
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmine-bearing cyclic ketene acetals for pH-responsive and degradable polyesters through radical ring-opening polymerisation†
Yiyi
Deng
 ab,
Anaïs
Frezel
ab,
Fabian
Mehner
ab,
Peter
Friedel
a and
Jens
Gaitzsch
*a
ab,
Anaïs
Frezel
ab,
Fabian
Mehner
ab,
Peter
Friedel
a and
Jens
Gaitzsch
*a
aLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Straße 6, 01069 Dresden, Germany. E-mail: gaitzsch@ipfdd.de
bTechnische Universität Dresden, Berg Straße 66, 01069 Dresden, Germany
First published on 6th September 2023
Abstract
A series of previously unreported amine-bearing cyclic ketene acetals (CKAs) furnished with different alkyl-substituents were prepared. These CKAs pave the way for a new range of pH-responsive and degradable polyesters from radical ring-opening polymerisation (RROP). CKA 2-methylene-1,3,6-trioxocane (MTC) was copolymerised with the amine-bearing CKAs to obtain a range of new degradable smart copolymers. Varying the type and amount of amine-bearing CKA allowed to fine-tune the 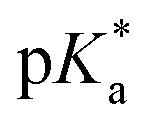 value of the polymers, showcasing the versatility of this approach.
value of the polymers, showcasing the versatility of this approach.
Following the increasing issues with plastic waste, polyesters like poly(caprolactone) (PCL) or poly(lactic acid) (PLA) and similar materials have become increasingly important materials owing to the biodegradable ester units in the polymer backbone. They found numerous applications in tissue engineering, drug delivery or commodity materials.1 Traditional synthetic routes towards polyesters are either by polycondensation or ring-opening polymerisation.2 However, the former tends to generate polyesters with low molecular weight and the latter method requires rigorous synthetic procedures (especially from ionic polymerization) to afford pure polymers.3 Proposed by Bailey et al., radical ring-opening polymerisation (RROP) of cyclic ketene acetals (CKAs) has received considerable attention and is regarded as an alternative tool to generate polyesters.4 RROP possesses the robustness of radical polymerisations, yields degradable polyesters2,5 and allows for functional groups (e.g. amines) within the polymer backbone.6
Homopolymerisations of CKAs are known to generate polyesters (bio)degrading under basic or enzymatic conditions.7,8 Until now, 2-methylene1,3-dioxepane (MDO), 2-methylene-1,3,6-trioxocane (MTC) and 2-methylene-4-phenyl-1,3-dioxolane (MPDL) are the CKAs with be highest publication record.9–13 These CKAs are generally characterised by an almost complete degree of ring-opening and a known structure in terms of the main repeating unit and branching positions from inter- or intramolecular H-transfer.13–16 Some of these polymers already have an additional functionality like the semi-crystallinity of poly(MDO) and the higher hydrophilicity of poly(MTC) as compared to other polyesters from RROP.12–14 However, the lack of responsive groups limits their range of potential applications like drug delivery.17
One strategy to circumvent this issue is the copolymerisation of CKAs with various responsive vinylic monomers.5,18–20 This strategy introduced ester linkages into the vinyl polymer backbone to confer (bio)degradability2 and responsive vinylic monomers endowed responsive properties.21,22 CKAs including MDO and BMDO have been successfully combined with oligoethylene glycol methacrylate and N,N-dimethylacrylamide for water solubility,23–25N-isopropylacrylamide for temperature responsiveness26,27 and 2-(diethylamino)ethyl methacrylate for pH-responsivness.23,28 CKA-bearing copolymers have been developed in the field of functional and degradable nanoparticles,29–31 hydrogels,32 3D printing materials,33 and coatings.34,35 However, the unfavorable CKA/vinylic copolymerisation parameters caused a composition drift towards gradient copolymer, which prohibited the randomly distributed ester units in the polymer backbone. This will therefore lead to an incomplete degradation into a mixture of oligomers.36,37 Hence, a reproducible method towards responsive and fully degradable polyesters from RROP is still a challenge.
Our group has focused on fully degradable polyesters purely based on CKAs,6,8,15,38 including amine-bearing CKAs for pH-responsive polyesters. This includes the amine-bearing N-iso-propyl-2-methylene-1,3,6-dioxazocane (iPr-MAC, 7c), allowing for pH-responsive and biodegradable nanoparticles.6 With a cloud point of 6.0, the pKa of the polymerised iPr-MAC was similar to the well-known pH-responsive poly(diisopropyl aminoethyl methacrylate) (PDPA).39 Copolymerisation of 7ciPr-MAC with 4,7-dimethyl-2-methylene-1,3-dioxepane (DMMDO) led to copolymers with an increased molecular weight, but the pH-responsive behaviour shifted from 6 for P(iPr-MAC) to below 4 for the copolymer. Taking advantage of these merits, i.e., pH-sensitivity and biodegradability, these purely CKA-based polyesters and nanoparticles accessed potentials in biomedical field, but lacked a pKa value closer to neutral pH. Inspired by this notion, we adopted the reported synthetic route to prepare new amine-bearing CKAs with varying substituents (Alk-MACs), including the ethyl (Et-MAC, 7a), n-propyl (nPr-MAC, 7b), n-butyl (nBu-MAC, 7d), sec-butyl (sBu-MAC, 7e), iso-butyl (iBu-MAC, 7f) and tert-butyl (tBu-MAC, 7g) derivatives (Fig. 1a). Following monomer synthesis and calculations on the molecule structure, we subsequently copolymerised the Alk-MACs with the more hydrophilic MTC as it ought to have a pKa closer to neutral pH (Fig. 1b). Hydrolytic degradation will be briefly discussed as well (Fig. 1c).
