
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA “turn-off” photoluminescent sensor for H2O2 detection based on a zinc oxide–graphene quantum dot (ZnO–GQD) nanocomposite and the role of amine in the development of GQD†
Rolando Efraín Ramírez Garza a,
Sara Luisa Rodríguez de Lunaab,
Genoveva Hernández Padrónc and
Idalia Gómez de la Fuente*a
a,
Sara Luisa Rodríguez de Lunaab,
Genoveva Hernández Padrónc and
Idalia Gómez de la Fuente*a
aUniversidad Autónoma de Nuevo León, Facultad de Ciencias Químicas, Ciudad Universitaria, Lab. de Materiales 1, Av. Pedro de Alba s/n, C.P. 66455, San Nicolás de los Garza, Nuevo León, Mexico. E-mail: idaliagomezmx@yahoo.com.mx; Tel: +52-81-83-294010 ext. 6240
bEscuela de Ingeniería y Ciencias, Tecnologico de Monterrey, Av. Eugenio Garza Sada 2501 Sur, Monterrey, Nuevo León C.P. 64849, Mexico
cDepartamento de Nanotecnología, Centro de Física Aplicada y Tecnología Avanzada, Universidad Nacional Autónoma de México. Campus Juriquilla, Boulevard Juriquilla no. 3001, Santiago de Querétaro, Querétaro, Mexico
First published on 19th July 2023
Abstract
In this work, graphene quantum dots (GQD) were prepared through a hydrothermal process. The photoluminescence (PL) emission spectrum for GQD prepared with high NH4OH concentration (sample D1-t) was attained at lower wavelength (406 nm), compared to GQD synthesized with low NH4OH concentration (sample D2-t attained at 418 nm). From these results, a smaller particle size for D1-t was deduced; according to TEM images the GQD particles are around 5 nm. The Raman ID3/IG ratio which is related to C–O groups at the edges of GQD and the full width at half maximum was lower for D1-t than D2-t. This was ascribed to the amine group incorporation at the edges and at the basal planes in D1-t, whilst in D2-t they prefer principally the edges of the GQD structure. The ZnO nanoparticles bonded to GQD (ZnO–GQD, nanocomposites) enhance the PL emission intensity. The H2O2 detection tested by photoluminescence spectroscopy, was found to occur thanks to the ZnO from the nanocomposite and its interaction with H2O2, producing a quenching effect. This quenching was accentuated by the increase of the H2O2 concentration. Such properties suggest the ZnO–GQD nanocomposite as a candidate to be used as a sensor material.
1 Introduction
Graphene oxide is obtained by the oxidation of graphene, and this process is essential for this material to be used in transistors, photodetectors, electronic displays, among others.1 The particle size reduction of graphene oxide leads to obtain what are known as graphene quantum dots (GQD). These GQD have lateral dimensions from 3 nm up to 100 nm having attributes of edge effects, quantum and electron confinements.2,3 These characteristics allow the GQD to be potentially used as materials for fluorescence imaging of organic tissues,4 drug delivery5 and sensors.6,7 The hydrothermal (HT) process is a recurring method to prepare GQD. The HT method has been used with water, chemical bases and acids, but one of the most effective solvents is the H2O2 which decomposes in ˙OH and ˙O− radicals, producing the cut and the development of epoxy and carbonyl groups, respectively.8 This method also induces the functional groups to link at the edges of GQD which serve as anchorage sites for polymers or metal oxides.9 In particular, when nitrogen functional groups are aggregated in the GQD structure, the photoluminescent properties are enhanced (i.e. increment in emission intensity).10Moreover, the incorporation of metallic particles to the graphene oxide conduces to the enhancement when its sensor properties are evaluated.11 Several research works for sensor applications have examined GQD along with metals as gold and platinum,12 silver13 and titanium oxide.14 Particularly, these composites (metals and metals oxides–GQD) have been used in H2O2 detection. The H2O2 is a molecule related to the degradation of some physiological functions which conduces to cardiovascular diseases, Alzheimer, atherosclerosis, cellular oxidative stress, cellular ageing and cancer.15–17 For this reason, the early and precise H2O2 detection is essential for an opportune diagnosis.
Numerous sensors have been developed for the H2O2 detection and they are mainly based in analytic methods as electrochemical (enzymatic and non-enzymatic), high resolution chromatography, chromatography mass spectroscopy, colorimetry, capillary electrophoresis, infrared absorption spectroscopy and photoluminescence spectroscopy.15,18–20 The photoluminescent sensors offer a myriad of advantages as operative simplicity, high sensibility, high selectivity and fast response.21,22
Recently nanomaterials (nanoclusters, nanorods, nanospheres) of noble metals as gold and silver, metal–ligand complexes, organic colourants, metals oxides nanoparticles and quantum dots of CdSe, carbon, ZnS and graphene, have been assessed as photoluminescent sensors.21–25 There are also some reports over the H2O2 detection by means of photoluminescence using GQD. For instance, He et al. developed a photoluminescent biosensor of GQD functionalized with hemine to monitor H2O2 and glucose in human serum.26 A TiO2–GQD nanocomposite was used for H2O2 detection by photoluminescence spectroscopy, using an excitation wavelength of 360 nm having well detection in a range from 10−14 M to 10−2 M.14
Photoluminescence studies for the detection of H2O2 still are scarce and to the best of our knowledge there are not works concerning ZnO–GQD nanocomposites for this purpose. This study contributes with a general knowledge from the synthesis of GQD with amine groups and the ZnO–GQD nanocomposite which is important since some of its advantages are the simplicity of the synthesis and the economic feasibility compared to noble metals. Therefore, in this study, it is presented an analysis over the role of NH4OH during the synthesis of GQD and ZnO–GQD and their performance in the H2O2 detection.
2 Experimental
2.1 Chemical substances and materials
In this investigation the following substances and materials were used. From Sigma-Aldrich: synthetic graphite with particle size < 20 μm, 30% H2O2 and dialysis tubing benzoylated, 32 mm width. From Fermont (Monterrey Mexico): 99.7% KMnO4, 28% NH4OH, 99.3% Zn(CH3COO)2·2H2O. Concentrated H2SO4 (DEQ, Monterrey, Mexico). 85% H3PO4 (CTR, Monterrey, Mexico). Deionized water (Karyeth reactivos, Monterrey, Mexico) used was 0.76 μS cm−1.2.2 Graphene oxide synthesis
Graphene oxide was synthesized by the modified method of Hummers similar to Yadav et al.27 and it is briefly described below. In a beaker set on an ice bath, 0.4 g of graphite, a volume of 53 mL of concentrated H2SO4, and 13.5 mL of H3PO4 were placed and gentle stirred. After one hour, 2.9 g of KMnO4 were carefully added maintaining the stirring during 15 min. The beaker was removed from the ice bath and taken to heat at 50 °C keeping a moderately stirring for 10 h. Upon completion, the mixture was prudently decanted and the residual solid was filled with 300 mL of deionized water and 4 mL of H2O2. After 15 min of stirring this mixture was taken to centrifugation at 6000 rpm for 30 min. The solid obtained was washed with deionized water until the rinsing water pH was 6. The solid then was placed in a dialysis tubing to eliminate the excess of ions. Finally, this sample was taken to centrifugation, decanted and the solid was dried in an oven at 40 °C during 5 h to later being stored in a glass container. The final sample was identified as GOx.2.3 Synthesis of graphene quantum dots
For the sake of simplicity the Table 1 summarizes the synthesis routes followed in this section, still the detailed synthesis is described below. Graphene quantum dots (GQD) were prepared by hydrothermal process and 3 routes were used. In all the experiments deionized water and a GOx mass of 0.0300 g were used for each route.| Route | GQD precursor | Solvent used | Solvent concentration | Samples obtained | |
|---|---|---|---|---|---|
| Without treatment | After treatment | ||||
| 1 | GOx | NH4OH | High | D1-w | D1-t |
| 2 | GOx | NH4OH | Low | D2-w | D2-t |
| 3 | GOx | H2O2 | Low | H-GOx | — |
| 3 | H-GOx | NH4OH | Low | D3-w | — |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 volume ratio) were added to a beaker. Later, the content was transferred into a 50 mL of Teflon autoclave and this was introduced to an oven at 170 °C over 6 h. Once the autoclave was at room temperature the product was filtered and the liquid was named D1-w and stored, whilst the solid was kept for ulterior characterization. A volume ratio of 1:1 from D1-w and water, was heated at 55 °C during 40 minutes to remove the excess of NH4OH and the product obtained was identified as D1-t. In this way, the samples without heat treatment are identified by “w” suffix and those treated with “t” suffix.:10 volume ratio). This mixture was transferred to a 50 mL Teflon autoclave and introduced into an oven at 170 °C during 6 h. When the autoclave reached room temperature the product was filtered and the liquid, named D2-w, was stored. A heat treatment, as described in route 1, was also carried out on this sample to finally obtain the D2-t.:5 volume ratio). This mixture was introduced into the Teflon autoclave and placed into an oven at 170 °C for 6 h. The reactor cooled down and the solid was found to be completely dissolved. The obtained product was identified as H-GOx.
