
Nebulization assisted molecular LIBS for sensitive and fast fluorine determination in aqueous solutions†
Cristina
Méndez-López
 ,
Luis Javier
Fernández-Menéndez
,
Cristina
González-Gago
,
Jorge
Pisonero
and
Nerea
Bordel
*
,
Luis Javier
Fernández-Menéndez
,
Cristina
González-Gago
,
Jorge
Pisonero
and
Nerea
Bordel
*
University of Oviedo, Department of Physics, Federico García Lorca 18, 33007, Oviedo, Asturias, Spain. E-mail: bordel@uniovi.es
First published on 11th November 2022
Abstract
The determination of halogen content in liquid samples by conventional laser induced breakdown spectroscopy (LIBS) is a difficult task due to the high excitation threshold of these elements. The indirect detection of these elements via molecular emission, product of recombination with an alkali-earth metal, is a well-known method to overcome this problem. However, current LIBS methods for halogen detection in liquid samples are scarce and based on the deposition of solutions on a CaCO3 substrate using a pipetting gun, followed by an annealing process, resulting in a relatively slow sample preparation method. In this work, a novel approach based on online nebulization of different concentrations of an aqueous solution of NaF towards a CaCO3 pellet is employed to obtain fluorine calibration curves from molecular emission of CaF. Operating parameters were optimized considering signal intensity, repeatability and analysis time (which determines the amount of consumed sample). Moreover, three fluorine calibration curves were built using different operating configurations and then tested by carrying out fluorine determination of mouthwash samples diluted in water. The optimized microanalysis configuration provided LODs of 10 mg kg−1 with a linear range of 900 mg kg−1 as well as accurate determination of the F-content in the mouthwash samples, which were validated by ion chromatography.
1. Introduction
Laser-induced breakdown spectroscopy (LIBS) is an analytical technique that relies on the use of a high-intensity pulsed laser that is focused on a sample to induce a fast-evolving, transient plasma. The emission of the excited species in the plasma provides a source for qualitative and quantitative information about the sample composition. One of the most attractive features of LIBS is the multi-elemental analysis capability. Nevertheless, the determination of halogens is complicated due to the high excitation and ionization potentials of these elements (over 10 eV) and their resonant emission lines being located in the VUV. In fact, a calculation of the relative intensity of selected emission lines from fluorine and other elements (alkali and alkali-earth metals, non-metals) as a function of excitation temperature shows that in order to maximize the emission from such a halogen, very high temperatures (∼1.5 eV) have to be reached within the plasma and therefore low acquisition delays should be used.1 Different approaches were developed to overcome these problems such as the use of NIR lines (less intense) enhanced by working with a helium atmosphere2,3 and the detection of molecular emission of molecules resulting from the recombination of halides and alkali-earth metals.4 Specifically, determination of F by means of CaF in Ca-containing samples showed a significant improvement with respect to infrared emission from the atomic species.5,6 Moreover, the methodology was extended to Ca-free samples using a nebulized solution to externally provide the alkali earth element.7 The limits of detection were practically identical in both schemes (∼50 mg kg−1).LIBS can be used with solid, liquid and gas samples but it is most usually applied to the former. Liquids are challenging samples whose analysis usually involves procedures such as liquid-to-solid conversion (adsorption, micro-extraction, freezing, drop coating deposition, electrodeposition…), avoidance of ablation on a static liquid target by either producing flowing jets or aerosol conversion over which the laser is then focused or double pulse systems.8–10 These strategies imply significant sample preparation and/or greater complexity of the experimental setup.
The detection of fluorine in aqueous samples by LIBS is, therefore, a complicated task subjected to the conjoint problematics of halogen determination and liquid samples nature in a context where there is an increasing interest on these elements. For example, the World Health Organization cites fluoride, a chemical commonly added to drinking water with the purpose of protecting against dental caries, as one of the chemicals that has been proved to cause adverse health effects when present in excessive quantities.11 Its concentration on drinking water can be naturally increased due to the presence of fluoride-containing minerals such as fluorite or apatite but also anthropogenically by the use of fertilizers and industrial activity, such as the manufacturing of fluorocarbon surfactants.12,13
The challenge of halogen determination in liquids via LIBS was firstly evaluated by Rusak et al. using substrates utilized in surface-enhanced Raman spectroscopy (SERS) as an approach to nanoparticle-enhanced LIBS (NE-LIBS) to enhance F I emission in the IR region.14 More recently, Tang et al. demonstrated a different methodology based on the drop coating deposition of the liquid sample on a CaCO3 substrate, which was afterwards heated to evaporate the liquid solvent before LIBS analysis was carried out detecting CaF emission.15 The first of these works was a proof of concept and therefore provided no LODs; the experiments were performed with 20 μL of a 0.1% F liquid sample. The latter provided remarkable LODs of 0.38 mg kg−1 with a usage of 100 μL of liquid sample. This methodology provides high sensitivity but requires a relatively long sample preparation protocol, which is not appropriate for fast or online analysis.