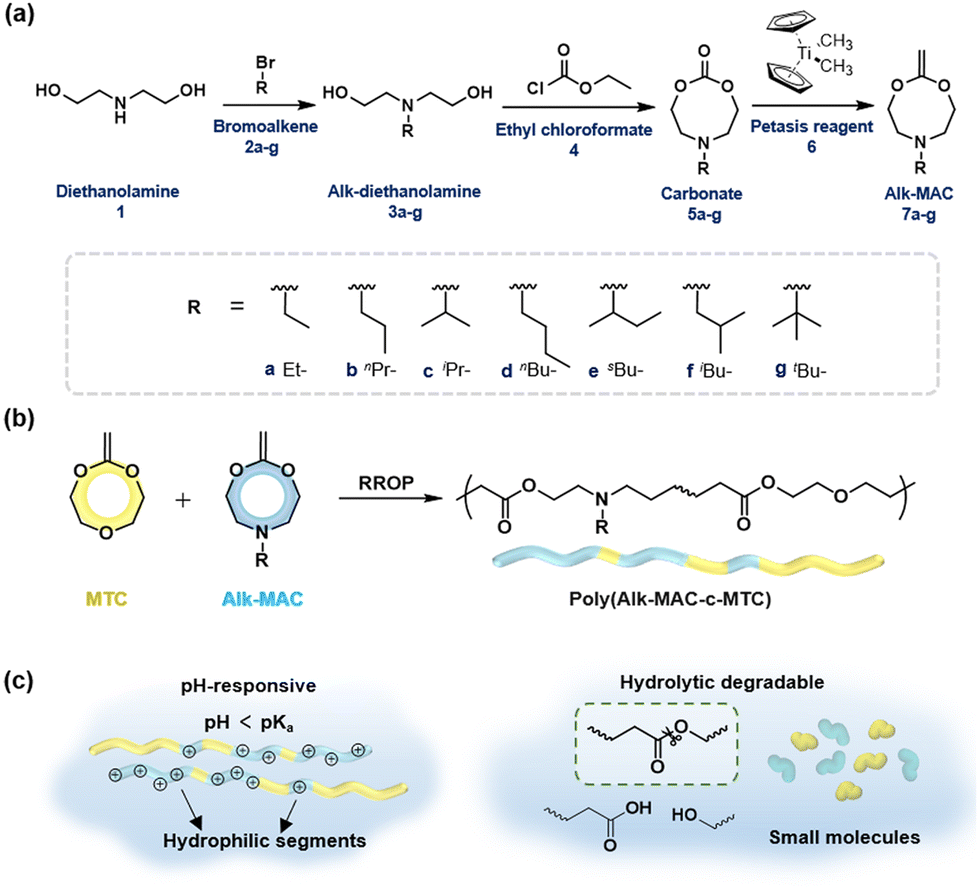 | ||
| Fig. 1 (a) Synthetic route towards Alk-MACs through an intermediate carbonate. Labels 3a–g, 5a–g, 7a–g represent alkyl-substituted diethanol amines, intermediate carbonates and CKAs, respectively. (b) Copolymerisation of Alk-MACs with the conventional CKA MTC yields pH-responsive and degradable copolymers. (c) The amines in the Alk-MAC segments get protonated upon acidification and copolymers become hydrophilic and upon hydrolytic degradation the ester bonds get cleaved to yield small molecules. | ||
The first goal of this work was to synthesise the new Alk-MACs following a similar protocol (carbonate route) as for the published iPr-MAC. Starting from diethanolamine 1, alkyl-substituted diols 3 were prepared by alkylation with the corresponding bromoalkene agents 2. Following some optimisation, the previously reported N-propyldiethanolamine 3b, N-iso-propyldiethanolamine 3c, N-sec-butyldiethanolamine 3e and also N-iso-butyldiethanolamine 3f were synthesised (Tables S2–S5 and Fig. S3†). The substances 3a, 3d, and 3g were sourced commercially.
These alkyl-modified diols 3a–g and ethyl chloroformate 4 then reacted in the presence of triethylamine (TEA) to yield the intermediate cyclic carbonates 5a–g. Reaction conditions, including reaction temperature, feeding speed of TEA, feed ratio of reagents, were fine-tuned individually to ensure optimal yield of each carbonate. For instance, the best yield of ethyl carbonate 5a (23%) was achieved with a low feeding speed of TEA (15 μL min−1). Conversely, a faster feeding speed of TEA (40 μL min−1) gave the best yield for n-propyl carbonate 5b and tert-butyl carbonate 5g with 29% each. An even higher feeding speed of 100 μL min−1 then gave the optimal yield of 45% for sec-butyl carbonate 5e and was increased further to 400 μL min−1 for the formation of n-butyl carbonate 5d (25% yield) and iso-butyl carbonate 5f (45% yield, summarized in Tables S6–11†). Hence, larger alkyl substituents mostly demanded a higher feeding speed of TEA for optimal carbonate formation. 1H NMR and 13C NMR spectroscopy with full signal assignment could be achieved for all carbonates 5a–g, always including the signal at around 155 ppm in the 13C NMR, (carbonate-bearing carbon atom). HR-ESI-MS measurements with positive mode were used to confirm the successful synthesis of all cyclic carbonates 5a–g (Fig. S4–10†). Molecular models were calculated to get insights into the geometry of the structures (Fig. S20†) and will be discussed separately later.
Methylenation of all cyclic carbonates 5a–g (1.0 equiv.) with the Petasis reagent 6 (2.3 equiv.) were performed in THF at 65 °C for 22 h. All the new CKAs were obtained in a low yield (from 6% for 7d over 11–15% for 7a–b, 7e–g to 25% for 7c (Table S1†)), representing the delicacy of the carbonate route. 1H, 13C NMR and HR-ESI-MS measurements confirmed the successful preparation of these new molecules (Fig. 2 and Fig. S12–18†). As depicted in Fig. 2, signals a–d could be assigned to the CKA ring. From the core CKA functionality, the tertiary carbon atom c between the oxygen atoms was positioned at 164 ppm for the 13C NMR spectrum (Fig. S12–S18†). The intense singlet d in the range of 3.53–3.61 ppm in the 1H NMR spectra and new signal at 69 ppm in the 13C NMR spectra represented exo-methylene group and hence proved the formation of the MACs. Signals 1–4 were attributed to the protons on the alkyl-substituents and remained mainly unchanged from the carbonates for all substituents. As compared to other chemical shifts of the starting diols and intermediate substances, protons a, a′ and 2, 2′ of the chiral sBu-MAC 7e and the corresponding carbonate 5e showed an additional splitting (see in Fig. 2 and S8†), which is typical for chiral molecules.
