumor biomarkers (carcinoembryonic antigen and alpha-fetoprotein).131Summary
The field of aptamer-based biosensors is expanding rapidly with a wide variety of application strategies. E-ABs take advantage of the facile chemistries and small size of aptamers for real-time, label-free, sensitive detection of analytes. Despite these advantages, E-ABs have yet to reach their full potential in clinical translation. In future work, streamlining of E-AB manufacturing and miniaturization of the equipment required may make E-ABs more amenable to real-world use cases. As evident in the COVID-19 pandemic, LFAs are a powerful tool for home diagnostics as healthcare shifts focus to mobile care instead of facility-based care.132 Aptamer-based LFAs can retain the sensitivity of antibody-based ones while also reducing production costs and increasing shelf life. However, challenges still remain for non-LFA colorimetric and fluorescence biosensors because they often rely on instruments to readout sensitive detection. Another concern for many biosensors is decreased sensitivity in non-optimal buffers. For example, aptamer binding is sensitive to ionic strength,1 and many biosensor have lower detection signals in biological fluids and complex matrices (saliva,112 viral transport media,110,115 nasal swab56) compared to buffer optimized for the aptamer. Some aptamer biosensor platforms dilute the sample at the cost of losing sensitivity,113 while others perform well with environmental and biological samples with no pretreatment or dilution.59,116 Aside from improving aptamer technology, other factors can improve point-of-care COVID-19 tests, including internet integration, expanded testing sites, and big data analysis.133The SARS-CoV-2 aptamer biosensors discussed have future applications beyond the immediate needs of the COVID-19 pandemic. Because aptamers are modular, the aptamers used in biosensors can be replaced with other sequences without needing new chemistries or bioconjugation strategies. Aptamers that bind another SARS-CoV-2 variant or another virus target can be substituted in, rapidly re-tooling the biosensor for other variants or diseases. Overall, ongoing investigation into aptamer biosensors can improve their cost effectiveness and ease of use for clinical translation.
Separations
Affinity chromatography is a partitioning method widely used to purify molecules and targets from complex mixtures (Fig. 5). Due to their high specificity, high yield, and ease of operation, techniques in affinity chromatography have successfully been used in biomedical, clinical, chemical, and environmental applications.134,135 These techniques rely on selective and adjustable interactions between immobilized affinity ligands and their target analytes. Affinity ligands range from small molecules to large biomolecules. Antibodies are commonly used as affinity ligands due to their high affinity and specificity to target analytes. However, aptamers are promising alternatives to antibodies as affinity ligands for the extraction, separation, and purification of cells, proteins, and small molecules.136 The smaller size of aptamers (∼20 kDa for a 60-nt aptamer compared to 150 kDa for an antibody) allows higher grafting densities to solid supports, thereby increasing throughput. The synthetic nature of aptamers allows easier chemical modification and conjugation to chromatographic supports. In addition, increased stability and less batch-to-batch variability facilitates more robust protocols and higher purity end products. Aptamers also can withstand harsh cleaning treatments and more variable elution conditions (e.g. pH and temperature).96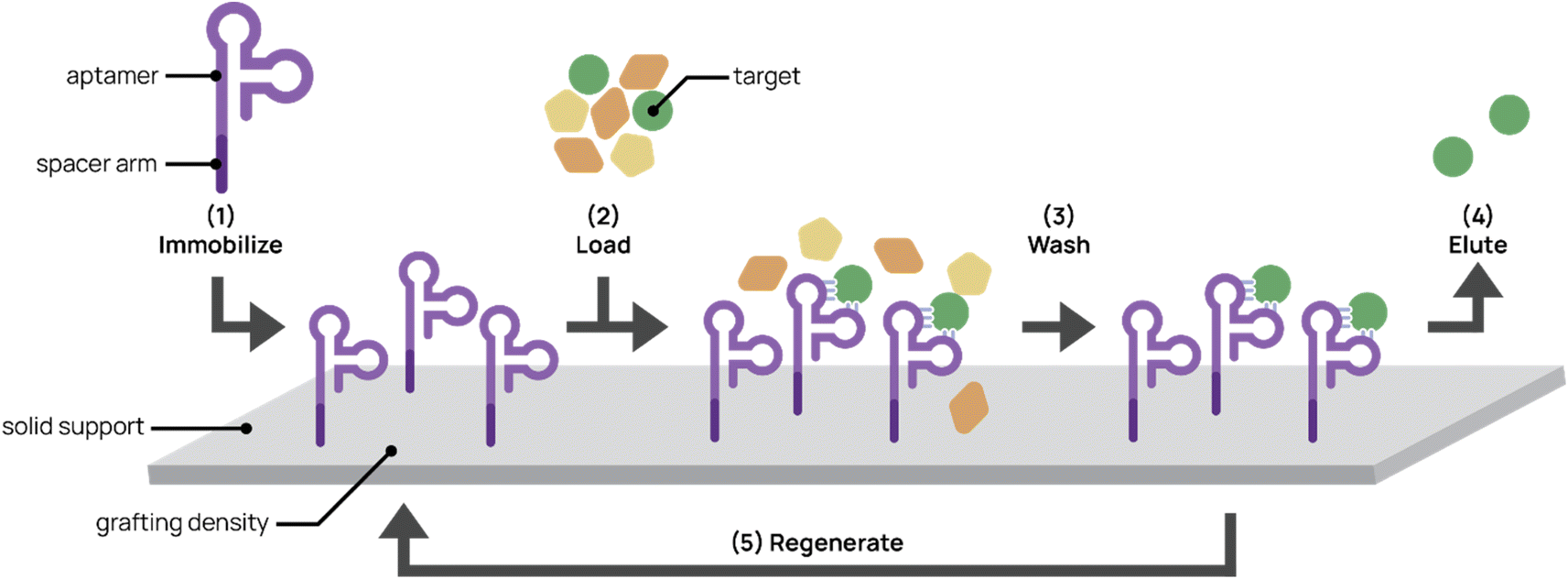 | ||
| Fig. 5 Aptamers for affinity chromatography. (1) Aptamers designed with spacer arms are immobilized onto a solid support. (2) Mixed input is loaded onto the platform and aptamer captures the target. (3) The platform is washed to remove non-targets. (4) The target is eluted from the support. (5) In some cases, the platform can be regenerated for repeated use. | ||
Optimization of aptamer-based affinity chromatography platform
There are several factors to consider in generating aptamer-bonded stationary phases for chromatography. First, aptamer ligands usually require a spacer arm modification prior to grafting to maximize specific binding interactions and reduce steric hindrance between the aptamer and its target. Commonly used spacer arms include PEG, carbon chains, and oligothymidine spacers between the aptamer and solid support surface. Lao et al. demonstrated through microarray aptamer detection studies that aptamers with oligothymidine or dodecyl spacers resulted in 100- to 1000-fold binding signal increases over aptamers without spacers.137In addition to the spacer arm, aptamer grafting densities require optimization. Aptamer concentrations that are too low can result in low binding efficiency. On the other hand, Lönne et al. showed that aptamer densities that are too high impaired binding for aptamer-based affinity purification of vascular endothelial growth factor from a complex protein mixture.138 High aptamer grafting density may interfere with proper aptamer folding and restrict steric accessibility to the target.