In this work, quantification of fluorine content in an aqueous solution is achieved through the emission of CaF produced by continuous nebulization of the solution directly towards a CaCO3 target, on which the laser is focused to induce the plasma. The quantification method is investigated, and the different operating parameters are evaluated and optimized. Moreover, an illustrative application of the developed methodology is demonstrated by using commercial mouthwash samples with fluorine content, which is also determined by ion exchange chromatography (IC), chosen as well-established and highly sensitive reference technique in order to validate our LIBS results. Other conventional, high-performance techniques utilized for fluorine determination aside from IC include molecular absorption spectrometry (MAS) and ion-selective electrode (ISE).16
2. Experimental setup
The main instrumental of the nebulization-assisted LIBS experimental set-up has been shown in an earlier work17 and it's described below.2.1. LIBS set-up
A Q-switched Nd:YAG laser (NL301HT, EKSPLA) with a pulse duration of 3–6 ns, operating at the infrared fundamental wavelength, 1064 nm, with an energy of 100 mJ per pulse, set by means of an attenuator (Lotis Tii) was focused onto a solid target with a 35 mm effective working-distance objective (LMH-5×1064, Thorlabs). A system of two orthogonal motorized linear stages and a manual micrometric vertical stage allowed to control the target positioning; objective-sample distance was monitored with a laser-meter (ILD1420-100, Micro-Epsilon). Light collection was carried out with a system of two 2′′ (50.8 mm) plano-convex lenses of 150 and 300 mm focal lengths (LA4904-UV and LA4855-UV, Thorlabs) that collimated and focused a two-times magnified image of the plasma on the 100 μm-wide entrance slit of a Czerny–Turner spectrometer (Andor Shamrock SR-500i-D1, with a focal length of 500 mm and f/6.5). A 1200 grooves per mm diffraction grating was utilized, which together with the iCCD detector (Andor iStar DH734-25f-03), provided spectral windows of approximately 33 nm; the central wavelength was 533.00 nm for all experiments, covering the 516.42–549.60 nm spectral region.2.2. Nebulization set-up
Samples were introduced in the microflow concentric nebulizer (Teledyne CETAC Technologies) by means of a syringe pump (Thermo Fisher Scientific), at a rate of 4 μL min−1 (value optimized in ref. 7) and with an Ar flow of 1 L min−1 that was set by a mass flow controller (MKS instruments). The nebulizer angle and position (50° with respect to the sample surface, at a height of ∼10 mm) was set with a manual rotation stage (MSRP01/M from Thorlabs) and a system of manual XYZ micrometric stages, respectively. Fig. 1 shows the distribution of the main elements in the experimental set-up.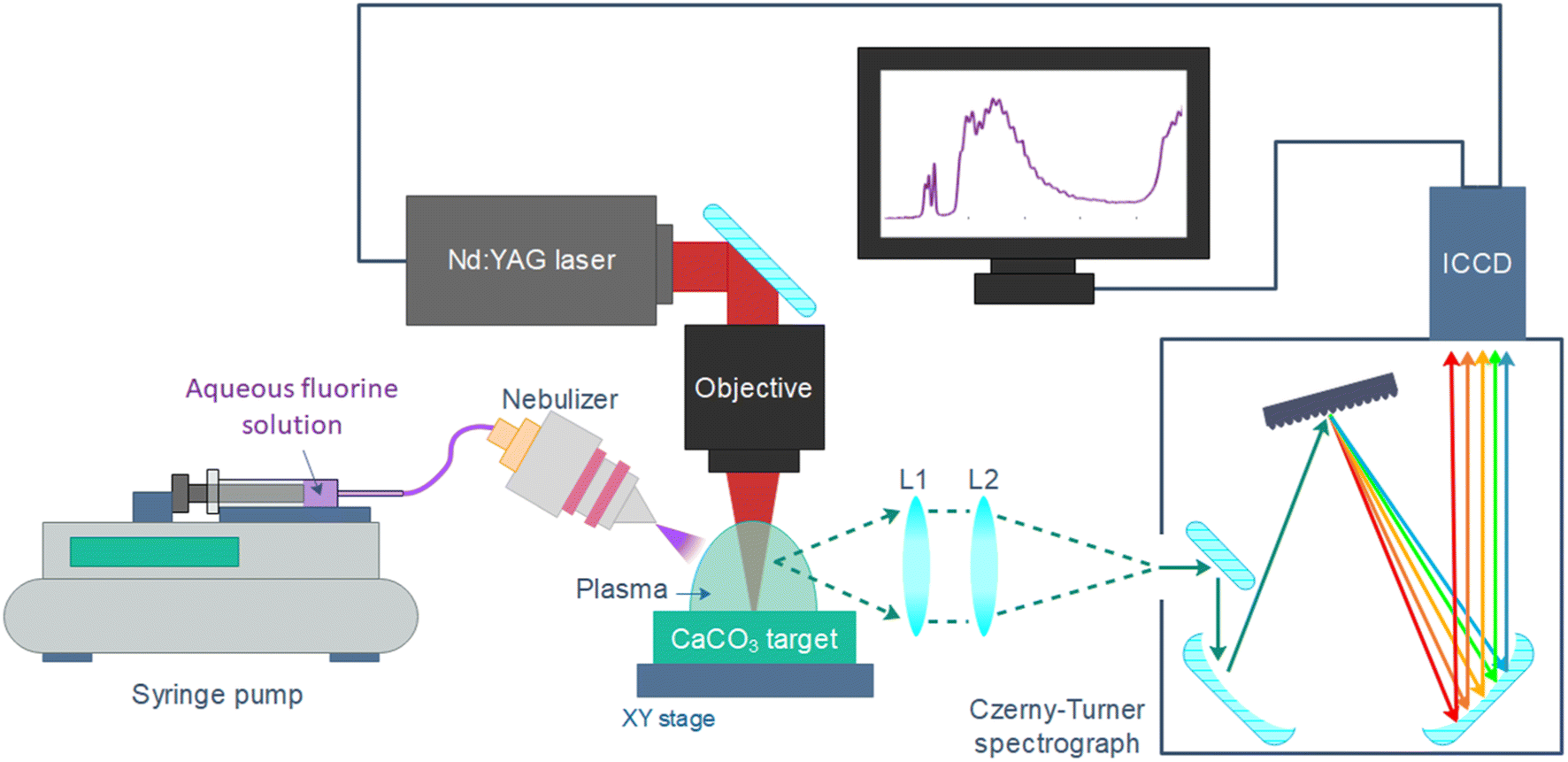 | ||
| Fig. 1 Main elements of the experimental setup. L1 = 150 mm focal length, L2 = 300 mm focal length. | ||
2.3. Liquid samples and CaCO3 pellets preparation
Liquid samples with different fluorine concentrations were prepared as calibration samples. A high-concentration solution was initially prepared by dissolving 2 g of NaF powder (purity 99%, Alfa Aesar) in 200 mL of Milli-Q water. Mixing was done with a magnetic stirrer heated up to 75 °C and 2 drops of 1 M NaOH were also added to ease solubility. After precipitation of some NaF, the supernatant was collected and its F− concentration was measured by means of ion chromatography (883 Basic IC plus, Metrohm) and determined to be 4287.4 ± 0.3 mg kg−1. This solution was diluted to produce various solutions with different concentrations in order to build a calibration curve; in particular, concentrations were prepared ranging from 12 mg kg−1 to 2146 mg kg−1, as listed in Table 1. Ultrapure water was used as a blank.| Sample | [F] (mg kg−1) | Sample | [F] (mg kg−1) |
|---|---|---|---|
| 1 | 12 | 7 | 342 |
| 2 | 25 | 8 | 481 |
| 3 | 54 | 9 | 613 |
| 4 | 112 | 10 | 860 |
| 5 | 161 | 11 | 1429 |
| 6 | 252 | 12 | 2146 |
Three commercial mouthwashes with different fluorine content were selected as testing samples. The original concentrations declared by the manufacturers as well as other relevant components of the mouthwash matrices are listed on Table 2. Mouthwashes were conveniently diluted in ultrapure water to minimize matrix effects (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 w/w for the glycerin-based mouthwash and 1:1 for the remaining two, respectively) and final fluoride concentration was then determined by ion chromatography, a sensitive and accurate technique standardly utilized for the analysis of liquids, in order to obtain an accurate reference value to validate the results from the LIBS methodology. Table 3 summarizes the chromatography results, where the concentration of chloride is also indicated due to its potential relevancy on the analysis, as this element is also prone to recombination with Ca.
:4 w/w for the glycerin-based mouthwash and 1:1 for the remaining two, respectively) and final fluoride concentration was then determined by ion chromatography, a sensitive and accurate technique standardly utilized for the analysis of liquids, in order to obtain an accurate reference value to validate the results from the LIBS methodology. Table 3 summarizes the chromatography results, where the concentration of chloride is also indicated due to its potential relevancy on the analysis, as this element is also prone to recombination with Ca.
| Mouthwash | Declared [F] (ppm) | Glycerine | Alcohol | Cl-containing compounds |
|---|---|---|---|---|
| 1 | 113 | No | Yes | No |
| 2 | 225 | Yes | No | Cetylpyridinium chloride |
| 3 | 900 | Unk. | No | Zinc chloride |
| Sample | [F−] (mg kg−1) | [Cl−] (mg kg−1) |
|---|---|---|
| M1 | 54.8 ± 0.2 | 1.88 ± 0.04 |
| M2 | 46.4 ± 0.4 | 14.0 ± 0.2 |
| M3 | 370.8 ± 0.9 | 403.7 ± 0.5 |
Due to the lack of Ca or any other alkali-earth metal in the samples to be nebulized, Ca-containing pellets were employed as targets. Each of them was prepared by mixing and homogenizing 5 g of CaCO3 low in alkali (AnalaR NORMAPUR, VWR Chemicals) and 0.5 g of micronized amide wax (Ceridust 3910), followed by a one-minute long hydraulic pressing (Specac, T-40, Atlas Series Evacuable Pellet Dies) at 10 metric tons. Resulting pellets were 32 mm in diameter with a thickness of roughly 4 mm. It is worth noting that the calcium carbonate reagent composition might contain up to 50 mg kg−1 of chloride.