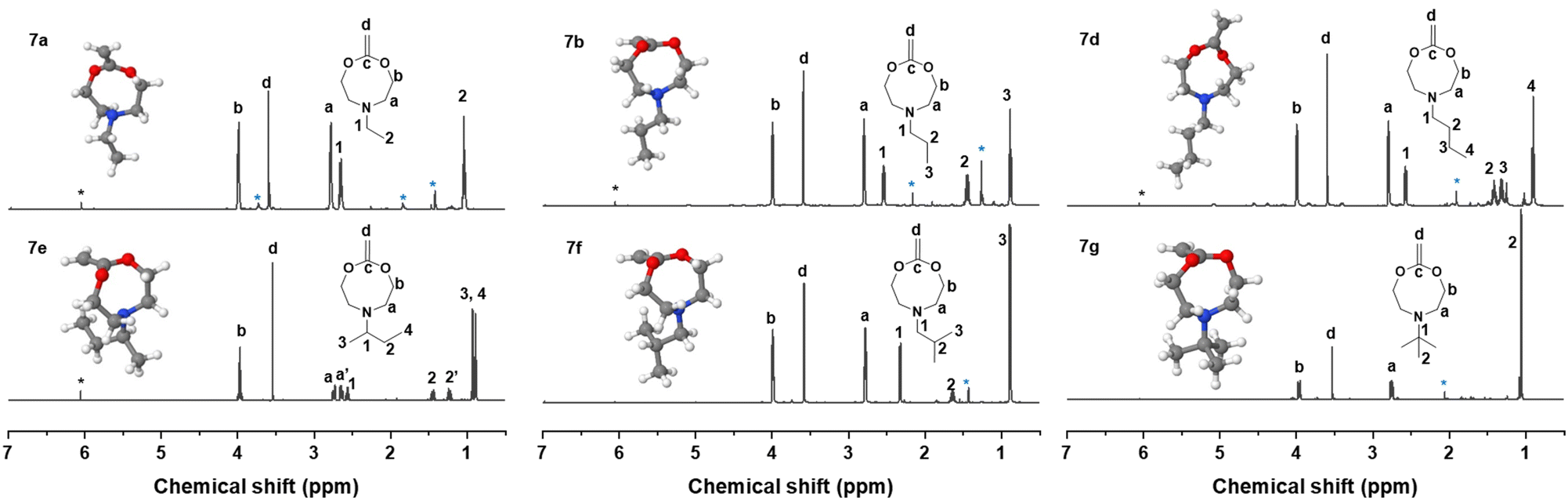 | ||
| Fig. 2 1H NMR spectra of all new Alk-MACs 7a–b, 7d–g with their respective alkyl side groups. The inserts are energetic optimal structures of CKAs calculated at 0 K, under vacuum; red balls represent oxygen atoms, grey balls represent carbon atoms, white balls represent hydrogen atoms, blue balls represent nitrogen atoms. The asterisked signals in the 1H NMR spectra represent the trace of Petasis reagent (black, 6.05 ppm), solvent and impurities (blue, tert-butanol: 1.28 ppm, acetone: 2.17 ppm, THF: 1.85 and 3.76 ppm). | ||
Additionally, theoretical calculations of the molecular structures of all intermediate carbonate and corresponding CKAs were performed using software package GAMESS with basis set STO-6G,40 simulating conditions of 0 K and vacuum. These helped us in understanding the energetically optimal structure of the carbonates 5a–g and CKAs 7a–g (see in the insets in Fig. 2 and Fig. S20†). They revealed that, except for the chiral sBu derivatives 5e and 7e, the side chain on the amine stretched away from the carbonate or CKA ring. Both chiral derivatives, however, presented an additional structural distinctiveness. Here, the sBu-side group tended to bend inwards to the eight-membered ring, which might lead to significant steric hindrance around the tertiary amine. However, as the calculations were performed at simulated 0 K, the effect on the molecule at room temperature remained unknown.
Copolymerisation of the previously reported iPr-MAC 7c and conventional CKA MTC was the starting point to fabricate the pH-responsive polyesters through free RROP. By varying feed ratio of 7c to MTC from 5/95, 10/90, 25/75 to 50/50 (mol%), copolymer composition was modulated. After polymerising either thermally or under UV light for 24 h, purified copolymers were obtained through dialysis. All copolymers were named according to the feed ratio of Alk-MACs and the polymerisation method. For instance, iPr-MAC-5T represents a 7c/MTC mixture with a feed ratio of 5/95 that was subjected to thermal polymerisation.
To determine the monomer conversion and the final molar fractions, crude and purified copolymers were analysed by 1H NMR spectroscopy (Fig. S21–24†). Signals attributed to protons on the P(MTC) parts were consistent with reported signal assignments.14 For the P(iPr-MAC) segment, the signal centered at 2.93 ppm appeared in all the samples, which is attributed to the α-proton on the iPr side chain. This signal was significant, as the chemical shift remained virtually unchanged for the monomer and the polymer and confirmed the successful incorporation of the iPr group through all attempts. After workup, the ester signal of P(MTC) (4.20 ppm) and amine signal of P(iPr-MAC) (2.93 ppm) were integrated to calculate the molar fractions in the copolymers (Fig. S23 and section 3.3 of the ESI†). Intriguingly, the final molar fraction of iPr-MAC in the copolymers was always higher than the feed ratio, indicating that iPr-MAC was more reactive than MTC. Additionally, UV-irradiated polymerisation consistently led to a higher iPr-MAC content in the copolymers as compared to thermal polymerization, implying an even greater imbalance of reactivities (Table 1). Copolymers of iPr-MAC and MTC could hence be gradient copolymers, which means copolymers first enriched with iPr-MAC. Considering the omnipresent dispersity within polymers from free radical polymerisation, the partial presence of homopolymers could not be excluded. DOSY measurements of the bimodal copolymer iPr-MAC-5T was carried out (Fig. S26†), and showed that the self-diffusion coefficients related to iPr-MAC and MTC motifs within the main diffusion peak and thus substantiated the successful formation of a copolymer. Additionally, MTC branching units occurred by intramolecular (MTC back-biting) or intermolecular H-abstraction reactions can also be observed. The characteristic signals around 1.20 ppm are related to the methyl end groups from each MTC branching unit (Fig. S24†).13,41 The SEC chromatograms of the resultant copolymers showed that with increasing iPr-MAC content, copolymers exhibited a broader distribution ranging from 1.3 for 5 mol% to over 11 for 50 mol% (Fig. S29† and Table 1). While thermal initiation showed a consistent increase in dispersity with rising iPr-MAC content, tendency was less clear for polymerizations under UV irradiation, again implying a more reliable reaction under purely thermal conditions. The corresponding results should be treated with caution owing to the complex polymeric structures including ring-opened and ring-retaining structures as well as branched and linear units in all possible combinations from MTC and iPr-MAC. 2D NMR measurements of sample copolymers (Fig. S27 and 28†) still allowed for allocating some major signals. As the ester bonds of MTC and the amines of iPr-MAC could be confirmed, both structural arguments for the targeted degradability and pH-responsiveness were present.