Aptamer ligands are compatible with a wide variety of solid matrices and immobilization techniques. Aptamers have traditionally been non-covalently attached to solid supports through biotin-labeling and binding to streptavidin-bonded supports.136,139 Romig et al. immobilized human L-selectin-binding DNA aptamer biotinylated at the 5′ end to a streptavidin-linked resin in an affinity column. By applying this aptamer column to Chinese hamster ovary cell-conditioned medium, they successfully achieved 1500-fold purification and 83% recovery of L-selectin. Although the biotin–streptavidin immobilization interaction is strong and easy to employ, its use is limited by a narrow range of ionic strength, temperature, and pH conditions and requires the use of recombinant proteins, increasing production costs.140 Thus, recent groups have taken advantage of the synthetic nature of aptamers to chemically conjugate aptamers to solid supports. Chemical conjugation confers higher stability on the aptamer chromatography platform, which can then be used with harsher elution conditions. The Scheper group modified anti-His-tag aptamers with terminal NH2 groups, activated the aptamers with cyanuric chloride, and covalently immobilized them onto amino-modified magnetic beads.141,142 Conjugation to amino-modified surfaces achieved more controlled aptamer orientation than that to aldehyde- or epoxide-modified surfaces because it minimized interactions within the aptamer sequence that interfered with proper folding. This aptamer platform successfully purified His-tagged proteins from crude E. coli lysates with purities between 58–84%, which was comparable to conventional immobilized metal affinity chromatography techniques with purities between 77–85%. More importantly, this covalent aptamer immobilization platform demonstrated potential regeneration ability and maintained functionality after six months in storage.
Aptamers have been immobilized to various solid support matrices including resin, magnetic nanoparticles (MNPs), monolithic capillary columns, and microfluidic channels. Resin-packed columns are traditionally used in affinity chromatography. For example, Forier et al. immobilized three different plasma-protein-binding, amine-functionalized aptamers to NHS-activated Sepharose and packed the resin beads into a column.96 With higher surface areas leading to higher binding capacities, the platform achieved high purity of target proteins. For example, coagulation FVII was separated from modified human plasma with 98% purity and 80% recovery, and human factor IX expressed in transgenic swine was isolated from milk at 98% purity in a single step. Resin platforms yield good performance due to higher surface areas compared to monoliths, membranes, and continuous-flow protocols, but can be subject to bed compaction, high pressure drops, and long processing times.143 Najafabadi et al. conjugated amino-modified, adenosine-binding DNA aptamers to glutaraldehyde-activated, amino-modified MNPs for adenosine partitioning.144 The MNP platform extracted adenosine from urine samples with an efficiency of 94% and a detection limit of 0.02 μg mL−1. In clinical applications to urine samples of lung cancer patients, the MNP platform achieved similar adenosine levels comparable to HPLC-UV analyses. Despite the high extraction efficiency of this technique, MNPs require magnets for washes and extraction, which becomes costly in industrial scale-up.
In contrast to packed resin columns, monolithic columns are composed of a single rigid porous element and offer greater mass transfer rate and lower back pressure. Zhao et al. developed an aptamer-based monolithic capillary platform for sensitive detection of thrombin in serum samples.145 The researchers performed in situ polymerization of the monolith inside a silanized capillary, coupled streptavidin to the monolithic column, and immobilized two different thrombin-binding, biotinylated aptamers to the streptavidin-modified monolithic column. With laser-induced fluorescent detection, the platform achieved a detection limit of 0.1 nM of thrombin pre-spiked into serum. Due to the high porosity of monoliths, the platform achieved faster convective flow than packed columns. However, tuning monolithic morphology is complex and ensuring specificity of monoliths for sample enrichment is not always consistent.146
Aptamers can also be used for partitioning in microfluidic devices, which offer advantages including rapid analysis, decreased sample and chemical consumption, and portability. Cho et al. developed a microfluidic chip with immobilized RNA aptamer on beads to detect hepatitis C virus (HCV) RNA polymerase from serum.147 Using a photocleavable linker elution strategy to attach the aptamer to the beads run through PDMS microchannels, the researchers successfully eluted and detected as low as 9.6 fmol of HCV RNA polymerase from patient serum.
An important design consideration in aptamer-based affinity chromatography are the wash and elution buffers. Wash buffers must preserve the structure and binding specificity of the aptamer. For instance, salt-containing SELEX wash buffers are commonly used as the affinity chromatography wash buffer. Elution buffers used at separation must be tested to preserve stability of the target molecule and facilitate dissociation of the aptamer-target complex. If aptamers were selected in buffers with metal ions (Ca2+, Mg2+), elution buffers containing EDTA, a biocompatible metal chelator, can disrupt aptamer structure and release the target without compromising target stability.
In 1999, the Drolet group first reported aptamer affinity chromatography for protein separations by using biotinylated DNA aptamer immobilized on streptavidin–polyacrylamide beads to purify human L-selectin-receptor globulin fusion protein from Chinese hamster ovary cell culture.139 Since then, aptamer affinity chromatography has been used widely in biomolecular separations. The Wang group successfully used DNA aptamer to extract lysozyme from a chicken egg white mixture with high precision and reproducibility.151 Zhao et al. also employed biotinylated DNA aptamers to separate cytochrome c and thrombin from human serum samples.145,152
One recent advance in aptamer affinity chromatography is streamlining separation procedures through single-step purification or fast, “one-pot” column syntheses,153,154 minimizing time and personnel effort and improving ease of separation. For example, Zhao et al. developed an aptamer-based hybrid affinity monolith at room temperature using photopolymerization and thiol–ene click chemistry that took only 7 minutes for complete synthesis.153 This platform detected ochratoxin A analytes in beer and red wine samples with yields >99% and lowered nonspecific adsorption up to 25-fold compared to monoliths formed by thermal polymerization.
