
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu@C/Pt core@shell/satellite supra-nanostructures: plasmonic antenna–reactor hybrid nanocatalysts†
Zixin
Wang
 and
Hui
Wang
*
and
Hui
Wang
*
Department of Chemistry and Biochemistry, University of South Carolina, Columbia, South Carolina 29208, USA. E-mail: wang344@mailbox.sc.edu; Fax: +1-803-777-9521; Tel: +1-803-777-2203
First published on 23rd August 2023
Abstract
Integration of plasmonic nanoantennas with catalytically active reactors in deliberately designed hybrid supra-nanostructures creates a dual-functional materials platform, based upon which precise modulation of catalytic reaction kinetics becomes accomplishable through optical excitations of plasmon resonances. Here, we have developed a multistep synthetic approach that enables us to assemble colloidal Au@C/Pt core@shell/satellite supra-nanostructures, in which the Au core functions as a light-harvesting plasmonic nanoantenna, the Pt satellites act as catalytically active reactors, and the C shell serves as a nanoscale dielectric spacer separating the reactors from the antenna, respectively. By adjusting several synthetic parameters, the size of the Au core, the thickness of the C shell, and the surface coverage of Pt satellites can all be tuned independently. Choosing Pt-catalyzed cascade oxidation of 3,3′,5,5′-tetramethylbenzidine in an aerobic aqueous environment as a model reaction, we have systematically studied the detailed kinetic features of the catalytic reactions both in the dark and under visible light illumination over a broad range of reaction conditions, which sheds light on the interplay between plasmonic and catalytic effects in these antenna–reactor nanohybrids. The plasmonic antenna effect can be effectively harnessed to kinetically modulate multiple crucial steps during the cascade reactions, benefiting from plasmon-enhanced interband electronic transitions in the Pt satellites and plasmon-enhanced intramolecular electronic excitations in chromogenic intermediate species. In addition to the plasmonic antenna effect, photothermal transduction derived from plasmonic excitations can also provide significant contributions to the kinetic enhancements under visible light illumination. The knowledge gained from this work serves as important guiding principles for rational design and structural optimization of plasmonic antenna–reactor hybrid nanomaterials, endowing us with enhanced capabilities to kinetically modulate targeted catalytic/photocatalytic molecule-transforming processes through light illumination.
Introduction
Metallic nanoparticles play crucial roles in catalyzing a diverse range of important chemical reactions directly relevant to fine chemical manufacture, environmental remediation, and energy conversion.1–10 Metallic nanocatalysts not only modulate activation energy barriers for chemical transformations under thermal conditions but can also interact with incident photons to trigger solar-to-chemical energy conversion processes through light-driven or light-enhanced heterogeneous catalysis.11–21 Nanoparticles made of well-behaving plasmonic metals, such as Au,22 Ag,23 Cu,24 and Al,25,26 may function as light-concentrating nanoantennas when the collective oscillations of their free electrons in the conduction band, known as plasmons, are resonantly excited by incident photons. Localized plasmon resonances sustained by metallic nanoparticles may decay either radiatively through elastic photon scattering, which leads to large local-field enhancements near the nanoparticle surfaces, or nonradiatively to generate hot electrons and holes, which can be injected into molecular adsorbates to drive photocatalytic reactions.12,14,18–21 Plasmonic hot carriers, if not harvested by molecular adsorbates, may become thermalized by interacting with phonons to generate heat, a process known as photothermal transduction.14,20,27–29 Local-field enhancements,30 hot carriers,12,31,32 and photothermal heating33–35 derived from excitation and decay of plasmons may all provide critical contributions to the kinetic enhancements of photocatalytic reactions. Although the great promise of plasmon-mediated photocatalysis has been well-recognized by both nanoplasmonics and heterogeneous catalysis communities, nanocatalysts composed of ideal plasmonic metals can only efficiently catalyze a limited number of industrially relevant chemical reactions.36–39 In contrast, nanoparticulate Pt-group noble metals, such as Pt, Pd, Rh, and Ru, are catalytically more versatile than plasmonic metals and thus have been more widely utilized for industrial applications. However, these catalytic metal nanoparticles are poorly behaving plasmonic materials displaying weak optical responses dominated by electronic interband transitions spanning the entire visible and near-infrared ranges of the solar spectrum.16,36–41 Although photoexcited interband hot carriers in these catalytic metals can also trigger interfacial molecular transformations,16,41–46 kinetic enhancements under photo-illumination are typically rather limited in comparison to those achievable through plasmon-mediated photocatalysis. Desired plasmonic and catalytic properties are often mutually exclusive in monometallic nanostructures because a deep-lying valence d-band significantly below the Fermi level is a prerequisite for strong plasmonic responses, whereas optimal metal–adsorbate interactions for catalysis typically require the center of the d-band to be located fairly close to the Fermi level.47The past decade has witnessed rapidly growing interest in hybrid nanocatalysts integrating plasmonic light-harvesting functions with desired catalytic properties.36–39,47–57 Such plasmonic–catalytic dual-functionalities can be realized by directly interfacing a plasmonic metal with a catalytic metal to form heteronanostructures, most commonly consisting of a plasmonic metal core either decorated with discrete catalytic metal nanoparticles or coated with a continuous catalytic metal shell.47,58–64 The plasmonic and catalytic metal elements can also be atomically intermixed to form alloy nanoparticles.47,65–67 Both the heterostructured and alloyed nanoparticles may exhibit catalytic/photocatalytic behaviors remarkably different from those of their monometallic counterparts because of strong optical and electronic coupling effects.47 The catalytic metal components may drastically modify the optical responses of the plasmonic metals, causing significant frequency shifts and damping of localized plasmons. On the other hand, the inherent behaviors of the catalytic metals can also be profoundly influenced by the plasmonic components due to the energy shift of electronic bands and interdomain transfer of charge carriers. Such mechanistic complexity makes it challenging to fully elucidate detailed structure–property relationships underpinning the remarkable photocatalytic tunability achievable in these plasmonic–catalytic nanohybrids.
A more straightforward strategy for constructing plasmonic–catalytic dual-functional nanocatalysts relies on the arrangement of plasmonic and catalytic metals in close proximity while keeping them separated from each other in deliberately designed supra-nanostructures, referred to as antenna–reactor complexes.36–38,48,50–52,56 In a hybrid antenna–reactor system, the plasmonic metal functions as the light-harvesting nanoantenna, while the reactors consisting of catalytic metals serve as active sites for reactions. Strikingly distinct from atomically intermixed alloys and directly interfaced heteronanostructures, each constituent metal domain in an antenna–reactor complex retains its inherent optical and catalytic properties. The catalytic reactions can be kinetically boosted by the enhanced local fields surrounding the plasmonic antenna, which may induce optical polarizations in the reactors and reinforce intramolecular electronic transitions in molecular adsorbates.47 The photocatalytic performances of antenna–reactor complexes can be systematically optimized either through independent structural tailoring of each functionally distinct domain or by tuning the strengths of antenna–reactor interactions without modifying the intrinsic properties of each constituent. Over the past few years, a series of antenna–reactor nanohybrids with desired compositional combinations adopting various targeted configurations have been constructed either using lithography-based top-down methods37,47,52,55 or through bottom-up colloidal synthesis.36,38,48,50,51,56 Although top-down approaches offer precise control over the dimensions and spatial arrangements of the antennas and reactors as well as the size of the gaps between the antenna and reactors, large-scale fabrication of the materials remains challenging, and the resulting nanostructures are limited to hetero-dimers or oligomers on two-dimensional planar surfaces.37,47,52,55 In contrast, colloidal synthesis enables larger-scale manufacture of antenna–reactor nanohybrids that can be easily processed and readily integrated into real reactor systems to meet the need for industrial applications. In addition, colloidal synthesis provides access to three-dimensional supra-nanostructures that are structurally more sophisticated than their lithographically patterned counterparts. Furthermore, in colloidally synthesized supra-nanostructures, a large number of nanoparticulate reactors can be accommodated in plasmonic hot spots near the surfaces of each nanoantenna, enabling us to make full use of plasmonic antenna effects to kinetically boost the catalytic reactions.
This work reports a multistep synthetic approach toward colloidal Au@C/Pt core@shell/satellite supra-nanostructures, in which the Au core acts as the light-harvesting plasmonic antenna, the Pt satellites function as catalytically active reactors, and the C shell serves as a dielectric spacer separating the Pt reactors from the Au antenna. Our synthetic approach allows us to tune the size of the Au core, the surface coverage of Pt satellites, and the thickness of the C shell independently. The photocatalytic behaviors of the Au@C/Pt nanohybrids can be tuned by simply varying the C shell thickness without modifying the inherent properties of the Au antennas and the Pt reactors. Therefore, the Au@C/Pt supra-nanostructures represent a model antenna–reactor system with unique structural tunability ideal for investigating the interplay between the plasmonic and catalytic effects. The catalytic reaction we choose for detailed kinetic studies is the multistep cascade oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB), which produces chromogenic intermediate species that have been widely utilized for colorimetric biosensing applications.68–71 Employing Pt-catalyzed TMB oxidation in an aerobic aqueous environment as a model reaction, we show that plasmonic antenna effects can be harnessed not only to kinetically modulate crucial steps in a catalytic tandem reaction but also to trigger molecular transformations along emergent reaction pathways under light illumination.