:3 volume ratio) were added to a beaker. Later, the content was transferred into a 50 mL of Teflon autoclave and this was introduced to an oven at 170 °C over 6 h. Once the autoclave was at room temperature the product was filtered and the liquid was named D1-w and stored, whilst the solid was kept for ulterior characterization. A volume ratio of 1:1 from D1-w and water, was heated at 55 °C during 40 minutes to remove the excess of NH4OH and the product obtained was identified as D1-t. In this way, the samples without heat treatment are identified by “w” suffix and those treated with “t” suffix.:10 volume ratio). This mixture was transferred to a 50 mL Teflon autoclave and introduced into an oven at 170 °C during 6 h. When the autoclave reached room temperature the product was filtered and the liquid, named D2-w, was stored. A heat treatment, as described in route 1, was also carried out on this sample to finally obtain the D2-t.:5 volume ratio). This mixture was introduced into the Teflon autoclave and placed into an oven at 170 °C for 6 h. The reactor cooled down and the solid was found to be completely dissolved. The obtained product was identified as H-GOx.Subsequently, 8 mL of a NH4OH/H-GOx mixture (1:12 volume ratio) was introduced into Teflon autoclave and placed in an oven at 170 °C for 6 h. After completion, a small fraction of black solid was emerged. The liquid was kept and named as D3-w, which was not taken to posterior treatment.
2.4 Synthesis of ZnO nanoparticles
For the synthesis of ZnO nanoparticles (ZnNP), 50 mL of a 0.125 M solution of Zn(CH3COO)2·2H2O was placed into a beaker magnetically and vigorously stirred. The initial pH of this solution was 7. A NaOH solution (0.5 M) was added (drop by drop) to the zinc acetate solution. Once the pH reached 12, the solid had a bright white jelly aspect, and a strong stirring was maintained for 1 h. The solid was washed with deionized water and a centrifugation process was taken until a pH of 7 was measured from rinse water. The humid solid was dried at 80 °C for 20 min. Finally, this solid was identified as ZnNP and stored for future tests and to prepare the composite with sample D1.2.5 Synthesis of ZD1-w and ZD1-t nanocomposites
A suspension of approximately 0.5% w/v of ZnNP in water was placed in an ultrasonic bath for 30 min. From D1-w and D1-t, 3 mL were diluted in 10 mL of deionized water. A volume of 2 mL from the ZnNP mixture was transferred into each diluted samples and the final mixture was stirred for 25 min. The prepared nanocomposites were named as ZD1-w and ZD1-t.2.6 X-ray diffraction
X-ray diffraction (XRD) patterns for graphite and GOx were carried out in a Brucker D2 Phaser equipment, with a step size of 0.1° s−1 whilst for ZnO a Siemens D5000 equipment was used with a step size of 0.05° s−1. Both equipments employed a copper source with λ = 1.5418 Å.2.7 Fourier transform infrared-ATR
In order to verify the functional groups from GOx and D1 sample, the attenuated total reflectance (ATR) adapted to a Fourier transform infrared (FTIR) equipment, was measured. A PerkinElmer Spectrum One spectrometer was used for this objective.2.8 UV-Vis
Electronic transitions from several samples were identified by UV-Vis using a Shimadzu UV-1800 equipment.2.9 Photoluminescence spectra
Photoluminescence (PL) spectra were recorded in a PerkinElmer LS55 spectrophotometer. The following modalities were required.2.10 Raman spectroscopy
Raman spectra were taken on a dispersive Raman instrument Senterra from Bruker, equipped with a 785 nm laser and an Olympus microscope. A 20× objective was employed; the integration times varied from 2 to 8 s depending on the band intensity, and the number of co-added spectra was between 8 and 10. The spectral range of detection went from 70 to 3500 cm−1 with a spectral resolution of 9–15 cm−1.2.11 SEM
With the aim to highlight the modifications between graphite (GF), graphene oxide (GOx) and graphene quantum dots (GQD) from sample D2-w, a scanning electronic microscopy (SEM) analysis was achieved. For this purpose, a JEOL JSM 6701F equipment was used. The images are shown in Fig. S3 from the ESI.†2.12 TEM
The sample for transmission electron microscopy (TEM) was prepared by dispersing the solids in distilled water, placed and dried on a TEM grid. Afterwards, they were observed with a JEOL JEM 1010 with 60–80 kV microscope.3 Results
3.1 Chemical structure study
An important matter of this study was the synthesis effectiveness. Graphene oxide structure pattern was obtained by XRD, compared with graphite and these patterns are presented in Fig. 1.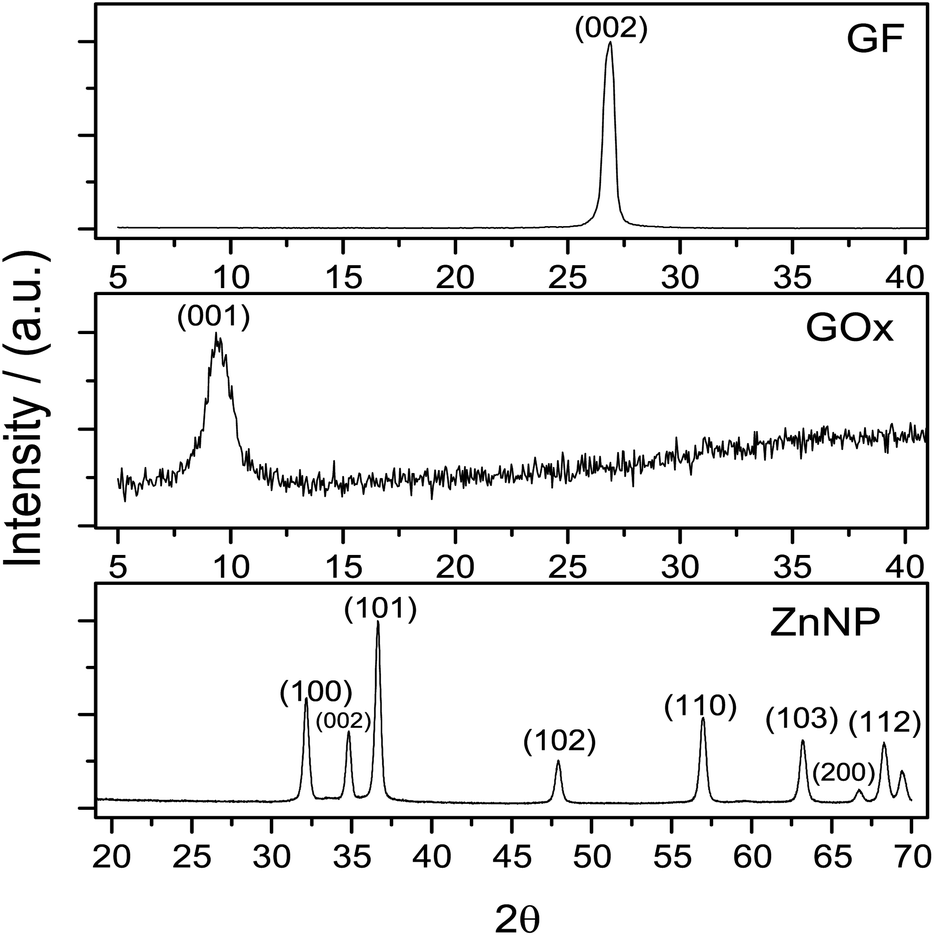 | ||
| Fig. 1 Normalized XRD diffraction patterns from graphite (GF), graphene oxide (GOx) and zinc nanoparticles (ZnNP). Graphite has a characteristic peak at 2θ = 26° whilst the GOx presents its peak at 2θ = 10°. XRD pattern from ZnNP exhibited characteristic peaks at the angles 2θ = 31, 34, 36, 47, 56, 62, 66, 68 and 69°. | ||
The diffraction pattern of graphite concur with JCPSD card 04-007-2076 where the peak of this solid was found at 2θ = 26°. In accordance with this card, the structure of graphite is rhombohedral which interplanar distance is d = 0.335 nm.28 After the synthesis of GOx a change in the graphite structure is evidenced (Fig. 1); this pattern, shows a peak around 2θ = 10° which normally is observed when the Hummers' method is used for the synthesis of graphene oxide.27,29 In Fig. 1 the diffraction pattern from ZnNP coincides with JCPSD card 01-007-2551.30 The diffraction patterns from GOx and ZnNP provide evidence that the required compounds were obtained.