3. Results and discussion
3.1. Time evolution of the emission spectra
Firstly, the calibrating sample with highest concentration of fluorine (2146 mg kg−1) was nebulized onto the Ca-containing pellets and simultaneously analyzed to determinate the optimum LIBS acquisition time window. Given the fungible nature of calcite targets (which once submitted to nebulization are not reused to ensure a clean surface in each analysis) simultaneous optimization of delay and integration times was performed in an exploratory manner. The integration times were increased with delay time due to the transient nature of the plasma emission, aiming for maximum signal as well as avoidance of detector saturation. Regarding the spectral signals, the sequence Δv = 0 of the B2Σ → X2Σ emission system of CaF,18 was considered as it presents only a moderate interference of CaO/CaOH emission. The collected spectral region also included Ca I emission lines (one line at 518.89 nm, a multiplet involving 5 lines between 526.17 and 527.03 nm and another line at 534.95 nm).As a first step of the data treatment, the spectra were normalized to the intensity at 517.2 nm, where no evident atomic nor molecular emission is observed, in order to reduce signal variability. Other potential normalization signals (atomic Ca I, molecular CaO/CaOH) were not used at this stage since their differing temporal behaviour would difficult a comparison between each time window. Afterwards, linear baselines were subtracted prior to signal integration of the atomic and molecular signals, respectively. The emission from CaF was integrated in the 528.8–538.0 nm region, excluding a narrow range (534.2–535.5 nm) in order to avoid Ca I emission at 534.9 nm. The Ca I multiplet was integrated in the 524.0–528.4 nm region. Fig. 2a shows the integrated intensities for CaF molecular emission and the joint atomic emission from the Ca I multiplet at all the considered time windows. A delay of 30 μs with an integration time of 5 μs was selected due to the high CaF emission at a relatively short integration time, avoiding the greater RSDs observed at longer delay times. This behaviour of signal uncertainty can be linked to a previous work regarding nebulization-modified LIBS,17 which showed increasing instabilities with longer delay times (ten microseconds onwards) of the plasma plume under a Ca-solution nebulization, as compared to a non-nebulized plume. Fig. 2b shows the LIBS spectrum obtained at the selected conditions.
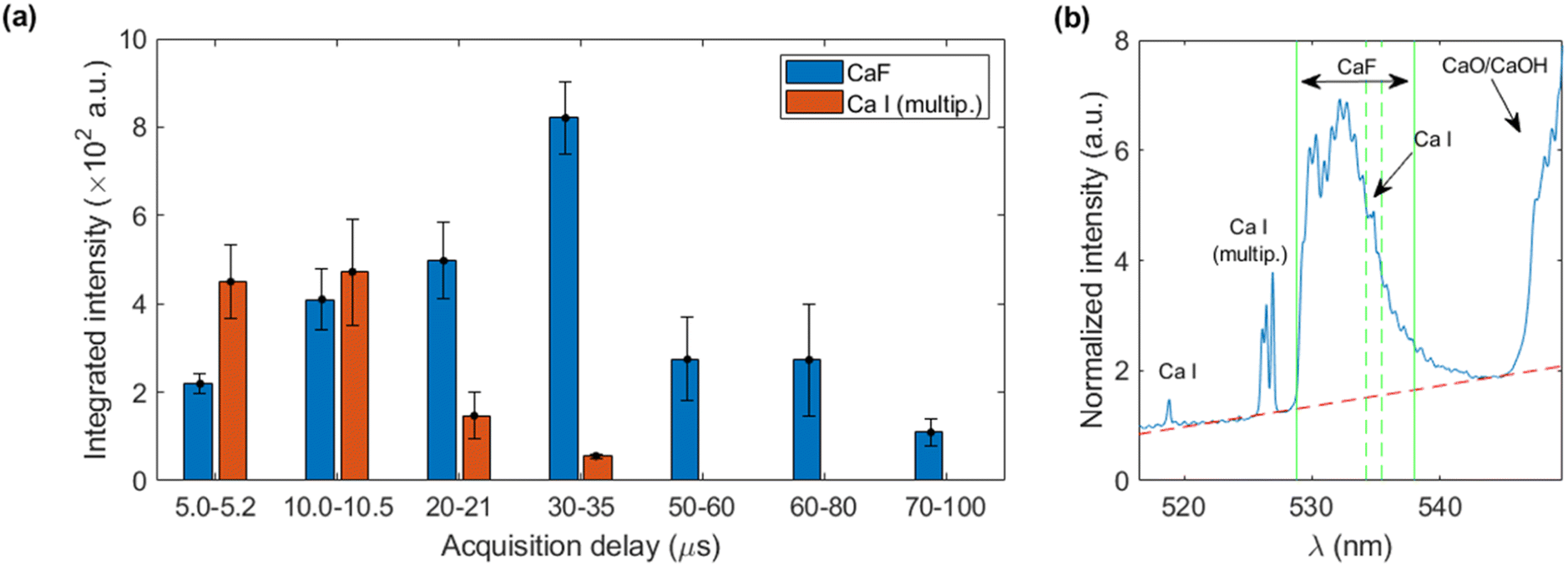 | ||
| Fig. 2 Results utilizing sample 12 (2146 mg kg−1 of F) (a) integrated intensity for CaF and Ca I at each acquisition window (b) average spectrum obtained at the optimum time window (30–35 μs) with red dotted line indicating baseline and green vertical lines, the integration region. Note a narrow interval is excluded from the integration due to Ca I interference. | ||
3.2. Optimization of operating conditions