Uniquely, aptamer ligands can be used with complementary reversal strands to separate target cells from a mixture in a label-free manner. Gray et al. immobilized EGFR-binding aptamer on magnetic beads or conjugated it to fluorescent dye to purify EGFR+ cells from a cell mixture using magnetic-activated or fluorescence-activated cell sorting, respectively (Fig. 6A).157 They achieved 99.9% purity and 33% recovery in EGFR+ cell purification from a 5% EGFR+ cell-spiked blood mixture with the complementary reversal strand. Importantly, they demonstrated that the reversal strand elution restored native EGFR phosphorylation levels, preserving cell viability and function. In another example, we discovered and applied an CD8+ T cell-binding aptamer with high affinity and specificity (KD ∼14.7 nM) to traceless T cell isolation for CAR T cell manufacturing (Fig. 6B).33 We selected CD8+ T cells from human peripheral blood mononuclear cells (PMBCs) through magnetic activated cell sorting with >95% purity and >72% yield. Compared to the standard isolation protocol, aptamers not only reduce manufacturing costs, but also create a less immunogenic product because aptamers can be reversibly denatured. Building on this work, we report in Cheng et al. a reversal strand for our CD71-binding aptamer, which we previously used to successfully deplete malignant cells from PBMC product in CAR T cell manufacturing (Fig. 6C).94 Upon combining CD71-binding aptamer and CD8-binding aptamer with their respective reversal strands in a single cell isolation, we successfully sorted out activated CD4+ and resting CD8+ T cells separately from the same bulk PBMCs with 93% and 85% purity, respectively.158 This aptamer-based multiplexed cell isolation system could decrease processing times and manufacturing costs and be applied to a diverse set of targets.
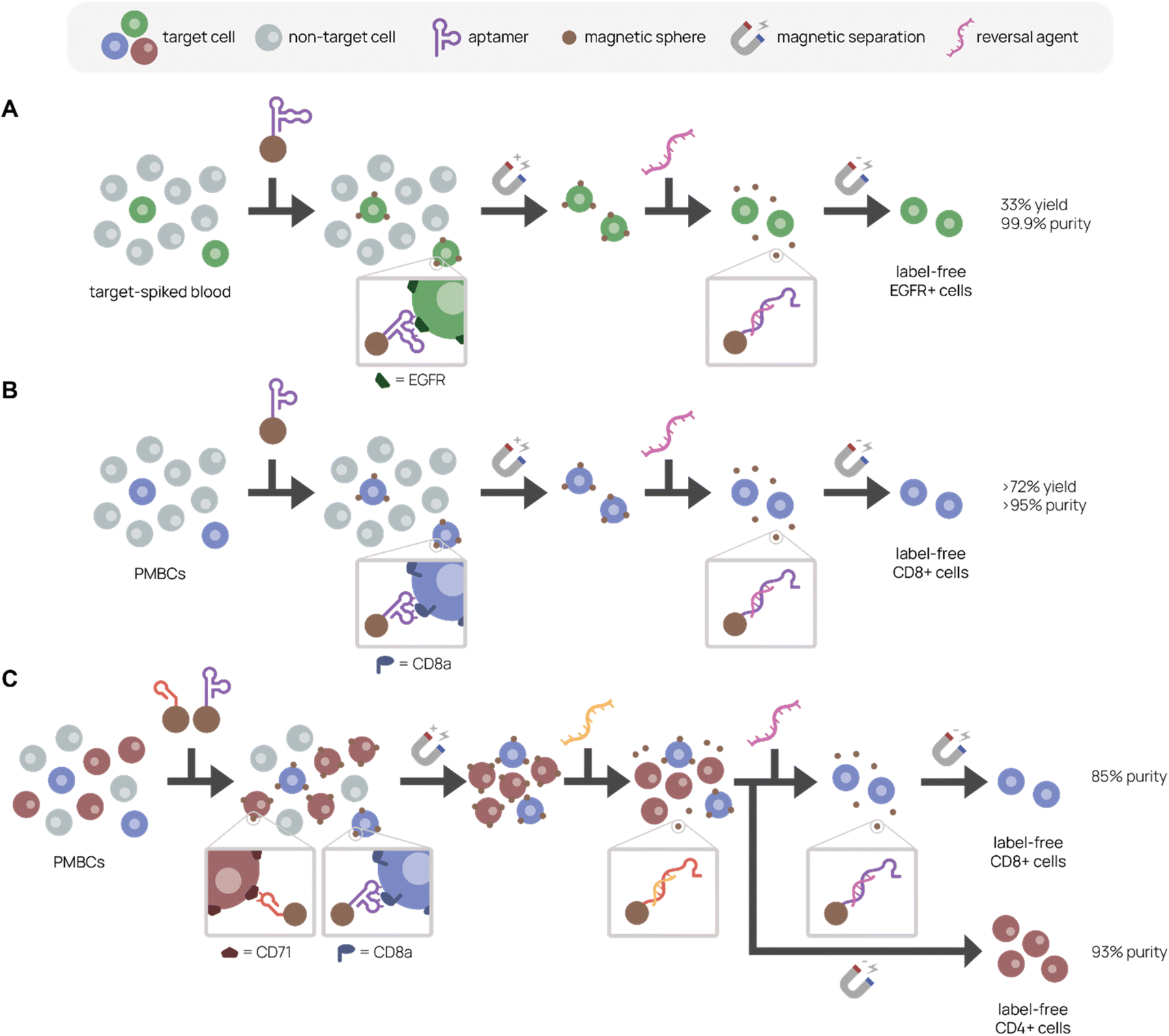 | ||
| Fig. 6 Aptamer-based platforms for label-free cell isolation. A mixed cell population is incubated with aptamer-labeled magnetic spheres, which bind specifically to the target cell. After magnetic separation to remove non-target cells (gray), labeled target cells remain. A reversal agent, or oligonucleotide complementary to a sequence within the aptamer, is added to release aptamer binding, resulting in a label-free target cell population. (A) Separation of EGFR+ cells (green) spiked in blood. Adapted from Gray et al.157 (B) Separation of CD8+ cells (purple) from PMBCs. Adapted from Kacherovsky et al.33 (C) Serial elution of CD4+ (red) and CD8+ (purple) cells from PBMCs. Adapted Cheng et al.94 | ||
Summary
Aptamers show great promise as affinity ligands in chromatographic separations of small molecules, proteins, and cells. With numerous advantages over antibody affinity ligands, such as greater stability, longer shelf-life, cheaper cost of manufacture, and ease of customizability, aptamers could be adapted in the future to industrial downstream purification processes. With complementary reversal sequences, aptamers can also achieve traceless and multiplexed target selection, thus improving product quality and decreasing processing times and manufacturing costs.Conclusion
Aptamers have a wide range of binding targets and applications, making them excellent tools among molecular recognition agents. Researchers use SELEX to identify new aptamers that bind cells, proteins, viruses, and other targets of interest. However, recent investigation into SARS-CoV-2 spike protein binding aptamers revealed the difficulty of isolating aptamers against a target that mutates faster than researchers can perform selection and create commercializable biosensors.60 Improving selection speed or easing the labor-intensive process of traditional SELEX are potential solutions to this problem. For instance, automation of benchwork or using computational methods to accelerate the selection process could improve selection speed.Compared to antibodies, aptamers lag behind in commercial support and development. For example, antibodies specific to SARS-CoV-2 variants were identified and characterized much faster than their aptamer counterparts. The antibodies were also disseminated more quickly, becoming commercially available for research purchase in the matter of weeks, despite their cell- or animal-based synthesis methods. Possible explanations for this delay in aptamer commercialization include SELEX being a rate-limiting step, nuclease degradation concerns for clinical samples, and lack of standard aptamer handling protocols.159 Given the surge of interest in aptamer-based biosensors due to COVID-19, the aptamer field is poised to quickly provide a greater range of biosensor strategies for the next global health crisis. Similarly, the rapid growth of immune cell therapy field for cancer treatment increases the need for aptamer-based cell separation technology, which can potentially provide high efficiency at a lower cost.