3.2 Functional groups of GQD and their interaction with ZnNP
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band is shown at ≈1733 cm−1, the CC band at ≈1629 cm−1, the C–O–C at ≈1230 cm−1 and lately the band assigned to C–O is shown at ≈1077 cm−1.31–33
O band is shown at ≈1733 cm−1, the CC band at ≈1629 cm−1, the C–O–C at ≈1230 cm−1 and lately the band assigned to C–O is shown at ≈1077 cm−1.31–33
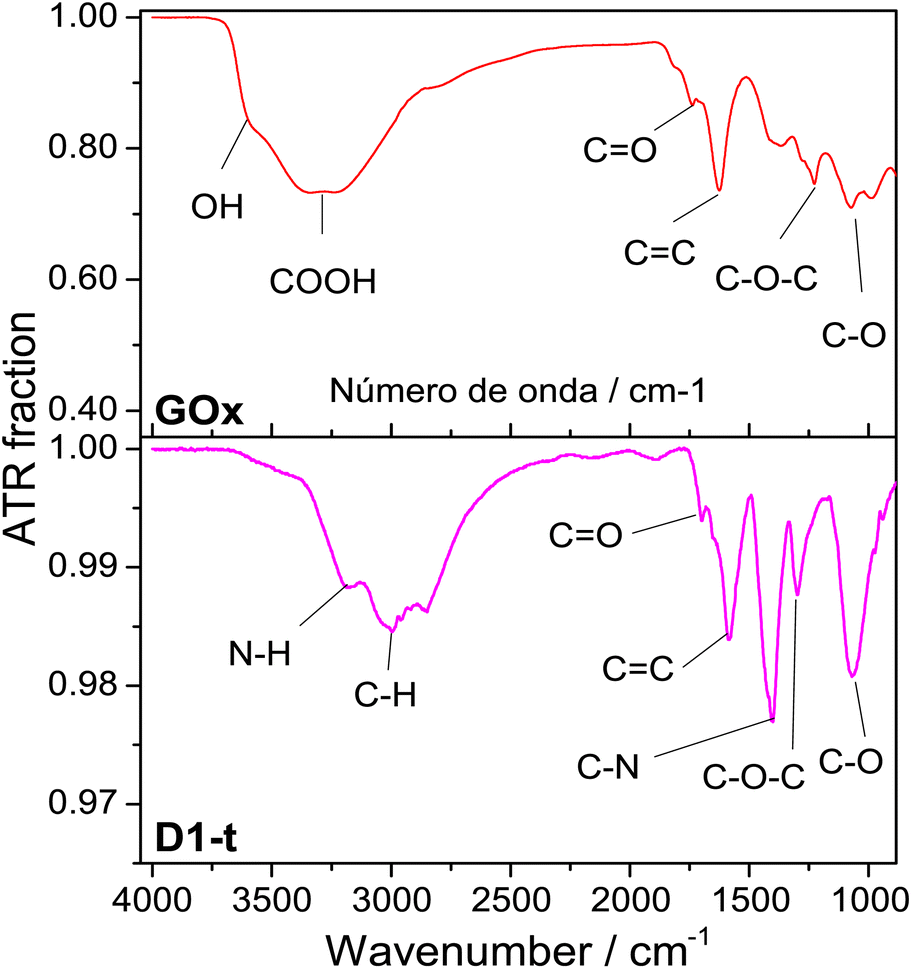 | ||
| Fig. 2 ATR normalized spectra from GOx and D1-t. Measurements were done at room temperature. Bands assignments in each sample are identified below spectra. | ||
From D1-t spectra, two new bands which are associated to the nitrogen incorporation to the GQD structure, are highlighted. One of these is shown at ≈1407 cm−1 and is assigned to the C–N bond and other at ≈3180 cm−1 from N–H bond. Both are characteristic of the amine groups.32 Note that the band at ≈3600 cm−1 attributed to O–H has vanished in the GQD spectra.
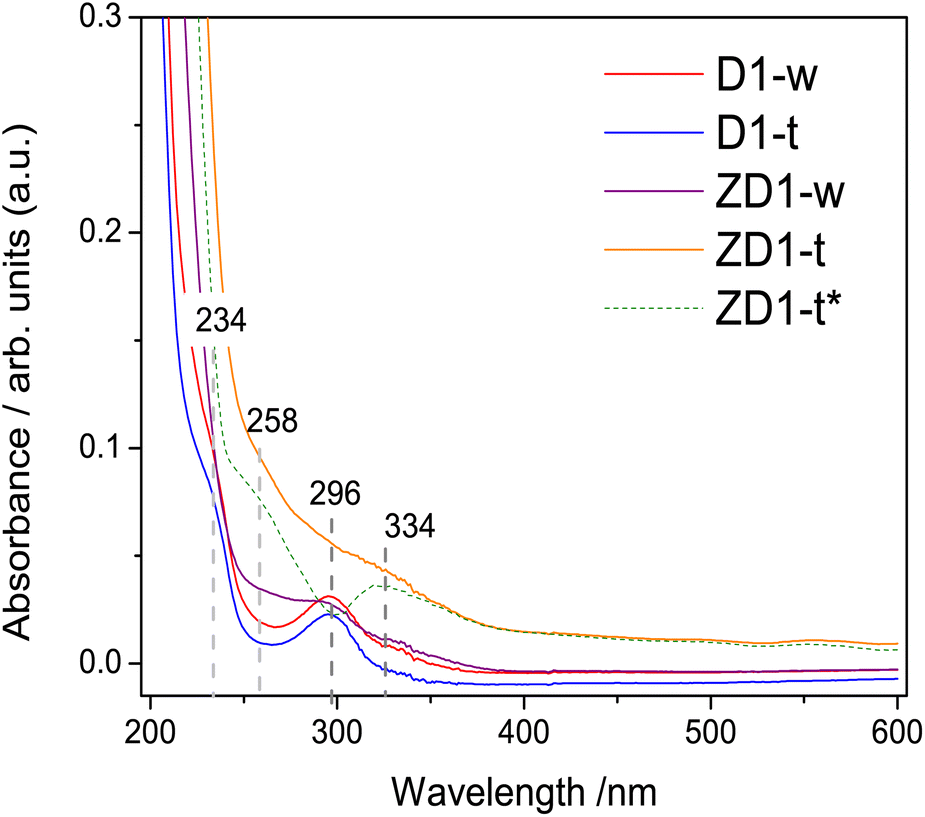 | ||
| Fig. 3 UV-Vis absorbance spectra from non-treated samples: D1-w and ZD1-w and treated: D1-t and ZD1-t. Spectra were taken in aqueous phase and at room temperature. Data shown over dashed gray lines indicate the corresponding values of bands in nanometers. For ZD1-t* (dashed green line) the shown spectrum is the subtraction of D1-t from ZD1-t. | ||
The band centred at 234 nm, corresponds to the electronic transitions from π → π* orbitals of the CC bonds which possesses principally sp2 hybridization.34,35 A band centred at 296 nm attributed to n → π* transition of the CO bond, emerges in all samples. This high intensity in D1-w and ZD1-w might be influenced by a grater amount in amine groups;35 whilst in D1-t and ZD1-t, even though the amine group are evidenced in the FTIR, this band has diminished and is not visible, respectively. At 258 nm and 334 nm, two bands are displayed in ZD1-t and very tenuous in ZD1-w which are assigned to ZnNP.36 With the aim to observe the absorbance spectrum from the ZnNP in the aqueous phase, the D1-t spectrum was subtracted from ZD1-t and the bands centred at 258 and 334 nm are more clearly noticed (Fig. 3 ZD1-t*).