Experimental
All chemicals and reagents were commercially available (see details in the ESI†) and used as received without further purification. Ultrapure deionized water (18.2 MΩ cm−1 resistivity) purified using a Millipore Milli-Q® water purification system was used for nanoparticle synthesis and sample preparation. Colloidal Au nanoparticles synthesized through seed-mediated nanocrystal growth were used as the seeds to mediate the growth of Ni3C in a polyol solvent, tetraethylene glycol (TEG), at a reaction temperature of 300 °C, to form Au@Ni3C core@shell nanoparticles, which further evolved into Au@C core@shell nanoparticles upon exposure to an acidic etchant solution. The detailed procedures for synthesizing Au, Au@Ni3C, and Au@C nanoparticles are described in the ESI.† To synthesize Au@C/Pt core@shell/satellite supra-nanostructures, 100 μL of the stock colloidal suspensions of the Au@C core@shell nanoparticles were mixed with x μL of 10 mM K2PtCl4 (x = 5, 10, 30, 50) in ethylene glycol (EG). The total volume of the reactant mixtures was kept at 1 mL. After the reactant mixtures were maintained at 70 °C for 20 min, the resulting colloidal nanoparticles were rapidly cooled down in an ice bath, collected through centrifugation, thoroughly washed with H2O through three cycles of redispersion/centrifugation, and finally stored in 1 mL of H2O. The as-synthesized nanoparticle samples were characterized by transmission electron microscopy (TEM), energy dispersive spectroscopy (EDS), powder X-ray diffraction (PXRD), UV-vis-near infrared extinction spectroscopy, X-ray photoelectron spectroscopy (XPS), and electrochemical impedance spectroscopy (EIS). Detailed information about the instruments used for the nanoparticle characterizations is presented in the ESI.†We studied the reaction kinetics of TMB oxidation catalyzed by the Au@C/Pt nanohybrids in an aqueous phosphate buffer solution at a pH of 8 both in the dark and under light illumination. All kinetic measurements were repeated three times under each reaction condition, and the standard deviations were represented by the error bars in the kinetic results. The reactant–catalyst mixtures were kept under magnetic stirring (300 rpm) in a 1 cm × 1 cm × 4 cm quartz cuvette and the reactions occurred in ambient air at room temperature (25 °C) unless otherwise mentioned. The initial concentration of TMB was 100 μM, and the total volume of the reactant mixtures was kept at 2.0 mL. The Au@C/Pt catalysts added to the reactant mixtures contained 2.33 μg Au. During the catalytic reactions, the temporally evolving concentrations of chromogenic intermediates were monitored through UV-vis absorption spectroscopic measurements using a Beckman Coulter Du 640 spectrophotometer. The extinction spectra of colloidal Au@C/Pt catalysts containing 2.33 μg Au dispersed in 2 mL of water were measured and used for baseline subtraction.
Photo-enhanced catalytic reactions were carried out under continuous laser illumination at various excitation wavelengths and powers. Au@C/Pt catalysts containing 2.33 μg Au were mixed with 100 μM TMB in 2 mL of water in a quartz cuvette. Continuous-wave (CW) diode lasers purchased from Lasever Inc. (Ningbo, Zhejiang, China) with emission wavelengths at 638 nm (model no. LSR638CP8), 520 nm (model no. LSR520CPD), and 785 nm (model no. LSR785NL) were used as light sources. The laser powers were adjustable in the range of 0–3.0 W. The laser beams were collimated with a 4 mm × 4 mm square cross-section and incident vertically through the open side of the cuvette. The reactant–catalyst mixtures were kept under magnetic stirring (300 rpm) to facilitate heat dissipation and the solution-phase temperature of the laser-illuminated samples was controlled at 25 ± 1 °C using a circulating water bath. The temperature evolution under laser illumination was monitored using a digital thermocouple (Therma Waterproof Thermometer for Type K Thermocouples, Priggen Special Electronic) immersed in the solutions.
Results and discussion
Our synthetic approach to the Au@C/Pt core@shell/satellite supra-nanostructures involves several major steps, as schematically illustrated in Fig. 1A. Colloidal Au nanoparticles with fine-controlled average diameters in the range of ∼30–200 nm were synthesized through seed-mediated nanocrystal growth following a previously published protocol.72 The as-synthesized Au nanoparticles were then used as colloidal seeds to mediate the growth of thin shells of Ni3C in TEG at a reaction temperature of 300 °C to form Au@Ni3C core@shell nanoparticles.73 In this seed-mediated growth process, nickel nitrate and polyvinylpyrrolidone (PVP) served as the nickel precursor and surface-capping agent, respectively, while TEG played triple roles as the solvent, a mild reducing agent, and the C precursor simultaneously. The Ni component in Ni3C could be selectively etched when exposing the Au@Ni3C core@shell nanoparticles to an etchant solution containing 0.5 M HCl, which led to the formation of Au@C core@shell nanoparticles. Pt nanoparticles were deposited onto each Au@C core@shell nanoparticle to form Au@C/Pt core@shell/satellite supra-nanostructures by thermally reducing K2PtCl4 in EG at a reaction temperature of 70 °C in the absence of any additional reducing agent or surface-capping ligands. TEM images revealing the overall particle morphologies of the Au nanoparticles, Au@Ni3C core@shell nanoparticles, Au@C core@shell nanoparticles, and Au@C/Pt supra-nanostructures are shown in Fig. 1B–E, respectively. Higher-magnification TEM images highlighting the detailed structures of individual Au@C and Au@C/Pt particles are shown in Fig. 1F and G, respectively. In this set of samples, Au nanoparticles with an average diameter of 114 nm were used as the cores, and the average thickness of C shells and the average size of Pt satellites were determined to be 20.4 and 2.8 nm, respectively. The size distributions of Au cores, C shells, and Pt satellites are shown in Fig. 1H. | ||
| Fig. 1 (A) Scheme illustrating the multistep procedure for synthesizing Au@C/Pt nanocatalysts. TEM images of (B) Au nanoparticles, (C) Au@Ni3C core@shell nanoparticles, (D) Au@C core@shell nanoparticles, and (E) Au@C/Pt nanocatalysts. High-magnification TEM images of an individual (F) Au@C and (G) Au@C/Pt particle. (H) Distributions of Au core sizes (left panel), C shell thicknesses (middle panel), and Pt particle sizes (right panel). (I) PXRD patterns of Au@Ni3C core@shell nanoparticles, Au@C core@shell nanoparticles, and Au@C/Pt nanohybrids. The diffraction patterns were offset for clarity. The standard diffraction patterns of bulk Ni3C, Au, and Pt are shown at the bottom, with the characteristic peak positions labeled using vertical dashed lines. (J) Optical extinction spectra of colloidal Au, Au@C, and Au@C/Pt nanoparticles. All colloidal samples for extinction spectroscopic measurements had nominally the same Au mass concentration at 21.3 μg mL−1. | ||
We used PXRD to characterize the crystalline structures of various nanostructures (Fig. 1I). In the Au@Ni3C core@shell nanoparticles, the Au atoms in the cores and the Ni atoms in the Ni3C shell formed face-centered cubic (fcc) and hexagonal close-packed (hcp) lattices, respectively. Etching of Ni from the Ni3C shells resulted in the formation of amorphous C shells, while the fcc structure of the Au cores remained unchanged. After depositing Pt nanoparticles onto the Au@C core@shell nanoparticles, the characteristic peaks of fcc Pt emerged in the PXRD pattern. The diffraction peaks of Pt were significantly broadened in comparison to those in the standard pattern of bulk Pt because of the small particle size of Pt.74 The diffraction peaks of Pt were relatively weak in comparison to those of Au primarily due to low Pt/Au mass ratios in the Au@C/Pt nanohybrids and the small sizes of the Pt satellites. The chemical compositions of the Au@C/Pt supra-nanostructures were further confirmed by EDS elemental analysis (Fig. S1 in the ESI†) and the Pt/Au atomic ratio of the sample shown in Fig. 1E and G was quantified to be 0.0526 ± 0.0053. The Au core-Pt satellite structure was confirmed by correlated TEM-EDS imaging (Fig. S2 in the ESI†). Because of the presence of C and Cu elements in the TEM grid, the EDS signals of C and Cu were not analyzed when quantifying the compositional stoichiometries of Pt/Au and collecting the EDS elemental maps. We further calculated the value of θ, which was defined as the average number of Pt satellites on each Au@C core@shell nanoparticle, using the following equation:
 | (1) |
Colloidal Au nanoparticles (114 ± 6.3 nm in size) suspended in water exhibited a well-defined light extinction peak centered at 592 nm (Fig. 1J), which is the spectral feature of the dipole plasmon resonance. After coating the Au core with a C shell, the plasmon peak redshifted to 650 nm and became more intense (Fig. 1J) because the effective dielectric constant of the C shell was higher than that of water. Such a spectral redshift with minimal further damping of the plasmons indicated that the C shells were dielectric in nature rather than being conductive. Deposition of Pt nanoparticles onto the Au@C core@shell nanoparticles led to increased extinction in the short wavelength range below ∼450 nm due to the d-to-sp interband electronic transitions in Pt.16 However, we did not observe any noticeable shift or broadening of the plasmon resonance peak (Fig. 1J), suggesting that the Pt nanoparticles were deposited on the C surfaces without creating any direct contact with the Au core. In contrast, direct deposition of Pt on Au nanoparticle surfaces to form Au@Pt core@shell heteronanostructures not only shifted the plasmon resonance wavelengths but also further dampened the Au plasmons, as featured by significant weakening and broadening of the plasmon resonance peaks in the extinction spectra.75,76 In the Au@C/Pt supra-nanostructures, the amorphous C shell served as a dielectric barrier that effectively suppressed the electron transfer between the Au core and the Pt satellites. We compared the electron-transfer resistance (Ret) of a redox probe, [Fe(CN)6]3/[Fe(CN)6]3−, on Au, Au@C, and Au@C/Pt particle-modified glassy carbon electrodes through EIS measurements (Fig. S4 in the ESI†). For a Randles equivalent circuit including the Ret of the redox probe, solution resistance (Rs), the constant phase element (Cdl), and Warburg impedance (W), the radii of the semicircles in the Nyquist diagrams reflected the Ret of the redox probe on the electrodes at certain applied potentials. When switching from Au nanoparticles to Au@C core@shell nanoparticles, a drastic increase in the Ret was clearly observed, further verifying the dielectric nature of the C shells.