| Name |
f
MAC,0/fMTC,0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
% Conv. MAC/MTCb |
F
MAC/FMTCc |
M
nd (kg mol−1) |
M
wd (kg mol−1) |
Đ |
e |
|---|---|---|---|---|---|---|---|
a Feed ratios of Alk-MACs to MTC.
b Conversion of Alk-MACs and MTC, calculated by 1H NMR spectra.
c Final molar fraction of Alk-MACs to MTC in the resulting copolymers, calculated from 1H NMR spectra.
d
M
n, Mw and polymer dispersity were measured by SEC at 25 °C with a PolarGel-M column (300 × 7.5 mm), using HPLC-Pump 1200 pump system (Agilent technologies) equipped with a multi angle laser light scattering (MALLS) detector (MiniDAWN TREOS II, Wyatt Technology) and a RI detector (K-2301 from KNAUER). DMAc with 3 g L−1 LiCl was used as an eluent and the flow rate was 1 mL min−1.
e 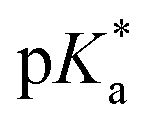 was obtained through UV-vis method, the transition point was determined when transmittance decreased 50%. It should be noted that was obtained through UV-vis method, the transition point was determined when transmittance decreased 50%. It should be noted that 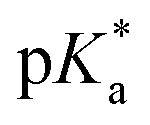 shown in the Table 1 was the average value obtained from three measurements, including the corresponding error margin.
f Conversion of Alk-MACs was quite high and no presence of vinyl protons can be found by 1H NMR spectra. shown in the Table 1 was the average value obtained from three measurements, including the corresponding error margin.
f Conversion of Alk-MACs was quite high and no presence of vinyl protons can be found by 1H NMR spectra.
|
|||||||
| i Pr-MAC-5T | 5/95 | 67/35 | 7/93 | 25.8 | 32.9 | 1.3 | — |
| i Pr-MAC-10T | 10/90 | 40/28 | 14/86 | 26.6 | 43.5 | 1.6 | — |
| i Pr-MAC-25T | 25/75 | 48/25 | 34/66 | 32.2 | 155 | 4.8 | 7.0 ± 0.1 |
| i Pr-MAC-50T | 50/50 | 35/37 | 69/31 | 18.2 | 193 | 11 | 6.5 ± 0.1 |
| i Pr-MAC-5U | 5/95 | 80/67 | 10/90 | 16.3 | 47.4 | 2.9 | — |
| i Pr-MAC-10U | 10/90 | 100f/38 | 19/81 | 29.3 | 68.2 | 2.3 | — |
| i Pr-MAC-25U | 25/75 | 64/15 | 55/45 | 9.2 | 31.5 | 3.4 | 6.9 ± 0.2 |
| i Pr-MAC-50U | 50/50 | 100f/44 | 80/20 | 16.5 | 248 | 15 | 6.7 ± 0.2 |
| Et-MAC-25T | 25/75 | 22/17 | 37/63 | 17.8 | 44.8 | 2.5 | 5.7 ± 0.3 |
| n Pr-MAC-25T | 25/75 | 27/26 | 26/74 | 15.3 | 42.4 | 2.8 | 6.1 ± 0.2 |
| n Bu-MAC-25T | 25/75 | 22/17 | 36/64 | 11.8 | 126 | 11 | 6.6 ± 0.3 |
| s Bu-MAC-25T | 25/75 | 29/13 | 45/55 | 49.1 | 420 | 8.6 | 6.6 ± 0.3 |
| i Bu-MAC-25T | 25/75 | 30/17 | 30/70 | 21.5 | 183 | 8.5 | 6.4 ± 0.2 |
| t Bu-MAC-25T | 25/75 | 60/19 | 50/50 | 10.8 | 65.4 | 6.1 | 7.2 ± 0.3 |
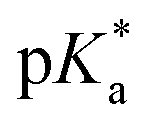
Following the more reliable reaction, thermal polymerisation was applied to other Alk-MAC/MTC pairs. A feed ratio of 25/75 (mol%, Alk-MAC/MTC) was applied to yield an Alk-MAC content between 25 and 50 mol% as this would ensure pH-responsiveness and also the MTC content necessary to keep structural diversity within reason. The purified copolymers were analysed by 1H- and 13C NMR spectroscopy and are comprised in Fig. S32–S33.† Similar to the iPr-MAC/MTC copolymers, the high amount of possible substructures prohibited complete signal assignment and hence also quantification of the degree of ring-opening. Branching from MTC units could be confirmed in all cases from the aforementioned signals around 1.20 ppm, but was not quantified due to signal overlap (Fig. S32†). Ester linkages within the copolymer were further confirmed by an exemplary ATR-FTIR spectrum of Et-MAC-25T (Fig. S34†). It showed a strong band 1734 cm−1, showcasing the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of ester units. The Alk-MAC/MTC content could still be determined as characteristic signals for MTC and the alkyl-substituents on the MACs could be attributed (Table 1 and Fig. S30–31†). Final molar ratios of Alk-MACs/MTC were higher than the feed ratio, highlighting the higher reactivity of all investigated Alk-MACs compared to MTC, again suggesting the formation of gradient copolymers. SEC measurements of all copolymers showed a somewhat constant Mn of 15.4 ± 4.5 kg mol−1 (Fig. S35† and Table 1). Only sBu-MAC-25T showed a higher Mn of 49.1 kg mol−1. While Et-MAC-25T and nPr-MAC-25T copolymers had relatively reasonable dispersity values of 2.5 and 2.8, these values went up to 4.8 for iPr-MAC-25T and rose further to 6–11 for the butyl derivatives. It indicated a lack of control over dispersity with increasing length and complexity of the side chain on the amine. A suspected reason for this could be the additional branching sites on the side chain, especially close to the amines. These new propagation centres could then themselves host new polymer chains with branches from all different branching points, leading to a plethora of possible sub-structures and also increased dispersities.