3.3 Fluorescence analysis
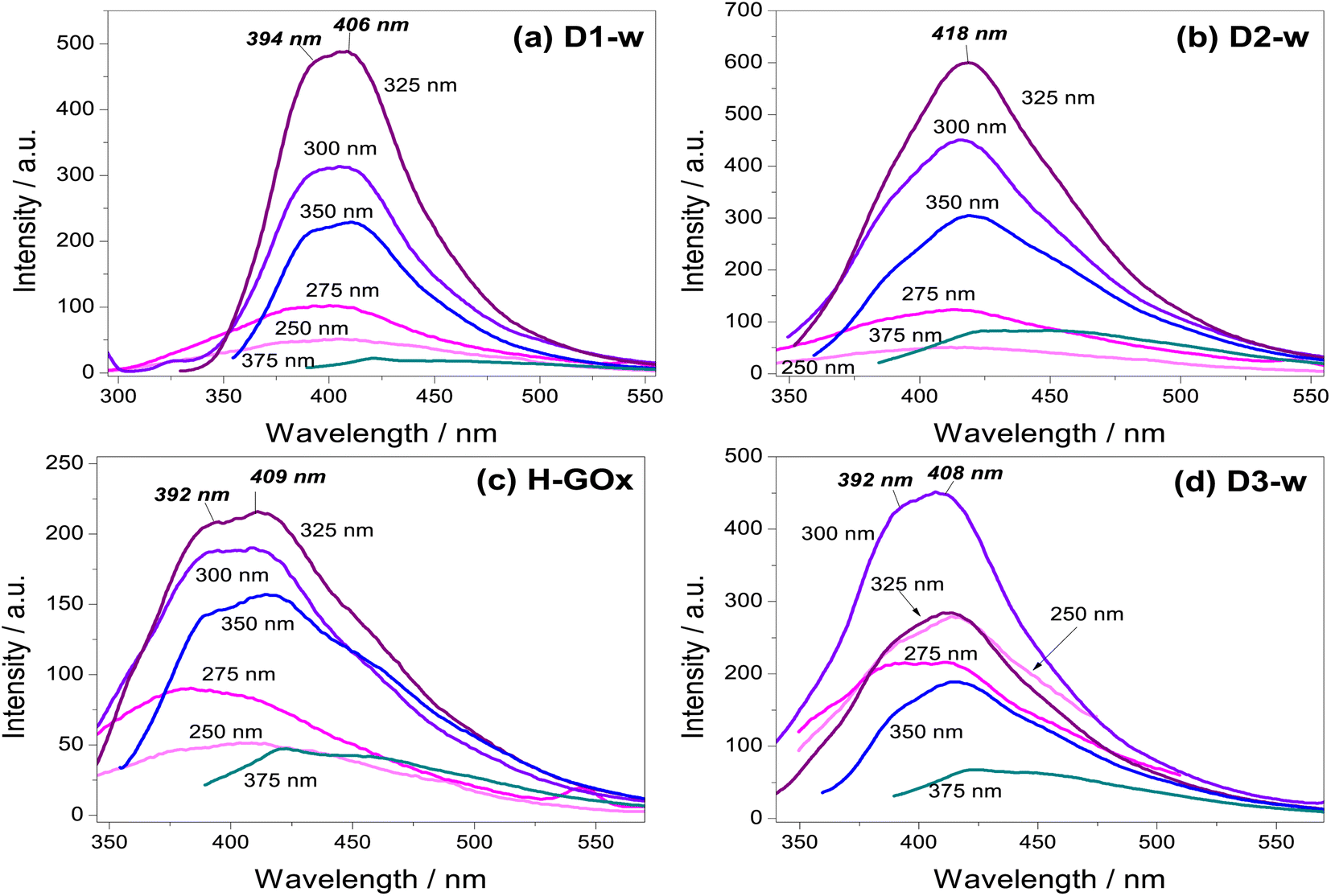 | ||
| Fig. 4 Emission spectra taken in aqueous phase to compare samples (a) D1-w (b) D2-w, and to observe the effect from (c) H-GOx in (d) D3-w. Values inside each graph for a particular peak of emission are the excitations wavelengths (λex). In cursive and bold are shown the peaks position of the maximum emission, only at the λex = 300 nm for D3-w and at the λex = 325 nm for the rest. | ||
Photoluminescence measurements started with an excitation wavelength (λex) of 250 nm and its variation, leaded to gradually increase the emission intensity to a maximum at the λex = 300 nm for D3-w and at the λex = 325 nm for the rest of the samples. From λex = 350 nm the emission intensity begins to decline. Therefore, for any of the routes the emission intensity of GQD, is in function of the excitation wavelength and the variation in the emission is similar for all routes. Another similar aspect in those samples is the peak position with respect to the excitation wavelength parameter; generally, as this parameter increases there is a red-shift. This behaviour is known as the giant red-edge effect which is a particular feature of graphene oxide and arises when polar molecules from solvents as water, are not aligned with fluorophores. As these fluorophores are exciting, water molecules align producing an additional step in the energy emission during the relaxation.37 In this regard, some differences might be noted from all the routes, by the analysis of the maxima intensity peaks. In D1-w, D2-w and H-GOx (Fig. 4a–c) the peak of maximum intensity is present at the λex = 325 nm. This peak shows a blue-shift which is linked to the higher concentrations of ammonium hydroxide and hydrogen peroxide used during the synthesis. For instance, for D2-w the maximum peak is centred around 418 nm while for D1-w is at 406 nm. This blue-shift might be caused due to a change in the particle size from GQD; this follows a similar trend to that theoretically deduced by Robertson and O'Reilly (1987) whose found that the energy gap between states π and π*, grows owing to a diminishing in particle size with sp2 hybridization.38 Therefore, in D1-w the formation of smaller particles is favoured and the same it is devised for H-GOx (Fig. 4).
The purpose of the H-GOx synthesis, was for hydrogen peroxide to split the sheets from graphene oxide reducing its size to a maximum. Comparing H-GOx with D1-w, for λex = 325 nm, there is a slight blue shift at 392 nm (Fig. 4c). Returning to hypothesis regarding the change in particle size, it is deduced that a reduction in particle size occurred during the H-GOx synthesis. However, the emission intensity from H-GOx considerably decreased, indicating that the introduced functional groups have weak photoluminescent properties; thus, it is inferred that the size does not influences in the intensity, as much as the incorporated nitrogenous ligands. This can be corroborated from the subsequent incorporation of amine group, through the reaction with NH4OH (route 3) in H-GOx. Apparently, the amine group now are linked to different particles that unveil its maxima emission when are excited at 300 nm (Fig. 4d). Therefore for the evolution of GQD, besides the particle size reduction of GOx, the reaction with ammonium hydroxide is necessary to generate this amine group linked to GQD structure in D3-w. It is worth mentioning that interaction among GQD and the amines ligands, is essential for a maximum emission intensity to exist, due to neither the graphene oxide nor ammonium hydroxide present this characteristic (see Fig. S1 in ESI†).
It has been found that the hydroxyl ions from graphene oxide interact with polar molecules of water (giant red-edge effect), whilst the carboxyls can be protonated with hydrogen ions.39 Because for this λex = 375 nm the giant red-edge effect is barely noted, it is deduced that in all cases the hydroxyl and carboxyl ions would be scarce. However, since the D1-w has the lowest intensity, it is claimed that in D1-w not only a decrement in particle size has occurred, but a facile incorporation from amine groups resulted.
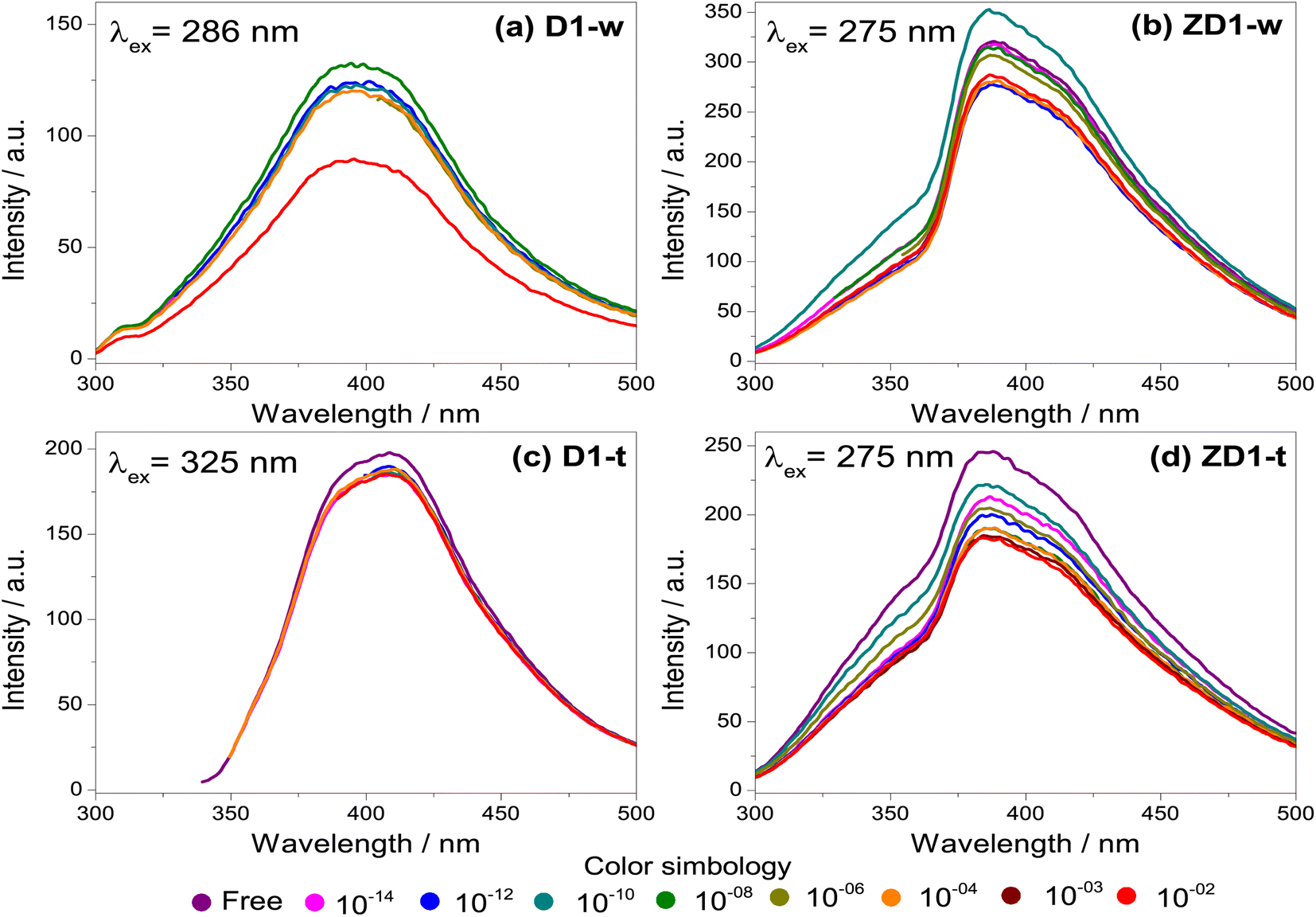 | ||
| Fig. 5 Emission spectra for the H2O2 detection. The spectra were measured in aqueous phase for the samples without thermal treatment (a) D1-w and (b) ZD1-w and for the thermal treated (c) D1-t and (d) ZD1-t. For each sample the λex which produced the maximum emission spectrum, was selected. The different concentrations of H2O2 are identified by the coloured dots marked at the bottom of the graphs and they are varied from free of H2O2 (fresh samples), up to a concentration of 10−2 M. | ||
The fresh samples D1-w and ZD1-w, initially present an emission intensity of 120 and 300 a.u. respectively (Fig. 5a and b). When the H2O2 concentration rises, there is not a trend in D1-w and its emission intensity remains almost the same. For the 10−8 M concentration, there is a slightly increment in the intensity regarding the previous. In ZD1-w a mild trend to decrease the intensity is exhibited as the H2O2 concentration rises. Nevertheless, this occurs until the concentrations of 10−6 M and 10−10 M are analysed; for these solutions there is also an increase in the emission intensity, reaching almost the 350 a.u. These emission increments are related with the chemical environment surrounding the particles of GQD. An excess of NH4OH produces a chemical reduction from GQD carboxylates (see ID1/IG ratio in Table 2). This reduction, increases the availability of the GQD as individuals fragments with sp2 domains, which are susceptible of being enveloped by greater particles40 (see explanation in section 4.2).