The size of the Au core, the thickness of the C shell, and the surface coverage of Pt satellites could all be fine-tuned independently through judicious adjustment of the synthetic conditions. Using Au nanoparticles with an average diameter of 114 nm as the cores, we synthesized a series of Au@C and Au@C/Pt samples with various C shell thicknesses and Pt/Au atomic ratios, which are listed in Table 1. When depositing Pt satellites onto a given Au@C sample, higher surface coverage of Pt was achieved by simply increasing the initial concentration of K2PtCl4 in the reactant mixtures. Both the Pt/Au atomic ratio and the θ value increased with the concentration of K2PtCl4, while the particle sizes of Pt appeared almost independent of K2PtCl4 concentration (Fig. S5 in the ESI†). The thickness of the C shells was essentially predetermined by the thickness of the Ni3C shells in the parental Au@Ni3C core@shell nanoparticles, which could be tuned by adjusting the relative amount of Ni(NO3)2 with respect to Au in the reactant mixtures (Fig. S6 in the ESI†). For a given Au core sample, the plasmon resonance peak redshifted and the optical extinction at the plasmon resonance wavelengths increased as the C shells became thicker (Fig. S7 in the ESI†) due to the increased effective dielectric constant of the medium surrounding the Au cores. The size of the Au core could be tuned simply by choosing Au nanoparticles with different sizes as the core materials. For example, we successfully synthesized an Au@C core@shell nanoparticle sample with an average core size of 39.6 nm and shell thickness of 9.7 nm (Fig. S8 in the ESI†). The optical extinction of an Au nanoparticle larger than ∼100 nm is dominated by light scattering at plasmon resonant wavelengths, whereas Au nanoparticles smaller than ∼50 nm are strong light absorbers with rather weak light scattering and local-field enhancements.77,78 To take full advantage of the plasmonic nanoantenna effect, we chose Au nanoparticles with an average diameter of 114 nm as the cores to construct Au@C/Pt core@shell/satellite nanohybrids.
| Sample label | Size of the Au core/nm | Thickness of the C shell/nm | Pt/Au atomic ratio |
|---|---|---|---|
| Au@C-1 | 114 ± 6.3 | 8.5 ± 1.2 | 0 |
| Au@C-2 | 20.4 ± 2.4 | 0 | |
| Au@C-3 | 34.2 ± 3.8 | 0 | |
| Au@C/Pt-1 | 20.4 ± 2.4 | 0.0164 ± 0.0023 | |
| Au@C/Pt-2 | 20.4 ± 2.4 | 0.0255 ± 0.0031 | |
| Au@C/Pt-3 | 20.4 ± 2.4 | 0.0526 ± 0.0053 | |
| Au@C/Pt-4 | 20.4 ± 2.4 | 0.104 ± 0.0088 | |
| Au@C/Pt-5 | 8.5 ± 1.2 | 0.0245 ± 0.0030 | |
| Au@C/Pt-6 | 34.2 ± 3.8 | 0.0858 ± 0.0081 | |
| Au@C/Pt-7 | 8.5 ± 1.2 | 0.0113 ± 0.0041 | |
| Au@C/Pt-8 | 34.2 ± 3.8 | 0.0193 ± 0.0067 |
The C shells derived from chemical etching of Ni3C were highly porous in nature rather than being nonporous solid shells. When reacting a nominally fixed number of Au@C core@shell nanoparticles with a certain amount of K2PtCl4, the Pt/Au atomic ratio in the resulting Au@C/Pt nanohybrids increased significantly as the thickness of the C shells increased, while the Pt particle sizes appeared essentially independent of the C shell thickness (Fig. S9 in the ESI†), which suggested that the Pt satellites were distributed not only on the outer surface of the C shells but also in the pore channels inside the C shells. Another piece of evidence verifying the porous structures of the C shells was that small etchant molecules or ions could penetrate through the C shells to selectively etch the Au cores. Upon exposure to an etchant solution containing I2 and KI,79 the Au@C core@shell nanoparticles gradually evolved into Au@C yolk@shell structures and eventually transformed into C nanoshells with hollow interiors (Fig. S10 in the ESI†). The nanoporous C shells became structurally more compact and less porous after thermal annealing at 450 °C in an N2 atmosphere, resulting in decreased shell thicknesses (Fig. S11 in the ESI†). The structural changes of the C shells caused by thermal annealing led to decreased surface areas available for Pt deposition, as reflected by lower Pt surface coverages on the annealed Au@C core@shell nanoparticles in comparison to those on their unannealed counterparts (Fig. S12 in the ESI†). After thermal annealing of a preformed Au@C/Pt sample (Au@C/Pt-6) at 450 °C in an N2 atmosphere for 2 h, we observed a significant increase in the Pt particle sizes by ∼30%, while the Pt/Au atomic ratio remained almost unchanged (Fig. S13 in the ESI†). Such a size increase of Pt satellites was caused by thermally induced particle sintering, further suggesting that mass transport of Pt occurred within the pore channels in the C shells.
We systematically investigated the catalytic behaviors of the Au@C/Pt core@shell/satellite supra-nanostructures both in the dark and under light illumination using the cascade aerobic oxidation of TMB as a model reaction. As schematically illustrated in Fig. 2A, the aerobic oxidation of TMB is a multistep process, forming two chromogenic intermediate species with well-defined characteristic light absorption peaks in the visible spectral region. While TMB is colorless, its one-electron oxidation product, denoted as oxTMB-i, is a cationic free radical that undergoes a dimerization process to produce a blue-colored diimine–diamine charge-transfer complex. oxTMB-i has two characteristic absorption peaks centered at 375 and 652 nm, respectively, in the visible region. An oxTMB-i molecule can further lose one electron to form a yellow-colored diimine compound, denoted as ox-TMB-ii, which has a characteristic absorption band centered at 450 nm. ox-TMB-ii can react with O2 to form a series of further oxidized products, all of which are colorless. From a thermodynamic point of view, TMB can eventually get fully oxidized to generate inorganic products, including CO2, N2, and H2O, in the presence of excessive O2. In our kinetic studies, we used UV-vis absorption spectroscopy as an in situ spectroscopic tool to track the formation and consumption of oxTMB-i and oxTMB-ii during the catalytic reactions based on the temporal evolutions of absorbance at 652 nm for oxTMB-i (molar absorption coefficient: 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) and 450 nm for oxTMB-ii (molar absorption coefficient: 59000 M−1 cm−1), respectively.80 Although thermodynamically spontaneous, the oxidation of TMB is kinetically sluggish without any detectable reaction progress over several days in the absence of catalysts and TMB exhibits long-term chemical stability when dissolved in an aqueous solution in ambient air. While Au@C core@shell nanoparticles appeared catalytically inactive, Au@C/Pt nanohybrids could effectively catalyze the oxidation of TMB (Fig. S14 in the ESI†), verifying the role of Pt satellites as the catalytically active reactors. The temporal evolution of UV-vis spectra collected during TMB oxidation catalyzed by Au@C/Pt-3 in the dark at room temperature is shown in Fig. 2B and snapshot spectra captured at several reaction times are highlighted in Fig. 2C. In a deoxygenated environment purged with N2, the reactions became drastically slower (Fig. S15 in the ESI†), indicating that the molecular O2 dissolved in the aqueous solutions served as the oxidant driving the catalytic oxidation of TMB.
000 M−1 cm−1) and 450 nm for oxTMB-ii (molar absorption coefficient: 59000 M−1 cm−1), respectively.80 Although thermodynamically spontaneous, the oxidation of TMB is kinetically sluggish without any detectable reaction progress over several days in the absence of catalysts and TMB exhibits long-term chemical stability when dissolved in an aqueous solution in ambient air. While Au@C core@shell nanoparticles appeared catalytically inactive, Au@C/Pt nanohybrids could effectively catalyze the oxidation of TMB (Fig. S14 in the ESI†), verifying the role of Pt satellites as the catalytically active reactors. The temporal evolution of UV-vis spectra collected during TMB oxidation catalyzed by Au@C/Pt-3 in the dark at room temperature is shown in Fig. 2B and snapshot spectra captured at several reaction times are highlighted in Fig. 2C. In a deoxygenated environment purged with N2, the reactions became drastically slower (Fig. S15 in the ESI†), indicating that the molecular O2 dissolved in the aqueous solutions served as the oxidant driving the catalytic oxidation of TMB.