O bonds of ester units. The Alk-MAC/MTC content could still be determined as characteristic signals for MTC and the alkyl-substituents on the MACs could be attributed (Table 1 and Fig. S30–31†). Final molar ratios of Alk-MACs/MTC were higher than the feed ratio, highlighting the higher reactivity of all investigated Alk-MACs compared to MTC, again suggesting the formation of gradient copolymers. SEC measurements of all copolymers showed a somewhat constant Mn of 15.4 ± 4.5 kg mol−1 (Fig. S35† and Table 1). Only sBu-MAC-25T showed a higher Mn of 49.1 kg mol−1. While Et-MAC-25T and nPr-MAC-25T copolymers had relatively reasonable dispersity values of 2.5 and 2.8, these values went up to 4.8 for iPr-MAC-25T and rose further to 6–11 for the butyl derivatives. It indicated a lack of control over dispersity with increasing length and complexity of the side chain on the amine. A suspected reason for this could be the additional branching sites on the side chain, especially close to the amines. These new propagation centres could then themselves host new polymer chains with branches from all different branching points, leading to a plethora of possible sub-structures and also increased dispersities.
All copolymers contained the aimed-for amount of Alk-MAC and pH-responsiveness was then characterised by dissolving each copolymer in a HCl solution, titrating it from pH 2 towards pH 10. This process deprotonated the ammonium units and gave tertiary amines, changing the solubility of the copolymers from hydrophilic (transparent solution) to hydrophobic (turbid solution) (Fig. 3a). Turbidity pointed towards aggregation and DLS measurement of iPr-MAC-25T/HCl at different pH values confirmed this (Fig. S46†). Volume size distribution traces showed significant increases in hydrodynamic size and polydispersity index (PDI) upon deprotonation, proofing the formation of agglomerates during this process. Turbidity measurements then showed a reversible deprotonation–protonation and hence transparent-turbidity switch of iPr-MAC-25T over three cycles between pH 3 and 10 (Fig. 3b). The titration plots of % transmittance against pH value were then measured at a wavelength of 500 nm and then fitted with a Boltzmann equation (in Fig. S36–45†). When transmittance decreased to 50% of the original value, this pH was defined as 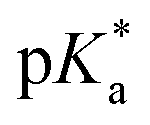 cloud point. Copolymer solutions of all substituents were measured as triplicates and first evaluated separately in order to get mean values and error margins (Fig. S36–45† left side, values in Table 1). In a second instance, the values of all three measurements were fitted together in a single Boltzmann fit in order to compare different substituents against each other (Fig. S36–45† right side, comparison in Fig. 3c–e). Within these studies, Mn values were within reason and dispersity values were found to have no measurable impact on this value. Subsequently, impact on the
cloud point. Copolymer solutions of all substituents were measured as triplicates and first evaluated separately in order to get mean values and error margins (Fig. S36–45† left side, values in Table 1). In a second instance, the values of all three measurements were fitted together in a single Boltzmann fit in order to compare different substituents against each other (Fig. S36–45† right side, comparison in Fig. 3c–e). Within these studies, Mn values were within reason and dispersity values were found to have no measurable impact on this value. Subsequently, impact on the 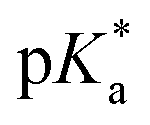 was analysed for (i) amount of Alk-MAC in the polymer, (ii) length of Alk-MAC substituent, (iii) bulkiness of Alk-MAC substituent. For the first category, copolymers with different amounts of iPr-MAC were compared (Fig. 3c). Within this series, the
was analysed for (i) amount of Alk-MAC in the polymer, (ii) length of Alk-MAC substituent, (iii) bulkiness of Alk-MAC substituent. For the first category, copolymers with different amounts of iPr-MAC were compared (Fig. 3c). Within this series, the 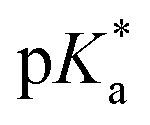 dropped from 7.1 for 34% of iPr-MAC (iPr-MAC-25T) to 6.6 for 69% of iPr-MAC (iPr-MAC-50T). Only the
dropped from 7.1 for 34% of iPr-MAC (iPr-MAC-25T) to 6.6 for 69% of iPr-MAC (iPr-MAC-50T). Only the 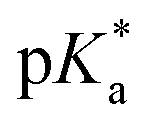 of 6.7 for 80% iPr-MAC-50U does not perfectly fit this trend, but this could be explained with increasing structural irregularities following the low MTC content. In our previous research, the iPr-MAC homopolymer exhibited a pKa value around 6.0.6 All
of 6.7 for 80% iPr-MAC-50U does not perfectly fit this trend, but this could be explained with increasing structural irregularities following the low MTC content. In our previous research, the iPr-MAC homopolymer exhibited a pKa value around 6.0.6 All 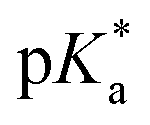 cloud-point values are within the same region, but cannot be compared directly following the different measurement principles. Owing to the higher hydrophilicity of MTC, all
cloud-point values are within the same region, but cannot be compared directly following the different measurement principles. Owing to the higher hydrophilicity of MTC, all 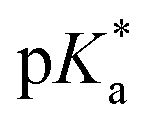 values were considerably higher than the one recorded for the iPr-MAC/DMMDO copolymer (found at pH 3.5),6,13,14 meeting a major goal of this study.
values were considerably higher than the one recorded for the iPr-MAC/DMMDO copolymer (found at pH 3.5),6,13,14 meeting a major goal of this study.