| Sample | Band characteristicsa | Ratio intensities | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D1 | D | D2 | D3 | G | D′ | ID/IG | ID1/IG | ID2/IG | ID3/IG | ID′/IG | |||||||
| p | w | p | w | p | w | p | w | p | w | p | w | ||||||
| a p = position of band peak in cm−1; w = full width at half maximum (FWHM) in cm−1. | |||||||||||||||||
| ZD1-t | 1303 | 39 | 1345 | 59 | 1402 | 94 | 1520 | 139 | 1627 | 91 | 1677 | 64 | 0.92 | 0.37 | 1.21 | 1.50 | 0.41 |
| D1-t | 1312 | 49 | 1353 | 59 | 1407 | 89 | 1517 | 119 | 1610 | 86 | 1662 | 69 | 1.03 | 0.71 | 1.20 | 1.38 | 1.01 |
| D2-t | 1312 | 49 | 1355 | 63 | 1414 | 91 | 1516 | 120 | 1615 | 83 | 1664 | 66 | 1.17 | 0.64 | 1.17 | 1.52 | 0.74 |
| D3-w | 1310 | 48 | 1351 | 62 | 1404 | 83 | 1489 | 98 | 1565 | 100 | 1647 | 95 | 1.40 | 0.82 | 1.32 | 1.23 | 2.22 |
Likewise as D1-w, the sample D1-t does not show a notable trend to diminish the emission intensity as the H2O2 increases, however it is not observed any increment in the emission for the subsequent spectra respecting the fresh spectrum (Fig. 5c). The emission intensity in ZD1-t decreases gradually as the H2O2 grows, except for the concentrations of 10−14 M and 10−12 M which not show a normal performance. Notwithstanding, in ZD1-t there is a proper trend to decrease the emission and therefore an adequate H2O2 detection. This enhancement is attributed to: (1) thermal treatment which eliminates the presence of ammonia reducing the effect of the unusual increase in the intensity, as specifically that shown in Fig. 5b, and (2) the presence of zinc contributes to improve the chemical stability of GQD.
3.4 Analysis of structure and inspection of the GQD morphology
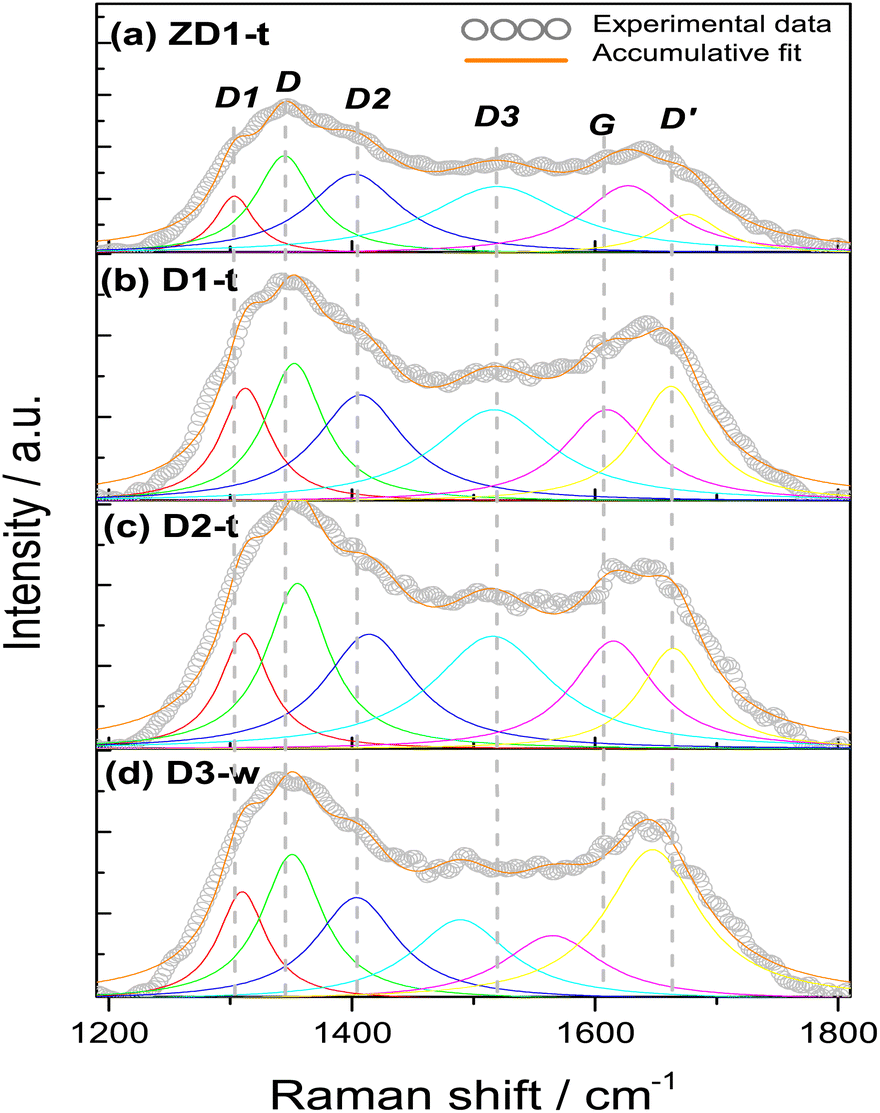 | ||
| Fig. 6 Raman spectra from samples (a) ZD1-t, (b) D1-t, (c) D2-t and (d) D3-w. All spectra were taken on aqueous phase using a laser of λex = 785 nm. The deconvolution produced 6 bands modelled by the Lorentz equation and its accumulative fit the data with a coefficient R2 = 0.98. The deconvolution represent the vibrations modes from the bands D1 = red, D = green, D2 = blue, D3 = cyan, G = pink, D′ = yellow. | ||
All the samples show the characteristics D, G and D′ bands encountered in several research works.42–44 The D band around 1350 cm−1, which is assigned to sp3 hybridization from a disordered structure, it is found for simple graphene but most commonly in graphene oxide. Occasionally, the intensity of this band is employed to differentiate some carbon structures and it has been established that the intensity in the oxidizing samples is greater, due to the functionalization with oxygen groups where the amount of sp3 hybridization increases;45 this finding reinforces the slight increment in the FWHM in D2-t and D3-w, respective D1-t and ZD1-t. If this D band is associated to oxidation of graphene and ultimately to formation of GQD, it is worth to note that the ID/IG ratio for D3-w, is the highest with 1.40 (Table 2). This indicates that the particles conceived by the pretreatment of H-GOx, are more susceptible to be oxidized. Moreover, this might reflects that the epoxy groups, which are associated with defects onto the basal plane,46 are responsible for the weakness and the split into smaller particles.41,47 In D1-t the value of 1.03 for ID/IG, is in accordance with the NH4OH weakness to cause a substantial rupture in the GOx sheets. Notwithstanding in this process GQD were produced, but mainly, also conduced the amine groups to bond at the GQD structure. Owing to a similar value in the ID/IG ratio for ZD1-t, it is inferred that the ZnNP produced only a slight modification in the GQD structure, probably acquiring a more ordered character due to a chemical stability granted by the ZnNP. The D2-t is among D1-t and D3-w with ID/IG = 1.17 meaning that this structure was susceptible to be attacked by oxygen ions which increased its defects.
The G band which is related with a crystalline structure from graphene, is normally presented around 1580 cm−1.41 Theoretically and experimentally it is known that shifts in the position and changes in the FWHM of the G band, are related with graphene layers. A shift to lower wavenumbers and an increment in FWHM reveals a trend to generate a graphene multilayer structure.48,49 Analysing the positions and the FWHM of the G bands for all the samples it is inferred that for D3-w, which shift to lower wavenumber and show an increment in FWHM, tend to form a multilayer structure contrary to D1-t, D2-t and ZD1-t (Table 2). The G band shift in D2-t is owing to it was synthesized with a lower NH4OH concentration, which reduces the placement of amine groups onto the basal plane from the GQD; in the same way, the removal of NH4OH leads to a lesser attraction among the GQD planes and this inhibits the formation of layers. For ZD1-t there might be occurring a similar situation as in D2-t. As mentioned, the amine groups from GQD in the sample D1-t interact with ZnNP and in this way despite the FWHM grows the band shift to the right, suggesting that the attraction between GQD is null; this provoke the trend to produce thin layers (monolayers-like) mainly for those GQD in the aqueous solution (or not incorporated to ZnNP). Nevertheless, the close chemical interaction between the ZnNP and the GQD is indispensable, owing to neither NH4OH nor ZnNP have strong photoluminescent properties (Fig. S1 and S2†).