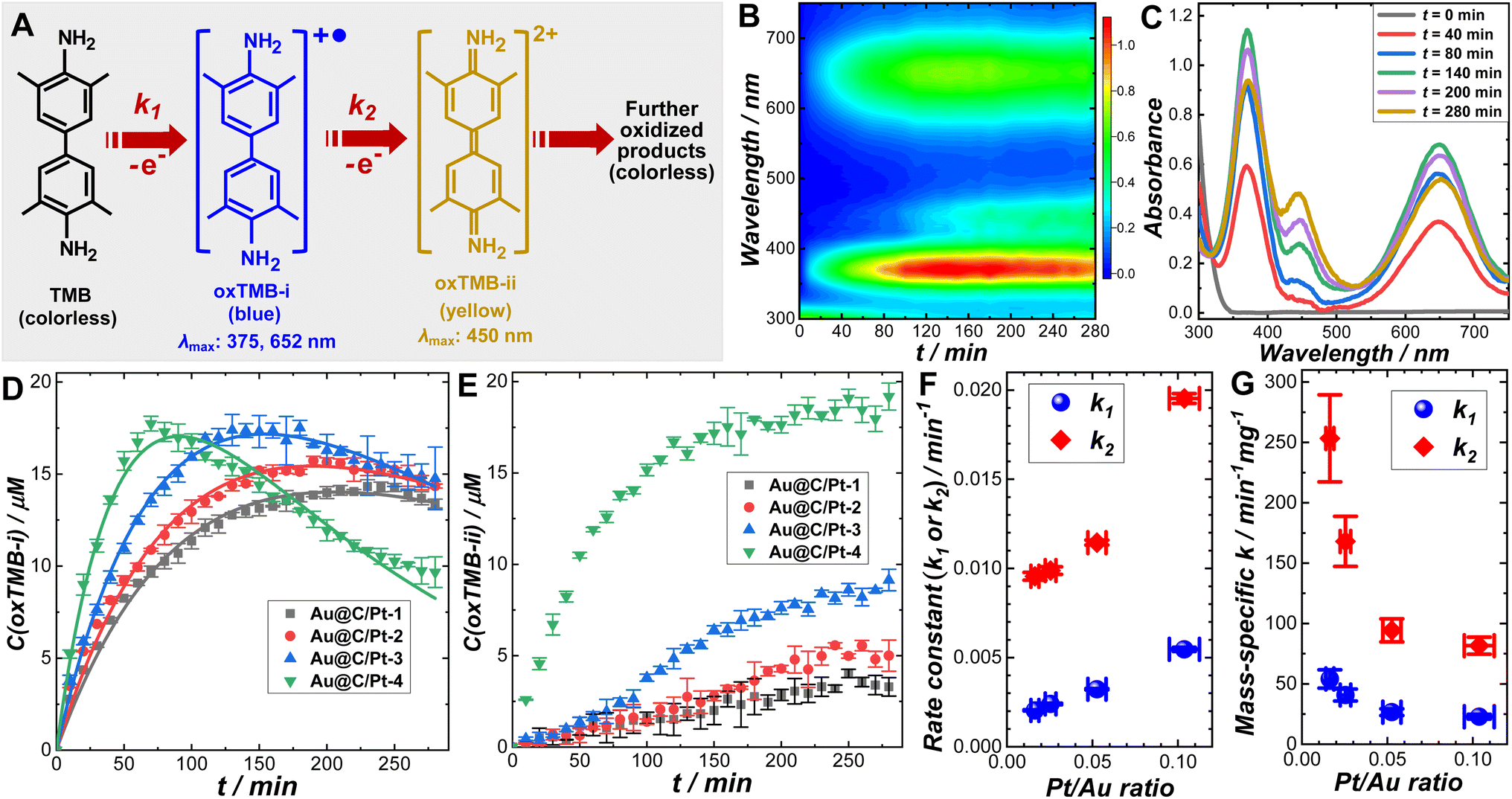 | ||
| Fig. 2 (A) Schematic illustration of multistep cascade oxidation of TMB. (B) Time-resolved UV-vis absorption spectra and (C) several snapshot spectra captured at various reaction times during TMB oxidation reactions catalyzed by Au@C/Pt-3. Temporal evolutions of (D) oxTMB-i and (E) oxTMB-ii concentrations during TMB oxidation catalyzed by Au@C/Pt-1, Au@C/Pt-2, Au@C/Pt-3, and Au@C/Pt-4. All reactions occurred at room temperature (25 °C) with an initial TMB concentration of 100 μM. The pH of the reaction medium was 8, and the total volume of the reactant mixtures was kept at 2.0 mL. The Au@C/Pt nanocatalysts contained 2.33 μg Au in all cases. The error bars in panels (D) and (E) represent the standard deviations of triplicate kinetic measurements under each reaction condition. The curve fitting results are shown as solid curves in panel (D). Pt/Au ratio-dependence of (F) apparent rate constants and (G) Pt-mass-specific rate constants for catalytic TMB oxidation reactions. The error bars of the rate constants represent the standard deviations associated with the least squares curve fitting of the kinetic results shown in panel (D). | ||
To fully unravel the relationships between the particle structures and catalytic behaviors of the Au@C/Pt nanohybrids, we conducted a series of comparative kinetic studies under ambient reaction conditions in the dark. We first selected four Au@C/Pt samples with the same Au core size and C shell thickness but different Pt coverages (Au@C/Pt-1, Au@C/Pt-2, Au@C/Pt-3, and Au@C/Pt-4) for comparison. The temporal evolution of the oxTMB-i concentrations, C(oxTMB-i), during the catalytic reactions could be well-fitted using the first-order rate law for a two-step consecutive reaction (Fig. 2D):
 | (2) |
Optical excitations of the plasmons sustained by the Au cores introduced pronounced modifications to both the catalytic behaviors of the Pt satellites and the kinetic profiles of the catalytic reactions. To investigate the plasmonic antenna effect with minimal interference from plasmonic photothermal heating, the reactant–catalyst mixtures under continuous laser illumination were immersed in a circulating water bath. The reactant–catalyst mixtures were also kept under constant magnetic stirring (300 rpm) to facilitate heat dissipation, which effectively minimized the temperature difference between the nanoparticle surfaces and the bulk solution.81 The solution-phase temperature of a colloidal suspension of Au@C/Pt-3 catalysts under continuous laser illumination at an excitation wavelength (λex) of 638 nm and an excitation power (Pex) of 3.0 W increased by less than 1 °C under our current reaction conditions (Fig. S18 in the ESI†). When illuminated using a 638 nm CW laser, both oxTMB-i and oxTMB-ii were produced more rapidly than during the dark reactions due to resonant excitation of the plasmons of Au@C/Pt-3 (Fig. 3A). As Pex increased, the maximal concentration of oxTMB-i achievable during the reactions decreased and it took a shorter time to reach the maximal oxTMB-i concentration (Fig. 3B) because of increased k2/k1 ratios. Meanwhile, the concentration of oxTMB-ii also increased more rapidly and reached a lower steady-state concentration upon increasing Pex (Fig. 3C). Here we used the ratio between the rate constant under light illumination, klight, and the rate constant in the dark, kdark, as a descriptor of photo-induced kinetic enhancement, which was found to be related to Pex through the following empirical power function:
 | (3) |
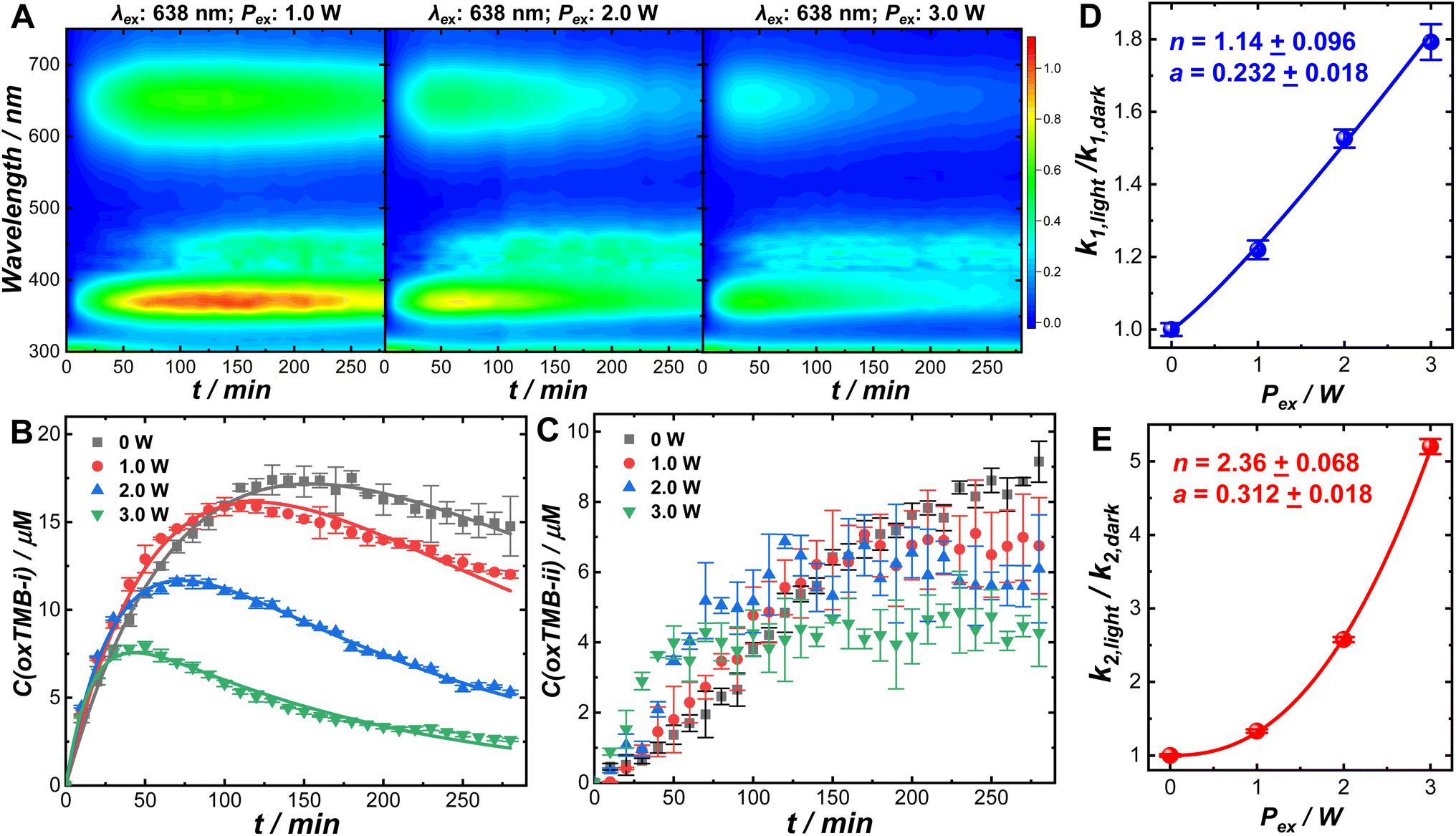 | ||
| Fig. 3 (A) Time-resolved UV-vis absorption spectra collected during TMB oxidation reactions catalyzed by Au@C/Pt-3 under continuous illumination using a 638 nm laser at Pexs of 1.0, 2.0, and 3.0 W. Temporal evolutions of (B) oxTMB-i and (C) oxTMB-ii concentrations during TMB oxidation catalyzed by Au@C/Pt-3 in the dark and under continuous illumination using a 638 nm laser at Pexs of 1.0, 2.0, and 3.0 W. The initial TMB concentration was 100 μM. The pH of the reaction medium was 8. The total volume of the reactant mixtures was kept at 2.0 mL. The Au@C/Pt-3 nanocatalysts contained 2.33 μg Au in all cases. The reaction temperatures under laser illumination were controlled at 25 ± 1 °C using a circulating water bath. The error bars in panels (B) and (C) represent the standard deviations of triplicate kinetic measurements under each reaction condition. The curve fitting results are shown as solid curves in panel (B). Pex-dependence of photo-induced kinetic enhancements (defined as the ratios between the rate constants under light illumination and in the dark) for (D) k1 and (E) k2. The error bars in panels (D) and (E) represent the standard deviations associated with the least squares curve fitting of the kinetic results shown in panel (B). The relationships between rate constants and Pex were fitted with a power function and the fitting results are shown as solid curves in panels (D) and (E). | ||
In control experiments conducted at λexs of 520 and 785 nm, no significant kinetic enhancements were observed (Fig. S19 in the ESI†) as the plasmons of Au@C/Pt-3 were off-resonance with the excitation lasers. In another set of control experiments, we compared the kinetics of the TMB oxidation reactions catalyzed by commercial Pt/C (2–4 nm Pt nanoparticles dispersed on carbon supports, 20 wt% of Pt, Alfa Aesar) in the dark and under laser illumination (λex: 638 nm; Pex: 3.0 W). Due to the lack of a plasmonic antenna in the Pt/C catalysts, light illumination gave rise to rather limited kinetic enhancement (Fig. S20 in the ESI†). The slight increase in reaction rates observed under light illumination was probably caused by the photoexcitation of interband hot carriers in Pt nanoparticles.16,36 When using the Au@C/Pt nanohybrids as the catalysts, the kinetic enhancements under laser illumination were observed to be substantially higher than those on Pt/C because of the plasmonic effects. The enhanced local fields in the vicinity of the Au nanoantennas played a crucial role in enhancing the electronic interband transitions in Pt, creating a higher abundance of hot carriers exploitable for boosting the catalytic reactions.36–38,50 The oxidation of oxTMB-i could be kinetically enhanced through an alternative pathway involving the optical excitations of the intramolecular electronic transitions82–86 in oxTMB-i. Even in the absence of Au@C/Pt antenna–reactor complexes, oxTMB-i could undergo a slow aerobic oxidation process to produce colorless products without forming oxTMB-ii when illuminated using the 638 nm laser, and elimination of the dissolved O2 by purging the solutions with N2 could effectively inhibit the oxidative photo-bleaching of oxTMB-i (Fig. S21 in the ESI†). When the excitation lasers were off-resonance with intramolecular electronic transitions in oxTMB-i (λex = 520 or 785 nm), oxTMB-i became chemically stable in an aerobic aqueous environment under continuous laser illumination (Fig. S22 in the ESI†). With the Au@C/Pt nanohybrids serving as the catalysts, the local-field enhancements