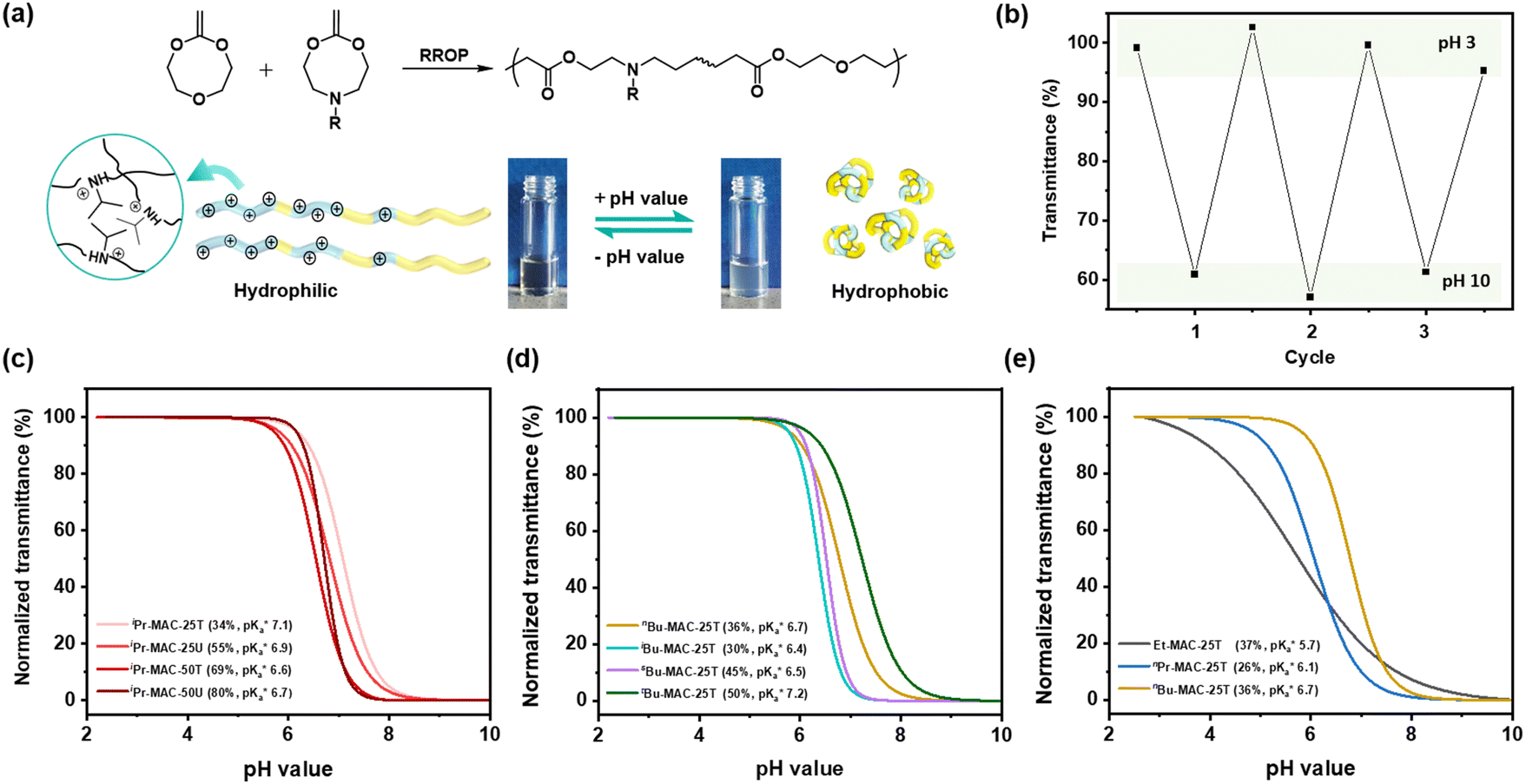 | ||
Fig. 3 (a) Transparent copolymer solutions became turbid when the pH value increased. (b) Reversible deprotonation–protonation process of iPr-MAC-25T copolymer solution detected by UV-Vis measurements over three cycles. (c)–(e) Evolution of % transmittance of copolymers solution over pH, showing dependencies of (c) amount of incorporated iPr-MAC, (d) bulkiness of the butyl substitute (nBu, iBu, sBu, tBu), (e) length of the n-alkyl substituent (Et, nPr, nBu). The 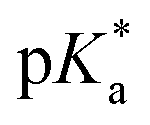 values shown in the figure caption were obtained from the single Boltzmann fit of all three measurements of respective copolymer, and therefore were slightly different from the average values shown in the figure caption were obtained from the single Boltzmann fit of all three measurements of respective copolymer, and therefore were slightly different from the average 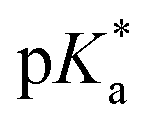 value listed in the Table 1 (for a comparison of both values, see Table S13 in the ESI†). value listed in the Table 1 (for a comparison of both values, see Table S13 in the ESI†). | ||
For all Bu-MAC copolymers (Fig. 3d), the MAC content ranged from 30% in iBu-MAC-25T to 50% in tBu-MAC-25T, being in a comparable range. The bulkiest residue of tBu-MAC-25T exhibited the highest 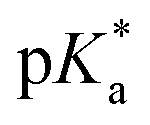 of 7.2 despite having the highest amount of Bu-MAC units. All three other Bu-MACs were relatively close to each other with
of 7.2 despite having the highest amount of Bu-MAC units. All three other Bu-MACs were relatively close to each other with 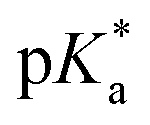 values of 6.4, 6.5 and 6.7 for iBu-MAC-25T, sBu-MAC-25T and nBu-MAC-25T, respectively. This implicated that the steric hindrance of the tBu residue caused this higher
values of 6.4, 6.5 and 6.7 for iBu-MAC-25T, sBu-MAC-25T and nBu-MAC-25T, respectively. This implicated that the steric hindrance of the tBu residue caused this higher 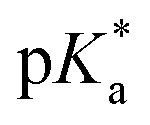 value.
value.