It is also clear that a standard for aptamer handling and validation is needed. Several recent publications reported that some published aptamer sequences do not bind to their targets as originally claimed.60,160 To that end, the Aptamer Consortium, which is part of the International Society on Aptamers, proposed minimum aptamer publication standards in order to address these concerns.161 For example, providing the complete details of the selection conditions in each aptamer publication allows the reader to determine whether the selection buffer or cell culture conditions were appropriate. Most importantly, scrambled sequence aptamer and non-target proteins or cells should be used as controls in binding experiments. If the proposed standards are followed, the aptamer research community can increase reproducibility of aptamer studies and promote the use of aptamers by a wider audience.
This review has focused on in vitro applications of aptamers for diagnostics and bioseparations, but aptamers also have great potential in vivo. Historically, in vivo application of aptamers have been hampered by their poor nuclease resistance and serum stability. However, several important advances have been reported in nucleic acid stabilization in recent years. Established chemical modification strategies for increased serum stability include 3′ inverted nucleotide cap, 2′-O-methyl, and 2′-fluoro modifications.162,163 Another strategy is to circularize (by complementary strand hybridization) or cyclize (by ligase or click chemistry) the aptamer to reduce misfolding, increase stability, and improve nuclease resistance.164–166 One recent strategy is to generate photochemical crosslink-locked (PCCL) aptamers to stabilize conformation.167 PCCL-aptamer Sgc8 had tighter binding affinity, longer in vivo circulation half-life, and improved therapeutic effect in animal studies compared to unmodified aptamer Sgc8. Nanomaterials can also be used to improve in vivo properties in a covalent modification-free method. For example, gold nanoclusters self-assemble with aptamers into clusters that resist nuclease degradation and enhance cell-specific delivery.168 With these advances in aptamer stabilization strategies, we anticipate a significant growth in biomedical applications of aptamers in the near future.
Author contributions
Lucy F. Yang and Melissa Ling were the primary writers for the review. Lucy F. Yang prepared artwork for the review. Suzie H. Pun contributed to overall concept and scope. All authors contributed to editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH 1U01AA029316. M. L. was supported by an NSF Graduate Research Fellowship.References
- I. Alves Ferreira-Bravo, C. Cozens, P. Holliger and J. J. DeStefano, Nucleic Acids Res., 2015, 43, 9587–9599 Search PubMed.
- L. Chen, F. Rashid, A. Shah, H. M. Awan, M. Wu, A. Liu, J. Wang, T. Zhu, Z. Luo and G. Shan, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10002–10007 CrossRef CAS PubMed.
- J. Zhou and J. Rossi, Nat. Rev. Drug Discovery, 2017, 16, 181–202 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Nat. Protoc., 2006, 1, 246–252 CrossRef CAS PubMed.
- S. Kosuri and G. M. Church, Nat. Methods, 2014, 11, 499–507 CrossRef CAS PubMed.
- M. H. Jazayeri, H. Amani, A. A. Pourfatollah, H. Pazoki-Toroudi and B. Sedighimoghaddam, Sens. Bio-Sens. Res., 2016, 9, 17–22 CrossRef.
- J. Y. Zheng and L. J. Janis, Int. J. Pharm., 2006, 308, 46–51 CrossRef CAS PubMed.
- V. Crivianu-Gaita and M. Thompson, Biosens. Bioelectron., 2016, 85, 32–45 CrossRef CAS PubMed.
- G. S. Baird, Am. J. Clin. Pathol., 2010, 134, 529–531 CrossRef PubMed.
- B. C. Stark, R. Kole, E. J. Bowman and S. Altman, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 3717–3721 CrossRef CAS PubMed.
- K. Kruger, P. J. Grabowski, A. J. Zaug, J. Sands, D. E. Gottschling and T. R. Cech, Cell, 1982, 31, 147–157 CrossRef CAS PubMed.
- C. Guerrier-Takada, K. Gardiner, T. Marsh, N. Pace and S. Altman, Cell, 1983, 35, 849–857 CrossRef CAS PubMed.
- B. R. Cullen and W. C. Greene, Cell, 1989, 58, 423–426 CrossRef CAS PubMed.
- D. L. Robertson and G. F. Joyce, Nature, 1990, 344(6265), 467–468 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- R. Green, A. D. Ellington and J. W. Szostak, Nature, 1990, 347, 406–408 CrossRef CAS PubMed.
- C. Wilson and J. W. Szostak, Nature, 1995, 374(6525), 777–782 CrossRef CAS PubMed.
- L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermaas and J. J. Toole, Nature, 1992, 355, 564–566 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1992, 355, 850–852 CrossRef CAS PubMed.
- M. Homann and H. U. Göringer, Nucleic Acids Res., 1999, 27, 2006–2014 CrossRef CAS PubMed.
- J.-H. Lee, M. D. Canny, A. De Erkenez, D. Krilleke, Y.-S. Ng, D. T. Shima, A. Pardi and F. Jucker, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18902–18907 CrossRef CAS PubMed.
- D. H. J. Bunka and P. G. Stockley, Nat. Rev. Microbiol., 2006, 4, 588–596 CrossRef CAS PubMed.
- L. I. Hernandez, I. Machado, T. Schafer and F. J. Hernandez, Curr. Top. Med. Chem., 2015, 15, 1066–1081 CrossRef CAS PubMed.
- A. V. Lakhin, V. Z. Tarantul and L. V. Gening, Acta Nat., 2013, 5, 34–43 CrossRef CAS.
- M. R. Dunn, R. M. Jimenez and J. C. Chaput, Nat. Rev. Chem., 2017, 1, 1–16 CrossRef.
- M. R. Gotrik, T. A. Feagin, A. T. Csordas, M. A. Nakamoto and H. T. Soh, Acc. Chem. Res., 2016, 49, 1903–1910 CrossRef CAS PubMed.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS PubMed.
- S. Tombelli, M. Minunni and M. Mascini, Biosens. Bioelectron., 2005, 20, 2424–2434 CrossRef CAS PubMed.
- T. Wang, C. Chen, L. M. Larcher, R. A. Barrero and R. N. Veedu, Biotechnol. Adv., 2019, 37, 28–50 CrossRef CAS PubMed.
- B. J. Thomas, D. Porciani and D. H. Burke, Mol. Ther.–Nucleic Acids, 2022, 27, 894–915 CrossRef CAS PubMed.
- K. Pobanz and A. Lupták, Methods, 2016, 106, 14–20 CrossRef CAS PubMed.