The vibration modes from adjacent D1, D2 and D3 bands are related to formation of the edge states in GQD during its synthesis. For instance, the D1 at 1260 cm−1 is assigned to the functional group COOH, the D2 at 1435 cm−1 band is attributed to the presence of functional groups CO,47 whilst the D3 band at 1515 cm−1, in this work, it was assigned to C–O bond situated at the edges from GQD. When the samples are being synthesized the presence of water might induces the incorporation of COOH. This is the reason that the FWHM of D1 band is very similar in all samples and the position shifts to higher wavenumbers, highlighting the characteristic deformation of the graphitic structure and promotion of sp3 hybridization. Notwithstanding, the slight decreasing in the FWHM of the D1 band for D3-w is in line with its FWHM values in the D2 and D3 bands; the FWHM of the D2 band for D3-w is lower than the rest of samples, however the rest have not excessively high values which indicates that all have CO groups at the edges. If it is compared the narrower FWHM and the shift to lower wavenumbers of the D3 band for D3-w, it can be inferred that it was more susceptible to be oxidized than D1-t and D2-t. Moreover, since ID1/IG and ID2/IG ratios for D3-w increased, these functional groups (CO, COOH) are more abundant at the edges of this sample (Table 2). In fact, in the UV-Vis spectra (Fig. 3) the bands assigned to CO are higher for the non-treated samples than for those treated. Nevertheless, the amine groups exist in D3-w due to it exhibits photoluminescent properties (Fig. 4d). In D1-t and D2-t, the ID1/IG and ID2/IG ratios, are lower than D3-w and this indicates that the amine groups were bonded more easily at the edges which is also corroborated with the decreasing of the FWHM in D3 band for D3-w; the amines bonding was marked in D2-t as it shows lower values in this instance. The values of the ID1/IG and ID2/IG ratios for ZD1-t are similar to D1-t but particularly in the ID2/IG ratio confirming the slight modifications promoted by the ZnNP. These modifications might be the chemical stability gained by GQD because of the CO and principally the COO functional groups losses. Analysing the ID3/IG ratio it is observed that D1-t is amid D2-t and D3-w in this instance. While in D1-t the amine incorporation was straightforward in D3-w, with the lower ID3/IG value, emphasizes the lack of amine groups at the edges (Table 2). As mentioned the trend from D3-w was to create multilayer of GQD; precisely the amine groups are responsible in the formation of this multilayer structure which for D3-w, act as ligand over the GQD planes, whilst there is not attraction at the edges between the same oxygen groups. Is reasonable the existence of epoxy and hydroxyl groups or ultimately (although less likely) π orbitals, all onto the GQD basal plane, in order to attract the amine groups. Therefore it is likely that in D3-w and moderately in D1-t, the amine groups principally are placed at the basal plane and in D1-t also at the edges. It is noteworthy that although the GQD from D1-t and D3-w start to stack in multilayer, there is only a mildly growth as is discussed in the PL studies (section 3.3) and as is observed in the micrograph from Fig. 7a.
 | ||
| Fig. 7 TEM images from (a) D1-t: the red squares inside the micrography enclose the accumulated GQD very close between them and (b) ZD1-t: the red circles enclose the ZnNP which are mostly lesser than 100 nm. | ||
The D′ band is related with the defects where the carbons tend to form pentagons or octagons instead of hexagons, besides to be associated with a graphitic structure.50 The formation of this type of defects is directly related with the aggressive synthesis conditions during the synthesis of D1-t and D3-w which present the higher ID′/IG ratios. Also the wide FWHM of D3-w and in less extent for D1-t might indicates higher predisposition to incorporate functional groups at the basal plane. On the contrary the rest of samples are prone to be attacked at the edges of the GQD. Moreover, D2-t and ZD1-t maintained its structure stable to chemical reaction induced by the ammonium hydroxide during the synthesis and due to the chemical stability by the ZnNP presence.
4 Discussion
4.1 GQD development and behaviour
According to the HT synthesis and to the characterization techniques used in this work, a scheme of GQD formation is presented in Fig. 8.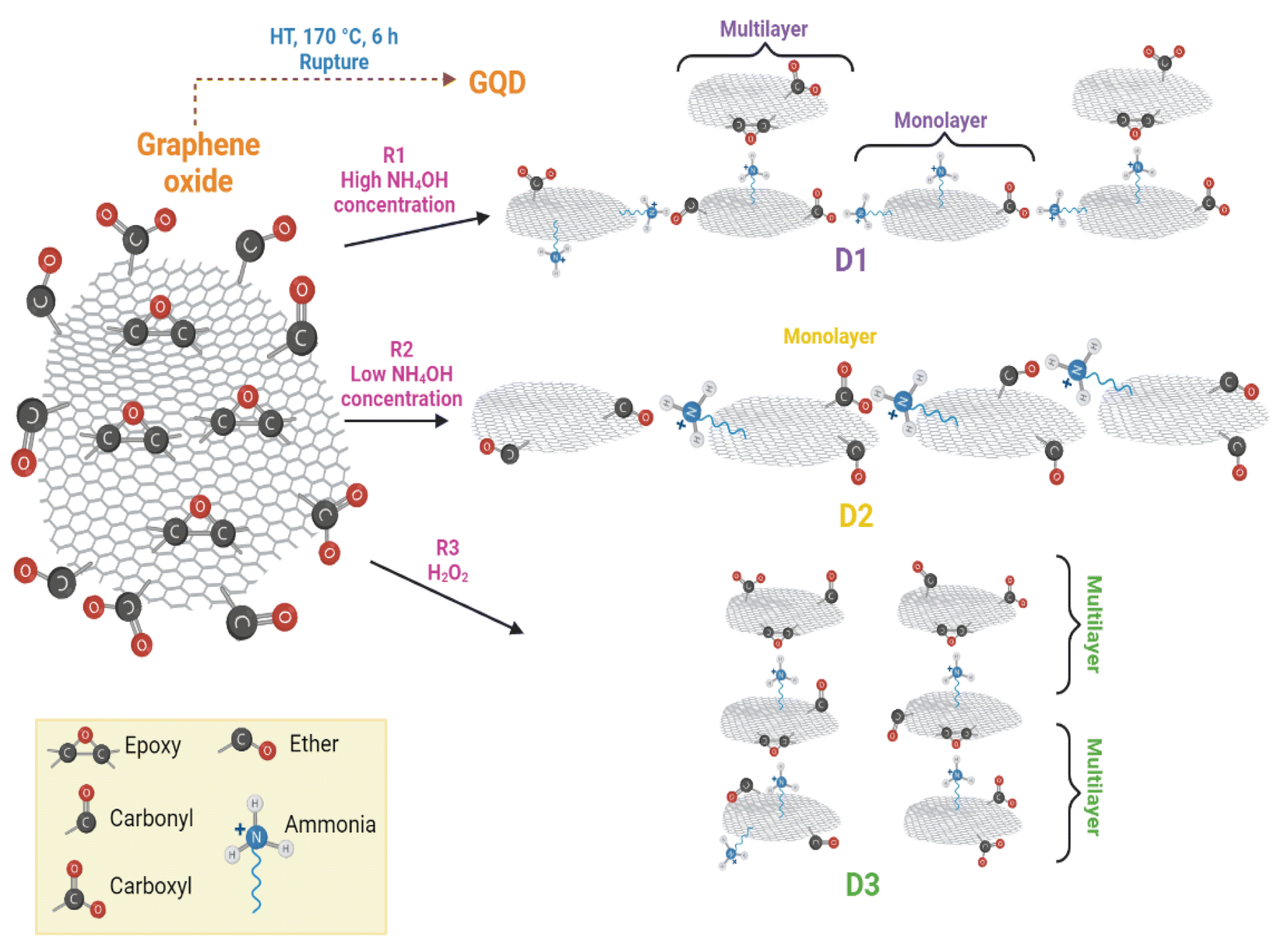 | ||
| Fig. 8 Reaction scheme for the GDQ formation by the three different routes described in section 2.3. | ||
The principal course of action in D2-t was the defects formation owing to the low concentration of NH4OH. These defects in D2-t were exploited by a slightly oxidant environment during the synthesis, maintaining epoxy groups at the basal plane and C–O at the edges (see the higher ID3/IG ratio in Table 2 for D2-t). The lower NH4OH concentration place some amines at the edges (substituting CO and mainly COO−1 groups) which with the presence of C–O from adjacent GQD sheets, slightly and laterally increased the GQD plane. This indicates that the development of GQD in D2-t is given by the weakness from the carboxyl which are attacked by amine groups incorporating them at the edges (see ID1/IG in Table 2 for D2-t and Fig. 8). On the other hand, the higher concentration of NH4OH in D1-t assures that no only a structure rupture to occur, but also facilitates the placement of amine groups both, at the basal plane as at the edges. Similarly, the band related to CO in Raman studies (see ID2/IG in Table 2) in D1-t and D3-w has a high ratio; thus the amines are not bonded at this sites but they prefer the basal plane. Since in D1-t and D3-w amine groups are principally at the basal plane, the attraction is given in a vertical fashion and ultimately producing a multilayer structure specially in D3-w (Fig. 8). Such situations, lateral growth and multilayer structure are responsible of the slightly PL variations.