could further enhance the intramolecular electronic excitations in oxTMB-i, as the characteristic absorption band of oxTMB-i centered at 652 nm overlapped with the Au plasmon resonance band of Au@C/Pt catalysts. Therefore, the apparent rate constant associated with consumption of oxTMB-i under light illumination, k2,light, reflected the overall rates of branched reactions including both one-electron oxidation of oxTMB-i into oxTMB-ii and multi-electron oxidation of oxTMB-i into further oxidized colorless products. Because of the emergence of a new reaction pathway involving oxidative photo-bleaching of oxTMB-i, optical excitation of plasmons at a λex of 638 nm resulted in a decreased steady-state concentration of oxTMB-ii (Fig. 3C). The origin of the superlinear Pex-dependence of k2,light observed in this work could be most reasonably interpreted as the synergistic effects resulting from both the plasmon-enhanced electronic interband transitions in Pt and plasmon-enhanced intramolecular electronic excitations in oxTMB-i. Because the local-field enhancements decayed rapidly as the distance from the nanoantenna surfaces increased, the photo-induced kinetic enhancements were expected to be sensitive to the variation of the C shell thicknesses. As shown in Fig. S23 and S24 in the ESI,† the photo-induced kinetic enhancements decreased as the C shell thickness increased, further verifying that the kinetic enhancements of the catalytic reactions were directly related to the local fields in the vicinity of the plasmonic nanoantennas. The kinetic enhancements observed under light illumination were unlikely to be related to the Au-to-Pt transfer of photoexcited hot carriers because in each Au@C/Pt hybrid particle, the Au antenna and the Pt reactors were separated by a dielectric C shell. The working mechanisms of the Au@C/Pt antenna–reactor nanohybrids also differ fundamentally from those of Au@Pt core–shell and Au–Pt alloy nanoparticles, in which the electronic interactions between Au and Pt play a critical role in determining the overall catalytic and photocatalytic behaviors of the materials.87
The remarkably different Pex-dependence of k1,light and k2,light (linear Pex-dependence for k1,light and superlinear Pex-dependence for k2,light) made it possible for us to modulate the relative concentrations of the two chromogenic intermediates, oxTMB-i and oxTMB-ii, during the catalytic reactions using light illumination. We used UV-vis absorption spectroscopy to monitor the temporal evolutions of oxTMB-i and oxTMB-ii concentrations during Au@C/Pt-3-catalyzed TMB oxidation over multiple cycles of laser illumination (λex: 638 nm; Pex: 3.0 W) and dark reactions. The time-resolved absorption spectra collected during the reactions are shown in Fig. 4A and temporal evolutions of oxTMB-i and oxTMB-ii concentrations are shown in Fig. 4B, respectively. We used a circulating water bath to maintain the reaction temperatures within 25 ± 1 °C under laser illumination. We first let the catalytic reactions proceed in the dark for 1 h to produce certain amounts of oxTMB-i and oxTMB-ii. Then we turned on the excitation laser and set the reaction time to zero. We exposed the reactant–catalyst mixtures to continuous laser illumination for certain time periods and then turned off the laser to let the catalytic reaction continue in the dark. We repeated this procedure multiple times and the time periods during which the laser was on and off are labeled in Fig. 4A and B. Laser illumination led to a decrease in the oxTMB-i concentration primarily due to plasmon-enhanced intramolecular excitations in oxTMB-i, whereas the oxTMB-i concentration continued to increase during dark reactions. In contrast, the temporal evolution of the oxTMB-ii concentration was less affected by the laser illumination, continuing to increase until reaching the steady-state concentration because the excitation laser (λex: 638 nm) was off-resonance with the electronic transitions in oxTMB-ii (absorption band centered at 450 nm).
 | ||
| Fig. 4 (A) Time-resolved UV-vis absorption spectra and (B) temporal evolutions of oxTMB-i (upper panel) and oxTMB-ii (lower panel) concentrations during TMB oxidation reactions catalyzed by Au@C/Pt-3 over multiple cycles of laser illumination (λex: 638 nm; Pex: 3.0 W) and dark reactions. The initial TMB concentration was 100 μM. The pH of the reaction medium was 8. The total volume of the reactant mixtures was kept at 2.0 mL. The Au@C/Pt-3 nanocatalysts contained 2.33 μg Au. The reaction temperatures under laser illumination were controlled at 25 ± 1 °C using a circulating water bath. The catalytic TMB oxidation reaction first occurred in the dark for 1 h. Then the laser was turned on and the time was set to zero. After laser illumination for certain time periods, the laser was turned off and the catalytic reaction continued in the dark. This procedure was repeated multiple times and the time periods during which the laser was on or off are labeled in each panel. The error bars in panel (B) represent the standard deviations of triplicate kinetic measurements. | ||
Besides the plasmonic antenna effect, plasmonic photothermal heating could also profoundly influence the overall reaction rates. When 2 mL of colloidal suspension of Au@C/Pt-3 (mass concentration of Au was 1.17 μg mL−1) were continuously illuminated using the 638 nm laser at a Pex of 3.0 W in ambient air (without the circulating water bath), the solution-phase temperatures were elevated by ∼38 °C within 10 min (Fig. S25D in the ESI†), reaching a steady-state temperature around 63 °C upon establishment of the thermal equilibrium. In contrast, the temperature was elevated by only 13 °C when illuminating the buffer solution without colloidal Au@C/Pt-3 particles (Fig. S25A in the ESI†). In this hybrid antenna–reactor material system, the plasmonic Au cores acted as primary photothermal transducers, whereas the C shells and Pt satellites provided relatively minor contributions to the photothermal heating (Fig. S25B–D in the ESI†). In Fig. 5, we compare the reaction rates in the dark and under continuous laser illumination at two different reaction temperatures, 25 and 63 °C. Elevation of the reaction temperature in the dark resulted in significantly increased k1 and k2 values, enabling oxTMB-i to reach its maximal concentration over shorter time periods (Fig. 5A). Meanwhile, the steady-state concentration of oxTMB-ii also increased with the reaction temperature (Fig. 5B). At both reaction temperatures, optical excitations of the plasmons led to decreases in both the maximal oxTMB-i concentrations (Fig. 5A) and the steady-state oxTMB-ii concentrations (Fig. 5B). In Fig. 5C and D, we compare the apparent rate constants in the dark and under light illumination at the two different reaction temperatures. Our results clearly revealed that both the plasmonic antenna effect and the photothermal effect could be effectively harnessed to kinetically boost Pt-catalyzed TMB oxidation reactions when deliberately designed antenna–reactor hybrid super-nanostructures were employed as catalyst/photocatalyst materials.