- A. Schmitz, A. Weber, M. Bayin, S. Breuers, V. Fieberg, M. Famulok and G. Mayer, Angew. Chem., Int. Ed., 2021, 2–9 Search PubMed.
- N. Kacherovsky, I. I. Cardle, E. L. Cheng, J. L. Yu, M. L. Baldwin, S. J. Salipante, M. C. Jensen and S. H. Pun, Nat. Biomed. Eng., 2019, 3, 783–795 CrossRef CAS PubMed.
- M. Legiewicz, C. Lozupone, R. Knight and M. Yarus, RNA, 2005, 11, 1701–1709 CrossRef CAS PubMed.
- N. Kacherovsky, L. F. Yang, H. V. Dang, E. L. Cheng, I. I. Cardle, A. C. Walls, M. McCallum, D. L. Sellers, F. DiMaio, S. J. Salipante, D. Corti, D. Veesler and S. Pun, Angew. Chem., Int. Ed., 2021, 60, 21211–21215 CrossRef CAS PubMed.
- E. W. Choi, L. V. Nayak and P. J. Bates, Nucleic Acids Res., 2010, 38(5), 1623–1635 CrossRef CAS PubMed.
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef CAS PubMed.
- C. Roxo, W. Kotkowiak and A. Pasternak, Molecules, 2019, 24, 3781 CrossRef CAS PubMed.
- P. J. Bates, D. A. Laber, D. M. Miller, S. D. Thomas and J. O. Trent, Exp. Mol. Pathol., 2009, 86, 151–164 CrossRef CAS PubMed.
- J. P. Elskens, J. M. Elskens and A. Madder, Int. J. Mol. Sci., 2020, 21, 4522 CrossRef CAS PubMed.
- C. M. McCloskey, Q. Li, E. J. Yik, N. Chim, A. K. Ngor, E. Medina, I. Grubisic, L. Co Ting Keh, R. Poplin and J. C. Chaput, ACS Synth. Biol., 2021, 10, 3190–3199 CrossRef CAS PubMed.
- F. Pfeiffer, F. Tolle, M. Rosenthal, G. M. Brändle, J. Ewers and G. Mayer, Nat. Protoc., 2018, 13, 1153–1180 CrossRef CAS PubMed.
- S. Klußmann, A. Nolte, R. Bald, V. A. Erdmann and J. P. Fürste, Nat. Biotechnol., 1996, 14, 1112–1115 CrossRef PubMed.
- K. P. Williams, X.-H. Liu, T. N. M. Schumacher, H. Y. Lin, D. A. Ausiello, P. S. Kim and D. P. Bartel, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 11285–11290 CrossRef CAS PubMed.
- S. Ren, S. Cho, R. Lin, V. Gedi, S. Park, C. W. Ahn, D.-K. Lee, M.-H. Lee, S. Lee and S. Kim, Biosensors, 2022, 12, 864 CrossRef CAS PubMed.
- T. Bing, N. Zhang and D. Shangguan, Adv. Biosyst., 2019, 3, 1900193 CrossRef PubMed.
- B. J. Hicke, C. Marion, Y.-F. Chang, T. Gould, C. K. Lynott, D. Parma, P. G. Schmidt and S. Warren, J. Biol. Chem., 2001, 276, 48644–48654 CrossRef CAS PubMed.
- N. Lin, L. Wu, X. Xu, Q. Wu, Y. Wang, H. Shen, Y. Song, H. Wang, Z. Zhu, D. Kang and C. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 9306–9315 CrossRef CAS PubMed.
- C. Kolm, I. Cervenka, U. J. Aschl, N. Baumann, S. Jakwerth, R. Krska, R. L. Mach, R. Sommer, M. C. DeRosa, A. K. T. Kirschner, A. H. Farnleitner and G. H. Reischer, Sci. Rep., 2020, 10, 20917 CrossRef CAS PubMed.
- K. Sefah, D. Shangguan, X. Xiong, M. B. O'Donoghue and W. Tan, Nat. Protoc., 2010, 5, 1169–1185 CrossRef CAS PubMed.
- W. H. Thiel, K. W. Thiel, K. S. Flenker, T. Bair, A. J. Dupuy, J. O. McNamara, F. J. Miller and P. H. Giangrande, in RNA Interference: Challenges and Therapeutic Opportunities, ed. M. Sioud, Springer, New York, NY, 2015, pp. 187–199 Search PubMed.
- D. Shangguan, Z. Cao, L. Meng, P. Mallikaratchy, K. Sefah, H. Wang, Y. Li and W. Tan, J. Proteome Res., 2008, 7, 2133–2139 CrossRef CAS PubMed.
- T. Bing, D. Shangguan and Y. Wang, Mol. Cell. Proteomics, 2015, 14, 2692–2700 CrossRef CAS PubMed.
- M. Khedri, K. Abnous, H. Rafatpanah, M. S. Nabavinia, S. M. Taghdisi and M. Ramezani, Immunol. Invest., 2020, 49, 535–554 CrossRef CAS PubMed.
- S. E. Wilner, B. Wengerter, K. Maier, M. de Lourdes Borba Magalhães, D. S. Del Amo, S. Pai, F. Opazo, S. O. Rizzoli, A. Yan and M. Levy, Mol. Ther.–Nucleic Acids, 2012, 1, e21 CrossRef PubMed.
- L. F. Yang, N. Kacherovsky, N. Panpradist, R. Wan, J. Liang, B. Zhang, S. J. Salipante, B. R. Lutz and S. H. Pun, Anal. Chem., 2022, 94, 7278–7285 CrossRef CAS PubMed.
- Z. Zhang, J. Li, J. Gu, R. Amini, H. D. Stacey, J. C. Ang, D. White, C. D. M. Filipe, K. Mossman, M. S. Miller, B. J. Salena, D. Yamamura, P. Sen, L. Soleymani, J. D. Brennan and Y. Li, Chem. - Eur. J., 2022, 28, e202200078 CAS.
- J. Kwon, C. Narayan, C. Kim, M. J. Han, M. Kim and S. K. Jang, J. Biomed. Nanotechnol., 2019, 15, 1609–1621 CrossRef CAS PubMed.
- A. S. Peinetti, R. J. Lake, W. Cong, L. Cooper, Y. Wu, Y. Ma, G. T. Pawel, M. E. Toimil-Molares, C. Trautmann, L. Rong, B. Mariñas, O. Azzaroni and Y. Lu, Sci. Adv., 2021, 7, eabh2848 CrossRef CAS PubMed.
- L. F. Yang, N. Kacherovsky, J. Liang, S. J. Salipante and S. H. Pun, Anal. Chem., 2022, 94, 12683–12690 CrossRef CAS PubMed.
- S. D. Mendonsa and M. T. Bowser, J. Am. Chem. Soc., 2004, 126, 20–21 CrossRef CAS PubMed.