Copolymers with linear substituents showed that Et-MAC-25T exhibited a significantly broader reduction region of % transmittance than nPr-MAC-25T and nBu-MAC-25T. As molecular weights remained within reason, this change could be attributed to the length of the side chain. Corresponding 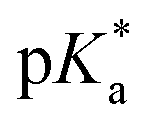 values increased from 5.7 for Et-MAC-25T over 6.1 for nPr-MAC-25T to 6.7 for nBu-MAC-25T. It can hence be concluded that longer alkyl substituents promoted a higher
values increased from 5.7 for Et-MAC-25T over 6.1 for nPr-MAC-25T to 6.7 for nBu-MAC-25T. It can hence be concluded that longer alkyl substituents promoted a higher 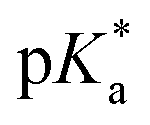 value. This could again be attributed to the increasing steric hindrance of a larger substituent, which goes in-line with the shift of the
value. This could again be attributed to the increasing steric hindrance of a larger substituent, which goes in-line with the shift of the 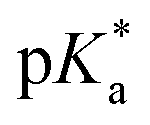 values observed within the series of butyl-substituents.
values observed within the series of butyl-substituents.
Selected copolymers were then subjected to hydrolytic degradation under accelerated condition. The copolymers were dissolved in THF/MeOH (67/33 vol%) with 5 wt% KOH and kept stirring at room temperature for 24 h to ensure complete degradation. Degradation was then proven in multiple ways: (i) 1H NMR measured in CDCl3 and D2O: signals around 4.2 ppm ascribed to the protons adjacent to ester bond of P(MTC) disappeared completely after degradation (Fig. S47 and S49†). (ii) 13C NMR: signals around 174 ppm (Fig. S48†), related to carbon atoms in ester bond, also disappeared. (iii) SEC: similar to other RROP studies,13,29 chromatograms before and after degradation showed a significant shift towards higher retention time after degradation for 24 h (Fig. 4 and Fig. S50†). Altogether, this confirmed the degradation of the copolymers.
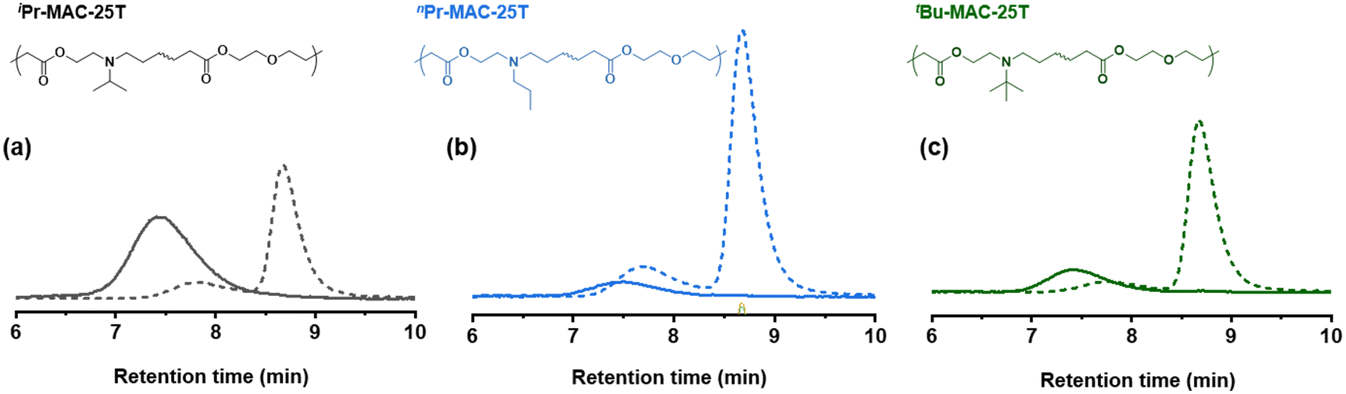 | ||
| Fig. 4 SEC analysis of copolymers before (solid lines) and after hydrolytic degradation (dashed lines) under accelerated conditions. Light scattering signals of (a) iPr-MAC-25T, (b) nPr-MAC-25T and (c) tBu-MAC-25T are depicted. | ||
Conclusions
We hence successfully synthesized a series of alkyl-substituted Alk-MACs and provided their calculated molecular structures. Copolymerisation of all Alk-MACs with MTC yielded pH-responsive polyesters best under thermal conditions. The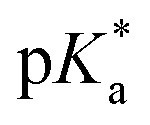 cloud point values varied from 5.7 to 7.2 with all the amount, length and bulkiness of the alkyl residue impacting the cloud points. Degradation of selected polyesters could then be proven by the disappearance of characteristic 1H and 13C NMR signals as well as characteristic shifts in the SEC chromatograms. All presented Alk-MACs are thus a valuable addition to heteroatom-bearing CKA library and copolymerisation with MTC is a facile platform to gain pH-responsive and degradable polyesters.
cloud point values varied from 5.7 to 7.2 with all the amount, length and bulkiness of the alkyl residue impacting the cloud points. Degradation of selected polyesters could then be proven by the disappearance of characteristic 1H and 13C NMR signals as well as characteristic shifts in the SEC chromatograms. All presented Alk-MACs are thus a valuable addition to heteroatom-bearing CKA library and copolymerisation with MTC is a facile platform to gain pH-responsive and degradable polyesters.
Author contributions
Yiyi Deng: conceptualisation, formal analysis, investigation, data curation, writing – original draft, writing – review and editing, visualisation, funding acquisition. Anais Frezel: validation, formal analysis, investigation, writing – review and editing, visualisation. Fabian Mehner: validation, formal analysis, investigation, writing – review and editing, visualisation. Peter Friedel: methodology, software, investigation, data curation, writing – review and editing, visualisation. Jens Gaitzsch: conceptualisation, methodology, formal analysis, resources, data curation, writing – original draft, writing – review and editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The manuscript was written by contributions from all authors and all authors gave their approval for the final version. We would like to thank Alissa Seifert for the SEC measurements and Albena Lederer for helpful discussions of the results. The authors thank the Chinese Scholarship Council for funding Y. D., the Evonik Foundation for funding F. M. and the Deutsche Forschungsgemeinschaft (GA2051/7-1) for financial support. We thank the Howdle group at the University of Nottingham for granting access to their NMR facilities.References
- D. Goldberg, J. Environ. Polym. Degrad., 1995, 3, 61–67 CrossRef CAS.
- S. Agarwal, Polym. Chem., 2010, 1, 953–964 RSC.
- J. N. Hoskins and S. M. Grayson, Polym. Chem., 2011, 2, 289–299 RSC.