 | ||
| Fig. 5 Temporal evolutions of (A) oxTMB-i and (B) oxTMB-ii concentrations during TMB oxidation reactions catalyzed by Au@C/Pt-3 in the dark and under continuous laser illumination (λex: 638 nm; Pex: 3.0 W) at 25 and 63 °C. The curve fitting results are shown as solid curves in panel (A). Apparent (C) k1 and (D) k2 values for reactions in the dark and under laser illumination (λex: 638 nm; Pex: 3.0 W) at 25 and 63 °C. For all reactions, the initial TMB concentration was 100 μM and the pH of the reaction medium was 8. The total volume of the reactant mixtures was kept at 2.0 mL. The Au@C/Pt-3 nanocatalysts contained 2.33 μg Au. The reaction temperatures under laser illumination were controlled at 25 ± 1 °C using a circulating water bath. Without temperature control using the circulating water bath, the solution-phase temperature reached a steady-state temperature of 63 °C within 10 minutes under continuous laser illumination (λex: 638 nm; Pex: 3.0 W). The dark reaction at 63 °C was carried out using a water bath with a preset temperature at 63 °C. The error bars in panels (A) and (B) represent the standard deviations of triplicate kinetic measurements. The error bars in panels (C) and (D) represent the standard deviations of least squares curve fitting. | ||
Conclusions
We have developed a colloidal synthesis method for the stepwise assembly of Au@C/Pt core@shell/satellite supra-nanostructures. In this multicomponent hybrid material system, the Au core and the Pt satellites, which are separated by a thin layer of dielectric carbon, function as light-harvesting plasmonic antenna and catalytically active reactors, respectively. With our synthetic approach, the size of the Au core, the thickness of the C shell, and the surface coverage of Pt satellites can all be tuned independently, enabling us to use the Au@C/Pt nanohybrids as a unique model antenna–reactor system with structural tunability ideal for detailed mechanistic studies. With Pt-catalyzed TMB oxidation in an aerobic environment serving as a model catalytic reaction, we have found that the plasmonic antenna effect can be judiciously harnessed as a leverage to kinetically modulate multiple crucial steps during the cascade reactions. The amplified local fields in the vicinity of the plasmonic antennas effectively enhance not only the interband electronic transitions in the Pt satellites but also the intramolecular electronic excitations in oxTMB-i, both of which provide important contributions to the kinetic enhancements of the catalytic reactions. Besides the plasmonic antenna effect, thermal energy generated from plasmonic photothermal transduction can also be exploited to further accelerate the catalytic reactions. Although we focus on Au@C/Pt nanohybrids in this work, our synthetic strategy can be adopted to assemble core@shell/satellite nanohybrids accommodating a diverse range of combinations of plasmonic and catalytic metals. The synthetic success and mechanistic understanding achieved in this work create a solid foundation for deliberate design and construction of plasmonic antenna–reactor nanohybrids with optimized catalytic/photocatalytic properties toward targeted molecule-transforming processes.Author contributions
Z. W. performed the experiments. Z. W. and H. W. analyzed the data. H. W. designed the project, acquired funding support, supervised the research, and wrote the paper. Both authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was primarily supported by the American Chemical Society Petroleum Research Fund through award 62579-ND5 and in part by an Advanced Support for Innovative Research Excellence (ASPIRE) Award (130200-22-60077) sponsored by the University of South Carolina (UofSC) Office of Vice President for Research. Z. W. was partially supported by a UofSC SPARC Graduate Research Grant (130200-21-55846). This work made use of the X-ray diffraction facilities at the South Carolina SAXS Collaborative and the electron microscopy facilities at the UofSC Electron Microscopy Center. The South Carolina SAXS Collaborative was supported by a United States National Science Foundation (NSF) Major Research Instrumentation grant (DMR-1428620). The Hitachi HT7800 transmission electron microscope used in this work was purchased using funds provided by an NSF EPSCoR RII Track-I Award (OIA-1655740). The authors thank Andrew B. Greytak for the use of the atmosphere-controlled tube furnace in his laboratory.References
- Y. H. Bing, H. S. Liu, L. Zhang, D. Ghosh and J. J. Zhang, Nanostructured Pt-Alloy Electrocatalysts for PEM Fuel Cell Oxygen Reduction Reaction, Chem. Soc. Rev., 2010, 39(6), 2184–2202 RSC.
- K. D. Gilroy, A. Ruditskiy, H. C. Peng, D. Qin and Y. N. Xia, Bimetallic Nanocrystals: Syntheses, Properties, and Applications, Chem. Rev., 2016, 116(18), 10414–10472 CrossRef CAS PubMed.
- P. Herves, M. Perez-Lorenzo, L. M. Liz-Marzan, J. Dzubiella, Y. Lu and M. Ballauff, Catalysis by Metallic Nanoparticles in Aqueous Solution: Model Reactions, Chem. Soc. Rev., 2012, 41(17), 5577–5587 RSC.
- R. C. Jin, C. J. Zeng, M. Zhou and Y. X. Chen, Atomically Precise Colloidal Metal Nanoclusters and Nanoparticles: Fundamentals and Opportunities, Chem. Rev., 2016, 116(18), 10346–10413 CrossRef CAS PubMed.
- G. Li and R. C. Jin, Atomically Precise Gold Nanoclusters as New Model Catalysts, Acc. Chem. Res., 2013, 46(8), 1749–1758 CrossRef CAS PubMed.
- L. C. Liu and A. Corma, Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles, Chem. Rev., 2018, 118(10), 4981–5079 CrossRef CAS PubMed.
- R. Narayanan and M. A. El-Sayed, Catalysis with Transition Metal Nanoparticles in Colloidal Solution: Nanoparticle Shape Dependence and Stability, J. Phys. Chem. B, 2005, 109(26), 12663–12676 CrossRef CAS PubMed.
- D. S. Wang and Y. D. Li, Bimetallic Nanocrystals: Liquid-Phase Synthesis and Catalytic Applications, Adv. Mater., 2011, 23(9), 1044–1060 CrossRef CAS PubMed.
- Z. W. Quan, Y. X. Wang and J. Y. Fang, High-Index Faceted Noble Metal Nanocrystals, Acc. Chem. Res., 2013, 46(2), 191–202 CrossRef CAS PubMed.
- B. H. Wu and N. F. Zheng, Surface and Interface Control of Noble Metal Nanocrystals for Catalytic and Electrocatalytic Applications, Nano Today, 2013, 8(2), 168–197 CrossRef CAS.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Catalytic Conversion of Solar to Chemical Energy on Plasmonic Metal Nanostructures, Nat. Catal., 2018, 1(9), 656–665 CrossRef.
- P. Christopher, H. L. Xin and S. Linic, Visible-Light-Enhanced Catalytic Oxidation Reactions on Plasmonic Silver Nanostructures, Nat. Chem., 2011, 3(6), 467–472 CrossRef CAS PubMed.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Photochemical Transformations on Plasmonic Metal Nanoparticles, Nat. Mater., 2015, 14(6), 567–576 CrossRef CAS PubMed.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Plasmon-Induced Hot Carrier Science and Technology, Nat. Nanotechnol., 2015, 10(1), 25–34 CrossRef CAS PubMed.
- W. B. Jiang, B. Q. L. Low, R. Long, J. X. Low, H. Loh, K. Y. Tang, C. H. T. Chai, H. J. Zhu, H. Zhu, Z. B. Li, X. J. Loh, Y. J. Xiong and E. Y. Ye, Active Site Engineering on Plasmonic Nanostructures for Efficient Photocatalysis, ACS Nano, 2023, 17(5), 4193–4229 CrossRef CAS PubMed.
- N. Zhang, C. Han, Y. J. Xu, J. J. Foley, D. T. Zhang, J. Codrington, S. K. Gray and Y. G. Sun, Near-Field Dielectric Scattering Promotes Optical Absorption by Platinum Nanoparticles, Nat. Photonics, 2016, 10(7), 473–482 CrossRef CAS.
- K. Watanabe, D. Menzel, N. Nilius and H. J. Freund, Photochemistry on Metal Nanoparticles, Chem. Rev., 2006, 106(10), 4301–4320 CrossRef CAS PubMed.
- A. Gelle, T. Jin, L. de la Garza, G. D. Price, L. V. Besteiro and A. Moores, Applications of Plasmon-Enhanced Nanocatalysis to Organic Transformations, Chem. Rev., 2020, 120(2), 986–1041 CrossRef CAS PubMed.
- M. J. Kale, T. Avanesian and P. Christopher, Direct Photocatalysis by Plasmonic Nanostructures, ACS Catal., 2014, 4(1), 116–128 CrossRef CAS.
- Y. C. Zhang, S. He, W. X. Guo, Y. Hu, J. W. Huang, J. R. Mulcahy and W. D. Wei, Surface-Plasmon-Driven Hot Electron Photochemistry, Chem. Rev., 2018, 118(6), 2927–2954 CrossRef CAS PubMed.
- P. Christopher and M. Moskovits, Annu. Rev. Phys. Chem., 2017, 68, 379–398 CrossRef CAS PubMed.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Marago and M. A. Iati, Surface Plasmon Resonance in Gold Nanoparticles: A Review, J. Phys.: Condens.Matter, 2017, 29(20), 203002 CrossRef PubMed.
- D. D. Evanoff and G. Chumanov, Synthesis and Optical Properties of Silver Nanoparticles and Arrays, ChemPhysChem, 2005, 6(7), 1221–1231 CrossRef CAS PubMed.
- Y. Xin, K. F. Yu, L. T. Zhang, Y. R. Yang, H. B. Yuan, H. L. Li, L. B. Wang and J. Zeng, Copper-Based Plasmonic Catalysis: Recent Advances and Future Perspectives, Adv. Mater., 2021, 33(32), 2008145 CrossRef CAS PubMed.
- M. W. Knight, N. S. King, L. F. Liu, H. O. Everitt, P. Nordlander and N. J. Halas, Aluminum for Plasmonics, ACS Nano, 2014, 8(1), 834–840 CrossRef CAS PubMed.
- M. W. Knight, L. F. Liu, Y. M. Wang, L. Brown, S. Mukherjee, N. S. King, H. O. Everitt, P. Nordlander and N. J. Halas, Aluminum Plasmonic Nanoantennas, Nano Lett., 2012, 12(11), 6000–6004 CrossRef CAS PubMed.