- C. Zhu, X. Wang, L. Li, C. Hao, Y. Hu, A. S. Rizvi and F. Qu, Biochem. Biophys. Res. Commun., 2018, 506, 169–175 CrossRef CAS PubMed.
- Z. Luo, H. Zhou, H. Jiang, H. Ou, X. Li and L. Zhang, Analyst, 2015, 140, 2664–2670 RSC.
- A. T. H. Le, S. M. Krylova, M. Kanoatov, S. Desai and S. N. Krylov, Angew. Chem., 2019, 131, 2765–2769 CrossRef.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Anal. Bioanal. Chem., 2005, 383, 83–91 CrossRef CAS PubMed.
- Y. Song, Z. Zhu, Y. An, W. Zhang, H. Zhang, D. Liu, C. Yu, W. Duan and C. J. Yang, Anal. Chem., 2013, 85, 4141–4149 CrossRef CAS PubMed.
- M. Khati, M. Schüman, J. Ibrahim, Q. Sattentau, S. Gordon and W. James, J. Virol., 2003, 77, 12692–12698 CrossRef CAS PubMed.
- T. S. Misono and P. K. R. Kumar, Anal. Biochem., 2005, 342, 312–317 CrossRef CAS PubMed.
- H. Kaur, M. Shorie and P. Sabherwal, Biosens. Bioelectron., 2020, 167, 112498 CrossRef CAS PubMed.
- M. Mukherjee, P. Appaiah, S. Sistla, B. Bk and P. Bhatt, J. Agric. Food Chem., 2022, 70, 6239–6246 CrossRef CAS PubMed.
- S. Saito, Anal. Sci., 2021, 37, 17–26 CrossRef CAS PubMed.
- D. Wu, C. K. L. Gordon, J. H. Shin, M. Eisenstein and H. T. Soh, Acc. Chem. Res., 2022, 55, 685–695 CrossRef CAS PubMed.
- R. G. Higuchi and H. Ochman, Nucleic Acids Res., 1989, 17, 5865 CrossRef CAS PubMed.
- U. B. Gyllensten and H. A. Erlich, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 7652–7656 CrossRef CAS PubMed.
- K. P. Williams and D. P. Bartel, Nucleic Acids Res., 1995, 23, 4220–4221 CrossRef CAS PubMed.
- J. G. Bruno, M. P. Carrillo, A. M. Richarte, T. Phillips, C. Andrews and J. S. Lee, BMC Res. Notes, 2012, 5, 633 CrossRef CAS PubMed.
- L. Zhang, X. Fang, X. Liu, H. Ou, H. Zhang, J. Wang, Q. Li, H. Cheng, W. Zhang and Z. Luo, Chem. Commun., 2020, 56, 10235–10238 RSC.
- K. K. Alam, J. L. Chang and D. H. Burke, Mol. Ther.–Nucleic Acids, 2015, 4, e230 CrossRef CAS PubMed.
- T. L. Bailey, M. Boden, F. A. Buske, M. Frith, C. E. Grant, L. Clementi, J. Ren, W. W. Li and W. S. Noble, Nucleic Acids Res., 2009, 37, W202–W208 CrossRef CAS PubMed.
- O. Kikin, L. D'Antonio and P. S. Bagga, Nucleic Acids Res., 2006, 34, W676–W682 CrossRef CAS PubMed.
- A. Rambaut, FigTree, http://tree.bio.ed.ac.uk/software/figtree/, accessed January 19, 2023 Search PubMed.
- J. N. Zadeh, C. D. Steenberg, J. S. Bois, B. R. Wolfe, M. B. Pierce, A. R. Khan, R. M. Dirks and N. A. Pierce, J. Comput. Chem., 2011, 32, 170–173 CrossRef CAS PubMed.
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406–3415 CrossRef CAS PubMed.
- K. Varadaraj and D. M. Skinner, Gene, 1994, 140, 1–5 CrossRef CAS PubMed.
- N. A. Ahmad, R. Mohamed Zulkifli, H. Hussin and M. H. Nadri, J. Mol. Graphics Modell., 2021, 105, 107872 CrossRef CAS PubMed.
- D. Sun, M. Sun, J. Zhang, X. Lin, Y. Zhang, F. Lin, P. Zhang, C. Yang and J. Song, TrAC, Trends Anal. Chem., 2022, 157, 116767 CrossRef CAS.
- G. Hybarger, J. Bynum, R. F. Williams, J. J. Valdes and J. P. Chambers, Anal. Bioanal. Chem., 2006, 384, 191–198 CrossRef CAS PubMed.
- H. Jiang, X.-F. Lv and K.-X. Zhao, Chin. J. Inorg. Anal. Chem., 2020, 48, 590–600 CrossRef CAS.
- A. Sinha, P. Gopinathan, Y.-D. Chung, H.-Y. Lin, K.-H. Li, H.-P. Ma, P.-C. Huang, S.-C. Shiesh and G.-B. Lee, Biosens. Bioelectron., 2018, 122, 104–112 CrossRef CAS PubMed.
- R. A. Langan, S. E. Boyken, A. H. Ng, J. A. Samson, G. Dods, A. M. Westbrook, T. H. Nguyen, M. J. Lajoie, Z. Chen, S. Berger, V. K. Mulligan, J. E. Dueber, W. R. P. Novak, H. El-Samad and D. Baker, Nature, 2019, 572, 205–210 CrossRef CAS PubMed.
- A. Quijano-Rubio, H.-W. Yeh, J. Park, H. Lee, R. A. Langan, S. E. Boyken, M. J. Lajoie, L. Cao, C. M. Chow, M. C. Miranda, J. Wi, H. J. Hong, L. Stewart, B.-H. Oh and D. Baker, Nature, 2021, 591, 482–487 CrossRef CAS PubMed.
- O. Rabal, F. Pastor, H. Villanueva, M. M. Soldevilla, S. Hervas-Stubbs and J. Oyarzabal, Mol. Ther.–Nucleic Acids, 2016, 5, e376 CrossRef PubMed.
- Y. Song, J. Song, X. Wei, M. Huang, M. Sun, L. Zhu, B. Lin, H. Shen, Z. Zhu and C. Yang, Anal. Chem., 2020, 92, 9895–9900 CrossRef CAS PubMed.
- E. L. Cheng, I. I. Cardle, N. Kacherovsky, H. Bansia, T. Wang, Y. Zhou, J. Raman, A. Yen, D. Gutierrez, S. J. Salipante, A. des Georges, M. C. Jensen and S. H. Pun, J. Am. Chem. Soc., 2022, 144, 13851–13864 CrossRef CAS PubMed.
- A. B. Iliuk, L. Hu and W. A. Tao, Anal. Chem., 2011, 83, 4440–4452 CrossRef CAS PubMed.
- C. Forier, E. Boschetti, M. Ouhammouch, A. Cibiel, F. Ducongé, M. Nogré, M. Tellier, D. Bataille, N. Bihoreau, P. Santambien, S. Chtourou and G. Perret, J. Chromatogr. A, 2017, 1489, 39–50 CrossRef CAS PubMed.