Operating parameters (such as laser frequency and raster speed) and nebulization protocols (such as pre-nebulization or just simultaneous nebulization) were also optimized using the 2146 mg kg−1 sample. A total number of 25 accumulated shots per acquisition and a length of 10 mm for the raster line were taken as fixed parameters, ensuring a distance of 400 μm between shots (roughly the spot diameter). Distance between raster lines was 1 mm. Since the optical axis is parallel to the target surface, rasters were always carried out along that direction, covering a half of the pellet. Fig. 3a summarizes the location of raster lines and their relative position on the target with respect to detection and nebulization (see Fig. 3b). This aimed to avoid a systematic error due to the positions at which each condition is tested, since less light is collected the further the spot is located with respect to the sample border due to the light collection geometry. Once the first half of the surface was fully consumed, the pellet was rotated, and the same procedure was carried out.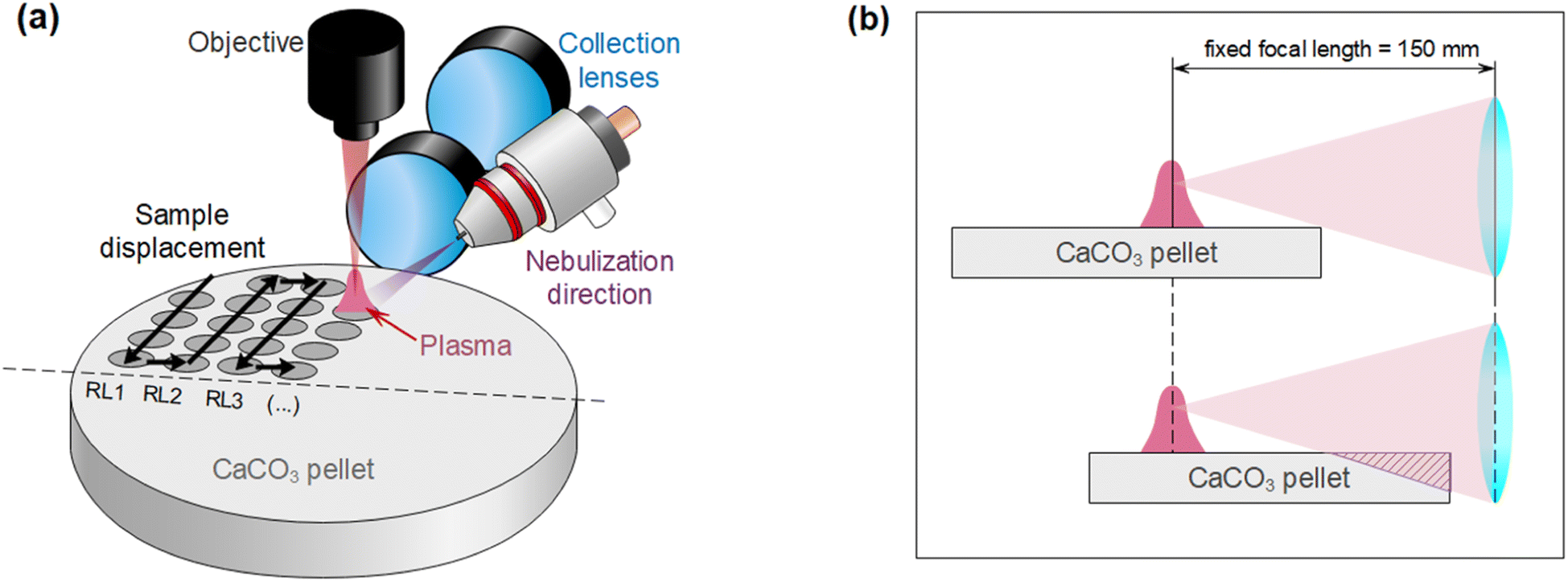 | ||
| Fig. 3 (a) Relative distribution of sample displacement, detection and nebulization. Raster lines are denoted RL. (b) Loss of light due to sample position with respect to the detection, given that the plasma is always generated at the focal plane. | ||
Three frequency rates (10, 5 and 2.5 Hz) with corresponding target displacement speeds (4, 2 and 1 mm s−1) were evaluated. Moreover, for each of these three cases, two nebulization strategies, schematized in Fig. 4, were considered. The simplest strategy “N1” consisted in simultaneously nebulizing and firing the laser at all times while the target is being displaced. A total of 4 rasters were carried out on the same line, obtaining 4 acquisitions of 25 accumulations each. On the other hand, for strategy “N2” the sample was firstly displaced under nebulization without firing the laser so that the surface got slightly wet with the nebulized substance (pre-pass). Afterwards, the sample was moved back to the initial position while simultaneously nebulizing and firing the laser (raster). “N2” strategy needed 8 displacements to perform 4 rasters (a single line), since a pre-pass is done each time. It is worth stressing that the nebulizer was continuously working and only turned off in order to change of the nebulized substance (i.e. in-between samples but never in-between repetitions). Fig. 4 summarizes “N1” and “N2” procedures.
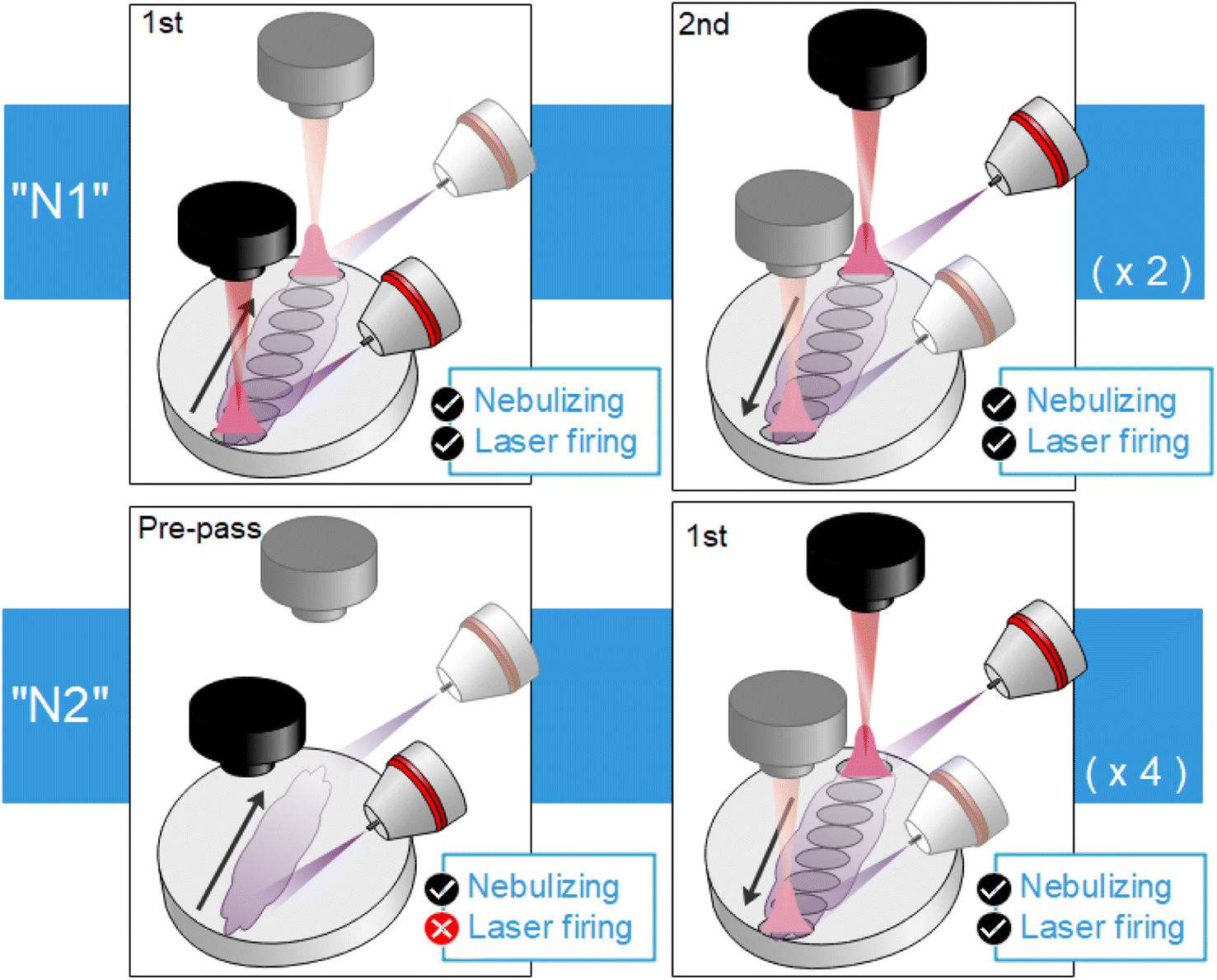 | ||
| Fig. 4 Diagrams showing both considered strategies. For “N1” (top), the laser is always firing whereas for “N2” (bottom), there is a previous displacement where the surface is nebulized on and a second one where the laser starts firing. | ||
Fig. 5 shows the integrated CaF intensity obtained using the 6 possible combinations of the evaluated conditions. In each case, the 4 rasters successively done over one line were individually detected (i.e. 4 spectra were obtained per line). Additionally, 10 repetitions were performed to obtain a properly representative average value. The uncertainty was obtained as the standard deviation of the 10 repetitions and the 2σ criterion (95% confidence interval) was utilized to identify and remove outliers. Lastly, detector gain was set to maximize signal but otherwise data treatment procedure was the same as previously described on Section 3.1.