The excess of NH4OH also varies the size of the GQD clusters, as it is shown in the SEM and TEM images from Fig. S3f† and 7a, and also influences the photoluminescent properties. This situation, is similar as that presented when higher concentrations of GOx are used to evaluate the PL spectra; a shift to lower energies is observed because the accumulation of some GOx particles (Fig. S4†). The influence of NH4OH not only is limited to the structure rupture, availability of amine groups and the circle-shape of GQD, but also independently of the route, it can chemically reduce the GOx even though a low NH4OH concentration was used (Fig. S5†).
4.2 GQD and ZnNP interaction
Photoluminescence studies (Fig. 5 and S4†) proved that slightly changes in the particles forming clusters might influence on the photoluminescent properties; changes in particle size can also impact on the interaction between GQD and ZnNP and finally with the H2O2 detection. Precisely, when NH4OH is in excess stacking or lateral growth of the GQD might be occurring. For the detection of H2O2 with photoluminescence spectroscopy, initially is prepared a mixture of this oxidant and the samples containing GQD; just at the beginning, particles from GQD with size around 5 nm (fragments of GQD) with sp2 domains, have a PL emission lower than the H2O2 free spectrum. After 5 minutes, these fragments creates clusters of sp2 domains contacting sp3 carbon structures starting an alteration in the intensity; this alteration is the growth of the spectrum, for instance for the 10−10 M H2O2 solution, higher than the H2O2 free PL spectrum (see Fig. 5b). Due to the amine groups there is an increase in the particles size, this is the contact of sp2–sp3 which reduces the emission intensity with the course of time as recombination of small clusters.40 If the particle size increases the band gap decreases and this might be also presenting in the GOx (Fig. S4†). As was proved, in spite that GQD posses photoluminescent properties, they do not detect H2O2 properly. When ZnNP is incorporated (GQD–ZnO nanocomposite) the fragments of GQD are attracted by the ZnO surface; principally where the oxygen from ZnO can trap the amine groups. The ZnO confer the GQD chemical stability avoiding the unnecessary reduction; in other words, the small clusters now are not in contact with this carbon-oxygen matrix with sp3 hybridization. Moreover, the removal of NH4OH enhances this interaction between ZnO and GQD, and now, only the spectra for the H2O2 concentrations of 10−14 and 10−12 (Fig. 5c and d) are not properly performed. However the for the rest of solutions concentrations, a gradual diminishing of the intensity is registered.4.3 Steps on the H2O2 detection
In order to prove the involved steps for the H2O2 detection, a simple alternative test was carried out. For this test, the wasted solid from D1-w and H2O2 were lead to react and thus compare if it had a similar effect as in the H-GOx synthesis. A scheme concerning this experiment is shown in Fig. S6.† After HT reaction (between the wasted solid and H2O2), the liquid was filtered and named sD1-w. It was found in this test, that the residual solid was slightly dissolved. The PL spectra (Fig. S7†) compares sD1-w with D1-w and H-GOx. The peaks intensities in sD1-w and D1-w are very similar and both are higher than H-GOx, although the position from sD1-w it is very similar to H-GOx. However if the PL spectrum from Fig. 4d, is compared with the sD1-w the λex in both spectra is different, suggesting that H2O2 only induces the rupture from the solids but the PL emission is caused from the amine functional group. Therefore, due to the previous amines groups incorporation in D1-w and because the wasted solid from D1-w was not dissolved, it is inferred that once the amine groups are as ligands into the GQD structure it is very difficult to remove them. Even though a minor hydroxyls might be linked to GQD structure due to the peaks positions in sD1-w and H-GOx are similar. This proves that the followed steps for the detection are given via the ZnNP and H2O2 interaction. The presence of H2O2 might induces a slight polarization in the ZnNP structure perturbing the environment from the amine and the GQD. In the Fig. 9 a scheme from this reaction is given. In this way, the quenching in the H2O2 detection it is easier when its concentration is augmented, likely perturbing the polarity of the ZnO structure. In fact, it is feasible that the presence of H2O2 oxidize the ZnO owing to its high reduction standard potential. Finally, the quenching caused by the H2O2 over the ZD1-t particles can be described by fit the results with a quadratic equation; this equation represents the tendency of diminishing in the emission intensity as the oxidant concentration is increased and it is properly accomplished for the interval of H2O2 concentrations from 0.008 to 0.1 M (Fig. S8†).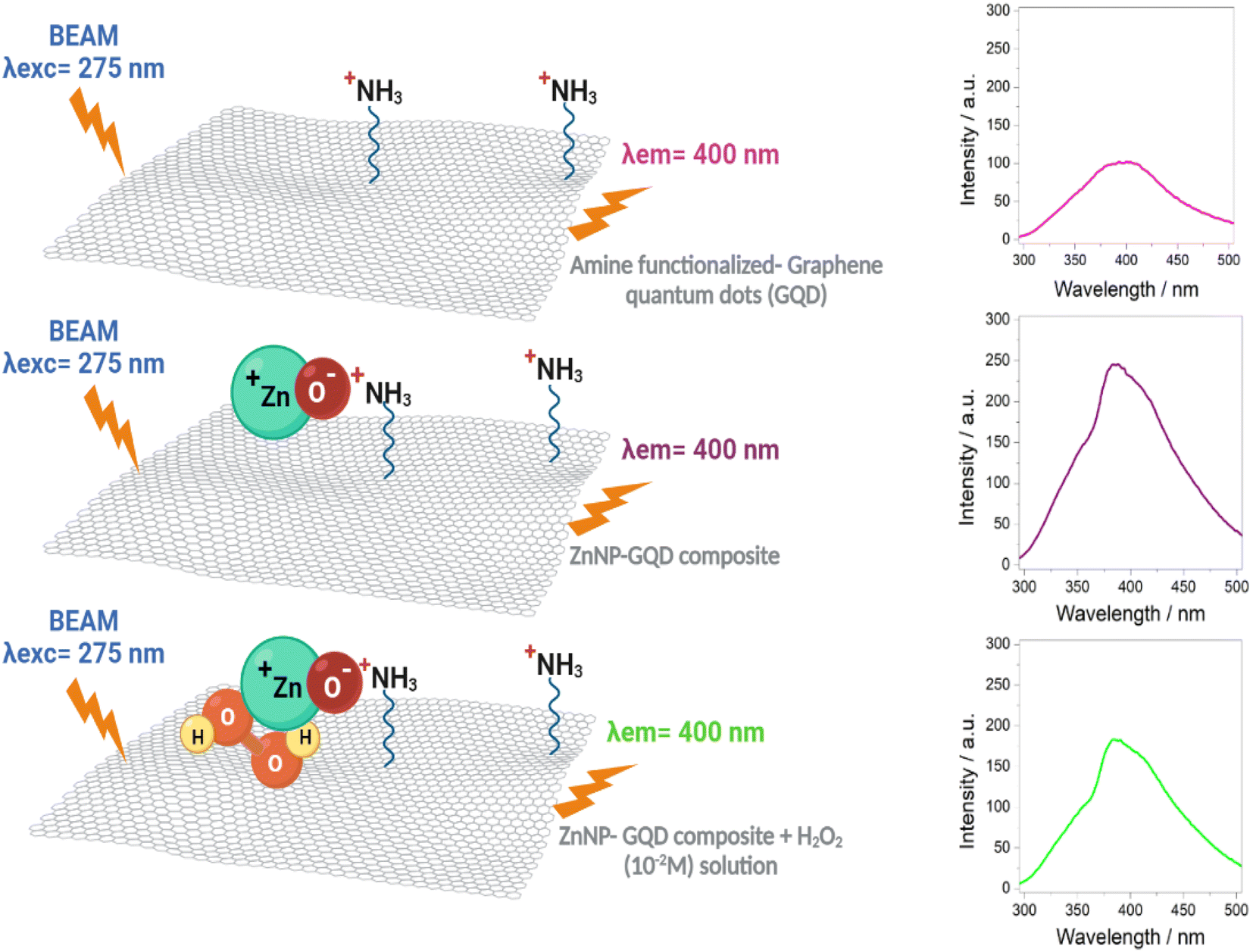 | ||
| Fig. 9 Reaction scheme for the H2O2 detection in the ZD1-t sample shown in the Fig. 5d. | ||
5 Conclusions
The photoluminescence (PL) technique was sensible to GQD functionalized with amines. PL spectra suggested that GQD from sample D1-t (synthesized with high NH4OH concentration) in general, resulted with lower particle size (emission peak centred at 406 nm) than D2-t synthesized with low NH4OH concentration (emission peak centred at 418 nm). GQD produced with H2O2 in general were also smaller, however they presented weak PL emission intensity, until they were functionalized with amine groups (sample D3-w), indicating that emission was due to this ligand.Because the ID3/IG ratios concerning C–O groups at the edges of GQD in D1-t and D3-t are lower than in D2-t the amines groups were incorporated at the basal planes in smaller GQD particles while higher particle size tend to place amines at the edges.
The ZnO nanoparticles in the GQD (ZnO–GQD, nanocomposite) enhance the PL emission intensity. Since once the amine groups are bonded with GQD cannot be replaced by hydroxyl or oxygen ions, the H2O2 detection tested by photoluminescence studies, is achieved thanks to the interaction between ZnO nanocomposite and H2O2, producing a quenching effect. This quenching is accentuated by the increasing of the H2O2 concentration and this behaviour can be described properly with a quadratic equation covering a concentration range from 0.008 M to 0.1 M.