- G. Baffou, I. Bordacchini, A. Baldi and R. Quidant, Simple Experimental Procedures to Distinguish Photothermal from Hot-Carrier Processes in Plasmonics, Light: Sci. Appl., 2020, 9(1), 108 CrossRef PubMed.
- G. Baffou, F. Cichos and R. Quidant, Applications and Challenges of Thermoplasmonics, Nat. Mater., 2020, 19(9), 946–958 CrossRef CAS PubMed.
- L. Mascaretti and A. Naldoni, Hot Electron and Thermal Effects in Plasmonic Photocatalysis, J. Appl. Phys., 2020, 128(4), 041101 CrossRef CAS.
- B. Seemala, A. J. Therrien, M. H. Lou, K. Li, J. P. Finzel, J. Qi, P. Nordlander and P. Christopher, Plasmon-Mediated Catalytic O2 Dissociation on Ag Nanostructures: Hot Electrons or Near Fields?, ACS Energy Lett., 2019, 4(8), 1803–1809 CrossRef CAS.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Hot Electrons Do the Impossible: Plasmon-Induced Dissociation of H2 on Au, Nano Lett., 2013, 13(1), 240–247 CrossRef CAS PubMed.
- L. A. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Q. Zhao, L. Henderson, L. L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Quantifying Hot Carrier and Thermal Contributions in Plasmonic Photocatalysis, Science, 2018, 362(6410), 69–72 CrossRef CAS PubMed.
- Y. Dubi, I. W. Un and Y. Sivan, Thermal Effects - an Alternative Mechanism for Plasmon-Assisted Photocatalysis, Chem. Sci., 2020, 11(19), 5017–5027 RSC.
- K. X. Chen and H. Wang, Origin of Superlinear Power Dependence of Reaction Rates in Plasmon-Driven Photocatalysis: A Case Study of Reductive Nitrothiophenol Coupling Reactions, Nano Lett., 2023, 23(7), 2870–2876 CrossRef CAS PubMed.
- X. Zhang, X. Q. Li, M. E. Reish, D. Zhang, N. Q. Su, Y. Gutierrez, F. Moreno, W. T. Yang, H. O. Everitt and J. Liu, Plasmon-Enhanced Catalysis: Distinguishing Thermal and Nonthermal Effects, Nano Lett., 2018, 18(3), 1714–1723 CrossRef CAS PubMed.
- D. F. Swearer, H. Q. Zhao, L. N. Zhou, C. Zhang, H. Robatjazi, J. M. P. Martirez, C. M. Krauter, S. Yazdi, M. J. McClain, E. Ringe, E. A. Carter, P. Nordlander and N. J. Halas, Heterometallic Antenna–Reactor Complexes for Photocatalysis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(32), 8916–8920 CrossRef CAS PubMed.
- C. Zhang, H. Q. Zhao, L. A. Zhou, A. E. Schlather, L. L. Dong, M. J. McClain, D. F. Swearer, P. Nordlander and N. J. Halas, Al-Pd Nanodisk Heterodimers as Antenna–Reactor Photocatalysts, Nano Lett., 2016, 16(10), 6677–6682 CrossRef CAS PubMed.
- K. Li, N. J. Hogan, M. J. Kale, N. J. Halas, P. Nordlander and P. Christopher, Balancing Near-Field Enhancement, Absorption, and Scattering for Effective Antenna–Reactor Plasmonic Photocatalysis, Nano Lett., 2017, 17(6), 3710–3717 CrossRef CAS PubMed.
- J. Quiroz, E. C. M. Barbosa, T. P. Araujo, J. L. Fiorio, Y. C. Wang, Y. C. Zou, T. Mou, T. V. Alves, D. C. de Oliveira, B. Wang, S. J. Haigh, L. M. Rossi and P. H. C. Camargo, Controlling Reaction Selectivity over Hybrid Plasmonic Nanocatalysts, Nano Lett., 2018, 18(11), 7289–7297 CrossRef CAS PubMed.
- J. H. Weaver, Optical-Properties of Rh, Pd, Ir, and Pt, Phys. Rev. B, 1975, 11(4), 1416–1425 CrossRef CAS.
- S. Sarina, H. Y. Zhu, Q. Xiao, E. Jaatinen, J. F. Jia, Y. M. Huang, Z. F. Zheng and H. S. Wu, Viable Photocatalysts under Solar-Spectrum Irradiation: Nonplasmonic Metal Nanoparticles, Angew. Chem., Int. Ed., 2014, 53(11), 2935–2940 CrossRef CAS PubMed.
- I. Ro, C. Sener, T. M. Stadelman, M. R. Ball, J. M. Venegas, S. P. Burt, I. Hermans, J. A. Dumesic and G. W. Huber, Measurement of Intrinsic Catalytic Activity of Pt Monometallic and Pt-MoOx Interfacial Sites over Visible Light Enhanced PtMoOx/SiO2 Catalyst in Reverse Water Gas Shift Reaction, J. Catal., 2016, 344, 784–794 CrossRef CAS.
- Y. Shiraishi, D. Tsukamoto, Y. Sugano, A. Shiro, S. Ichikawa, S. Tanaka and T. Hirai, Platinum Nanoparticles Supported on Anatase Titanium Dioxide as Highly Active Catalysts for Aerobic Oxidation under Visible Light Irradiation, ACS Catal., 2012, 2(9), 1984–1992 CrossRef CAS.
- Y. Shiraishi, H. Sakamoto, Y. Sugano, S. Ichikawa and T. Hirai, Pt-Cu Bimetallic Alloy Nanoparticles Supported on Anatase TiO2: Highly Active Catalysts for Aerobic Oxidation Driven by Visible Light, ACS Nano, 2013, 7(10), 9287–9297 CrossRef CAS PubMed.
- Y. Shiraishi, H. Sakamoto, K. Fujiwara, S. Ichikawa and T. Hirai, Selective Photocatalytic Oxidation of Aniline to Nitrosobenzene by Pt Nanoparticles Supported on TiO2 under Visible Light Irradiation, ACS Catal., 2014, 4(8), 2418–2425 CrossRef CAS.
- R. H. Li, W. X. Chen, H. Kobayashi and C. X. Ma, Platinum-Nanoparticle-Loaded Bismuth Oxide: An Efficient Plasmonic Photocatalyst Active under Visible Light, Green Chem., 2010, 12(2), 212–215 RSC.
- K. Sytwu, M. Vadai and J. A. Dionne, Bimetallic Nanostructures: Combining Plasmonic and Catalytic Metals for Photocatalysis, Adv. Phys.: X, 2019, 4(1), 1619480 CAS.
- H. Robatjazi, H. Q. Zhao, D. F. Swearer, N. J. Hogan, L. N. Zhou, A. Alabastri, M. J. McClain, P. Nordlander and N. J. Halas, Plasmon-Induced Selective Carbon Dioxide Conversion on Earth-Abundant Aluminum-Cuprous Oxide Antenna–Reactor Nanoparticles, Nat. Commun., 2017, 8, 27 CrossRef PubMed.
- D. F. Swearer, R. K. Leary, R. Newell, S. Yazdi, H. Robatjazi, Y. Zhang, D. Renard, P. Nordlander, P. A. Midgley, N. J. Halas and E. Ringe, Transition-Metal Decorated Aluminum Nanocrystals, ACS Nano, 2017, 11(10), 10281–10288 CrossRef CAS PubMed.
- D. F. Swearer, H. Robatjazi, J. M. P. Martirez, M. Zhang, L. N. Zhou, E. A. Carter, P. Nordlander and N. J. Halas, Plasmonic Photocatalysis of Nitrous Oxide into N2 and O2 Using Aluminum-Iridium Antenna–Reactor Nanoparticles, ACS Nano, 2019, 13(7), 8076–8086 CrossRef CAS PubMed.
- L. A. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. H. Lou, L. L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Light-Driven Methane Dry Reforming with Single Atomic Site Antenna–Reactor Plasmonic Photocatalysts, Nat. Energy, 2020, 5(1), 61–70 CrossRef CAS.
- P. D. Dongare, Y. G. Zhao, D. Renard, J. Yang, O. Neumann, J. Metz, L. Yuan, A. Alabastri, P. Nordlander and N. J. Halas, A 3d Plasmonic Antenna–Reactor for Nanoscale Thermal Hotspots and Gradients, ACS Nano, 2021, 15(5), 8761–8769 CrossRef CAS PubMed.
- C. H. Han, D. E. Gomez, Q. Xiao and J. S. Xu, Near-Field Enhancement by Plasmonic Antennas for Photocatalytic Suzuki-Miyaura Cross-Coupling Reactions, J. Catal., 2021, 397, 205–211 CrossRef CAS.
- D. F. Swearer, B. B. Bourgeois, D. K. Angell and J. A. Dionne, Advancing Plasmon-Induced Selectivity in Chemical Transformations with Optically Coupled Transmission Electron Microscopy, Acc. Chem. Res., 2021, 54(19), 3632–3642 CrossRef CAS PubMed.
- K. Sytwu, M. Vadai, F. Hayee, D. K. Angell, A. Dai, J. Dixon and J. A. Dionne, Driving Energetically Unfavorable Dehydrogenation Dynamics with Plasmonics, Science, 2021, 371(6526), 280–283 CrossRef CAS PubMed.
- X. B. Zhang, Y. Y. Fan, E. M. You, Z. X. Li, Y. D. Dong, L. I. Chen, Y. Yang, Z. X. Xie, Q. Kuang and L. S. Zheng, MOF Encapsulated Sub-nm Pd Skin/Au Nanoparticles as Antenna–Reactor Plasmonic Catalyst for Light Driven CO2 Hydrogenation, Nano Energy, 2021, 84, 105950 CrossRef CAS.