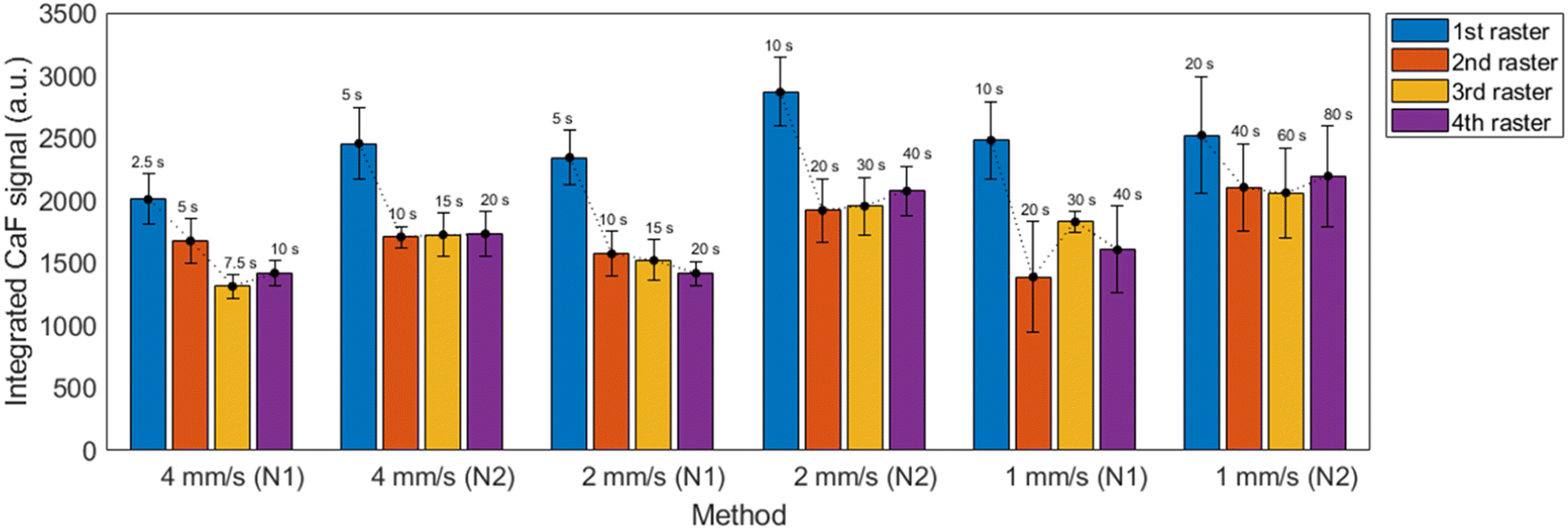 | ||
| Fig. 5 Integrated CaF signal for the 6 proposed methods at each raster line, averaged for 10 repetitions. The total time utilized for one repetition of the measurement is displayed above each bar. | ||
It is observed that nebulization protocol based on previous nebulization pass (“N2”) showed a moderate increase in signals for all considered cases, with no significant effect on the uncertainties. Additionally, a slight relative decrease of atomic Ca I signal with respect to the molecular emission was also produced. This can be seen in the spectra shown in the ESI.† Measurements carried out at 1 mm s−1 showed higher overall uncertainties, which were related to the increased accumulation of liquid on the CaCO3 pellet, making it more likely to damage the surface as shown in Fig. 6. As a general trend, a higher signal with ∼10% RSD (∼15% for 1 mm s−1 measurements) was obtained for the first raster carried out in a line, whereas the value corresponding to the next three rasters remained stable.
 | ||
| Fig. 6 Detail of ablated lines on CaCO3 pellets showing (a) undamaged surface and (b) effect of the excess of liquid on the sample surface typically observed at slower speeds. | ||
Another important aspect is that the distance between lines, which was limited by the size of the pellet, is small enough that the liquid nebulized while a line was being carried out had a noticeable effect on the next line. In fact, it was seen that the first repetition was systematically identified as an outlier, exceeding a difference greater of two times the standard deviation of the mean. Given this effect, it was considered that in order to perform N repetitions, N + 1 lines had to be carried out with the first one playing the role of merely pre-conditioning the adjacent surface. Ablation, and not only nebulization, was carried out in the pre-conditioning in order to maintain as most similarity among the N lines as possible (e.g. same “splashing” of the liquid).
3.3. Calibration curves
Two operating configurations based on the pre-pass methodology (“N2”) were considered in order to build calibration curves, which are summarized on Table 4. One of them focused on minimizing sample consumption and therefore utilized the fastest parameters (10 Hz, 4 mm s−1) and is referred as “speed config.” from now on. The second aimed to maximize signal (“signal config.”) and used the intermediate parameters (5 Hz, 2 mm s−1). One of the main differences between the selected configurations, aside from the aforementioned parameters, is that for the “speed” configuration only one raster (with a pre-pass) was done per line in order to minimize sample consumption. On the other hand, for the “signal” configuration, all four rasters (again, each of them with a pre-pass) were carried out. Since the difference in emission between the 1st pass and the subsequent three passes were obvious in all methods tested (Fig. 5), two calibrations for “signal config.” were built, using either just the first pass or a sum of the second to fourth rasters.| Config. | Raster speed (mm s−1) | Frequency (Hz) | Total rasters | Pre-pass before each raster | Analysis time fora 10 rep. (s) | Sample consumed fora 10 rep. (μL) |
|---|---|---|---|---|---|---|
| a Includes the first line, which is not useful but still involves sample consumption. Averages are obtained with 9 values. | ||||||
| Speed | 4 | 10 | 1 | Yes | 50 | 3.3 |
| Signal | 2 | 5 | 4 | Yes | 400 | 26.8 |
Given that nebulization of ultrapure water was carried out in order to obtain a blank signal, a modification of the data treatment procedure was included. As a first step, all spectra were normalized to the CaO/CaOH integrated signal (545.5–550.0 nm) and the subtraction of the average normalized spectrum of the blank was performed on each spectrum. This ensures not only the reduction of variability but also the precise removal of the blank signal. This additional step is particularly important in order to deal with lower concentrations of fluorine, for which the emission of CaF becomes quite moderate with respect to the CaO/CaOH emission and a simple baseline correction would not be enough to eliminate its contribution to the analyte signal, resulting in a non-negligible interference that would introduce a systematic error in the integration of CaF molecular emission. Afterwards, a linear baseline was subtracted and signal was integrated in the same spectral range as described on Section 3.1. The data treatment procedure is shown in Fig. 7(a–c), exemplified with a calibration sample containing 161 mg kg−1 of fluorine. Again, outliers were removed according to the 2σ criterion.