Author contributions
Rolando Ramírez: conceptualization, methodology, investigation, writing original draft. Sara Rodríguez: writing review and editing. Genoveva Hernández: investigation and resources. Idalia Gómez: conceptualization, supervision, resources and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the financial support from CONACYT (grant no. I1200/320/2022) and also from the UANL-FCQ through the PAICYT program. We also thanks to the LANCAM for the Raman measurements, Ms Ma. Lourdes Palma Tirado for the technical support with TEM micrographs and to Laboratorio de Materiales I from UANL-FCQ specially to Yolanda Peña and Dalmy Ochoa for their support with XRD measurements and to Fernanda Retana for her technical support with photoluminescence equipment and Hugo Salas for the SEM images.Notes and references
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- R. Kaimal, V. Vinoth, A. S. Salunke, H. Valdés, R. V. Mangalaraja, B. Aljafari and S. Anandan, Ultrason. Sonochem., 2022, 82, 105868 CrossRef CAS PubMed.
- L. A. Ponomarenko, F. Schedin, M. I. Katsnelson, R. Yang, E. W. Hill, K. S. Novoselov and A. K. Geim, Science, 2008, 320, 356–358 CrossRef CAS PubMed.
- H. Cao, W. Qi, X. Gao, Q. Wu, L. Tian and W. Wu, Nanotheranostics, 2022, 6, 205–214 CrossRef PubMed.
- A.-M. Croitoru, A. Moroşan, B. Tihăuan, O. Oprea, L. Motelică, R. Truşcă, A. I. Nicoară, R.-C. Popescu, D. Savu, D. E. Mihăiescu and A. Ficai, Nanomaterials, 2022, 12, 1943 CrossRef CAS PubMed.
- M. Facure, R. Schneider, L. Mercante and D. Correa, Mater. Today Chem., 2022, 23, 100755 CrossRef CAS.
- S. Akbari, Int. J. Environ. Anal. Chem., 2020, 102, 789–803 CrossRef.
- R. Tian, S. Zhong, J. Wu, W. Jiang, Y. Shen, W. Jiang and T. Wang, Opt. Mater., 2016, 60, 204–208 CrossRef CAS.
- Y. R. Kumar, K. Deshmukh, M. M. N. Ali, G. Abhijay, W. A. Al-Onazi, A. M. Al-Mohaimeed and S. K. Pasha, Environ. Res., 2022, 203, 111842 CrossRef CAS PubMed.
- S. M. Mousavi, S. A. Hashemi, A. Gholami, S. Mazraedoost, W.-H. Chiang, O. Arjmand, N. Omidifar and A. Babapoor, J. Sens., 2021, 2021, 1–14 CAS.
- P. Szustakiewicz, N. Kołsut, A. Leniart and W. Lewandowski, Nanomaterials, 2019, 9, 602 CrossRef CAS.
- M. L. Yola and N. Atar, J. Electrochem. Soc., 2016, 163, B718–B725 CrossRef CAS.
- G. Chaloeipote, J. Samarnwong, P. Traiwatcharanon, T. Kerdcharoen and C. Wongchoosuk, R. Soc. Open Sci., 2021, 8, 210407 CrossRef CAS PubMed.
- T. T. B. Quyen, N. N. T. My, D. T. Pham and D. V. H. Thien, Talanta Open, 2022, 5, 100103 CrossRef.
- J. Wen, D. He, T. Zhuo, W. Li, J. Liu, J. Wu, Y. Zhao and Y. Yuan, J. Environ. Chem. Eng., 2023, 11, 109333 CrossRef CAS.
- Y. Zhang, Y. Liu, F. Sun and N. Yang, Appl. Surf. Sci., 2023, 611, 155655 CrossRef CAS.
- Z. Zhang, H. Liu, L. Zhai, J. Wu and L. Li, Chem. Phys. Lett., 2023, 811, 140177 CrossRef CAS.
- A. S. Siddiqui, M. A. Ahmad, M. H. Nawaz, A. Hayat and M. Nasir, Catalysts, 2022, 12, 1105 CrossRef CAS.
- H. Shen, H. Liu and X. Wang, Appl. Surf. Sci., 2023, 612, 155816 CrossRef CAS.
- Z. Tan, C. Zhu, L. Han, X. Liao and C. Wang, Sens. Actuators, B, 2022, 373, 132770 CrossRef CAS.
- Z. Xu, C. Zeng, Y. Zhao, M. Zhou, T. Lv, C. Song, T. Qin, L. Wang, B. Liu and X. Peng, Food Chem., 2023, 410, 135381 CrossRef CAS PubMed.
- M.-J. Lee, J.-A. Song, J.-H. Choi, J.-H. Shin, J.-W. Myeong, K.-P. Lee, T. Kim, K.-E. Park and B.-K. Oh, Biosensors, 2023, 13, 289 CrossRef CAS PubMed.
- X. Deng, H. Tang and J. Jiang, Anal. Bioanal. Chem., 2014, 406, 6903–6916 CrossRef CAS.
- O. Adegoke, S. Khene and T. Nyokong, J. Fluoresc., 2013, 23, 963–974 CrossRef CAS PubMed.
- X. Liu, H. Tian, L. Yang, Y. Su, M. Guo and X. Song, Sens. Actuators, B, 2018, 255, 1160–1165 CrossRef CAS.
- Y. He, X. Wang, J. Sun, S. Jiao, H. Chen, F. Gao and L. Wang, Anal. Chim. Acta, 2014, 810, 71–78 CrossRef CAS PubMed.
- N. Yadav, V. Kallur, D. Chakraborty, P. Johari and B. Lochab, ACS Omega, 2019, 4, 9407–9418 CrossRef CAS.
- S. Hembacher, F. J. Giessibl, J. Mannhart and C. F. Quate, Proc. Natl. Acad. Sci., 2003, 100, 12539–12542 CrossRef CAS PubMed.
- K. Xuan Sheng, Y. Xi Xu, C. Li and G. Quan Shi, New Carbon Mater., 2011, 26, 9–15 CrossRef.
- W. Muhammad, N. Ullah, M. Haroon and B. H. Abbasi, RSC Adv., 2019, 9, 29541–29548 RSC.
- Q. Lu, C. Wu, D. Liu, H. Wang, W. Su, H. Li, Y. Zhang and S. Yao, Green Chem., 2017, 19, 900–904 RSC.
- X. Gao, Z. Ma, M. Sun, X. Liu, K. Zhong, L. Tang, X. Li and J. Li, Food Chem., 2022, 369, 130964 CrossRef CAS PubMed.
- D. L. Pavia, Introduction to spectroscopy, Harcourt College Publishers, 2001, p. 579 Search PubMed.
- Z. Benzait and L. Trabzon, ACS Omega, 2022, 7, 37885–37895 CrossRef CAS PubMed.
- S. Kang, H. Han, K. Lee and K. M. Kim, ACS Omega, 2022, 7, 2074–2081 CrossRef CAS PubMed.
- S. Talam, S. R. Karumuri and N. Gunnam, ISRN Nanotechnol., 2012, 2012, 1–6 CrossRef.
- P. Zheng and N. Wu, Chem.–Asian J., 2017, 12, 2343–2353 CrossRef CAS.
- J. Robertson and E. P. O'Reilly, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 2946–2957 CrossRef CAS PubMed.
- S. K. Cushing, M. Li, F. Huang and N. Wu, ACS Nano, 2013, 8, 1002–1013 CrossRef PubMed.
- G. Eda, Y.-Y. Lin, C. Mattevi, H. Yamaguchi, H.-A. Chen, I.-S. Chen, C.-W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505–509 CrossRef CAS.
- L.-M. Peng, Z. Zhang and S. Wang, Mater. Today, 2014, 17, 433–442 CrossRef CAS.
- S. H. Song, M.-H. Jang, J. Chung, S. H. Jin, B. H. Kim, S.-H. Hur, S. Yoo, Y.-H. Cho and S. Jeon, Adv. Opt. Mater., 2014, 2, 1016–1023 CrossRef CAS.
- E. Dervishi, Z. Ji, H. Htoon, M. Sykora and S. K. Doorn, Nanoscale, 2019, 11, 16571–16581 RSC.
- D. Liu, X. Chen, Y. Hu, T. Sun, Z. Song, Y. Zheng, Y. Cao, Z. Cai, M. Cao, L. Peng, Y. Huang, L. Du, W. Yang, G. Chen, D. Wei, A. T. S. Wee and D. Wei, Nat. Commun., 2018, 9, 1–10 CrossRef.
- M. Pumera, Nanoscale Res. Lett., 2007, 2, 87–93 CrossRef CAS.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2009, 114, 832–842 CrossRef.
- G. Rajender and P. K. Giri, J. Mater. Chem. C, 2016, 4, 10852–10865 RSC.
- R. Vuppuladhadium, H. E. Jackson and R. L. C. Wu, J. Appl. Phys., 1995, 77, 2714–2718 CrossRef CAS.
- H. Wang, Y. Wang, X. Cao, M. Feng and G. Lan, J. Raman Spectrosc., 2009, 40, 1791–1796 CrossRef CAS.
- O. Vryonis, T. Andritsch, A. S. Vaughan and P. L. Lewin, J. Mater. Sci., 2019, 54, 8302–8318 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02355a |
| This journal is © The Royal Society of Chemistry 2023 |