- W. Xie, C. Herrmann, K. Kompe, M. Haase and S. Schlucker, Synthesis of Bifunctional Au/Pt/Au Core/Shell Nanoraspberries for in situ SERS Monitoring of Platinum-Catalyzed Reactions, J. Am. Chem. Soc., 2011, 133(48), 19302–19305 CrossRef CAS PubMed.
- F. Wang, C. H. Li, H. J. Chen, R. B. Jiang, L. D. Sun, Q. Li, J. F. Wang, J. C. Yu and C. H. Yan, Plasmonic Harvesting of Light Energy for Suzuki Coupling Reactions, J. Am. Chem. Soc., 2013, 135(15), 5588–5601 CrossRef CAS PubMed.
- G. Q. Chen, N. M. Zou, B. Chen, J. B. Sambur, E. Choudhary and P. Chen, Bimetallic Effect of Single Nanocatalysts Visualized by Super-Resolution Catalysis Imaging, ACS Cent. Sci., 2017, 3(11), 1189–1197 CrossRef CAS PubMed.
- H. J. Chen, F. Wang, K. Li, K. C. Woo, J. F. Wang, Q. Li, L. D. Sun, X. X. Zhang, H. Q. Lin and C. H. Yan, Plasmonic Percolation: Plasmon-Manifested Dielectric-to-Metal Transition, ACS Nano, 2012, 6(8), 7162–7171 CrossRef CAS PubMed.
- Z. K. Zheng, T. Tachikawa and T. Majima, Plasmon-Enhanced Formic Acid Dehydrogenation Using Anisotropic Pd-Au Nanorods Studied at the Single-Particle Level, J. Am. Chem. Soc., 2015, 137(2), 948–957 CrossRef CAS PubMed.
- Z. Z. Lou, M. Fujitsuka and T. Majima, Pt-Au Triangular Nanoprisms with Strong Dipole Plasmon Resonance for Hydrogen Generation Studied by Single-Particle Spectroscopy, ACS Nano, 2016, 10(6), 6299–6305 CrossRef CAS PubMed.
- X. Z. Zhu, H. L. Jia, X. M. Zhu, S. Cheng, X. L. Zhuo, F. Qin, Z. Yang and J. F. Wang, Selective Pd Deposition on Au Nanobipyramids and Pd Site-Dependent Plasmonic Photocatalytic Activity, Adv. Funct. Mater., 2017, 27(22), 1700016 CrossRef.
- A. Joplin, S. A. H. Jebeli, E. Sung, N. Diemler, P. J. Straney, M. Yorulmaz, W. S. Chang, J. E. Millstone and S. Link, Correlated Absorption and Scattering Spectroscopy of Individual Platinum-Decorated Gold Nanorods Reveals Strong Excitation Enhancement in the Nonplasmonic Metal, ACS Nano, 2017, 11(12), 12346–12357 CrossRef CAS PubMed.
- F. A. A. Nugroho, B. Iandolo, J. B. Wagner and C. Langhammer, Bottom-up Nanofabrication of Supported Noble Metal Alloy Nanoparticle Arrays for Plasmonics, ACS Nano, 2016, 10(2), 2871–2879 CrossRef CAS PubMed.
- S. Kadkhodazadeh, F. A. A. Nugroho, C. Langhammer, M. Beleggia and J. B. Wagner, Optical Property-Composition Correlation in Noble Metal Alloy Nanoparticles Studied with EELS, ACS Photonics, 2019, 6(3), 779–786 CrossRef CAS.
- S. K. F. Stofela, O. Kizilkaya, B. T. Diroll, T. R. Leite, M. M. Taheri, D. E. Willis, J. B. Baxter, W. A. Shelton, P. T. Sprunger and K. M. McPeak, A Noble-Transition Alloy Excels at Hot-Carrier Generation in the Near Infrared, Adv. Mater., 2020, 32(23), 1906478 CrossRef CAS PubMed.
- J. Y. Zhang, S. H. Wu, X. M. Lu, P. Wu and J. W. Liu, Manganese as a Catalytic Mediator for Photo-Oxidation and Breaking the pH Limitation of Nanozymes, Nano Lett., 2019, 19(5), 3214–3220 CrossRef CAS PubMed.
- A. Asati, C. Kaittanis, S. Santra and J. M. Perez, pH-Tunable Oxidase-Like Activity of Cerium Oxide Nanoparticles Achieving Sensitive Fluorigenic Detection of Cancer Biomarkers at Neutral pH, Anal. Chem., 2011, 83(7), 2547–2553 CrossRef CAS PubMed.
- Y. Zhao, Y. C. Huang, H. Zhu, Q. Q. Zhu and Y. S. Xia, Three-in-One: Sensing, Self-Assembly, and Cascade Catalysis of Cyclodextrin Modified Gold Nanoparticles, J. Am. Chem. Soc., 2016, 138(51), 16645–16654 CrossRef CAS PubMed.
- J. Liu, L. J. Meng, Z. F. Fei, P. J. Dyson, X. N. Jing and X. Liu, MnO2 Nanosheets as an Artificial Enzyme to Mimic Oxidase for Rapid and Sensitive Detection of Glutathione, Biosens. Bioelectron., 2017, 90, 69–74 CrossRef CAS PubMed.
- P. N. Zhang, Y. J. Li, D. Y. Wang and H. B. Xia, High-Yield Production of Uniform Gold Nanoparticles with Sizes from 31 to 577 nm Via One-Pot Seeded Growth and Size-Dependent SERS Property, Part. Part. Syst. Charact., 2016, 33(12), 924–932 CrossRef CAS.
- G. G. Li, D. A. Blom, S. Pandey, R. J. Koch, S. T. Misture, S. R. Phillpot and H. Wang, Overcoming the Interfacial Lattice Mismatch: Geometry Control of Gold-Nickel Bimetallic Heteronanostructures, Part. Part. Syst. Charact., 2018, 35(5), 1700361 CrossRef.
- A. L. Patterson, The Scherrer Formula for X-Ray Particle Size Determination, Phys. Rev., 1939, 56(10), 978–982 CrossRef CAS.
- C. Zhang, B. Q. Chen, Z. Y. Li, Y. N. Xia and Y. G. Chen, Surface Plasmon Resonance in Bimetallic Core-Shell Nanoparticles, J. Phys. Chem. C, 2015, 119(29), 16836–16845 CrossRef CAS.
- L. L. Feng, X. C. Wu, L. R. Ren, Y. J. Xiang, W. W. He, K. Zhang, W. Y. Zhou and S. S. Xie, Well-Controlled Synthesis of Au@Pt Nanostructures by Gold-Nanorod-Seeded Growth, Chem. - Eur. J., 2008, 14(31), 9764–9771 CrossRef CAS PubMed.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, Calculated Absorption and Scattering Properties of Gold Nanoparticles of Different Size, Shape, and Composition: Applications in Biological Imaging and Biomedicine, J. Phys. Chem. B, 2006, 110(14), 7238–7248 CrossRef CAS PubMed.
- A. R. Shafiqa, A. Abdul Aziz and B. Mehrdel, Nanoparticle Optical Properties: Size Dependence of a Single Gold Spherical Nanoparticle, J. Phys.: Conf. Ser., 2018, 1083(1), 012040 CrossRef.
- E. C. Cho, J. Xie, P. A. Wurm and Y. Xia, Understanding the Role of Surface Charges in Cellular Adsorption Versus Internalization by Selectively Removing Gold Nanoparticles on the Cell Surface with a I2/KI Etchant, Nano Lett., 2009, 9(3), 1080–1084 CrossRef CAS PubMed.
- P. D. Josephy, T. Eling and R. P. Mason, The Horseradish Peroxidase-Catalyzed Oxidation of 3,5,3′,5′-Tetramethylbenzidine – Free-Radical and Charge-Transfer Complex Intermediates, J. Biol. Chem., 1982, 257(7), 3669–3675 CrossRef CAS PubMed.
- P. K. Jain, Taking the Heat Off of Plasmonic Chemistry, J. Phys. Chem. C, 2019, 123(40), 24347–24351 CrossRef CAS.
- E. Kazuma and Y. Kim, Mechanistic Studies of Plasmon Chemistry on Metal Catalysts, Angew. Chem., Int. Ed., 2019, 58(15), 4800–4808 CrossRef CAS PubMed.
- E. Kazuma, J. Jung, H. Ueba, M. Trenary and Y. Kim, Real-Space and Real-Time Observation of a Plasmon-Induced Chemical Reaction of a Single Molecule, Science, 2018, 360(6388), 521–525 CrossRef CAS PubMed.
- T. E. Tesema, C. Annesley and T. G. Habteyes, Plasmon-Enhanced Autocatalytic N-Demethylation, J. Phys. Chem. C, 2018, 122(34), 19831–19841 CrossRef CAS.
- T. E. Tesema, B. Kafle, M. G. Tadesse and T. G. Habteyes, Plasmon-Enhanced Resonant Excitation and Demethylation of Methylene Blue, J. Phys. Chem. C, 2017, 121(13), 7421–7428 CrossRef CAS.
- T. E. Tesema, B. Kafle and T. G. Habteyes, Plasmon-Driven Reaction Mechanisms: Hot Electron Transfer versus Plasmon-Pumped Adsorbate Excitation, J. Phys. Chem. C, 2019, 123(14), 8469–8483 CrossRef CAS.
- Q. Xue, X.-Y. Bai, Y. Zhao, Y.-N. Li, T.-J. Wang, H.-Y. Sun, F.-M. Li, P. Chen, P. Jin, S.-B. Yin and Y. Chen, Au Core-PtAu Alloy Shell Nanowires for Formic Acid Electrolysis, J. Energy Chem., 2022, 65, 94–102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and additional figures as noted in the main text, including TEM images, EDS results, XPS results, particle size distributions, optical extinction spectra, PXRD patterns, EIS results, temperature evolution under laser illumination, and detailed kinetic results. See DOI: https://doi.org/10.1039/d3na00498h |
| This journal is © The Royal Society of Chemistry 2023 |