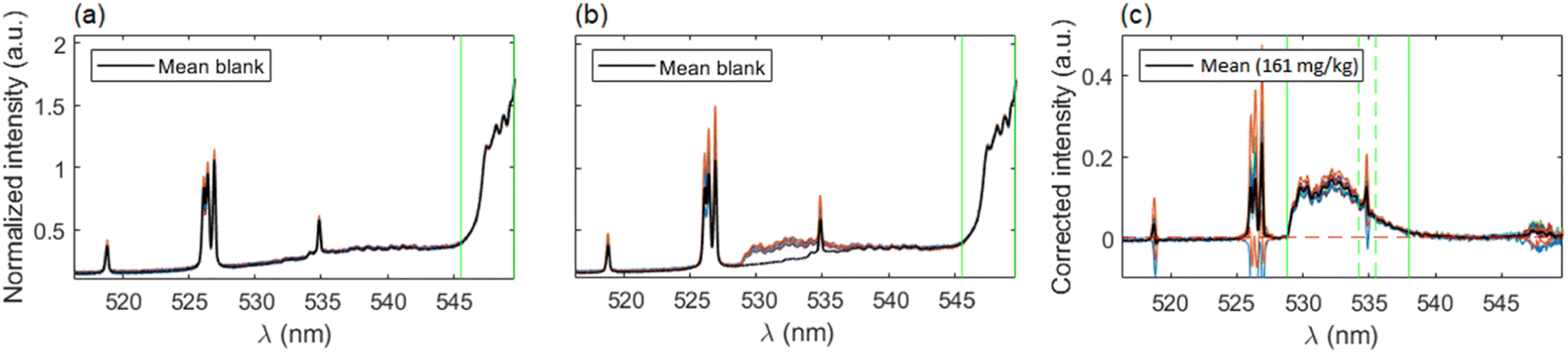 | ||
| Fig. 7 Examples of spectra processing for the first raster of signal configuration. (a) Blank signals after normalization to 545–550 nm region. Vertical lines indicate normalization region. (b) Emission spectra from the 161 mg kg−1 calibration sample, normalized and compared to mean blank spectrum. (c) Spectra from a calibration sample after blank subtraction and baseline correction. Vertical lines indicate integration region for the CaF signal. | ||
Fig. 8(a–c) shows the calibration curves obtained with each operating configuration. Linear fits were weighted according to the uncertainty of the data points 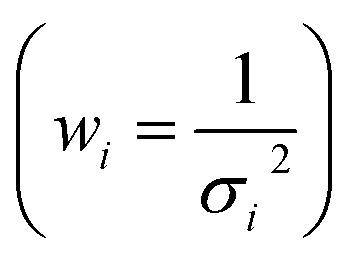 . As expected, a moderate increase (1.9 times) in sensitivity was observed for the 1st raster of signal configuration, whereas the 2nd–4th rasters calibration resulted in a decrease of the slope. Linearity was slightly better for the “speed” configuration, which may be due to the reduced amount of liquid that was deposited on the sample surface. The limits of detection, obtained as three times the ratio between the standard deviation of the blank and the calibration slope
. As expected, a moderate increase (1.9 times) in sensitivity was observed for the 1st raster of signal configuration, whereas the 2nd–4th rasters calibration resulted in a decrease of the slope. Linearity was slightly better for the “speed” configuration, which may be due to the reduced amount of liquid that was deposited on the sample surface. The limits of detection, obtained as three times the ratio between the standard deviation of the blank and the calibration slope 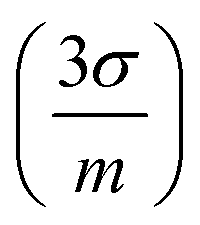 , were determined to be very similar (about 10 mg kg−1) for the “speed” and the “signal” 1st raster configurations. A lower value of 6 mg kg−1 was achieved with the subsequent rasters of “signal” configuration due to the low uncertainty of the blank signal. Similarly, the linear range in the former cases was the same, extending up to 900 mg kg−1 but it was severely decreased for the latter, reaching a concentration of approximately 500 mg kg−1.
, were determined to be very similar (about 10 mg kg−1) for the “speed” and the “signal” 1st raster configurations. A lower value of 6 mg kg−1 was achieved with the subsequent rasters of “signal” configuration due to the low uncertainty of the blank signal. Similarly, the linear range in the former cases was the same, extending up to 900 mg kg−1 but it was severely decreased for the latter, reaching a concentration of approximately 500 mg kg−1.
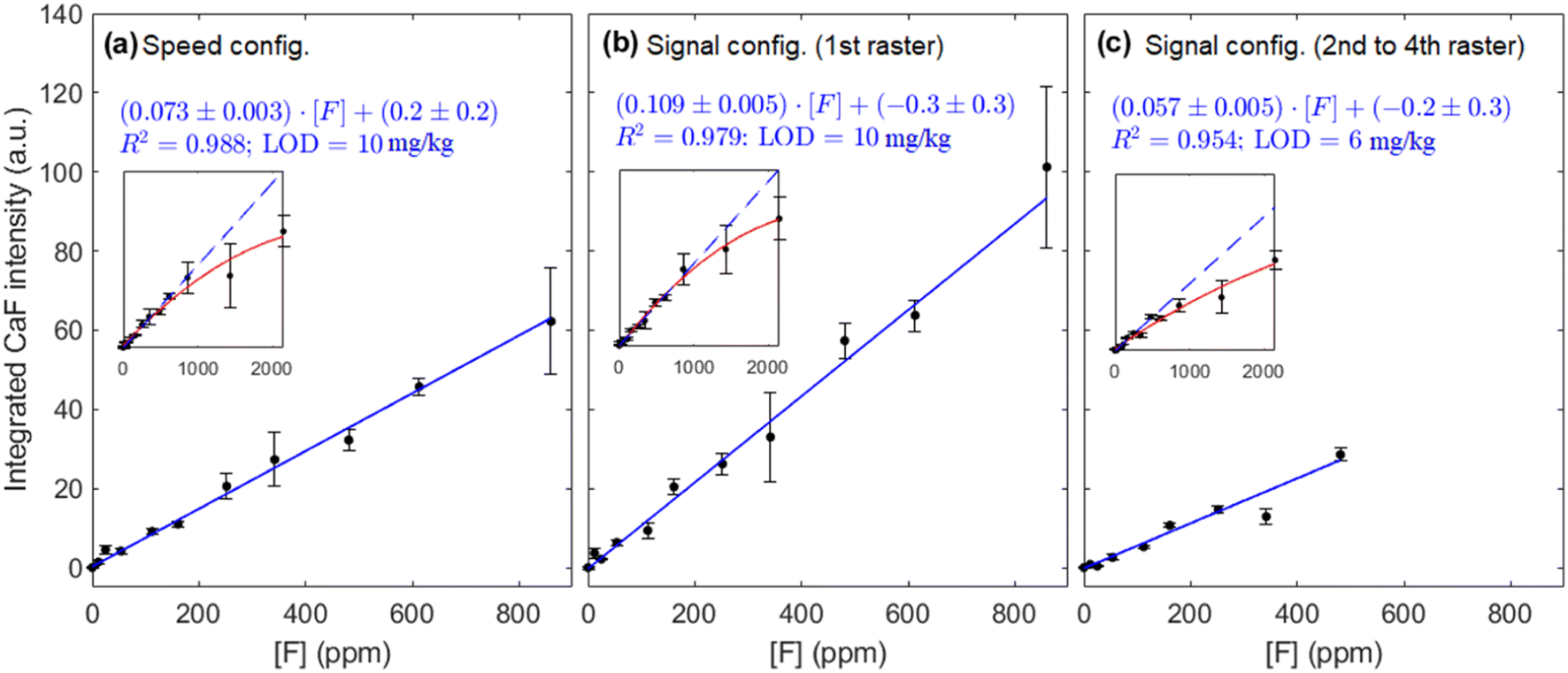 | ||
| Fig. 8 Calibration curve for the (a) speed and (b and c) signal configurations. Insets show the curve of growth for the complete concentration range considered. The same y-scale has been maintained for ease of comparison. | ||
Points from these calibration curves typically have an average relative uncertainty of 11% but, for some points in all methodologies considered, RSDs >25% are observed. Regarding these uncertainties, the shot-to-shot variations associated to LIBS as an analytical technique, which have been related to the modification of the plasma plume shape and the subsequent change of region that is detected with the collection optics,19 are likely to be an important factor. Particularly, it has been shown that these morphology fluctuations increase with delay time in general.19 In addition, it has also been observed in a previous work17 that the nebulization affects several parameters of the plasma plume including shape variability. Another source of uncertainty, which could explain the increased RSDs of some points in the calibrations, could be due to occasional undetected malfunction of the nebulizer. This device is continuously flowing, and its performance cannot be meticulously monitored with the spectral window that is being utilized, since just one nebulized element is being observed. However, this could be corrected if two elements from the nebulized sample (e.g. F and Na) could be simultaneously monitored, since any abnormal behaviour of the nebulizer could be easily identified by a simultaneous decrease/increase of both elements, with a corresponding increase/decrease of matrix emission (Ca I and/or CaO/CaOH), and the measurement, discarded.