- W. J. Bailey, Z. Ni and S.-R. Wu, J. Polym. Sci., Part A: Polym. Chem., 1982, 20, 3021–3030 CAS.
- T. Pesenti and J. Nicolas, ACS Macro Lett., 2020, 9, 1812–1835 CrossRef CAS PubMed.
- J. Folini, C.-H. Huang, J. C. Anderson, W. P. Meier and J. Gaitzsch, Polym. Chem., 2019, 10, 5285–5288 RSC.
- G. Hedir, C. Stubbs, P. Aston, A. P. Dove and M. I. Gibson, ACS Macro Lett., 2017, 6, 1404–1408 CrossRef CAS PubMed.
- J. Gaitzsch, P. C. Welsch, J. Folini, C.-A. Schoenenberger, J. C. Anderson and W. P. Meier, Eur. Polym. J., 2018, 101, 113–119 CrossRef CAS.
- M. R. Hill, T. Kubo, S. L. Goodrich, C. A. Figg and B. S. Sumerlin, Macromolecules, 2018, 51, 5079–5084 CrossRef CAS.
- J. Tran, E. Guégain, N. Ibrahim, S. Harrisson and J. Nicolas, Polym. Chem., 2016, 7, 4427–4435 RSC.
- M. R. Hill, E. Guégain, J. Tran, C. A. Figg, A. C. Turner, J. Nicolas and B. S. Sumerlin, ACS Macro Lett., 2017, 6, 1071–1077 CrossRef CAS PubMed.
- F. Mehner, M. Geisler, K. Arnhold, H. Komber and J. Gaitzsch, ACS Appl. Polym. Mater., 2022, 4, 7891–7902 CrossRef CAS.
- T. Pesenti, E. Gillon, S. Ishii, S. Messaoudi, Y. Guillaneuf, A. Imberty and J. Nicolas, Biomacromolecules, 2023, 24, 991–1002 CrossRef CAS PubMed.
- J. Undin, P. Plikk, A. Finne-Wistrand and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4965–4973 CrossRef CAS.
- Y. Deng, F. Mehner and J. Gaitzsch, Macromol. Rapid Commun., 2023, 44, 2200941 CrossRef CAS PubMed.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef CAS PubMed.
- A. Tardy, J.-C. Honoré, J. Tran, D. Siri, V. Delplace, I. Bataille, D. Letourneur, J. Perrier, C. Nicoletti, M. Maresca, C. Lefay, D. Gigmes, J. Nicolas and Y. Guillaneuf, Angew. Chem., Int. Ed., 2017, 56, 16515–16520 CrossRef CAS PubMed.
- Y. Zhang, M. Zheng, T. Kissel and S. Agarwal, Biomacromolecules, 2012, 13, 313–322 CrossRef CAS PubMed.
- T. Pesenti, C. Zhu, N. Gonzalez-Martinez, R. M. F. Tomás, M. I. Gibson and J. Nicolas, ACS Macro Lett., 2022, 11, 889–894 CrossRef CAS PubMed.
- J. Tran, T. Pesenti, J. Cressonnier, C. Lefay, D. Gigmes, Y. Guillaneuf and J. Nicolas, Biomacromolecules, 2019, 20, 305–317 CrossRef CAS PubMed.
- A. Bossion, C. Zhu, L. Guerassimoff, J. Mougin and J. Nicolas, Nat. Commun., 2022, 13, 2873 CrossRef CAS PubMed.
- U. Wais, L. R. Chennamaneni, P. Thoniyot, H. Zhang and A. W. Jackson, Polym. Chem., 2018, 9, 4824–4839 RSC.
- J.-F. Lutz, J. Andrieu, S. Üzgün, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40, 8540–8543 CrossRef CAS.
- S. Komatsu, T. Sato and A. Kikuchi, Polym. J., 2021, 53, 731–739 CrossRef CAS.
- D. J. Siegwart, S. A. Bencherif, A. Srinivasan, J. O. Hollinger and K. Matyjaszewski, J. Biomed. Mater. Res., Part A, 2008, 87, 345–358 CrossRef PubMed.
- T.-A. Asoh, T. Nakajima, T. Matsuyama and A. Kikuchi, Langmuir, 2015, 31, 6879–6885 CrossRef CAS PubMed.
- A. W. Jackson, L. R. Chennamaneni and P. Thoniyot, Eur. Polym. J., 2020, 122, 109391 CrossRef CAS.
- C. Zhu, S. Denis and J. Nicolas, Chem. Mater., 2022, 34, 1875–1888 CrossRef CAS.
- C. Zhu and J. Nicolas, Polym. Chem., 2021, 12, 594–607 RSC.
- E. Guégain, C. Zhu, E. Giovanardi and J. Nicolas, Macromolecules, 2019, 52, 3612–3624 CrossRef.
- Y. Hiraguri, K. Katase and Y. Tokiwa, J. Macromol. Sci., Part A: Pure Appl. Chem., 2006, 43, 1021–1027 CrossRef.
- M. Carlotti, O. Tricinci and V. Mattoli, Adv. Mater. Technol., 2022, 7, 2101590 CrossRef CAS.
- X. Ai, J. Pan, Q. Xie, C. Ma and G. Zhang, Polym. Chem., 2021, 12, 4524–4531 RSC.
- Q. Xie, Q. Xie, J. Pan, C. Ma and G. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 11213–11220 CrossRef CAS PubMed.
- D. Gigmes, P. H. M. Van Steenberge, D. Siri, D. R. D'hooge, Y. Guillaneuf and C. Lefay, Macromol. Rapid Commun., 2018, 39, 1800193 CrossRef PubMed.
- A. Tardy, N. Gil, C. M. Plummer, D. Siri, D. Gigmes, C. Lefay and Y. Guillaneuf, Angew. Chem., Int. Ed., 2020, 59, 14517–14526 CrossRef CAS PubMed.
- J. Folini, W. Murad, F. Mehner, W. Meier and J. Gaitzsch, Eur. Polym. J., 2020, 134, 109851 CrossRef CAS.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr., J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- F. Mehner, T. Meissner, A. Seifert, A. Lederer and J. Gaitzsch, J. Polym. Sci., 2023, 61, 1882–1892 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00929g |
| This journal is © The Royal Society of Chemistry 2023 |