- A. Scohy, A. Anantharajah, M. Bodéus, B. Kabamba-Mukadi, A. Verroken and H. Rodriguez-Villalobos, J. Clin. Virol., 2020, 129, 104455 CrossRef CAS PubMed.
- G. C. Mak, P. K. Cheng, S. S. Lau, K. K. Wong, C. Lau, E. T. Lam, R. C. Chan and D. N. Tsang, J. Clin. Virol., 2020, 129, 104500 CrossRef CAS PubMed.
- C. Daughton, Sci. Total Environ., 2020, 726, 138149 CrossRef CAS PubMed.
- M. J. Mina, R. Parker and D. B. Larremore, N. Engl. J. Med., 2020, 383, e120 CrossRef CAS PubMed.
- R. J. White, A. A. Rowe and K. W. Plaxco, Analyst, 2010, 135, 589–594 RSC.
- L. R. Schoukroun-Barnes, F. C. Macazo, B. Gutierrez, J. Lottermoser, J. Liu and R. J. White, Annu. Rev. Anal. Chem., 2016, 9, 163–181 CrossRef CAS PubMed.
- Y. Xiao, A. A. Lubin, A. J. Heeger and K. W. Plaxco, Angew. Chem., 2005, 117, 5592–5595 CrossRef.
- B. R. Baker, R. Y. Lai, M. S. Wood, E. H. Doctor, A. J. Heeger and K. W. Plaxco, J. Am. Chem. Soc., 2006, 128, 3138–3139 CrossRef CAS PubMed.
- Z. gang Yu, A. L. Sutlief and R. Y. Lai, Sens. Actuators, B, 2018, 258, 722–729 CrossRef.
- M. Jarczewska, J. Rębiś, Ł. Górski and E. Malinowska, Talanta, 2018, 189, 45–54 CrossRef CAS PubMed.
- Y. Wu, B. Midinov and R. J. White, ACS Sens., 2019, 4, 498–503 CrossRef CAS PubMed.
- S. Dolai and M. Tabib-Azar, Med. Devices Sens., 2020, 3, 1–9 Search PubMed.
- J. Kwon, Y. Lee, T. Lee and J. H. Ahn, Anal. Chem., 2020, 92, 5524–5531 CrossRef CAS PubMed.
- P. Lasserre, B. Balansethupathy, V. J. Vezza, A. Butterworth, A. Macdonald, E. O. Blair, L. McAteer, S. Hannah, A. C. Ward, P. A. Hoskisson, A. Longmuir, S. Setford, E. C. W. Farmer, M. E. Murphy, H. Flynn and D. K. Corrigan, Anal. Chem., 2022, 94, 2126–2133 CrossRef CAS PubMed.
- M. A. Martínez-Roque, P. A. Franco-Urquijo, V. M. García-Velásquez, M. Choukeife, G. Mayer, S. R. Molina-Ramírez, G. Figueroa-Miranda, D. Mayer and L. M. Alvarez-Salas, Anal. Biochem., 2022, 645, 114633 CrossRef PubMed.
- A. Idili, C. Parolo, R. Alvarez-Diduk and A. Merkoçi, ACS Sens., 2021, 6, 3093–3101 CrossRef CAS PubMed.
- Z. Zhang, R. Pandey, J. Li, J. Gu, D. White, H. D. Stacey, J. C. Ang, C.-J. Steinberg, A. Capretta, C. D. M. Filipe, K. Mossman, C. Balion, M. S. Miller, B. J. Salena, D. Yamamura, L. Soleymani, J. D. Brennan and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 24266–24274 CrossRef CAS PubMed.
- D. K. Ban, T. Bodily, A. G. Karkisaval, Y. Dong, S. Natani, A. Ramanathan, A. Ramil, S. Srivastava, P. Bandaru, G. Glinsky and R. Lal, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2206521119 CrossRef CAS PubMed.
- F. Curti, S. Fortunati, W. Knoll, M. Giannetto, R. Corradini, A. Bertucci and M. Careri, ACS Appl. Mater. Interfaces, 2022, 14, 19204–19211 CrossRef CAS PubMed.
- L. Shi, L. Wang, X. Ma, X. Fang, L. Xiang, Y. Yi, J. Li, Z. Luo and G. Li, Anal. Chem., 2021, 93, 16646–16654 CrossRef CAS PubMed.
- L. Wide and C. A. Gemzell, Eur. J. Endocrinol., 1960, XXXV, 261–267 Search PubMed.
- G. A. Posthuma-Trumpie, J. Korf and A. van Amerongen, Anal. Bioanal. Chem., 2009, 393, 569–582 CrossRef CAS PubMed.
- A. Chen and S. Yang, Biosens. Bioelectron., 2015, 71, 230–242 CrossRef CAS PubMed.
- Z. Chen, Q. Wu, J. Chen, X. Ni and J. Dai, Virol. Sin., 2020, 35, 351–354 CrossRef CAS PubMed.
- W. Zhao, M. A. Brook and Y. Li, ChemBioChem, 2008, 9, 2363–2371 CrossRef CAS PubMed.
- S. Aithal, S. Mishriki, R. Gupta, R. P. Sahu, G. Botos, S. Tanvir, R. W. Hanson and I. K. Puri, Talanta, 2022, 236, 122841 CrossRef CAS PubMed.
- R. Nutiu and Y. Li, Chem. - Eur. J., 2004, 10, 1868–1876 CrossRef CAS PubMed.
- M. N. Stojanovic, P. de Prada and D. W. Landry, J. Am. Chem. Soc., 2001, 123, 4928–4931 CrossRef CAS PubMed.
- X. Zhao, X. Dai, S. Zhao, X. Cui, T. Gong, Z. Song, H. Meng, X. Zhang and B. Yu, Spectrochim. Acta, Part A, 2021, 247, 119038 CrossRef CAS PubMed.
- N. Chauhan, Y. Xiong, S. Ren, A. Dwivedy, N. Magazine, L. Zhou, X. Jin, T. Zhang, B. T. Cunningham, S. Yao, W. Huang and X. Wang, J. Am. Chem. Soc. DOI:10.1021/jacs.2c04835.
- R. Liu, L. He, Y. Hu, Z. Luo and J. Zhang, Chem. Sci., 2020, 11, 12157–12164 RSC.
- K. M. Hansen and T. Thundat, Methods, 2005, 37, 57–64 CrossRef CAS PubMed.
- G. Zhang, C. Li, S. Wu and Q. Zhang, Sens. Actuators, B, 2018, 260, 42–47 CrossRef CAS.
- C. Li, M. Zhang, Z. Zhang, J. Tang and B. Zhang, Sens. Actuators, B, 2019, 297, 126759 CrossRef CAS.
- C. Li, X. Ma, Y. Guan, J. Tang and B. Zhang, ACS Sens., 2019, 4, 3034–3041 CrossRef CAS PubMed.