3.4. Mouthwash samples
In order to evaluate the potential application of this methodology to a sample of interest, nebulization of three mouthwashes previously diluted (Table 3) was carried out and spectra were obtained according to the three experimental configurations previously described. Data treatment procedure was the same as described in Section 3.3, relying on the normalized blank subtraction step. The calibration curves from Fig. 8 were then utilized to determine the concentrations of the mouthwashes. Fig. 9 summarizes the values determined in each case as compared to the validated concentrations.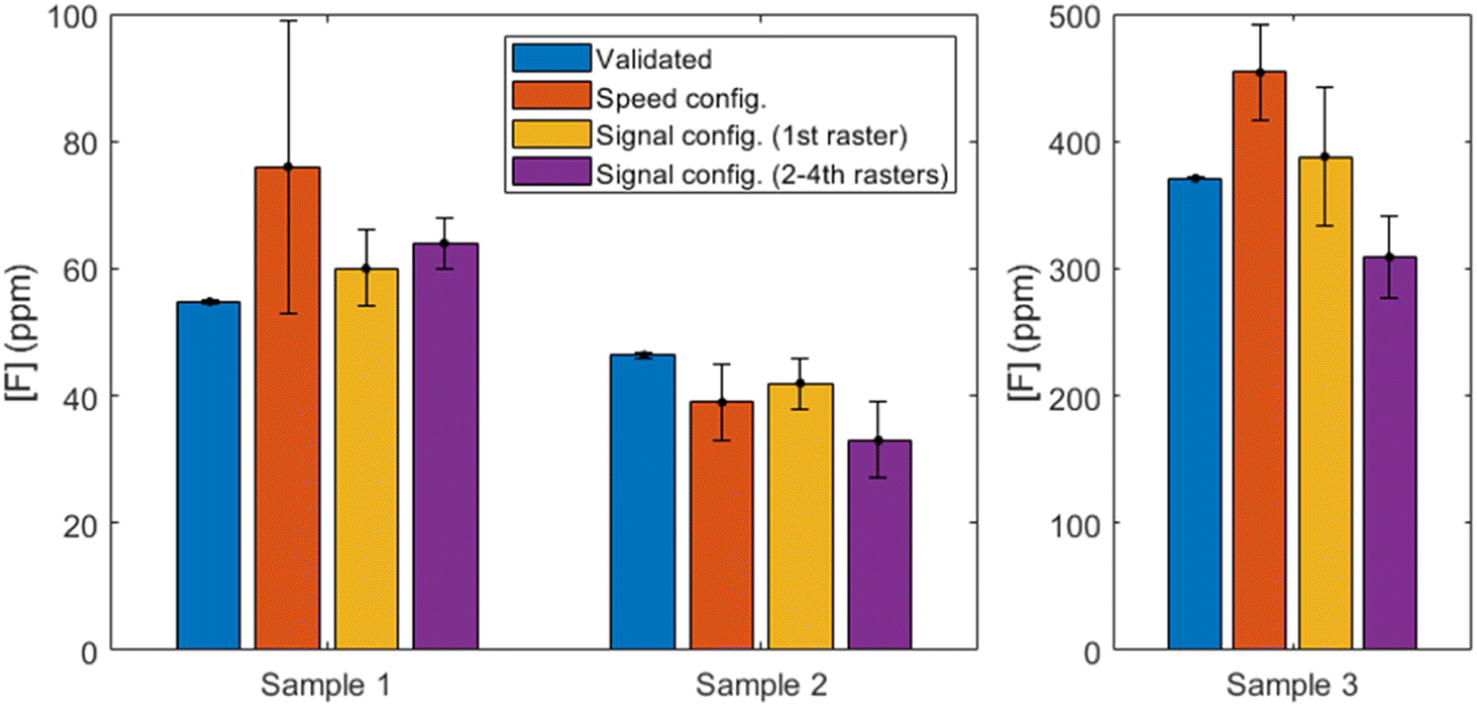 | ||
| Fig. 9 Fluorine concentrations determined with each configuration compared to the validated concentrations. Sample 3 is shown with a different y-scale for ease of visualization. Reader is referred to the online version of this article for color version of the figure. | ||
Firstly, if the results of the “speed” configuration are observed (shown in orange), we can see that accuracy is deficient. Two of the samples are overestimated (a deviation of 39% and 22% for samples M1 and M3, respectively) and the second is underestimated (by a 16%). The RSD for the first sample is very high (30%) and no outliers could be identified, whereas the F content in the rest of the mouthwashes was quantified with higher precision (15% and 8%, respectively).
On the other hand, it can be seen that the 1st raster of “signal” configuration (shown in yellow) provides the most accurate results for all mouthwashes. The relative uncertainties of the results, which are notably higher than those obtained by IC for the reasons previously discussed, vary between 10% and 14%. On the other hand, the relative deviation from the reference values (IC) are 9% (M1), 9% (M2) and 5% (M3), with all the determined concentrations comprising the reference value within their uncertainty ranges. According to the composition stated by the manufacturers, M2 is the only mouthwash known to contain glycerine in a significant amount, and the consistent underestimation that is observed in all experimental configurations might be due to a matrix effect. Since the composition of glycerine does not include any element that was not present in a significant amount for all other experiments (i.e. carbon, hydrogen and oxygen), no spectral interferences are expected to be present. However, the change in viscosity could be translated in slightly bigger deposited droplets of aerosol on the calcium carbonate pellet surface. It has been shown that droplets deposited on the surface of a metal solid can produce an increase in ablation rate, even for small aerosol droplets,20 and this effect has also been observed in nebulization experiments with a solid copper target.17 If the glycerin content is enough to increase the size of the accumulated droplets and a greater amount of pellet is ablated, the relative amount of clean bulk matter will be increased (since F is just at the most superficial layer of the pellet) and greater emission from the CaO/CaOH molecules will be expected. In such a case, since CaF emission is maintained, the normalization procedure to a now-higher signal of CaO/CaOH would be translated into an underestimated determination of CaF. This is consistent to what is observed for all methodologies, but since the solid target is not a metal but rather a somewhat permeable matrix, a thorough fundamental study outside the scope of the present work would be necessary to fully characterize the physical effects of having glycerine in the nebulized sample.
The following 2nd to 4th rasters of the “signal” configuration (shown in purple) maintain moderate RSDs (6–18%) but show a significant worsening of the accuracy (deviations of 17–29%) for all samples. It becomes evident than no benefit is obtained from performing these rasters, as the linear range has been practically shortened in half (see Fig. 8(b and c)) and despite a promising but moderate lowering of the LODs, the results with the mouthwash samples are unsatisfactory.
It could be useful to take the chlorine content into account, since Cl atoms could be competing to recombine with Ca to form CaCl. The low Cl content in M1 and M2 (see Table 2) are probably negligible but M3 has higher concentration of Cl than F and its effects, if any, should be evident. However, the results from each configuration vary between overestimation and underestimation of the fluorine concentration and no specific tendency can be pointed out. Therefore, no decreasing of CaF formation can be observed due to competing CaCl recombinations in light of our results. This strengthens the usefulness of the methodology for its application to samples such as mouthwashes, since active ingredients like zinc chloride or cetylpyridinium chloride can be present as well as fluorine.
The previous considerations show that “signal” configuration with a single raster is the experimental configuration that provides the best results for the study of real samples, without any benefit from adding further rasters on each line. The sample consumption for such a case would then be reduced to 0.67 μL per repetition, a fourth of that stated in Table 4. On the other hand, it is possible that an improvement for M2 could be achieved by performing a calibrate that includes glycerin as a matrix component. The results are, however, still far from the precision and accuracy that ion chromatography can provide, but they contribute to show that LIBS can be utilized to determine halogens in liquid samples in a simple, quick manner that minimizes sample consumption.
4. Conclusions
In the present work, a methodology for the determination of fluorine in aqueous samples by means of direct, online nebulization of the aqueous sample towards a CaCO3 pellet has been proposed and its potential application to real samples has been tested. It is shown that the optimized configuration consists in performing a nebulization pre-pass on the calcium target surface followed by a single raster with simultaneous nebulizing and laser ablation. This methodology allows detection of fluorine concentrations down to 10 mg kg−1 with a linear range extending up to 900 mg kg−1 while using a very reduced sample volume (0.67 μL per repetition). Considering this consumption, the absolute limit of detection for fluorine would be of 6.7 ng. Additionally, suitable aqueous dilution (depending on matrix and original F concentration) is the only sample preparation step that is required, if at all. This is a remarkable reduction of time, costs and residue production when compared to other techniques in which reagents have to be added to the liquid solution.In order to test the feasibility of this methodology in real samples, fluorine concentration in three commercial mouthwashes was determined. Results were adequately validated by ion chromatography, with deviations in accuracy below 10% for all samples, although the analytical performance of the methodology is still significantly lower than that of IC. Nevertheless, an improvement of outlier detection and removal, resulting in an improvement of precision, could be achieved by either utilizing a suitable, wider spectral window or even including an internal standard with an emission line in the studied region, given that it does not introduce a troublesome spectral interference. Another potential improvement of the methodology could be achieved by utilizing a more convenient solid target, less reagent- and time-consuming to prepare, so that a greater number of repetitions can be carried out.
This microanalysis technique could be implemented for online quality control of fluorinated mouthwashes and other suitable liquid samples whose concentration of fluorine remains in the linear range of our calibration curve, due to its simplicity and rapid analysis time, as well as its reduced sample consumption. Moreover, this methodology could be potentially utilized as a screening technique to determine total fluorine content in samples even if they contain fluorine complexes that require thorough sample preparation in order to dissociate the halogen for fluorine detection with other analytical techniques, since ns laser ablation does not preserve such bonds.
Author contributions
C. Méndez-López: conceptualization, methodology, investigation, formal analysis, visualization, writing – original draft, writing – review & editing. L. J. Fernández-Menéndez: conceptualization, methodology, writing – review & editing. C. González-Gago: supervision, writing – review & editing. J. Pisonero: conceptualization, supervision, writing – review & editing, funding acquisition. N. Bordel: conceptualization, supervision, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors gratefully acknowledge financial support provided by the Spanish Government through project MCI-21-PID-2020-113951GB-I00. Authors C. Méndez-López and L.J. Fernández-Menéndez also recognize financial support through Severo Ochoa (Principality of Asturias, project PA-21 PF-PB20-059) and FPI (Spanish Government, MINECO BES-2017-080768) predoctoral grants, respectively. The assistance provided by the Scientific-Technical Services (SCTs) of the University of Oviedo, particularly by the “Environmental Testing” (sample validations) and the “Thermal Testing and Elemental Analysis” (CaCO3 pellets preparation) units, is also thankfully acknowledged.References
- K. G. Myhre, H. B. Andrews, D. Sulejmanovic, C. I. Contescu, J. R. Keiser and N. C. Gallego, Approach to using 3D laser-induced breakdown spectroscopy (LIBS) data to explore the interaction of FLiNaK and FLiBe molten salts with nuclear-grade graphite, J. Anal. At. Spectrom., 2022, 37, 1629 RSC.
- M. Tran, Q. Sun, B. W. Smith and J. D. Winefordner, Determination of F, Cl, and Br in solid organic compounds by laser-induced plasma spectroscopy, Appl. Spectrosc., 2001, 55, 739–744 CrossRef CAS.
- D. A. Cremers and L. J. Radziemski, Detection of chlorine and fluorine in air by laser induced breakdown spectrometry, Anal. Chem., 1983, 55, 1252–1256 CrossRef CAS.
- M. Gaft, L. Nagli, N. Eliezer, Y. Groisman and O. Forni, Elemental analysis of halogens using molecular emission by laser-induced breakdown spectroscopy in air, Spectrochim. Acta, Part B, 2014, 98, 39–47 CrossRef CAS.
- C. Álvarez-Llamas, J. Pisonero and N. Bordel, Quantification of fluorine traces in solid samples using CaF molecular emission bands in atmospheric air Laser-Induced Breakdown Spectroscopy, Spectrochim. Acta, Part B, 2016, 123, 157–162 CrossRef.
- C. Alvarez, J. Pisonero and N. Bordel, Quantification of fluorine mass-content in powdered ores using a laser-induced breakdown spectroscopy method based on the detection of minor elements and CaF molecular bands, Spectrochim. Acta, Part B, 2014, 100, 123–128 CrossRef CAS.
- C. Álvarez-Llamas, J. Pisonero and N. Bordel, A novel approach for quantitative LIBS fluorine analysis using CaF emission in calcium-free samples, J. Anal. At. Spectrom., 2017, 32, 162–166 RSC.
- Y. Zhang, T. Zhang and H. Li, Application of laser-induced breakdown spectroscopy (LIBS) in environmental monitoring, Spectrochim. Acta, Part B, 2021, 181, 106218 CrossRef CAS.
- X. Yu, Y. Li, X. Gu, J. Bao, H. Yang and L. Sun, Laser-induced breakdown spectroscopy application in environmental monitoring of water quality: a review, Environ. Monit. Assess., 2014, 186, 8969–8980 CrossRef.
- J. Keerthi, S. D. George, S. D. Kulkarni, S. Chidangil and V. K. Unnikrishnan, Elemental analysis of liquid samples by laser induced breakdown spectroscopy (LIBS): Challenge and potential experimental strategies, Opt. Laser Technol., 2022, 147, 107622 CrossRef.
- World Health Organization, Guidelines for drinking-water quality: fourth edition incorporating the first, 2017 Search PubMed.
- S. Ali, S. K. Thakur, A. Sarkar and S. Shekhar, Worldwide contamination of water by fluoride, Environ. Chem. Lett., 2016, 14, 291–315 CrossRef CAS.
- X. Yu, N. Jiang, X. Miao, F. Li, J. Wang and R. Zong, et al., Comparative studies on foam stability, oil-film interaction and fire extinguishing performance for fluorine-free and fluorinated foams, Process Saf. Environ. Prot., 2020, 133, 201–215 CrossRef CAS.
- D. A. Rusak, T. P. Anthony and Z. T. Bell, Note: A novel technique for analysis of aqueous solutions by laser-induced breakdown spectroscopy, Rev. Sci. Instrum., 2015, 86, 116106 CrossRef CAS PubMed.
- Z. Tang, Z. Hao, R. Zhou, Q. Li, K. Liu and W. Zhang, et al., Sensitive Analysis of Fluorine and Chlorine elements in Water Solution using Laser-induced Breakdown Spectroscopy assisted with Molecular Synthesis, Talanta, 2021, 224, 121784 CrossRef CAS PubMed.
- P. A. Mello, J. S. Barin, F. A. Duarte, C. A. Bizzi, L. O. Diehl and E. I. Muller, et al., Analytical methods for the determination of halogens in bioanalytical sciences: A review, Anal. Bioanal. Chem., 2013, 405, 7615–7642 CrossRef CAS.
- C. Méndez-López, R. Álvarez-García, C. Alvarez-Llamas, L. J. Fernández-Menéndez, C. González-Gago and J. Pisonero, et al., Laser induced plasmas at different nebulization conditions: Spatio-temporal distribution of emission signals and excitation temperatures, Spectrochim. Acta, Part B, 2020, 170, 105906 CrossRef.
- R. W. B. Pearse and A. G. Gaydon, The Identification of Molecular Spectra, Chapman & Hall, 1976 Search PubMed.
- Y. Fu, Z. Hou, T. Li, Z. Li and Z. Wang, Investigation of intrinsic origins of the signal uncertainty for laser-induced breakdown spectroscopy, Spectrochim. Acta, Part B, 2019, 155, 67–78 CrossRef CAS.
- L. M. Cabalín, A. González, V. Lazic and J. J. Laserna, Laser induced breakdown spectroscopy of metals covered by water droplets, Spectrochim. Acta, Part B, 2012, 74–75, 95–102 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ja00319h |
| This journal is © The Royal Society of Chemistry 2023 |