- G. L. Tortorella, F. S. Fogliatto, A. Mac Cawley Vergara, R. Vassolo and R. Sawhney, Prod. Plan. Control, 2020, 31, 1245–1260 CrossRef.
- S. Ardalan and A. Ignaszak, Adv. Mater. Technol., 2022, 7, 2200208 CrossRef CAS PubMed.
- D. S. Hage, J. A. Anguizola, C. Bi, R. Li, R. Matsuda, E. Papastavros, E. Pfaunmiller, J. Vargas and X. Zheng, J. Pharm. Biomed. Anal., 2012, 69, 93–105 CrossRef CAS PubMed.
- D. S. Hage, Clin. Chem., 1999, 45, 593–615 CrossRef CAS.
- Q. Zhao, M. Wu, X. Chris Le and X.-F. Li, TrAC, Trends Anal. Chem., 2012, 41, 46–57 CrossRef CAS.
- Y.-H. Lao, K. Peck and L.-C. Chen, Anal. Chem., 2009, 81, 1747–1754 CrossRef CAS PubMed.
- M. Lönne, S. Bolten, A. Lavrentieva, F. Stahl, T. Scheper and J.-G. Walter, Biotechnol. Rep., 2015, 8, 16–23 CrossRef PubMed.
- T. S. Romig, C. Bell and D. W. Drolet, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 731, 275–284 CrossRef CAS.
- J.-G. Walter, F. Stahl and T. Scheper, Eng. Life Sci., 2012, 12, 496–506 CrossRef CAS.
- Ö. Kökpinar, J.-G. Walter, Y. Shoham, F. Stahl and T. Scheper, Biotechnol. Bioeng., 2011, 108, 2371–2379 CrossRef PubMed.
- J.-G. Walter, Ö. Kökpinar, K. Friehs, F. Stahl and T. Scheper, Anal. Chem., 2008, 80, 7372–7378 CrossRef CAS PubMed.
- E. Lalli, J. S. Silva, C. Boi and G. C. Sarti, Membranes, 2019, 10, 1 CrossRef PubMed.
- M. E. Najafabadi, T. Khayamian and Z. Hashemian, J. Pharm. Biomed. Anal., 2015, 107, 244–250 CrossRef CAS PubMed.
- Q. Zhao, X.-F. Li, Y. Shao and X. C. Le, Anal. Chem., 2008, 80, 7586–7593 CrossRef CAS PubMed.
- S. Eeltink, D. Meston and F. Svec, Anal. Sci. Adv., 2021, 2, 250–260 CrossRef.
- S. Cho, S.-H. Lee, W.-J. Chung, Y.-K. Kim, Y.-S. Lee and B.-G. Kim, Electrophoresis, 2004, 25, 3730–3739 CrossRef CAS PubMed.
- Q. Deng, I. German, D. Buchanan and R. T. Kennedy, Anal. Chem., 2001, 73, 5415–5421 CrossRef CAS PubMed.
- Q. Deng, C. J. Watson and R. T. Kennedy, J. Chromatogr. A, 2003, 1005, 123–130 CrossRef CAS PubMed.
- C. Ravelet, R. Boulkedid, A. Ravel, C. Grosset, A. Villet, J. Fize and E. Peyrin, J. Chromatogr. A, 2005, 1076, 62–70 CrossRef CAS PubMed.
- B. Han, C. Zhao, J. Yin and H. Wang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 903, 112–117 CrossRef CAS PubMed.
- Q. Zhao, X.-F. Li and X. C. Le, Anal. Chem., 2008, 80, 3915–3920 CrossRef CAS PubMed.
- T. Zhao, X. Ding, Y. Chen, C. Lin, G. Qi, X. Lin and Z. Xie, Anal. Chim. Acta, 2021, 1165, 338517 CrossRef CAS PubMed.
- C. Kuehne, S. Wedepohl and J. Dernedde, Sensors, 2017, 17, 226 CrossRef PubMed.
- J. A. Phillips, Y. Xu, Z. Xia, Z. H. Fan and W. Tan, Anal. Chem., 2009, 81, 1033–1039 CrossRef CAS PubMed.
- L. Wu, L. Zhu, M. Huang, J. Song, H. Zhang, Y. Song, W. Wang and C. Yang, TrAC, Trends Anal. Chem., 2019, 117, 69–77 CrossRef CAS.
- B. P. Gray, M. D. Requena, M. D. Nichols and B. A. Sullenger, Cell Chem. Biol., 2020, 27(2), 232–244 CrossRef CAS PubMed.
- E. L. Cheng, N. Kacherovsky and S. H. Pun, ACS Appl. Mater. Interfaces, 2022, 14, 44136–44146 CrossRef CAS PubMed.
- S. Liu, Y. Xu, X. Jiang, H. Tan and B. Ying, Biosens. Bioelectron., 2022, 208, 114168 CrossRef CAS PubMed.
- Y. Zhao, K. Yavari and J. Liu, TrAC, Trends Anal. Chem., 2022, 146, 116480 CrossRef CAS.
- M. McKeague, V. Calzada, L. Cerchia, M. DeRosa, J. M. Heemstra, N. Janjic, P. E. Johnson, L. Kraus, J. Limson, G. Mayer, M. Nilsen-Hamilton, D. Porciani, T. K. Sharma, B. Suess, J. A. Tanner and S. Shigdar, Aptamers, 2022, 6, 10–18 Search PubMed.
- C. Kratschmer and M. Levy, Nucleic Acid Ther., 2017, 27, 335–344 CrossRef CAS PubMed.
- R. R. White, S. Shan, C. P. Rusconi, G. Shetty, M. W. Dewhirst, C. D. Kontos and B. A. Sullenger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5028–5033 CrossRef CAS PubMed.
- Y. Jiang, X. Pan, J. Chang, W. Niu, W. Hou, H. Kuai, Z. Zhao, J. Liu, M. Wang and W. Tan, J. Am. Chem. Soc., 2018, 140, 6780–6784 CrossRef CAS PubMed.
- D. Ji, K. Lyu, H. Zhao and C. K. Kwok, Nucleic Acids Res., 2021, 49, 7280–7291 CrossRef CAS PubMed.
- H. Kuai, Z. Zhao, L. Mo, H. Liu, X. Hu, T. Fu, X. Zhang and W. Tan, J. Am. Chem. Soc., 2017, 139, 9128–9131 CrossRef CAS PubMed.
- F. Zhou, P. Wang, J. Chen, Z. Zhu, Y. Li, S. Wang, S. Wu, Y. Sima, T. Fu, W. Tan and Z. Zhao, Nucleic Acids Res., 2022, 50, 9039–9050 CrossRef CAS PubMed.
- F. Xia, A. He, H. Zhao, Y. Sun, Q. Duan, S. J. Abbas, J. Liu, Z. Xiao and W. Tan, ACS Nano, 2022, 16, 169–179 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |