DOI:
10.1039/D3EY00017F
(Review Article)
EES Catal., 2023,
1, 413-433
Electrocatalysts for value-added electrolysis coupled with hydrogen evolution
Received
26th January 2023
, Accepted 27th April 2023
First published on 29th April 2023
Abstract
A green and sustainable energy route to produce hydrogen and other oxidation products with maximized values deserves more attention than common water electrolysis. The search for alternative pathways is motivated not only by the need to reduce electricity consumption but also by the desire to produce valuable chemicals sustainably. Value-added anodic reactions (VAARs) can replace the kinetically sluggish oxygen evolution reaction (OER) in electrochemical energy conversion systems to pair with cathodic hydrogen evolution reaction (HER) and fulfil the growing demand for green hydrogen and chemicals. In this review, electrocatalysts for value-added conversion reactions such as iodide oxidation reaction (IOR), poly-alcohol oxidation reaction (AOR), chitin oxidation reaction (COR), urea oxidation reaction (UOR), and hydrazine oxidation reaction (HzOR) are systematically summarized. Electrocatalysts, such as single atom catalysts (SACs), transition metal oxides/hydroxides, heteroatom doped carbon support, and self-supported mixed alloy oxide, that promote various value-added anodic reactions are summarized. Finally, an outlook on the future challenge in new value-added electrolysis is proposed.
Broader context
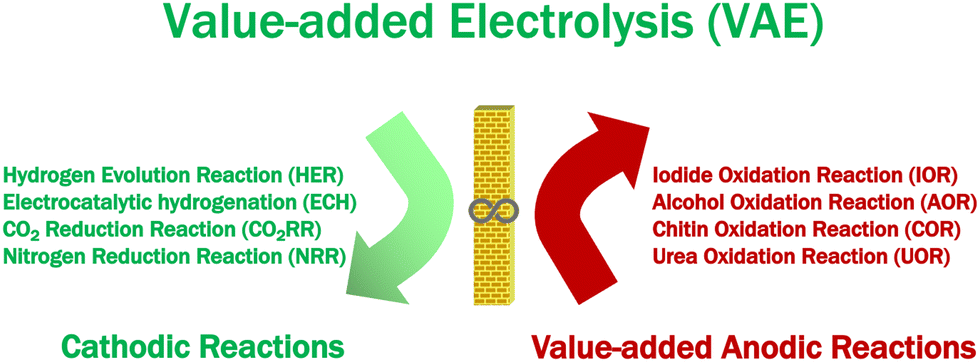
The increasing energy consumption and associated environmental pollution are a challenge for human society, triggering a global demand for clean energy in the future. Thus, hydrogen (H2) is expected to become an integral part of all sectors of the global economy because of its high density and lack of greenhouse gas emissions, and has attracted more and more attention. Hydrogen generation is paired with oxygen evolution reaction (OER) in a traditional water electrolyzer. However, its energy efficiency is low due to the sluggish kinetics of OER. To overcome this problem, a combination of value-added anodic reactions (VAARs) and hydrogen evolution reactions (HER) is regarded as one of the most appealing methods of lowering the production costs of green and sustainable hydrogen energy generation and value-added chemicals. There are various cathodic reactions, including HER, CO2 reduction reaction (CO2RR), nitrogen reduction reaction (NRR), and electrochemical hydrogenation (ECH), which can be paired with value-added anodic reactions (VAARs) to develop novel value-added electrolysis processes (VAEs). Moreover, the most important features in designing electrocatalysts are based on improving their electrocatalytic activity, selectivity, and stability, which could reduce the cost of clean hydrogen energy generation and value-added chemical production, and mitigate the need for further separation and purification of the target products. As a result, this review intends to address the most critical issue in harnessing the benefits of selective value-added anodic reactions and the common advantages of VAARs coupled with HER. Continued research efforts are required to reach the global goal of net-zero emissions by 2050 and to allow addressing the mitigation actions pushed ahead by the Paris Agreement.
|
1. Green and sustainable route to produce value-added chemicals
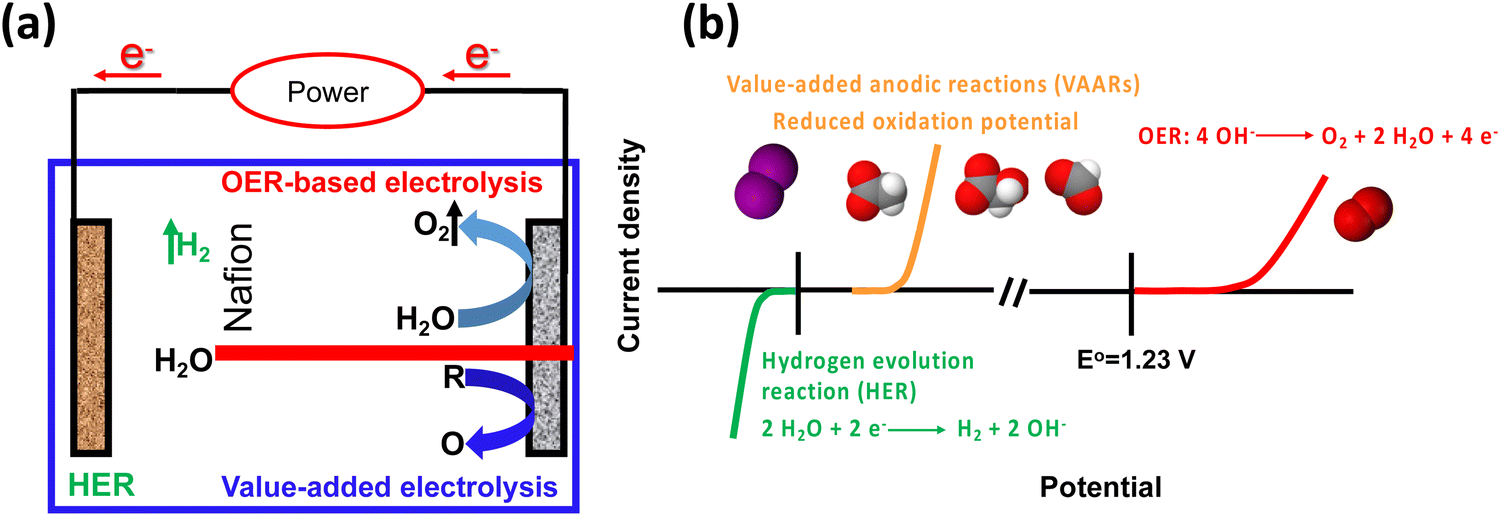
Rapid industrial and population growth, as well as the recent pandemic (COVID-19), has resulted in price spikes for a variety of commodities, as well as an increase in energy consumption and associated pollution, posing a challenge to human society, sparking demand for green and sustainable energy.1–3 A new strategy is urgently required to reduce the cost of commodities, energy consumption, and pollution. Hydrogen (H2) is expected to become an integral part of all sectors of the global economy because of its high density and lack of greenhouse gas emissions. It is predicted to play a significant role in the future in the green and sustainable energy sector.4–6 Moreover, hydrogen production by water electrolysis has been widely regarded as an advanced approach for addressing the energy crisis and environmental pollution issues. Hydrogen generation is paired with oxygen evolution reaction (OER) in a traditional water electrolyzer. However, its energy efficiency is low due to the sluggish kinetics of OER. Recently, the selective electrooxidation to substitute OER has become a promising strategy, because the voltage input of the electrolyzer can be reduced and the products generated at the anode possess greater value compared with the cheap oxygen (O2) product (Fig. 1a).7–10 Alternative value-added anodic reactions (VAARs) with lower oxidation potential can be achieved at a lower full-cell voltage to produce green and sustainable hydrogen with a lower energy supply by avoiding O2 generation.11 Thus, value-added conversion reactions have been developed that use easily oxidizable species such as alcohol oxidation,12 iodide oxidation,13 chitin oxidation,14 oxidative polymerization and plastic waste upgrading,15 urea oxidation16,17 and hydrazine oxidation.18 As a result, the value-added anodic reaction coupled with the hydrogen evolution reaction is an environmentally friendly and sustainable approach for producing value-added chemicals while improving energy conversion efficiency (Fig. 1b). This review summarizes the recent advances in the electrocatalytic performance of value-added anodic reactions towards various products. The driving forces, or motivation, for developing green, sustainable, safe, and durable electrolysis systems with low cell voltages, high value-added chemicals, and green hydrogen production efficiencies are discussed. An overview of the techno-economic analysis and environmental impact feasibility for value-added anodic reactions with specific cell designs are also included. Finally, an outlook on future challenges in developing electrocatalysts for new value-added electrolysis is proposed.
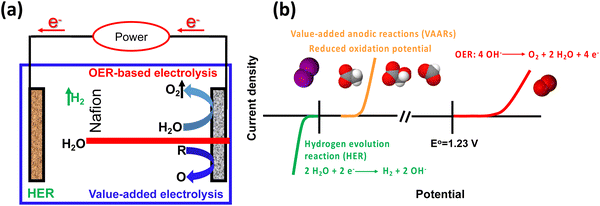 |
| | Fig. 1 (a) Scheme of applying a value-added anodic reaction (VARR) to replace OER-based electrolysis. (b) Comparison of the conventional water splitting and value-added anodic reaction performance (HER ||OER vs. HER||VAARs). | |
2. Challenges for value-added anodic reactions coupled with hydrogen evolution reaction (VAARs-HER)
The difficulty with VAARs-HER is that the overpotential of the anodic reaction increases significantly with increasing overall current density, wherein the competing anodic OER might occur and thus decrease the selectivity for the value-added product. Another challenge in VAARs is the selection of suitable raw materials and the search for efficient, selective, long-lived, and earth-abundant electrocatalysts. In some cases, separating and purifying the organic and inorganic products generated during electrolyte generation can be complicated and expensive. Most important is selecting suitable organic and inorganic substrates well soluble in water and having a lower theoretical potential than OER to avoid competition and reduce electrolytic efficiency.
The VAARs usually involve oxidation processes of organic and inorganic species with multiple electrons and intermediate transfer reactions, which inherently have sluggish kinetics and complex development paths. One criterium for successful VAARs is to react with lower theoretical potential than OER. Therefore, rational design and development of new catalysts with abundant and well-defined active sites for VAARs are critical for efficient production of green hydrogen energy and value-added products. Traditionally, noble metal-based catalysts (Pt, Pd, Ru, and others) have been used to oxidize organic and inorganic substrates.19–21 However, their scarcity and excessive cost limit their widespread use. In recent years, various design strategies for base metal electrocatalysts have been developed to improve their electrocatalytic activities and selectivity for value-added anodic reactions, such as hybrid material engineering, alloying, defect introduction (defect engineering), heteroatom doping (doping engineering), strain engineering, morphology regulation, etc. Typical electrocatalysts include transition metal nitrides, sulfides, phosphides, oxides/oxyhydroxides, and alloys. In addition, extensive research should be conducted to replace OER with organic oxygenation reactions in the future. Thus, oxygen from water can be transferred to organic substrates to produce value-added oxygenated chemicals at the anode and fully utilize the hydrogen and oxygen elements of water.
3. Techno-economic and environmental perspective
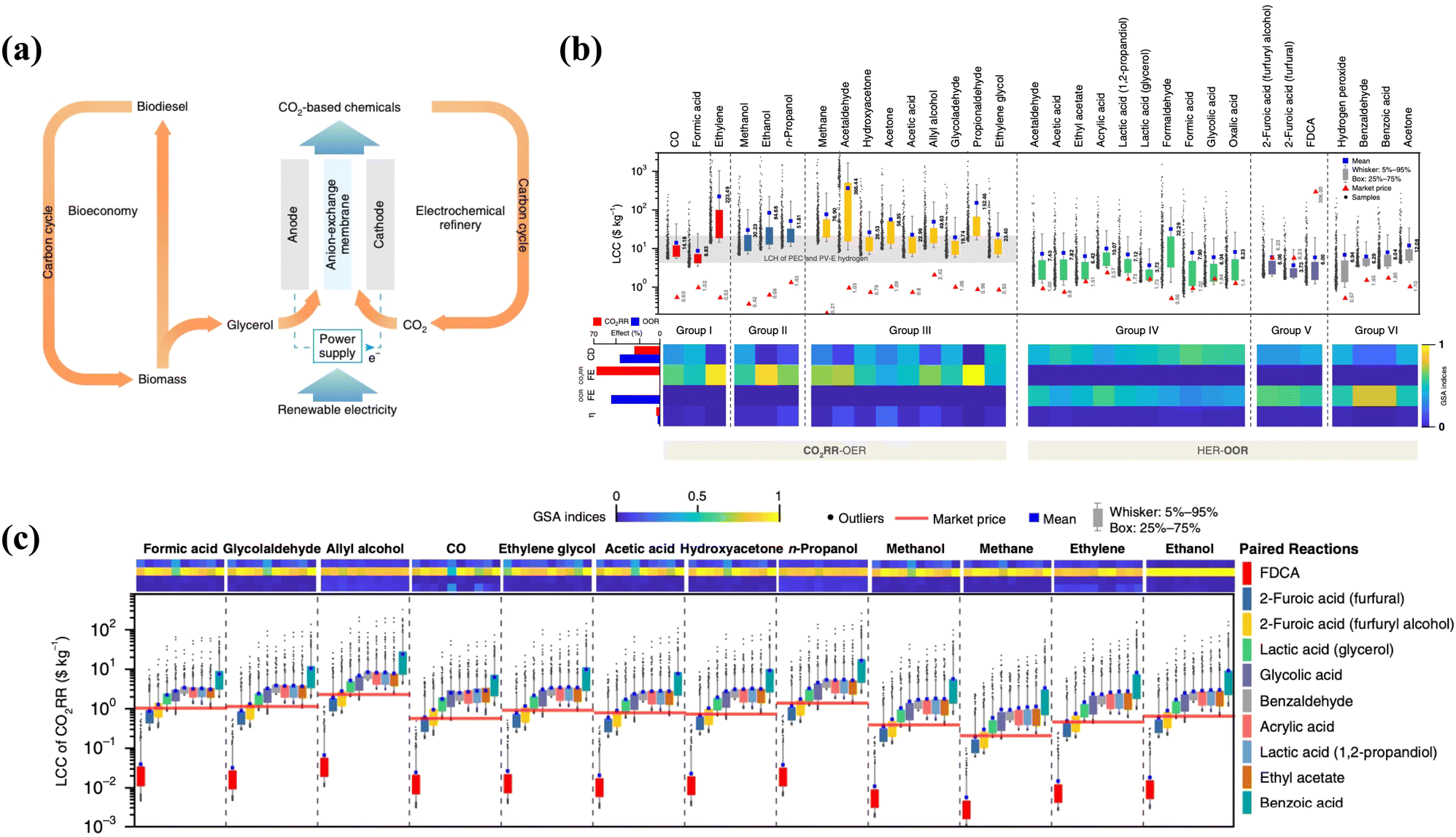
The value-added anodic reaction of organic and inorganic substrates (precursors) could yield valuable chemicals of significant economic importance, generating additional revenue and providing a more cost-competitive alternative. Thus, we reviewed some basic techno-economic assessments of effective coupling of VAARs-HER/CO2RR to provide some indicative information on the economic value and environmental impact of this process on a larger scale. Value-added anodic reactions coupling the HER are promising approaches for simultaneously producing chemicals and generating clean H2 with safety, high selectivity, and environmental friendliness.22 Furthermore, such electrochemical synthesis of organic and inorganic molecules (such as glycolate, formate, acetate, iodine, etc.) is less environmentally harmful than conventional non-electrochemical industrial processes. The electrochemical technique is typically performed at room temperature and pressure, minimizing the use of hazardous chemical oxidants. Nowadays, the techno-economic analysis emphasizes that combining the HER with value-added organic and inorganic oxidation has significant economic feasibility. Hwang et al.6 and Verma et al.23 demonstrated that anodic electro-oxidation of iodide and glycerol substituting O2 evolution reaction (OER) reduces electricity consumption by around 50%. Notably, the search for alternative pathways was motivated by the need to decrease electricity consumption and the desire to use an abundant or sustainable feedstock (biomass and its derivatives, chitin, etc.). For example, glycerol is particularly promising in this regard because it is a cheap byproduct of industrial biodiesel and soap production.24–26 Thus, the proposed co-electrolysis concept makes good use of this byproduct (anodic oxidation of glycerol) and, at some point, could enable both bio-based and valuable molecule-based economies (Fig. 2a). In 2019, Lee et al.27 explored the technical feasibility and economic viability by creating a fully automated synthesis framework to guide process simulations. They concluded that coupling the CO2 reduction process, which results in the production of formic acid, n-propanol, acetaldehyde, allyl alcohol, glycolaldehyde, and ethylene glycol, with alternative value-added anodic reactions such as biomass oxidation, which results in the formation of ethyl acetate, acetic acid, formic acid, glycolic acid, and oxalic acid, constitutes an excellent means for improving FE, current density, and overpotential. If both cathode and anode reactions are considered, the economic feasibility of CO2RR/HER coupled with VAAR technology could be assessed more profitably and reasonably. Profitability was calculated using the relative ratio of levelized cost of chemicals (LCC) to market price, where LCC represents the lowest selling price without a margin. Techno-economic analysis23,28 has suggested that substituting OER with VAARs can reduce full-cell voltage and produce salable products from both cathode and anode of the cell. In general, the levelized costs of chemicals (LCC) of HER/CO2RR coupled with OER are higher than the market prices due to low oxygen market price and higher cell potential. Meanwhile, there is a possibility that the HER process will be coupled, resulting in the generation of green hydrogen energy and value-added chemicals such as formic acid, acetate, glycolate, and iodine using the same parameters (FE, current density, and overpotential) and having lower levelized chemical costs (LCC) than the market price. However, a few VAARs-HER systems, because of the price of products being much lower than that of raw materials and due to the volatility of the products such as benzaldehyde, benzoic acid, and acetone, have proven to be economically not feasible. Therefore, the effective coupling of value-added electrolysis could not only improve the efficiency of H2 generation, but also lower value-added product levelized chemical costs (LCC) (Fig. 2b and c). As shown in Table 1, the market price for some value-added products of the anodic reaction has not been able to establish itself, as indicated by the wide range of reported market prices. Moreover, the price of both the product and the organic and inorganic substrates (raw materials) may change significantly if reliable technology is introduced in the future.
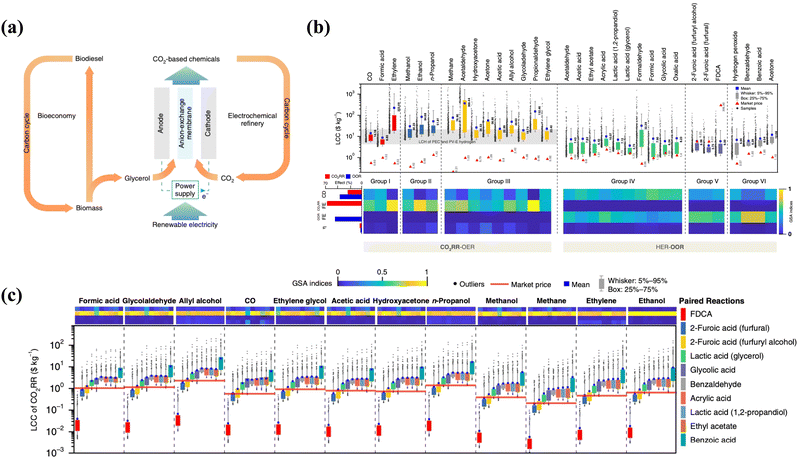 |
| | Fig. 2 (a) The use of glycerol from biodiesel production in electrochemical CO2 conversion to value-added chemicals.42 (b) Levelized costs of chemicals (LCC) of HER-OOR (coupled HER with organic oxidation reaction or HER-VAARs) and CO2RR-OER are compared with their market price determined via global sensitivity analysis through current density, FE, and overpotential. The light gray band represents the levelized cost of hydrogen with various types of PEC and PV-E technology. (c) Levelized cost of chemicals of CO2RR paired with 10 different OOR (organic oxidation reactions to produce value-added products) processes. Reproduced27 Copyright (2019), with permission from Springer Nature. | |
Table 1 Possible value-added anodic reactions that can be covered with HER/CO2RR, based primarily on the works of Na et al.28 and A. Vass et al.31
| Reactant |
Reactant price ($ per Kg) |
Product |
Product price ($ per Kg) |
E° (VSHE) |
Production (Mt per year) |
Ref. |
| Deionized water |
0.02–0.1 |
Oxygen |
0.024–0.04 |
1.23 |
2 |
32 and 33
|
| Hydrogen peroxide |
0.56–0.58 |
1.78 |
2.8 |
| Ethylene glycol |
0.83–1 |
Glycolic acid |
1.84 |
−0.334 |
0.04 |
34
|
| Oxalic acid |
1.4 |
−0.455 |
0.19 |
| Methanol |
0.34–0.49 |
Formic acid |
0.97–1.08 |
−0.258 |
0.95 |
35
|
| 0.3 |
Formaldehyde |
0.37–0.74 |
0.465 |
18 |
33
|
| Glycerol |
0.16–0.80 |
Glyceraldehyde |
2.11 |
0.35 |
|
33, 36 and 37 |
| Dihydroxyacetone |
2.0 |
0.33 |
0.004 |
38 and 39
|
| Benzyl alcohol |
1.92–3.47 |
Benzaldehyde |
1.18–2.11 |
0.193 |
0.09 |
40
|
| Ethanol |
0.4 |
Acetaldehyde |
1.0 |
0.193 |
1.7 |
33
|
| Acetic acid |
0.68–0.92 |
−0.334 |
10 |
41
|
| 1,3-Propanediol |
2.20 |
Acrylic acid |
2.25–2.88 |
0.248 |
5.2 |
41
|
Environmental security, energy resource conservation, and sustainable energy production are the most pressing issues confronting the world today. Power consumption is constantly increasing as a result of population growth and industrialization.29,30 Today, most hydrogen is “gray”, which could be from hydrocarbons, most commonly natural gas (steam reforming), which emits CO2. On the other hand, green hydrogen is created using renewable energy, typically through water electrolysis, and emits no pollutants. The issues are addressed by developing alternative methods for obtaining clean energy and value-added chemicals through the electrochemical synthesis of easily oxidized organic/inorganic compounds and generating H2 in a more environmentally friendly manner. Thus, the environmental impact of value-added anodic reactions paired with HER is lowering the capital costs and avoiding the use of fossil fuels as raw materials, thereby avoiding net greenhouse gas emissions into the atmosphere. In addition to designing anodic catalysts, coupling alternative value-added anodic reactions with HER is critical for improving full cell energy efficiency and economic feasibility, and advancing value-added electrolysis technology to a more practical level, where new systems, new electrocatalysts and cell-designs remain to be explored. Importantly, the goal of this section is to screen the potential viability of VAARs-HER/CO2RR processes for future deployment based on indicative values rather than to present comprehensive techno-economic modeling of electro-oxidation processes. This allows researchers to identify and develop the most relevant set of processes to achieve carbon-free emission in future.
4. Coupling energy-saving anodic oxidation for promoting hydrogen production
Developing more profitable chemicals to replace oxygen at the anode reduces the practical cost of producing green hydrogen energy. Selecting suitable organic and inorganic oxidative substrates (raw materials) is critical in developing design strategies for value-added chemical and energy-efficient H2 production. First, the organic and inorganic oxidative substrates should be soluble in water at room temperature and have lower theoretical potentials than OER, improving the overall electrolytic efficiency and avoiding interference from OER. Furthermore, these anodic oxidation reactions should be able to convert organic and inorganic oxidative substrates into value-added products or alleviate the problem of wastewater pollution (examples: urea, hydrazine, plastic waste, and chitin) while maintaining high selectivity and conversion efficiency, and achieving simultaneous cathode H2 production. In the meantime, these organic and inorganic oxidative substrates have abundant natural reserves (biomass and their derivatives). Neither these selective oxidative substrates nor their products should interfere with HER at the cathode.43,44 Enormous efforts have been made to reduce the energy barrier of the anodic oxidation reaction for energy-saving hydrogen production by developing low-cost, earth-friendly high-performance electrocatalysts for value-added anodic reaction coupled with HER.45 In the following sections, we will briefly review and discuss recently reported green and sustainable routes for value-added electrolysis coupled with HER, which could generate products at the anode that have a higher value than cheap O2 products.
4.1 Iodide oxidation reaction (IOR)
Environmental pollution and energy crisis have become increasingly important issues in recent years. Molecular hydrogen is a diatomic molecule and the two atoms are joined by a strong covalent bond (bond energy 436 kJ mol−1). Thus, molecular hydrogen is a safe (not explosive) and zero-carbon-emission energy carrier for the global future. Electrochemical water splitting is a simple and efficient technology that can occur with OER at the anode and HER at the cathode, but with high overpotentials due to the sluggish anodic OER, which results in low energy conversion efficiency. People have recently begun to look for innovative and long-term hybrid water electrolysis strategies. In this regard, the iodide oxidation with a relatively low equilibrium potential of 0.54 V is promising. Its anodic oxidation products, iodine and NaOH, have a wide range of industrial catalysis applications that are very similar to transition metal-catalyzed reactions.46,47 The oxidation product of iodide in an alkaline solution is always oxy-iodide, which is widely regarded as an effective additive to salt for iodine deficiency compensation and is used as a catalyst for electrochemical synthesis,46 as a pharmaceutical,48 as a food supplement,49 and in photography, medicine, and many others.50,51
The inorganic substrate (NaI) is the raw material for the anodic cell, which is separated by a Nafion membrane from the cathodic cell, where water splitting takes place. In this way, a two-electrode configuration is set up and coupled with the electrocatalyst as the anode and Pt/C as the cathode to oxidize iodide and supply electrons to the cathode side for water splitting. The iodide oxidation reaction coupled with HER produces iodine and delivers electrons to the cathode side to produce hydrogen. Finally, in the case of IOR-assisted HER electrolysis, the first step can be the iodine evolution reaction to deliver electrons at the cathode, and then hydrogen evolution reactions can take place. In the case of iodide oxidation reactions, the process consists of two half-reactions (from eqn (1)–(3)):
| | | Anodic reaction: 2I−(aq) → I2 + 2e− E° = 0.54 V | (1) |
| | | Cathodic reaction: 2H+(aq) + 2e− → H2(g) E0 = 0.00 V | (2) |
| | | Overall reaction: 2H+(aq) + 2I−(aq) → I2 + H2(g) | (3) |
Importantly, it is challenging to fabricate active and durable electrocatalysts for IOR-based electrolysis from the same low-cost precursor materials. After extensive research, our group discovered that electrocatalysts could be easily synthesized from metal oxides, single atoms, and metal-free carbon materials, such as ruthenium-tin surface alloy oxide (RuSn SAO),
13 self-supporting RuTiO oxide alloy,
52 defective carbon-supported single-atom Mo-N
4/d-C electrocatalyst,
53 and graphite-based carbon fiber paper (CFP).
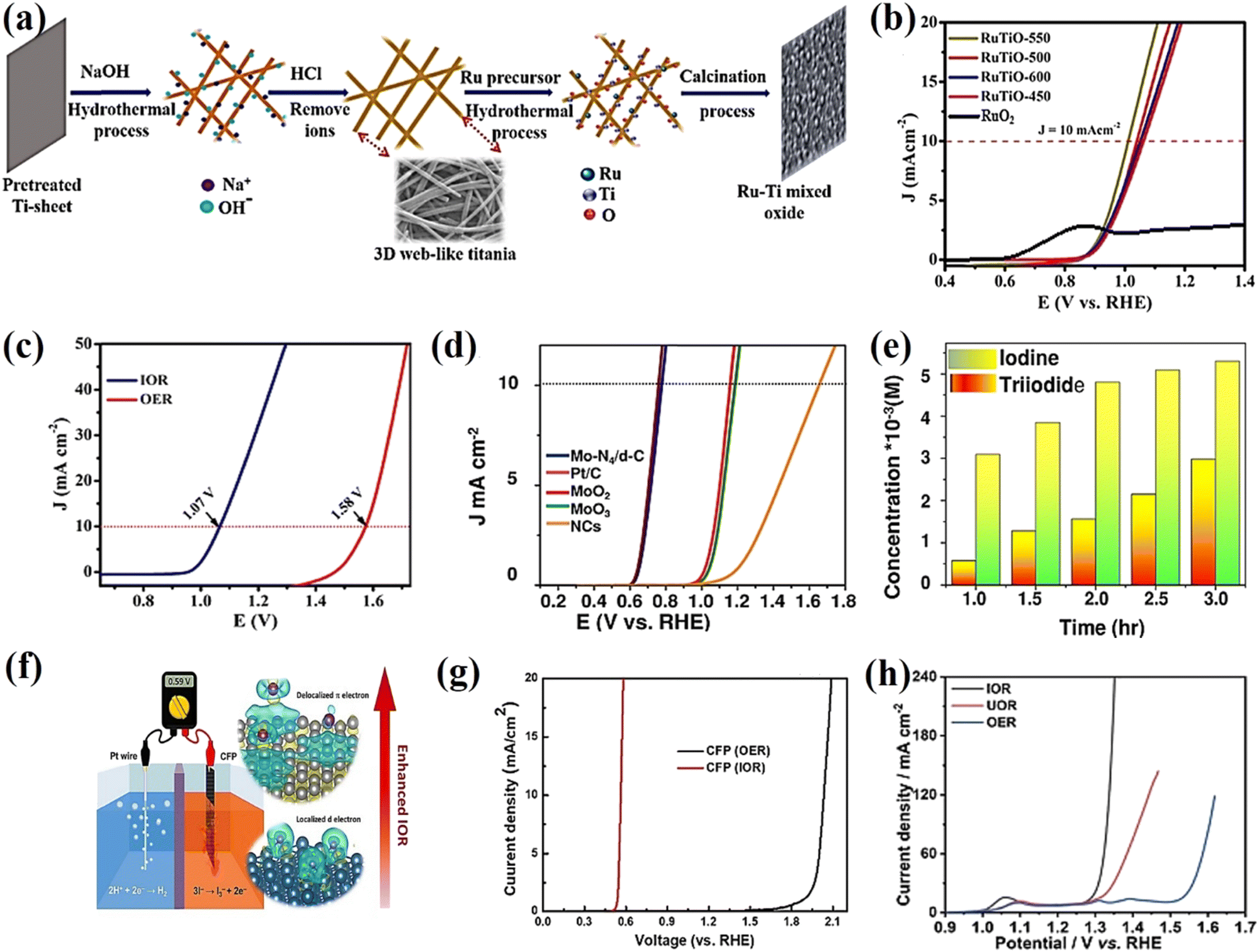
54 These electrocatalysts have abundant active sites and a delocalized electron system which play an important role in the adsorption, activation, and desorption of iodide ions at the catalytic sites, which are favorable for the production of valuable iodine and green hydrogen energy at a lower operating voltage. Especially another major advancement in this field of research has been reported – a systematic approach to developing a RuTiO oxide alloy on 3D web-like titania
via a hydrothermal route followed by a calcination process (
Fig. 3a). The self-supported RuTiO electrocatalyst requires an ultralow cell voltage of 1.01 V to achieve 10 mA cm
−2 and excellent durability in a 36 h test without degradation, indicating that the electrocatalyst could be used as a robust anode in IOR-based electrolysis for value-added H
2 production (
Fig. 3b). The excellent activity of RuSn SAO enables it to be the best catalyst for IOR toward energy-saving hydrogen production. Its two-electrode acidic electrolyzer requires a cell voltage of only 1.07 V to afford 10 mA cm
−2, which is 0.51 V less than that required for OER to reach the same current density (
Fig. 3c). Moreover, in 2022, the value-added anodic reaction of iodide and the hydrogen evolution reaction were coupled into an electrochemical cell using a defective carbon-supported single atom Mo-N
4 electrocatalyst to provide a record current density of 10 mA cm
−2 at 0.77 V and a Tafel slope of 25.58 mV dec
−1 with exceptional stability over time in acidic media and a higher hydrogen generation rate of 0.1063 mL g
cat−1 min
−1 (
Fig. 3d and e). Recently, a metal free-based carbon fiber paper (CFP) graphite structure with a delocalized π electron system exhibited fast charge transfer with physically adsorbed iodide ions, resulting in remarkable IOR performance and an onset potential of 0.54 V
RHE for the IOR, close to the theoretical iodide oxidation potential (
Fig. 3f and g).
54 As shown in
Fig. 3h, the polarization curves of the IOR, UOR, and OER, which have the potential to drive a current density of 20 mA cm
−2, are 1.30, 1.33, and 1.54 V
RHE, respectively. Interestingly, the single-atom (such as defective carbon-supported single-atom Mo-N
4 electrocatalysts) and metal-free carbon fiber paper (CFP)-graphite structures showed not only excellent IOR performance (requiring a potential of 0.77 and 0.54 V
RHE to achieve a current density of 10 mA cm
−2, respectively) but also 50–65% lower energy consumption compared to conventional OER-based water electrolysis. In addition, we believe that electrocatalysts/electrodes grown directly on conductive substrates (3D web-like titania and nickel foam) could ensure close contact between them and promote the penetration of the electrolyte and transfer of iodide, thereby leading to improved IOR activity.
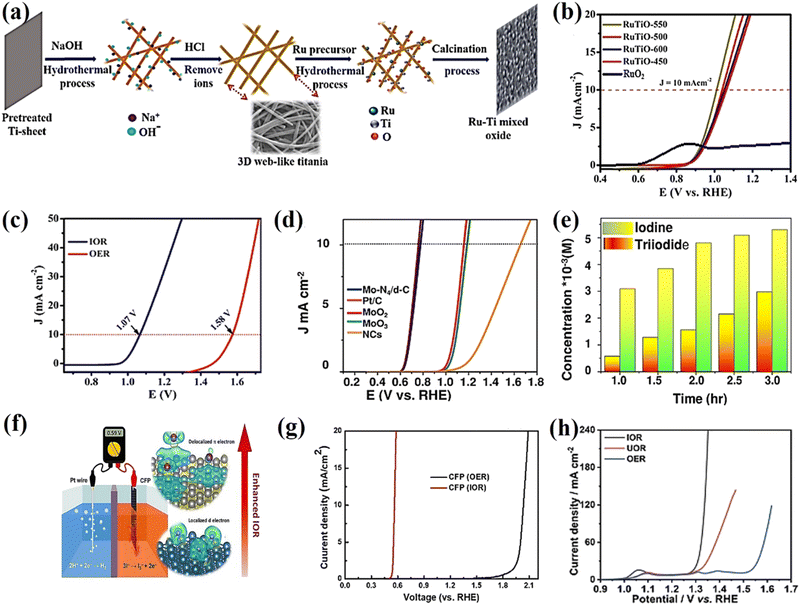 |
| | Fig. 3 (a) Schematic illustration of the synthesis self-supported ruthenium–titanium mixed alloy oxide on 3D web-like titania. (b) Comparison of the LSV of RuTO-550, 500, 600, 450 (550–450, stand for annealing temperature) and commercial RuO2.52 (c) LSV of RuSn SAO for IOR and OER.13 (d) Comparison of the LSV of the Mo-N4/d-C single-atom catalyst in 0.1 M HClO4 with Pt/C, MoO2·MoO3, and NCs. (e) Iodine and triiodide production of the Mo-N4/d-C single-atom catalyst.53 (f) LSV curves of CFP as an electrocatalyst for IOR and OER in a three-electrode system. (g) Schematic illustration of the IOR on a graphite structure with delocalized π electrons.54 (h) Polarization curves of the IOR, UOR, and OER over Ni-Co(OH)2 NSAs.55 | |
4.2 Alcohol oxidation reactions
In modern chemistry, renewable feedstocks have received a lot of attention as alternatives to petroleum-derived materials.56,57 Considering the limited reserves of fossil fuels, developing efficient technologies for biomass conversion to value-added chemicals and clean hydrogen energy is critical.58,59 Alcohol oxidation reaction (AOR) of diols and poly-alcohols is an excellent means for producing high-value-added chemicals at the anode with low electricity consumption on hydrogen generation at the cathode.23,60 Glycerol is low in price and more easily electrochemically oxidized than water.61 Glycerol oxidation reaction (GOR) is an important anodic reaction among alcohol oxidation reactions, since glycerol is an abundant byproduct from industrial biodiesel and soap production. One crucial point is the selective oxidation reaction, which can predict the reaction path to produce high-cost multiple oxygenated carbon used in the chemical and pharmaceutical industries. Thus, the US Department of Energy (DOE) has listed glycerol, a byproduct of the biodiesel industry, as one of the top ten biomass-derived platform molecules for producing high-value chemicals.62 In particular, glyceraldehyde, glyceric acid, and dihydroxyacetone are regarded as high-value chemicals of economic interest and are widely used in the cosmetics (as an ingredient in sunless skin tanning lotions), pharmaceutical, and food industries.63
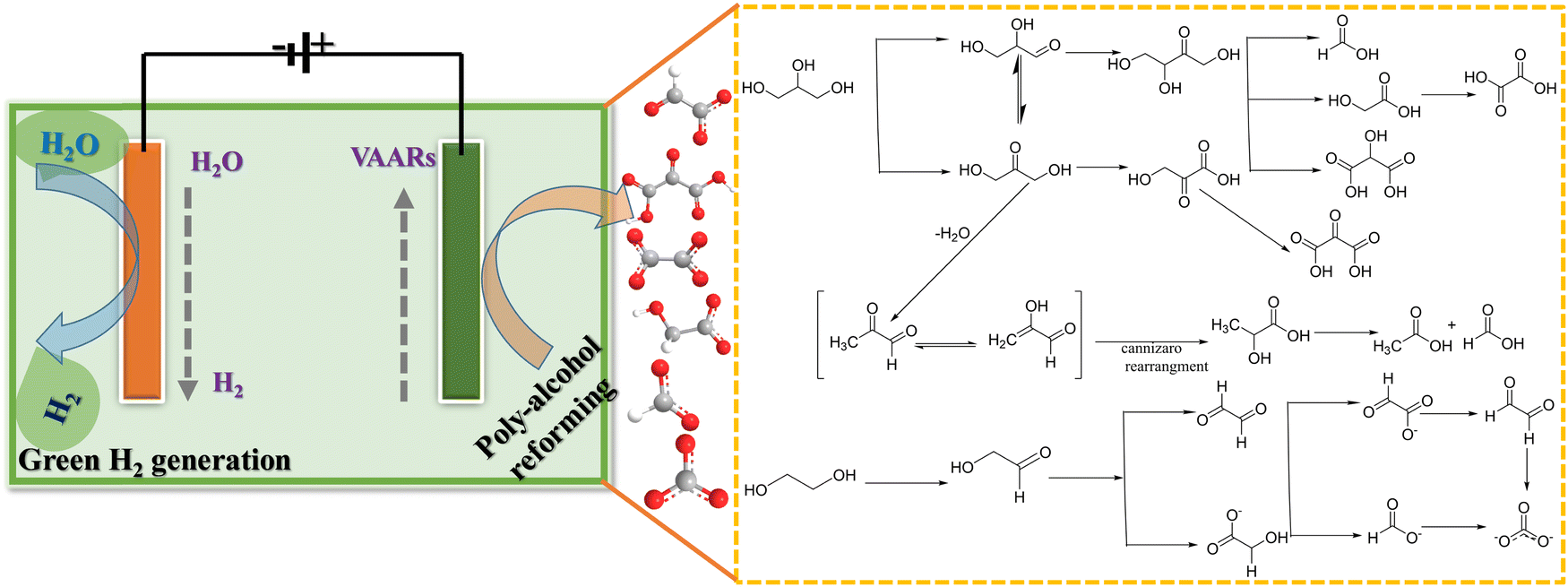
Ethylene glycol (EG) is a renewable material and a diol-alcohol produced by heterogeneous hydrogenation of cellulose (biomass-derived resources). It is a non-toxic, high boiling point, energy dense, and reactive material.64–66 Bimetallic catalysts or a mixture of catalysts (single-atom, dual-atom, and nanoclusters) seem to be suitable for the electrooxidation of EG into C2 and C1 products compared to Pd alone (Pd-based nanoparticles). For instance, Qin et al.67 demonstrated that Pd–PdSe heterostructure nanosheets have distinct electronic and geometrical structures in which unconventional P–d hybridization interactions and the tensile strain effect jointly reduce the activation energy of the C–C bond and favor C1 products. Bambagioni et al.68 and Marchionni et al.66 have explored the selective electrooxidation of EG by Pd-(Ni-Zn)/C or Pd-(Ni-Zn-P)/C and Pd nanoparticles to offer mixtures of value-added products in an alkaline medium. At the same time, the ternary catalyst PtPdBi/C demonstrated that adding foreign atoms to platinum reduced the catalyst's ability to break the C–C bond, most likely due to the dilution of the surface platinum atoms.69 Interestingly, Coutanceau et al. developed Bi-modified Pt and Pd nanocrystals for producing hydrogen and value-added chemicals from polyol solution.70 One of the most active catalysts reported, Pt9Bi/C, demonstrated an onset potential as low as 0.3 VRHE, with a hydrogen production rate of around 0.11 and 0.23 Nm3 H2 h−1 m−2 with an electrical energy consumption of 1.3 or 1.65 kW h (Nm3 H2)−1 at 0.55 V and 0.7 V, respectively. Compared to traditional water splitting systems, 56–66% electricity energy could be saved. Chen et al. stated that “the conversion of different bio-derived alcohols not only increases the versatility of the electrolysis process for H2 generation with energy saving, but also leads to generation of valuable chemicals from the large variety of products that can be obtained by the electrooxidation of such poly-alcohols” (Fig. 4).71 Moreover, Shi et al. developed selectively glycolic acid produced by electro-oxidation of EG in alkaline medium on an anode electrocatalyst of AgPd/NF (Pd acts as a catalytic site, while Ag acts as a structural site), for cathodic (Pt plate) hydrogen production with a faradaic efficiency of 100%.72
 |
| | Fig. 4 Electrooxidation of poly-alcohol (glycerol and ethylene glycol) via value-added anodic reactions (VAARs) and generation of green hydrogen energy at the cathode. | |
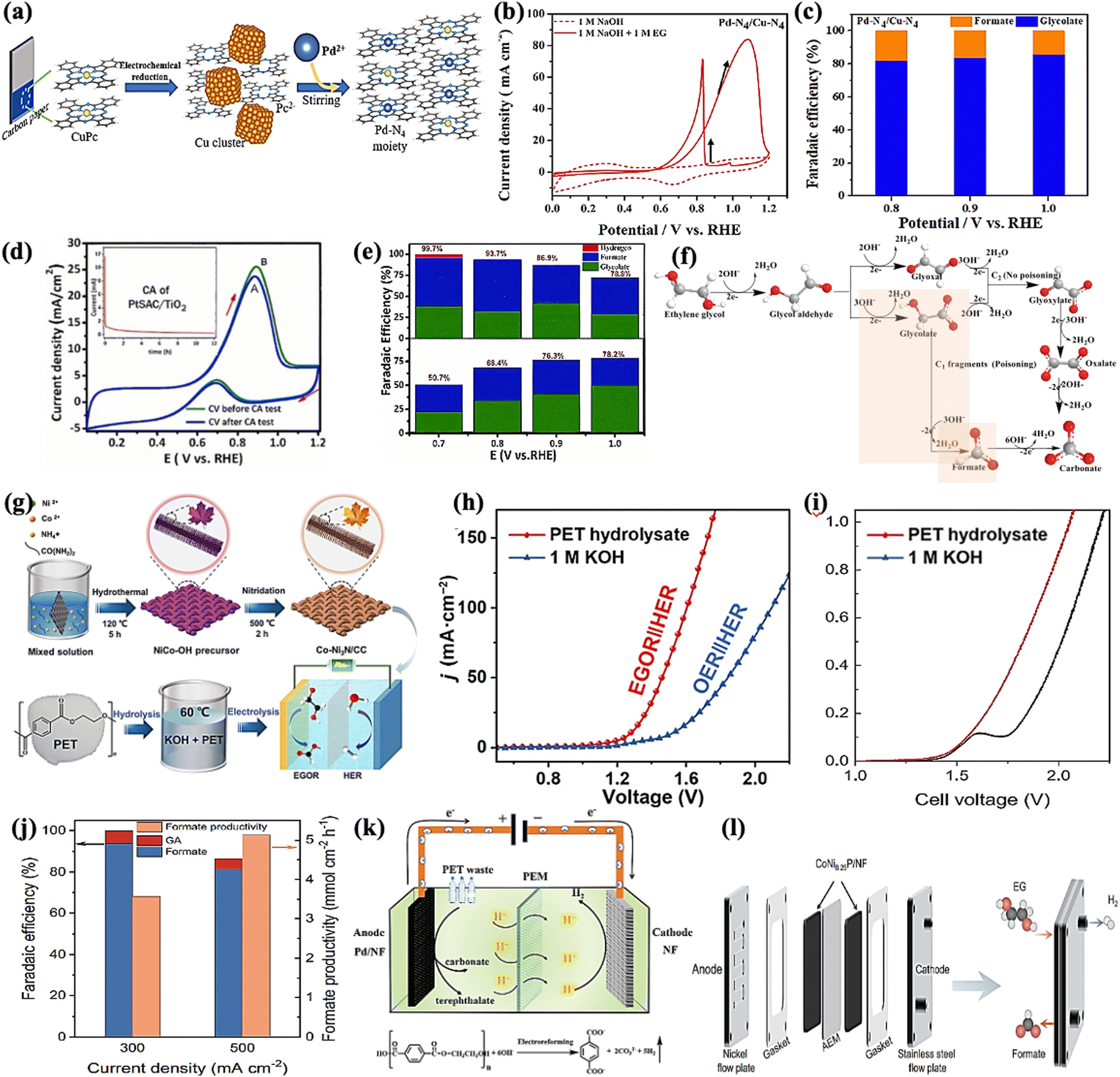
Our group recently synthesized a dual single-atom catalyst (DSAC) Pd-N4/Cu-N4 for EGO. It possessed excellent glycolate selectivity (above 88%) in an alkaline solution with an onset oxidation potential as low as 0.6 VRHE, compared to commercial Pd/C, as shown in Fig. 5a–c. The DSAC Pd-N4/Cu-N4 showed synergistically improved electrocatalytic performance, with a high current density of 83.92 mA cm−2 and a high faradaic efficiency value (>80%) for glycolate at 1.0 VRHE. Pt single atoms on a titanium oxide support (PtSAC/TiO2) were synthesized via a hydrothermal-assisted co-precipitation method, which is simple, fast, and efficient. The strong metal–support interaction (SMSI) between Pt and TiO2 and the availability of identical Pt active sites enhanced the catalytic selectivity of PtSAC/TiO2. PtSAC/TiO2 maintained a higher oxidation current density, thus providing better electrocatalytic activity and stability. More importantly, the faradaic efficiency of PtSAC/TiO2 reached a maximum of 99.7% at 0.7 VRHE and gradually decreased with increasing applied voltage, as shown in Fig. 5d and e. The EG oxidation reaction mechanism on the DSAC Pd-N4/Cu-N4 and PtSAC/TiO2 in alkaline media can be divided into two consecutive events: EG oxidation into glycolate and further oxidation of glycolate into formate, as shown in Fig. 5f.
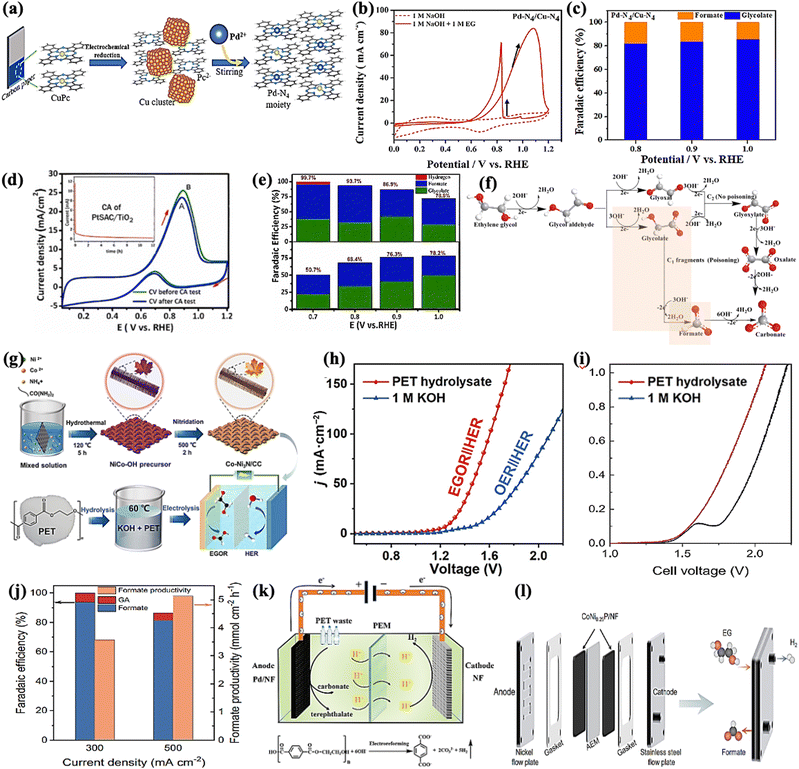 |
| | Fig. 5 (a) The schematic diagram showing the synthesis of the DSAC (dual single-atom catalyst) Pd-N4/Cu-N4 by electrochemical reduction. (b) CV curves of Pd-N4/Cu-N4 with/without EG in the electrolyte solution. (c) Faradaic efficiency of the glycolate (blue) and formate (orange) produced by the catalyst Pd-N4/Cu-N4.80 (d) CV curve before and after the stability test on PtSAC/TiO2. (e) Faradaic efficiency on PtSAC/TiO2 (upper one) and PtNP/TiO2 (lower one).81 (f) Reaction pathways of electrochemical oxidation of ethylene glycol in alkaline media. (g) Schematic illustration of the synthesis of the Co-Ni3N/CC electrode and its use for simultaneous PET upcycling and hydrogen production. (h) LSV curves to achieve different current densities for the coupled Co-Ni3N/CC electrodes in 1.0 M KOH or PET hydrolysate solution.82 (i) Polarization curves for water splitting and EG electrolysis in a MEA flow reactor at a scan rate of 10 mV s−1. (j) Faradaic efficiency and productivity as a function of current density for EG oxidation.15 (k) Schematic illustration of the electroreforming of PET into high value-added chemicals and H2 fuel.83 (l) A zero-gap membrane-electrode assembly (MEA) flow reactor developed to convert EG into formate.15 | |
Approximately 70 million tons of polyethylene terephthalate (PET) are produced annually for packing and textile,69 with only a small amount of plastic waste being recycled via the mechanical method. Chemical recycling methods such as hydrolysis,73 methanolysis,74 and glycolysis75 result in the recovery of monomers without structural deterioration. The recycled products can then be converted into value-added energy products via value-added anodic reactions (VAARs) coupled with hydrogen evolution reactions (HER). As a result, plastics have been overlooked as starting oxidative substrates, such as ethylene glycol (EG),76–78 for producing both value-added chemicals and green hydrogen (H2) in a single system. Interestingly, as the proverb says, “Kill two birds with one stone,” the electroreforming of waste plastics offers a green and sustainable route for electrocatalysis for the production of value-added chemicals and the generation of renewable hydrogen energy, as well as the concurrent elimination of plastic waste. Erwin and colleagues reported a photo-reforming strategy for converting PET waste into clean H2 fuel and oxygenates (formates, glyoxal, and acetate) under mild conditions.78,79 Compared to the conventional plastic recycling strategies (e.g., mechanical methods) and chemical reclaim methods draw more benefits from waste by catalytically converting it into high-quality monomer subunits or upcycling it into value-added products. The efficiency and selectivity of the catalysts, as well as the process's sustainability and profitability, would be critical for the success of these approaches.
CoNi0.25P/NF, CoNi3N/CC,17 and Pd-modified nickel foam electrodes can achieve electroreforming of EG into formate at the anode and hydrogen generation at the cathode. One must ensure that the operating stability of CoNi0.25P/NF is evaluated with different potential and time, which could be at 1.7 V RHE for 39 h and 1.5 VRHE for 33 h giving high faradaic efficiency (FE) and formate selectivity (80–90%). Furthermore, the FE of the Co-Ni3N/CC bifunctional electrocatalyst for the oxidation of EG to formic acid can reach over 92% with an optimized potential of 1.30 VRHE and a current density of 50 mA cm−2 in only 12 hours. However, CoNi0.25P/NF maintains a current of approximately 350 mA cm−2 at 1.7 VRHE and 180 mA cm−2 at 1.5 VRHE for 39 and 33 hours, respectively. This indicates that CoNi0.25P/NF has high stability for EG oxidation and maximum energy conversion efficiency (Fig. 5g–j). The Pd-modified Ni foam also exhibited excellent performance (400 mA cm−2 at 0.7 VRHE) and produced terephthalate and carbonate (Fig. 5k). Importantly, a zero-gap membrane-electrode assembly (MEA) flow reactor is being developed to convert EG into formate while also producing H2 using CoNi0.25P/NF as both cathodic and anodic catalyst (Fig. 5l). The most critical aspects of ethylene glycol-assisted HER are producing value-added chemicals and clean energy and reducing severe environmental and biological threats. Finally, the discussion concluded that a green and a sustainable route toward simultaneous plastic waste elimination (recycling of plastic waste), electrochemical synthesis of value-added chemicals, and production of green and renewable hydrogen energy should be pursued. Since the product distribution of electrocatalytic alcohol oxidation is always very complex, how to develop an electrocatalyst with a robust and highly selective feature to a targeted product is an important topic for anodic organic oxidation reactions (AOORs). It is suggested that dual single-atom or multi-metallic catalysts are promising candidates for highly selective AOORs.
4.3 Chitin oxidation reaction (COR)
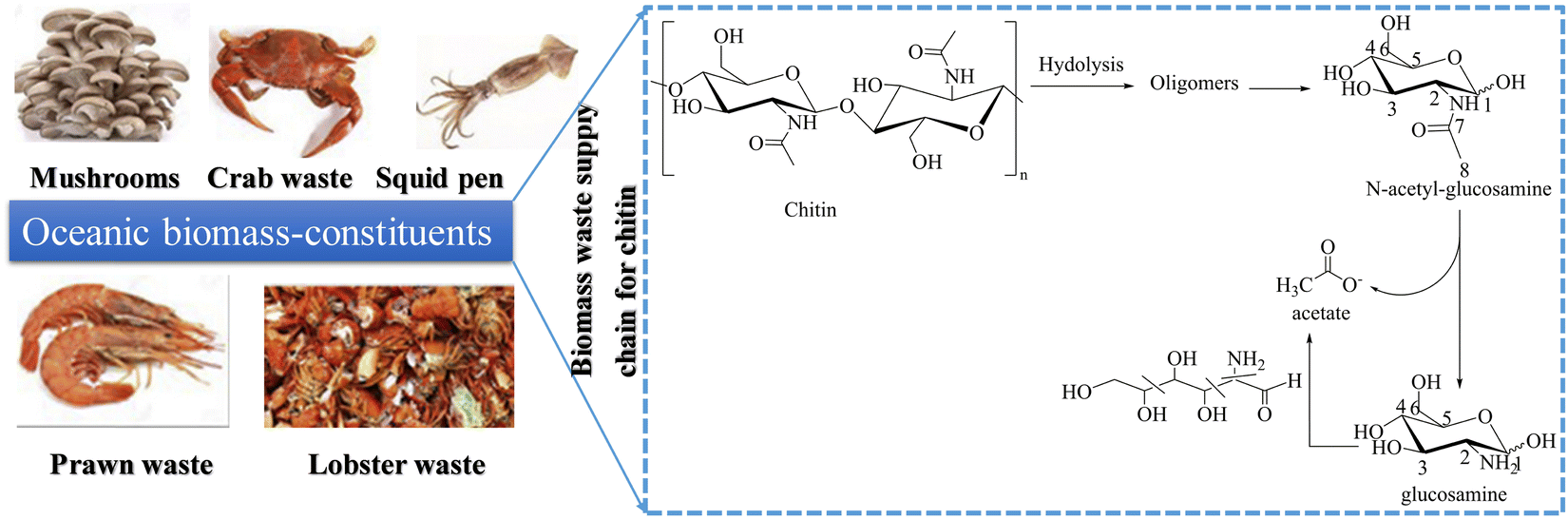
In the long run, various biomass components can be used as sustainable carbon resources for producing organic chemicals, and green chemistry has played a significant role in advancing this field.84 While cellulose, lignin, and lipids have all been extensively studied, the chemical transformation of chitin is still in its early stages.85,86–89 Chitin is the most abundant natural amino biopolymer and accounts for 20–30 wt% of crustacean shells.90
Chitin is used in electrooxidation coupled with HER for the cogeneration of acetate and green hydrogen energy, which can then be upgraded to a more valuable single-cell protein. Under alkaline conditions, chitin oxidation reaction (COR) is more favorable than OER in terms of thermodynamics and kinetics. Yan et al.91 reported that chitin produced 38.1% more acetic acid than cellulose, which was more than a two-fold increase. Furthermore, a 47.9% yield was obtained directly from crude shrimp shells. Interestingly, they achieved 100% conversion of chitin into acetic acid with no complicated purification and separation of the product on the anode since the product was almost pure acetate.92 The bottom line here is that COR offers significantly sustainable environmental benefits since chitin is a major component of solid waste93 and is coupled with HER to generate commodity chemicals. Li et al.14 reported that a hierarchical porous nickel (hp-Ni) electrocatalyst is more efficient towards chitin oxidation to produce acetate with a yield of over 90% at the anode and green hydrogen energy generated at the cathode. The electrode has a highly porous structure with pores ranging in size from nanometers to millimeters, which greatly facilitates mass transport, and gas diffusion in the exposed active sites (high-valent nickel species) that may be responsible for the high current density observed.94 The N-acetyl-glucosamine (NAG is a chitin oligomer) nucleophilic oxidation reaction (NOR) current with 2.2 g L−1 NAG as the reactant leaps at around 1.24 VRHE, which is lower than the OER onset potential of 1.49 VRHE. The low onset potential of the NOR indicates that Ni2+ can selectively oxidize NAG. The value-added electrolysis process is scalable and safe for renewable resource upcycling (ocean-based biomass) and green hydrogen production for a sustainable energy future (Fig. 6).
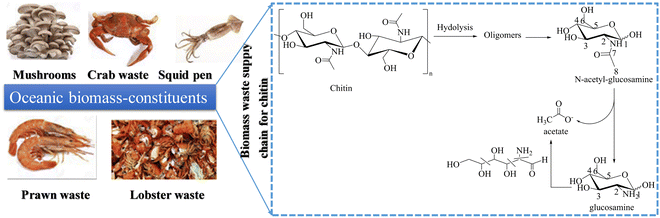 |
| | Fig. 6 The electrochemical scheme of chitin/NAG into acetate with a yield of over 90% in value-added electrolysis (a reaction pathway that produces acetate as the main product). | |
5. Pollutant oxidation reaction
Coupling pollutant-assisted oxidation (usually organic substrates) with hydrogen production might allow a more sustainable and economically viable solution. Although such reactions produce low-value-added products, they have shown great potential for decomposition of urea and hydrazine-rich wastewater in solving environmental problems and producing high green energy for energy conversion (direct urea fuel cell and direct hydrazine fuel cell). In this section, we will have a brief summary of the recent progress in urea and hydrazine oxidation reaction-assisted hydrogen production systems.
5.1 Urea oxidation reaction (UOR)
Environmental pollution and energy scarcity are two major issues that threaten sustainable human development in the coming decades.95 Urea is abundant in industrial wastewater and human and animal urine and has dual pollution and resource characteristics. Without prompt and reasonable treatment, ammonia, nitrite, nitrate, and nitrogen oxides produced by urea pollute the air and groundwater, endangering human health and safety. On top of that, with the rapid development of electrochemistry technology, urea has proven to be an effective hydrogen storage chemical due to its high gravimetric hydrogen content of 6.71 wt%.95,96 As a result, designing a device that can treat urea wastewater while utilizing urea's energy potential is promising. Pollutants may contribute to harmful cyanobacterial blooms, which are enhanced by nitrogenous wastes from protein metabolism of mammals and agricultural wastewater (urea).97 After treatment, wastewater (urea-containing) could form some flocculant species (hydroxy complexes, polynuclear hydroxy complexes, hydroxides) with strong pollutant adsorption capacity (adsorption of suspended solids, heavy metal ions, organic matter).98 However, it must be investigated whether other pollutants are also formed during the oxidation reaction of urea. In the anodic oxidation reaction, urea is directly oxidized to nitrogen. For instance, Stevenson et al.99 reported that the nanostructured LaNiO3 perovskite coated on a carbonaceous powder as an electrocatalyst tends to restructure and become poisoned by CO2 produced during prolonged cycling. This shows that urea is directly oxidized to N2 and CO2. However, CO2 causes carbon emissions, a greenhouse effect and poisoning of the catalyst by carbon species.
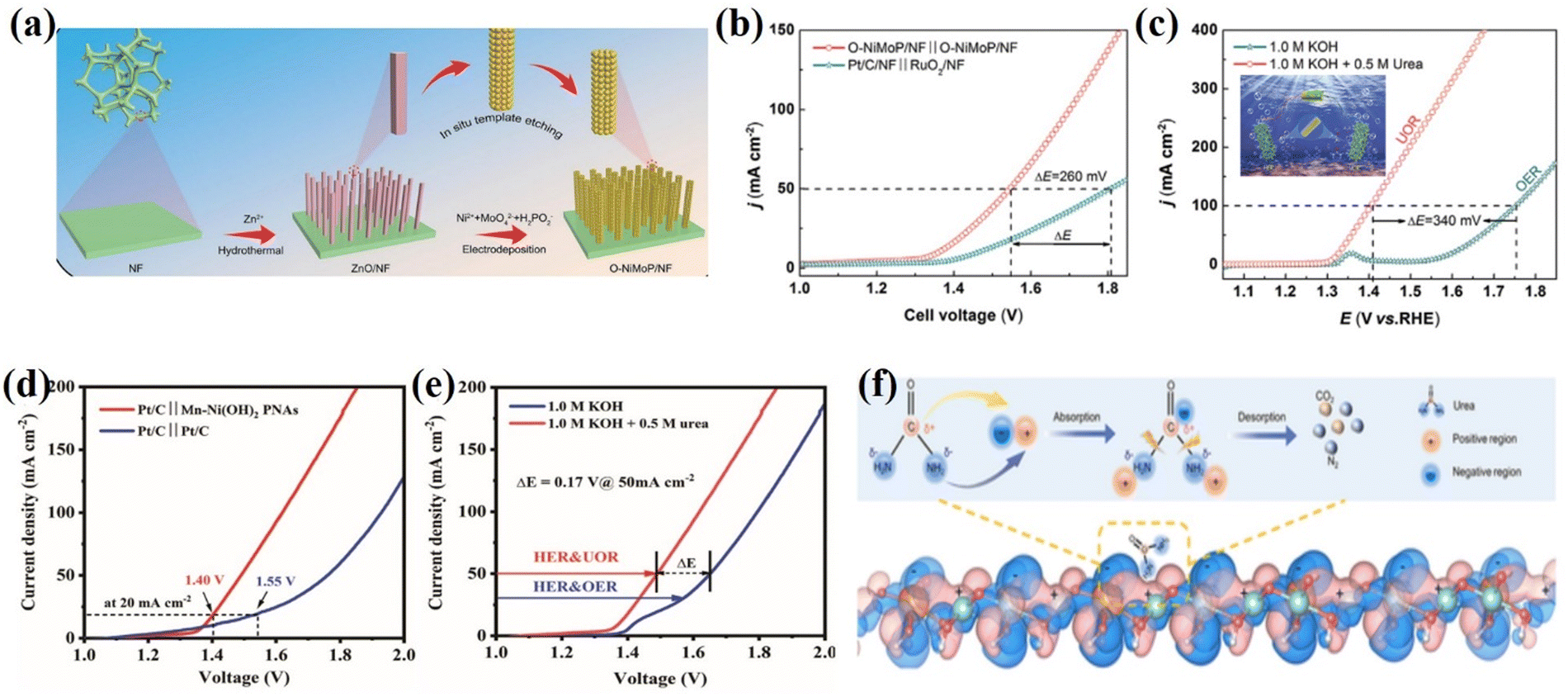
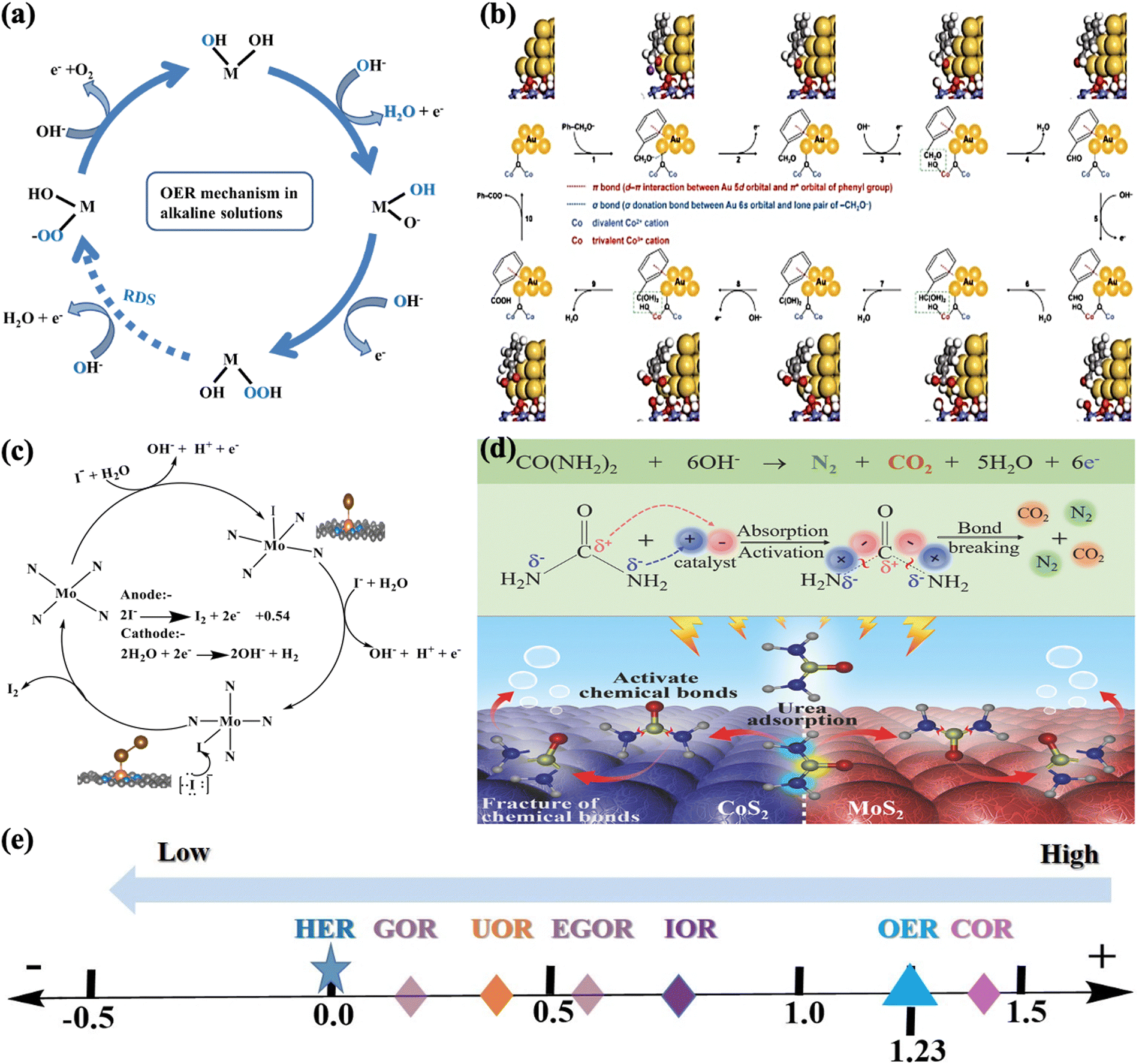
Urea oxidation reactions can use urea from free urea wastewater sources and have low thermodynamic potentials (0.37 VRHE). In the case of Ni2Fe(CN)6, for example, the cooperative action of the two active sites of Ni and Fe is responsible for the conversion of urea into ammonia and carbon dioxide.100 At the same time, Fe is responsible for transforming ammonia into nitrogen. This catalysis results from synergistic catalysis between the Ni and Fe sites, achieving a 100 mA cm−2 anodic current density at 1.35 V. Moreover, multichannel nanorods in situ grown on nickel foam (Ru/P-NiMoO4@NF) exhibit bifunctional activity with a working potential of 0.23 V for 3000 mA cm−2 HER and 1.46 V for 1000 mA cm−2 UOR.101 One advantage of Ru/P doping is that it can modulate the d-band center, allowing for simultaneous reactant adsorption and product desorption on the active materials on the exposed surface. As a result, the rate of urea purification-based hybrid seawater splitting, traditional water splitting, and low-energy hydrogen production is increased by the highly active doped Ru/P-NiMoO4@NF electrocatalyst. The self-supporting O-NiMoP/NF electrode exhibits highly efficient bifunctional activity towards HER and UOR due to the modulated electronic structure and nanotube array (i.e., enhanced electron/mass transfer) architecture of O-NiMoP (Fig. 7a–c).102 Furthermore, no additional binder (Nafion) or conductive agent (activated carbon) is required between the active material and the conductive substrate, ensuring maximum active material mass loading.103 Chen et al.104 developed novel hollow nanorod arrays (HNRAs) made from CoNiP nanosheets (NiCoPNSs@HNRAs), and the distinct HNRAs enable fast transport and short diffusion paths for electroactive species and high UOR catalytic activity. The catalyst's nature is the driving force behind the reduction in energy consumption for hydrogen and urea production from wastewater purification. As a result, the NiCoPNSs@HNRAs' unique nanostructure can improve its electronic structure and large surface area, potentially significantly increasing its electronic density and optimizing the energy barrier of the CO*/NH2* intermediates of UOR (Fig. 7d and e). A series of reactions occur on the electrocatalysts’ exposed surface, including adsorption, surface reaction, and desorption. Zhang et al. conducted computational calculation studies for charge distribution between W and Ni atoms to understand the benefit of charge-transfer interaction between W and Ni atoms in facilitating the UOR process. The most interesting aspect of UOR is that wastewater purification (reduce the urea content of wastewater) can be done at the anode via direct or mediated charge transfer, in addition to the production of low value-added products (N2, CO2, and H2O) and the great potential for highly efficient production of green hydrogen energy (Fig. 7f).105
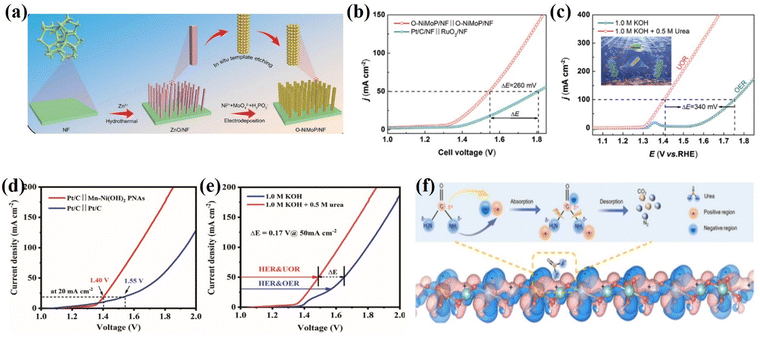 |
| | Fig. 7 (a) Schematic illustration of the preparation of O-NiMoP/NF. (b) Polarization curves for the OER and UOR of O-NiMoP/NF in 1.0 M KOH + 0.5 M urea with a scan rate of 5 mV s−1. (c) Polarization curves of O-NiMoP/NF electrodes in HER||UOR and HER||OER coupled systems.102 Reprinted with permission Copyright 2021, Wiley-VCH. (d) LSV plots of Pt/C||NiCoP NSs@HNRAs and Pt/C||Pt/C two-electrode systems for overall urea electrolysis in an alkaline solution. (e) LSV plots of overall electrolysis for Pt/C||NiCoP NSs@HNRAs in 1.0 M KOH with or without 0.5 M urea.106 Reprinted with permission Copyright 2021. (f) Charge density difference of Ni-WOx. Reprinted with permission.107 Copyright 2021, Wiley-VCH. | |
5.2 Hydrazine oxidation reaction (HzOR)
Hydrazine is highly toxic and carcinogenic, posing a significant risk to human health. The threshold of hydrazine in surface water should be strictly restricted to protect the ecosystem.108 However, hydrazine is also an important chemical reagent widely used as an industrial raw material for the synthesis of organic and inorganic compounds, a corrosion inhibitor, a component of photography chemicals, fuel for fuel cells, and rocket fuel.109–111 Recently, electrochemical oxidation of some active organic and inorganic substrates such as hydrazine, urea, polyalcohol, chitin, and iodine has shown great potential to replace the anodic OER.112 In particular, when compared to UOR the hydrazine oxidation reaction (HzOR: N2H4 + 4OH− → N2 + 4H2O + 4e−) has a lower theoretical voltage of 0.33 VRHE, a higher power density, and faster electrooxidation kinetics.113–115 Moreover, the HzOR provides a ground-breaking strategy for energy-saving hydrogen production while also adding extra functionality such as degrading N2H4 contaminated wastewater, and low value-added products like water (H2O) and nitrogen (N2), with no carbon dioxide green-house gas.108,116
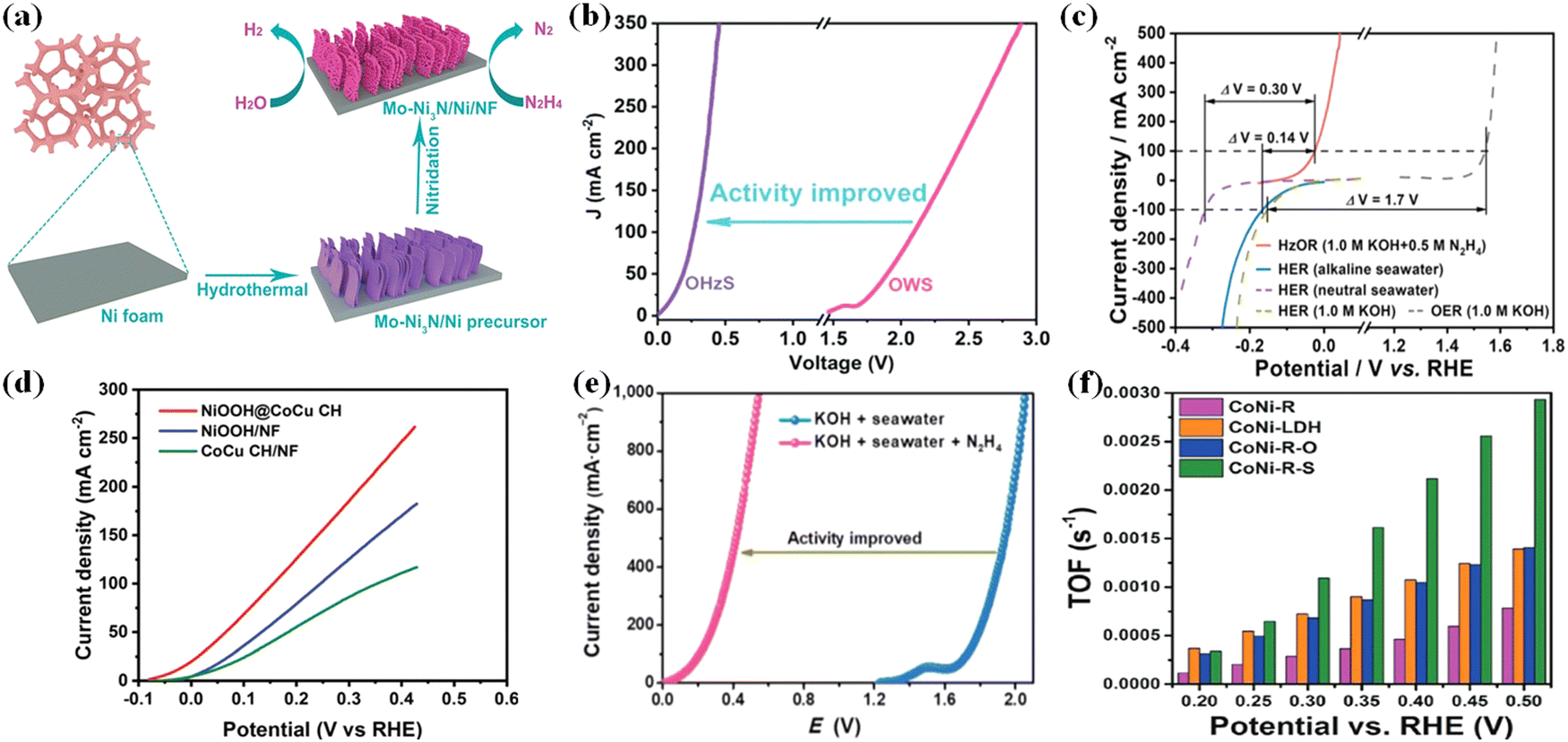
Recently, it was revealed that a topotactic transformation process of mixed alloy oxide, LDH (layered double hydroxide), and metal organic framework (MOF) materials to self-supported metal nanoparticles can occur via various techniques (hydrothermal, calcination, and so on) and these nanoparticles have unique properties of high dispersion and stability.117,118 These nanoparticles as electrocatalysts have shown significant electrochemical performance (activity, selectivity, and stability) in hydrazine degradation. For instance, the Cu1Ni2-N system described by Mai et al. demonstrated bifunctional activity, with an overpotential of 71.4 mV at 10 mA cm−2 for HER in 1 M KOH and a working potential of 0.5 mV for HzOR in 1 M KOH/0.5 M N2H4, which could be attributed to the rich Cu4N/Ni3N interface.119 Fan et al. reported a Co-Ni3N heterostructure array with increased HER capacity due to the interface transfer effect.120 Furthermore, a synergistic effect is demonstrated, which may be due to the conductive CoNi alloy as the core providing the highway for electron transfer and the CoNisulfide shell providing highly exposed active sites for hydrazine electrooxidation.118 On the other hand, heteroatom substitution (Fig. 8a and b), as exemplified by Mo doped Ni3N and Ni-heterostructure porous nanosheets grown on Ni foam (Mo-Ni3N/Ni/NF), could lead to outstanding bifunctional electrocatalytic performances toward both HzOR and HER.117 The hybrid seawater electrolyzer (HSE) achieved a high current density of 500 mA cm−2 under alkaline conditions when using NiCo@C/MXene/CF and an ultralow cell voltage of 0.31 V. An anion exchange membrane (AEM) separates the identical anode and cathode, allowing for energy-saving hydrogen production from seawater as well as simultaneous hydrazine degradation (Fig. 8c).108 In comparison to NiOOH, and CoCu CH grown directly on nickel foam, NiOOH@CoCu CH is the most promising self-support electrocatalyst for large-scale hydrogen production via water electrolysis using the hydrazine oxidation reaction (Fig. 8d).18Fig. 8e shows the comparison of LSV curves for overall value-added splitting (VAS) and overall seawater splitting, where the use of HzOR to assist hydrogen production can visually improve the value-added electrolysis efficiency after the addition of hydrazine (N2H4).121 Topo-transformation of CoNi-LDH arrays was used to create the CoNi-sulfide shell on the surface of the CoNi-alloy network. As a result, the resulting CoNialloy@CoNi-sulfide (CoNi-R-S) has the highest TOF (turnover frequency) value, confirming that the presence of CoNi-R-S significantly increases the rate of hydrazine decomposition when compared to CoNi-oxide and CoNi-LDH (Fig. 8f).110 Though the coupled value-added anodic reaction (UOR and HzOR) with HER which could produce less valuable chemicals, they have the potential to solve the problem of serious environmental pollution and eutrophication of water resources.
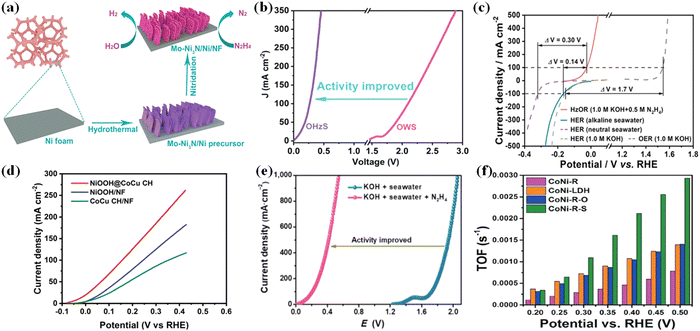 |
| | Fig. 8 (a) Schematic illustration of the formation process of Mo-Ni3N/Ni/NF through a facile two-step synthetic route. (b) Comparison of LSV curves and cell voltages for OHzS and OWS.117 (c) The voltage differences (ΔV) between HER and HzOR or OER on NiCo@C/MXene/CF in different electrolytes.108 (d) HzOR polarization curves of NiOOH@CoCu CH, NiOOH/NF and CoCu CH/NF.18 (e) LSV curves of the MoNi@NF couple in different solutions.121 (f) Comparison of TOF of hydrazine decomposition of CoNi–LDH, CoNi-R, CoNi-R-O, and CoNi-R-S.110 | |
6. Rational design of electrocatalysts for value-added anodic reactions
In general, electrode surface composition,122,123 surface structure,124 and electrolyte composition125–128 are vital for good electrocatalytic performance, as the reaction occurs on the exposed surface of electrocatalysts. The main value-added anodic reaction (VAAR) catalysts reported to date can be divided into single atom catalysts (SACs), transition metal (mixed metal/oxide heterojunction), hetero-elements including non-metal (S, P, N, and B) doped carbon-based support, and bimetallic alloy electrocatalysts. In addition, special attention has been given to self-supported three-dimensional electrocatalysts. The first approach involves introducing a second metal into the transition metal (TM) network to create selective intermetallic compounds with more selectivity toward specific anodic oxidation reactions. The coordination chemistry of the elemental metals changes after the formation of an intermetallic compound, resulting in an alteration of both metals' electronic environments, which could favor the electron/mass transfer due to the band overlap. Moreover, electrons are partially transferred to the element with lower orbital energy, causing the d band center to shift relative to the Fermi level, affecting the adsorption properties of intermediates and directly influencing the material's catalytic activity.129 The introduction of a second metal into the TM network could result in the formation of an intermetallic compound. Thus, the altered coordination structure of intermetallic compounds provides more active sites, changes the adsorption properties of intermediates, and improves material corrosion resistance. Recently, various intermetallic compounds have been shown in the methanol oxidation reactions without CO poisoning of the catalytic site for the production of formic acid and CO. An intermetallic catalyst avoiding CO poisoning depends on the chemical and electronic nature of the introduced metals.130,131 TM oxides (NiO, CoO, CuO) can efficiently catalyze the alcohol oxidation and water oxidation processes.132 TMs exist as oxides in either higher or lower valence states depending on the nature of the metals and electronic geometry. On top of that, heteroatom (S, P, N, and B) doped carbon-based supports could improve the electronic structure of electrocatalysts to produce value-added chemicals and hydrogen.133 The hetero-atom doped carbon-based electrocatalysts can facilitate the oxidation process of organic and inorganic substrates by generating electronegativity between carbon and hetero-atoms.134 The electronic structure of the active site, which could be regulated by introducing different hetero-atoms to coordinate with the metal atoms, affects the coordination configuration and charge density of the metal centers (active sites). For example, the N-doped substrates significantly increase the charge of the metal atoms (single/dual atoms), and B- and P-doped substrates provide multiple active sites on the exposed surface.135 Lu et al. reported an ultrafast method for successfully engineering the surface of Ni foam into a hydrophilic S-doped Ni/Fe (oxy)hydroxide layer with multiple levels of porosity and with a large surface area and numerous active sites. In terms of site-selectivity governed by the S-doped NiFe-LDH/NF, it could be regulated by the local charge distribution of the Ni atoms, leading to the formation of Ni2+ sites with excellent activity and thus favoring the interfacial catalytic reaction.136 Thus, the organic and inorganic substrates can easily access the active sites to produce value-added chemicals and green hydrogen energy. Recent studies have shown that 3D PdCu alloy nanosheets enable highly selective partial oxidation of ethanol into carboxylate coupled with HER.137 The Pt–Ru alloy-based catalyst showed remarkable CO tolerance as CO oxidizes on the Ru active surface due to its oxophillic nature.138 Our group developed a new approach to a self-supported ruthenium–titanium mixed alloy oxide electrocatalyst on 3D web-like titania.52 In the end, the most important issues in designing electrocatalysts are electrocatalytic activity, selectivity, and stability which could reduce the cost of production of value-added chemicals and hydrogen generation. Especially, if the electrocatalyst is more selective for one specific target product, then no further separation and purification of the product is needed. We briefly highlight the non-precious metal electrocatalysts that exhibit remarkable catalytic performance and even outperform the precious metal-based electrocatalysts for hydrogen production (Table 2). For instance, in 2023, a self-supported electrocatalyst was fabricated via an ultrafast solution combustion strategy which could be highly dispersed on FeNi oxide heterojunction anchored on nickel foam (Fe2O3/NiO is labeled Fe-NF-500) and required only a low potential of 1.472 V to achieve an industrial scale current density of 600 mA cm−2.139 Moreover, the current density of Co(OH)2@HOS/CP was 70 mA cm−2 at 1.66 V, which is 4.78 times higher than that of conventional water splitting (HER and OER), indicating the boosting of hydrogen generation and high selectivity for methanol to value-added formate oxidation (MFO).140 Besides, some other non-precious metal electrocatalysts also have excellent UOR, HzOR, and MOR faradaic efficiency, such as Ni3Se2/MoO2@Ni12P5,141 Mo-Ni3N/Ni/NF,117 and Mo-CoN4,142 respectively.
Table 2 Recently reported electrocatalysts for the value-added anodic reactions with HER including IOR (iodide oxidation reaction), EGOR (ethylene glycol oxidation reaction), COR (chitin oxidation reaction), MOR (methanol oxidation reaction), GOR (glycerol oxidation reaction), UOR (urea oxidation reaction), and HzOR (hydrazine oxidation reaction)
| Rxn |
Anode electrocatalysts |
Substrate Conc. |
Value-added products |
Anolyte |
Potential at 10 mA cm−2 (V) |
Tafel slope (mV dec−1) |
FE (%) |
Ref. |
| The numbers in parentheses are the current densities (mA cm−2). ≈ represents the value estimated from the LSV curves. FE: faradaic efficiency; NF: nickel foam; CF: copper foam; CP: carbon paper; CC: carbon cloth; CFC: carbon fiber cloth; uNSs: ultra nanosheets; HOS: hydroxysulfide nanosheet; HNRAs: hollow nanorod arrays; N/A: not available; PET: polyethylene terephthalate. |
| IOR |
RuTiO-550 |
0.1 M NaI |
Iodine (I2) |
0.1 M HClO4 |
1.01 |
N/A |
100 |
52
|
| RuSn SAO |
0.1 M NaI |
Iodine (I2) |
0.1 M HClO4 |
1.07 |
40 |
100 |
13
|
| Single atom Mo-N4/d-C |
0.1 M NaI |
Iodine (I2) |
0.1 M HClO4 |
0.77 |
25.58 |
99.8 |
53
|
| Carbon fiber paper (CFP) |
0.1 M NaI |
Iodine (I2) |
0.1 M HClO4 |
0.59 |
47.78 |
N/A |
54
|
| Ni-Co(OH)2 |
0.33 M KI |
Iodine (I2) |
1 M KOH |
1.36 |
47 |
N/A |
55
|
| EGOR |
Dual single atom Pd-N4/Cu-N4 |
1 M EG |
Glycolate (>80% selectivity) and formate |
1 M NaOH |
0.6 |
N/A |
N/A |
80
|
| Single atom Pt/TiO2 |
0.5 M EG |
Glycolate and formate |
0.1 M NaOH |
0.489 |
N/A |
99.7 |
81
|
| Pd-Ag/NF |
1 M EG |
Glycolate |
1 M KOH |
0.57 |
N/A |
100 |
72
|
| Modified Pd/NF |
1 M EG (PET) |
Carbonate (CO32−) |
1 M KOH |
0.7 |
N/A |
98 |
83
|
| CoNi0.25P/NF |
0.3 M EG (PET) |
Formate (>80% selectivity) |
1 M KOH |
1.8/100 mA cm−2 |
N/A |
N/A |
15
|
| CoNi3N/CC |
1 M EG (PET) |
Formic acid |
1 M KOH |
1.46/50 mA cm−2 |
78 |
N/A |
17
|
| COR |
hp-Ni/NF |
3.5 M chitin |
Acetate |
1 M KOH |
1.45 |
N/A |
100 |
14
|
| MOR |
Pt1/Ti0.8W0.2NxOy |
0.5 M methanol (MeOH) |
Formate and formaldehyde |
0.5 M KOH |
0.7 |
93 |
N/A |
144
|
| Fe2O3/NiO/NF |
1 M MeOH |
Formate |
1 M KOH |
1.65/500 mA cm−2 |
N/A |
98 |
139
|
| Co(OH)2@HOS/CP |
3 M MeOH |
Formate |
1 M KOH |
1.385 |
N/A |
100 |
140
|
| Mo-CoN4 |
3 M MeOH |
Formate |
1 M KOH |
1.356 |
42 |
100 |
142
|
| GOR |
Ni-Mo-N/CFC |
3 M glycerol |
Formate |
1 M KOH |
1.3 |
87 |
95 |
116
|
| MnO2/CP |
0.2 M glycerol |
Formate |
0.005 M H2SO4 |
1.36 |
N/A |
53 |
145
|
| Cu–Cu2O/CC |
0.5 M glycerol |
Formate |
1 M KOH |
1.21 |
N/A |
100 |
146
|
| UOR |
NiF3/Ni2P @CC |
0.33 M urea |
N2, CO2 and H2O |
1 M KOH |
1.36 |
33 |
N/A |
147
|
| Ni3Se2/MoO2@Ni12P5 |
0.5 M urea |
N2, CO2 and H2O |
1 M KOH |
1.277 |
53.8 |
97.4 |
141
|
| Ni@C-250 |
0.5 M urea |
N2, CO2 and H2O |
1 M KOH |
1.5/100 mA cm−2 |
43 |
N/A |
148
|
| Ni3N/Ni0.2Mo0.8N |
0.5 M urea |
N2, CO2 and H2O |
1 M KOH |
1.328 |
34.5 |
N/A |
149
|
| Ni3Se2@CuSex/CF |
0.5 M urea |
N2, CO2 and H2O |
1 M KOH |
1.49/100 mA cm−2 |
87 |
N/A |
150
|
| NiCoPNSs@HNRAs |
0.5 M urea |
N2, CO2 and H2O |
1 M KOH |
1.35/50 mA cm−2 |
47 |
N/A |
104
|
| CoS2-MoS2 |
0.5 M urea |
N2, CO2 and H2O |
1 M KOH |
1.29 |
N/A |
N/A |
151
|
| Ni2Fe(CN)6 |
0.33 M urea |
N2, CO2 and H2O |
1 M KOH |
1.35/100 mA cm−2 |
N/A |
90 |
100
|
| O-NiMoP/NF |
0.5 M urea |
N2, CO2 and H2O |
1 M KOH |
1.55 |
54 |
N/A |
102
|
| HzOR |
Mo-Ni3N/Ni/NF |
0.05 N2H4 |
N2 and H2O |
1 M KOH |
−0.3 |
45 |
100 |
117
|
| Rh2P uNSs |
50mM N2H4 |
N2 and H2O |
0.5 M H2SO4 |
0.377 |
33.4 |
N/A |
152
|
| NiOOH@CoCu CH |
0.5 M N2H4 |
N2 and H2O |
1 M KOH |
−0.031 |
108 |
N/A |
18
|
| NiCo@C/MXene/CF |
0.5 M N2H4 |
N2 and H2O |
1 M KOH |
0.31/500 mA cm−2 |
73 |
N/A |
108
|
| MoNi@NF |
0.5 M N2H4 |
N2 and H2O |
1 M KOH |
0.54/1000 mA cm−2 |
60.7 |
100 |
121
|
The benefits of selective value-added anodic reactions were briefly discussed above, and the common advantages of VAARs paired with HER are summarized below. The exploration of suitable value-added reactions at the anode would give additional advantages over the conventional water splitting process, while addressing other energy-related applications, such as high-efficiency hydrogen energy generation, high-value-added chemical production, and pollutant degradation. Moreover, products generated through value-added anodic reactions (VAARs) have higher values than cheap O2 gas. As a green and a sustainable route toward simultaneous plastic and global shellfish waste (rigid polymeric structure of raw biomass) elimination, electrochemical synthesis of value-added chemicals and production of green and renewable hydrogen energy should be pursued. The new direction to use the rigid polymeric structure of raw biomass as a raw material for value-added electrolysis is being investigated and coupled with hydrogen evolution reaction or CO2RR to reduce the full cell energy requirement at both sides of the value-added electrolyzer.7 Furthermore, electrocatalytic hydrogenation (ECH) has sparked tremendous interest in recent years as a viable alternative to the hydrogenation process involving dihydrogen (H2). Instead of producing hydrogen at the cathode, ECH uses water as a hydrogen source to produce high-value-added chemicals from unsaturated organics (alkene and alkyne substrates).143 As shown in Scheme 1, various cathodic reactions including HER, CO2RR, NRR, and ECH can be paired with value-added anodic reactions (VAARs) to develop novel value-added electrolysis processes (VAEs), which will be the necessary foundation to reach the global goal of net-zero emission by 2050. Research in developing energy-efficient, highly selective and durable electrocatalysts is extremely important for new value-added electrolysis processes but is still in the early stages. Understanding the reaction mechanism of an electrochemical reaction is essential to design and develop a novel electrocatalyst. However, most of the electrochemical reactions in a value-added electrolysis process are very complicated and involve multistep reaction mechanisms. Existing ex situ and in situ analytical techniques might not be sufficient to explore their mechanisms.
 |
| | Scheme 1 Concept of value-added electrolysis. | |
7. Proposed mechanism
Different reaction mechanisms may be involved in electrocatalysis systems with different reactants, electrocatalysts, and electrolytes. However, the oxygen evolution reaction (OER) is a process of electrocatalytic oxidation of water to molecular oxygen that involves four proton-coupled electron transfers and O–O bond formation, with unfavorable kinetics and requiring a significantly large overpotential to speed up the reaction. In the case of OER, water oxidation's primary goal is to extract electrons and transport them to the cathode side to produce hydrogen, which may be the most desirable byproduct of water splitting. As was confirmed by recent studies, the OER generally occurs on the hydroxide, (oxy)hydroxide, or oxide layer formed in situ on the exposed surface of the electrocatalyst.153,154 The main intermediates observed in the OER are O*, HO*, and HOO*. As for the OER mechanism of transition metal oxide (TMO) catalysts under alkaline electrolytes, M–OH species are first adsorbed and transformed into M–O species through oxidation at the metal sites to which they are coordinated. Following this, the mechanism can proceed by coupling two neighboring M–O species to form O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds or the nucleophilic attack of water by the M–O species to form M–OOH species. These M–OOH species formed can subsequently be oxidized to form M–OO species that are replaced by water in the subsequent process, allowing the release of oxygen (O2), and providing free surface sites (regenerating the catalytic active sites) for the next cycle, as illustrated in Fig. 9a.155 In particular, all of the reaction steps in water nucleophilic attack (WNA in eqn (6) and (7)) are assumed to involve proton-coupled electron transfer (PCET). Likewise, the path of interaction of intermolecular O–O coupling (12 M in eqn (11) and (12)) starts with the formation of the same metal-oxo species; however, the final O2 release is enabled through the interaction of two separate metal-oxo species under alkaline and acidic conditions.156–158
O bonds or the nucleophilic attack of water by the M–O species to form M–OOH species. These M–OOH species formed can subsequently be oxidized to form M–OO species that are replaced by water in the subsequent process, allowing the release of oxygen (O2), and providing free surface sites (regenerating the catalytic active sites) for the next cycle, as illustrated in Fig. 9a.155 In particular, all of the reaction steps in water nucleophilic attack (WNA in eqn (6) and (7)) are assumed to involve proton-coupled electron transfer (PCET). Likewise, the path of interaction of intermolecular O–O coupling (12 M in eqn (11) and (12)) starts with the formation of the same metal-oxo species; however, the final O2 release is enabled through the interaction of two separate metal-oxo species under alkaline and acidic conditions.156–158
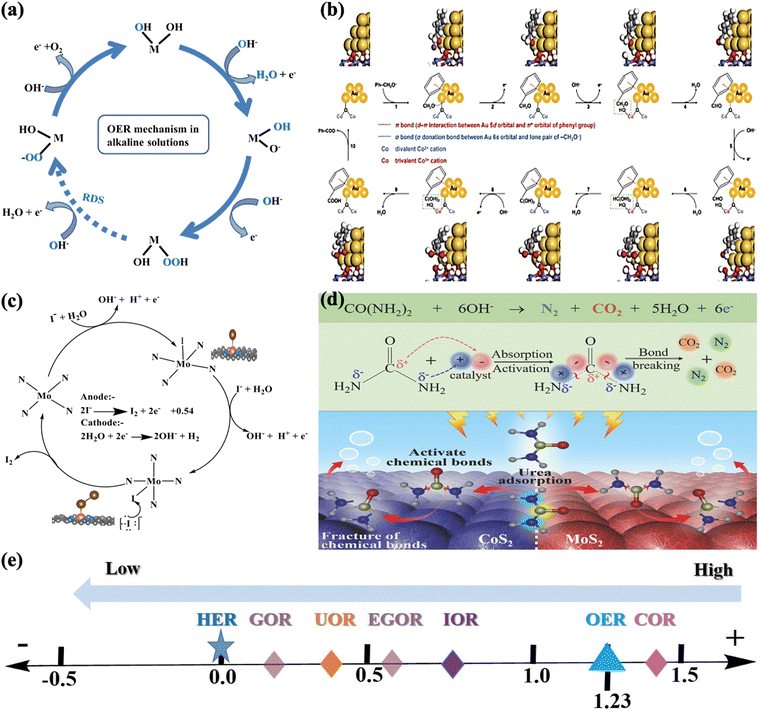 |
| | Fig. 9 (a) Proposed oxygen evolution reaction (OER) mechanisms for various transition metal oxides under alkaline conditions.155 (b) Proposed reaction mechanism of alcohol oxidation, example illustrations of the reaction processes for benzyl alcohol oxidation on Au/CoOOH. The optimized geometries of reaction intermediates on Au/CoOOH are also displayed.161 (c) Proposed reaction mechanism of iodide adsorption and iodine on the Mo-N4/d-C surface.53 (d) The schematic UOR catalytic mechanism using the CoS2-MoS2 Schottky electrocatalyst.151 (e) Thermodynamic equilibrium potential vs. reversible hydrogen electrode (RHE), for oxygen evolution reaction (OER), iodide oxidation reaction (IOR), urea oxidation reaction (UOR), glycerol oxidation reaction (GOR), ethylene glycol oxidation reaction (EGOR), chitin oxidation reaction (COR) and hydrogen evolution reaction (HER). | |
The proposed OER pathways under alkaline conditions are summarized as follows (from eqn (4) to (7)):
| | | MOH + OH− → MO + H2O + e− | (5) |
| | | MO + OH− → MOOH + e− (WNA) | (6) |
| | | MOOH + OH− → MOH + e− (WNA) | (7) |
Overall reaction: 4OH
− → O
2 + H
2O
2(1) + 4e
− (0.401 V
vs. SHE, pH 14)
The proposed OER pathways under acidic/neutral conditions are summarized as follows (from eqn (8) to (13)):
| | | M + H2O → MOH + H+ + e− | (8) |
| | | MO + H2O → MOOH + H+ + e− (WNA) | (10) |
| | | MOOH → M + O2 + H+ + e− (WNA) | (11) |
| | | MO + MO → M–O–O–M (12 M) | (12) |
| | | M–O–O–M → 2M + O2 (12 M) | (13) |
Overall reaction: 2H
2O
(1) → O
2 + 4H
+ + 4e
− (1.229 V
vs. SHE, pH 0)
According to the researchers, the rate-determining step (RDS) for OER is either the peroxide formation step or the step involving O2 evolution from the peroxide.159,160 However, the actual RDS in the OER mechanism is still obscure. As a result, additional research using in situ spectroscopic techniques or density functional theory (DFT) calculations is required.
On the other hand, there is no clear cut or general mechanism for coupled VAARs-HER systems. However, great effort has been made to develop value-added electrolysis processes by designing appropriate electrocatalysts for selective value-added anodic reaction coupled with HER to generate green hydrogen gas and value-added chemicals. Still, different issues could not be solved, especially when biomass fuel is used. We believe that a clear mechanism is vitally important for system optimization, electrocatalyst design, and raw material selection. This section illustrates three value-added electrolysis mechanisms: AOR, IOR, and UOR.
Firstly, the reaction begins with the adsorption of benzyl alkoxide on the exposed surface of Au (acts as a catalytic site), as shown in Fig. 9b. When the potential is raised, the adsorbed benzyl alkoxide (Ph-CH2O−*) is converted to nucleophilic Ph-CH2O*, and an electron is transferred to Au. Then, electrophilic OH*, generated at specific potentials (0.856 VRHE on Au and 1.216 VRHE on CoOOH), attacks Ph-CH2O* to generate benzaldehyde (Ph-CHO*) via the proton–electron transfer process. Then, by spontaneous hydration, Ph-CHO* is converted to Ph-CH(OH)2*, which is then reacted with OH* to give Ph-C(OH)2* and finally Ph-COOH*. The generation of OH* for both Au and Au/CoOOH is the rate-determining step (RDS) in benzyl alcohol oxidation, with Gibbs free energy changes (G) of 0.856 eV and 1.216 eV, respectively. Therefore, OH* can be generated on Au at a lower potential than that generated on CoOOH in Au/CoOOH.
Secondly, as shown in Fig. 9c, in the case of the proposed iodide oxidation reaction mechanism, all of the iodide ions are sequentially adsorbed before the surface reaction proceeds on the exposed surface of the active site (Mo). The charge density of the Mo atom is further tuned to a higher oxidation state, where it collaborates with the surrounding N atoms to boost iodine production. According to the DFT calculation results, the defective Mo-N4 model outperforms the reference Pt catalyst regarding iodine production consistent with the electrochemical results. As a result, the formation of defects on carbon supports is critical for molybdenum atom catalysis of IOR.
Finally, the mechanism of urea electrooxidation reaction (UOR) at the molecular level (Fig. 9d) indicates self-driven charge transfer across Co and Mo in enhancing the UOR-assisted HER system. Furthermore, the CoS2/MoS2 heterointerface promotes the formation of a local electrophilic/nucleophilic region that intelligently adsorbs chemical groups of the urea molecules, lowers kinetic barriers, and activates chemical bonds. When UOR occurs on the exposed surface of the electrocatalyst (CoS2/MoS2), the “electron-withdrawing” group of carbonyl (acts as an electrophilic region) is heavily absorbed on the negative region, whereas the “electron-donating” group of amino (acts as a nucleophilic region) is heavily absorbed on the positive region, facilitating urea molecule decomposition. As a result of the charge distribution on the surface regulated by Mo, high UOR activities and accelerated UOR kinetics were observed. This fascinating study shows how charge-distribution modulation can create high-efficiency electrocatalysts for green hydrogen energy production and pollutant degradation (generate safe gases such as N2 and CO2).
In the end, the VAARs-HER strategy allows the use of anodic reaction to produce chemicals of interest with added value and clean hydrogen at the cathode at a much lower overall applied voltage as shown in Fig. 9e.
7.1 Mechanism investigation for IOR-assisted HER
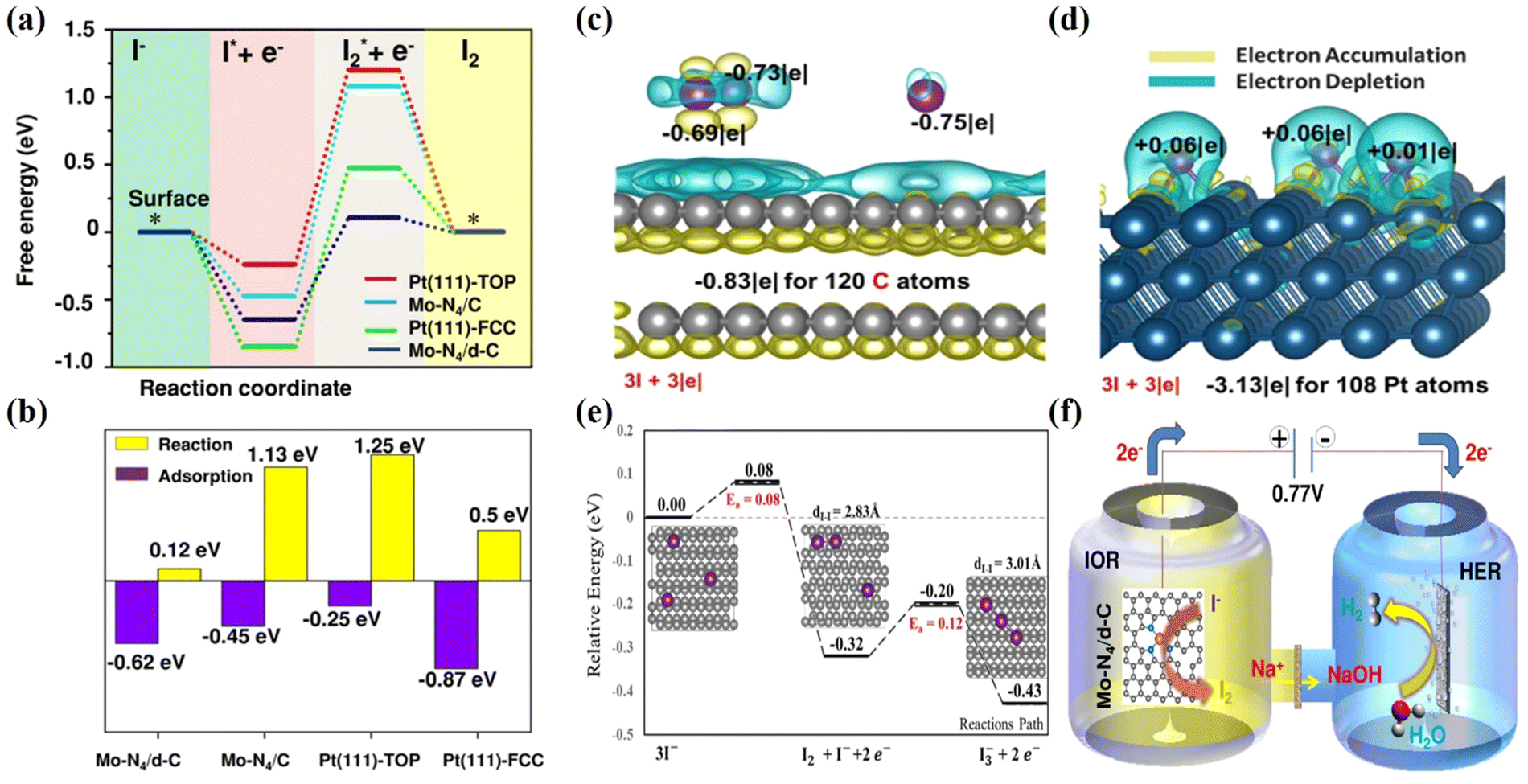
To illustrate the working mechanism of the Mo-N4/d-C SAC and non-metallic carbon fiber paper (CFP) graphite structure, electrocatalysts were employed to exhibit the free energy of IOR. We have designed two models for the Mo single-atom catalyst according to the experimentally observed structural features. First, the pyrrolic nitrogen (pyrrolic-N) dominates in the N-doped carbon material, and the coordination number of Mo-N is 4, resulting in an abundant and well-defined Mo single atom anchoring four pyrrolic-N in the model (Mo-N4/C). Second, the Pt (111) surface as the reference model was constructed based on its popular crystalline plane. ΔG1 and ΔG2 to the corresponding free-energy diagram were calculated to know the mechanism of different active sites for IOR. ΔG1 represents the reaction free energy of NaI to adsorbed I (I*), and ΔG2 is the reaction free energy of I* to I2*. Interestingly, the Mo-N4/d-C model reveals a slightly stronger ΔG1 than the Mo-N4/C model and Pt (111)-Top. Based on the DFT calculation the Mo-N4/d-C can significantly reduce ΔG2 (Fig. 10a and b), which could be due to the existence of the defect, to properly tune the charge density of the active site (Mo-N4 sites). In the end, the defective Mo-N4 mode has advantages in producing I2 in the IOR compared with the Pt catalyst, which agrees with the electrochemical results. Peng et al. adopted non-metallic carbon fiber paper with graphite structure has multiple adsorption modes to turn three iodide ions (3I−) into one triiodide ion and investigates the oxidation mechanism on the exposed surface of CFP graphite electrocatalyst.54 The charge analysis was introduced to describe the interaction between graphite and 3I− in detail, as shown in Fig. 10c and d. First, the 3I− of the most stable adsorption mode can undergo a proper charge transfer with the π orbital graphite system and it was found that the iodide ions can physically adsorb on graphite and donate a charge to an undeformed graphite structure (Fig. 10c). For comparison the charge analysis between metallic Pt (111) and iodide ions is considered. The Pt (111) surface and iodide ions showed strong electron transfer to form strong chemical bonds, which could inhibit I2 formation compared with graphite (Fig. 10d). Second, the formation reactions of I2 + I− and I3− on the exposed surface of graphite are examined via electron analysis. In the end, the adsorbed I2 molecule can further couple with another neighboring iodide ion to form a triiodide ion (I3−). Fig. 10e shows a profile of the potential energy surface (PES) describing 3I− → I2 + I− + 2e− → I3− + 2e− on the graphite surface. Based on the DFT analysis, the most stable adsorbed iodide ions (I−) can undergo dimerization to form the iodine (I2) molecule with a tiny barrier of 0.08 eV. Then, the adsorbed iodine is further coupled with the iodide ion to form the triiodide ion with an energy barrier of 0.12 eV. Moreover, the overall reaction is exothermic, with a reaction energy of −0.43 eV, indicating that it is a spontaneously favorable reaction. A schematic of the entire process is shown in Fig. 10f. In addition, there are two separate compartments in which iodine and hydrogen evolution reactions could occur. Thus, hydrogen gas is generated in the cathodic compartment, while iodine is generated in the anodic compartment via coupled VAARs-HER, which can be catalyzed by a single-atom Mo-N4/d-C electrocatalyst. For simultaneous iodide oxidation and HER, single-atom Mo-N4/d-C requires only 0.77 VRHE to reach the benchmark current density of 10 mA cm−2. This remarkable study has promoted research on green hydrogen generation via the IOR-assisted HER system.
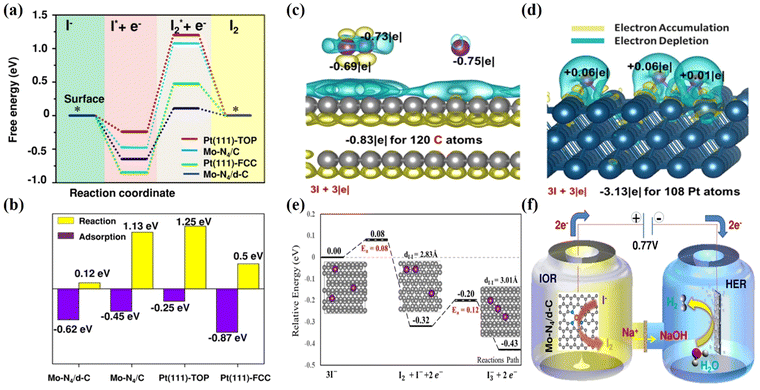 |
| | Fig. 10 Calculated DFT of the metal and metal free electrocatalysts for IOR. (a) The free energy diagram of Mo-N4/d-C. (b) Comparisons of iodide adsorption and iodine desorption on different catalyst surfaces.162 (c) Calaculated DFT of the metal-free based catalyst on IOR and the charge analysis between graphite and 3I− was performed in comparison with (d) that between Pt and 3I−. (e) The profiles of the potential energy surface (PES) for 3I−/I2 + I− +2e−/I3− + 2e− on the graphite surface.54 (f) Schematic diagram of IOR in a H-type double cell with a proton exchange Nafion membrane.162 | |
8. Summary and perspectives
This review has summarized and discussed recent advances in value-added electrolysis processes in which value-added anodic oxidation reactions are coupled with hydrogen evolution reaction for producing hydrogen and high-value chemicals while utilizing renewable energy. Meanwhile, recent advances in the rational design of electrocatalysts for value-added anodic oxidation reactions including iodide oxidation reaction (IOR), alcohol oxidation reaction (AOR), chitin oxidation reaction (COR), urea oxidation reaction (UOR), and hydrazine oxidation reaction (HzOR) are also reviewed and discussed.
The most pressing issue is to develop more value-added electrolysis processes and the corresponding electrocatalysts to help achieve the global goal of net-zero emissions by 2050. To bring the coupled system for value-added electrolysis (iodide oxidation reaction, poly-alcohol oxidation reaction, chitin oxidation reaction, urea oxidation reaction, and hydrazine oxidation reaction) with HER to a practical level, we must address the related technological, economic, and environmental issues, On top of that, the most important performance metrics, such as total current density, faradaic efficiency of different products, stability, and overpotential of value-added chemicals, should be reported for comparison of published works and predict the levelized cost of chemicals at various technology levels. Thus, in terms of technology and economics, a general techno-economic analysis model for coupled electrolysis is urgently required to include value-added anodic products and assess the economic feasibility of the developed system. Such a coupled electrolysis process drew more scientific attention in conjunction with cathodic HER, and we expect the same will be true for CO2RR, NRR, and ECH.
Another challenge is the rational design of value-added anodic electrocatalysts to improve the electroactivity, selectivity, and durability. This should include the electrocatalysts’ key thermodynamic and kinetic requirements and the costs for economically affordable large-scale applications. An optimized example could also minimize the subsequent separation costs and purification of byproducts from the targeted product. The activity could be achieved by designing multi-metallic single atom/nanoparticle electrocatalysts to provide more active and robust sites on the exposed surface. Bimetallic catalysts or a mixture of catalysts (single-atom, dual-atom, multimetallic single atom, and nanoclusters) could be promising candidates for various reactions in future value-added electrolysis processes, because their active sites are not easily poisoned by passivated oxides on the exposed surface due to the synergetic effect. Combining hetero-active sites and/or coordination environment design could provide insights into optimizing catalytic and structural active sites. Although value-added electrolysis is a green and sustainable approach, its research is still at the infant stage. Meanwhile, the anodic and cathodic reactions in value-added electrolysis processes are always very complex and involve many reaction pathways or intermediates. Improving their activity and targeted product selectivity is hard without understanding their reaction mechanisms. Many in situ spectroscopic and structural techniques such as X-ray absorption spectroscopy, Raman spectroscopy, IR spectroscopy, X-ray diffraction, etc. have been developed to investigate the reaction mechanisms of electrochemical reactions. However, a single in situ technique is probably insufficient to resolve their complex reaction mechanisms. The development of multiple in situ complementary techniques combined with computational approaches will be extremely important for investigating either anodic or cathodic reaction mechanisms for future value-added electrolysis processes.
The separation and purification of anodic value-added products is important for industrial applications. Generally, the target value-added product's separation and purification involves liquid–liquid extraction, distillation, recrystallization, and gas chromatography (GC). Importantly, one or multiple methods are usually used in combination during the practical operation process, according to the nature of the target value-added products (such as solubility, polarity, melting, and boiling points) required to be separated and purified. The most common separation strategies to separate the compounds of value-added products are liquid–liquid extraction163 and precipitation.54 Especially, the organic solvent extraction technique is based on the higher affinity of some compounds towards some solvents (depending on the polarity of the fraction and acid–base separation), which results in a phase transfer that allows the selective separation of value-added products. Most importantly, the cascade splitter is activated to use the products from the cathode as organic and inorganic raw materials for the anode. For example, methanol generated at the cathode may be used as a raw material for anode oxidation to produce formic acid. The solution is converted into target anode products through an oxidation reaction.
We believe that several approaches to achieve VAARs-HER/CO2RR coupling exist, including the incorporation of CO2RR into already existing technologies (for example, chloralkali and chlorate electrolysis, organic electrosynthesis (such as benzene to benzoquinone)), oxidation of industrial waste (“sacrificial agents”), the production of raw chemicals such as hydrogen peroxide in large quantities, and generation of fine chemicals. However, more effort is required to develop novel and optimized co-electrolysis systems capable of producing high-purity and high-concentration value-added products easily and economically.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the National Science and Technology Council (NSTC 111-2639-E-011-001-ASP, 111-2923-E-011-001; 111-2923-E-011-002; 111-3116-F-011-004, 111-3116-F-011-006), the Ministry of Education of Taiwan (MOE, U2RSC program 1080059), Academia Sinica (AS-KPQ-106-DDPP) as well as the supporting facilities from National Taiwan University of Science and Technology (NTUST), Instrumentation Center at National Taiwan Normal University (NTNU, NMR000400), and National Synchrotron Radiation Research Centre (NSRRC) are all gratefully acknowledged.
References
- I. Vincent and D. Bessarabov, Renew. Sust. Energ., 2018, 81, 1690–1704 CrossRef CAS.
- Y. Xu, M. Kraft and R. Xu, Chem. Soc. Rev., 2016, 45, 3039–3052 RSC.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo and L. A. Hackett, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- L. Chen, X. Dong, Y. Wang and Y. Xia, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1781 RSC.
- B. You and Y. Sun, ChemPlusChem, 2016, 81, 1045–1055 CrossRef CAS PubMed.
- Y. Zhao, M. Cai, J. Xian, Y. Sun and G. Li, J. Mater. Chem., 2021, 9, 20164–20183 RSC.
- X. Deng, X. Kang, M. Li, K. Xiang, C. Wang, Z. Guo, J. Zhang, X.-Z. Fu and J.-L. Luo, J. Mater. Chem., 2020, 8, 1138–1146 RSC.
- K. Xiang, Z. Song, D. Wu, X. Deng, X. Wang, W. You, Z. Peng, L. Wang, J.-L. Luo and X.-Z. Fu, J. Mater. Chem., 2021, 9, 6316–6324 RSC.
- N. P. Martínez, M. Isaacs and K. K. Nanda, New J. Chem., 2020, 44, 5617–5637 RSC.
- X. Liu, Y. Han, Y. Guo, X. Zhao, D. Pan, K. Li and Z. Wen, Adv. Sustain. Syst., 2022, 2200005 CAS.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- D. B. Adam, M.-C. Tsai, Y. A. Awoke, W.-H. Huang, Y.-W. Yang, C.-W. Pao, W.-N. Su and B. J. Hwang, ACS Sustainable Chem. Eng., 2021, 9, 8803–8812 CrossRef CAS.
- H. Zhao, D. Lu, J. Wang, W. Tu, D. Wu, S. W. Koh, P. Gao, Z. J. Xu, S. Deng and Y. Zhou, Nat. Commun., 2021, 12, 2008 CrossRef CAS PubMed.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- J. Wang, X. Li, M. Wang, T. Zhang, X. Chai, J. Lu, T. Wang, Y. Zhao and D. Ma, ACS Catal., 2022, 12, 6722–6728 CrossRef CAS.
- X. Liu, Z. Fang, D. Xiong, S. Gong, Y. Niu, W. Chen and Z. Chen, Nano Res., 2022, 1–9 Search PubMed.
- B. Li, K. Wang, J. Ren and P. Qu, New J. Chem., 2022, 46, 7615–7625 RSC.
- A. Serov and C. Kwak, Appl. Catal., B, 2010, 98, 1–9 CrossRef CAS.
- Y. Xu and B. Zhang, ChemElectroChem, 2019, 6, 3214–3226 CrossRef CAS.
- L. Chen and J. Shi, J. Mater. Chem., 2018, 6, 13538–13548 RSC.
- J. Xie, W. Liu, X. Zhang, Y. Guo, L. Gao, F. Lei, B. Tang and Y. Xie, ACS Mater. Lett., 2019, 1, 103–110 CrossRef CAS.
- S. Verma, S. Lu and P. J. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- S. S. Yazdani and R. Gonzalez, COBIOT, 2007, 18, 213–219 CAS.
- M. Simões, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106–2124 CrossRef PubMed.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 1–13 CrossRef CAS PubMed.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- L. Al-Ghussain, R. Samu, O. Taylan and M. Fahrioglu, Inventions, 2020, 5, 27 CrossRef.
- Z. Abdin and W. Mérida, Energy Convers. Manag., 2019, 196, 1068–1079 CrossRef.
- Á. Vass, B. Endrődi and C. Janáky, Curr. Opin. Electrochem., 2021, 25, 100621 CrossRef.
- B. Adhikari, C. J. Orme, J. R. Klaehn and F. F. Stewart, Sep. Purif. Technol., 2021, 268, 118703 CrossRef CAS.
- Sujata and Priyanka Kaushal, Biofuels, 2020, 11, 763–775 CAS.
-
T. M. Schmidt, Encyclopedia of microbiology, Academic Press, 2019 Search PubMed.
- D. A. Bulushev and J. R. Ross, ChemSusChem, 2018, 11, 821–836 CrossRef CAS PubMed.
- K.-E. Guima, L. M. Alencar, G. C. da Silva, M. A. Trindade and C. A. Martins, ACS Sustainable Chem. Eng., 2018, 6, 1202–1207 CrossRef CAS.
- M. Valter, M. Busch, B. R. Wickman, H. Gronbeck, J. Baltrusaitis and A. Hellman, J. Phys. Chem. C, 2018, 122, 10489–10494 CrossRef CAS.
- Y. Kwon, T. J. Hersbach and M. T. Koper, Top. Catal., 2014, 57, 1272–1276 CrossRef CAS.
- R. Ciriminna, A. Fidalgo, L. M. Ilharco and M. Pagliaro, ChemistryOpen, 2018, 7, 233–236 CrossRef CAS PubMed.
- S. Pugh, R. McKenna, I. Halloum and D. R. Nielsen, Metab. Eng. Commun., 2015, 2, 39–45 CrossRef PubMed.
- P. M. Lorz, F. K. Towae, W. Enke, R. Jäckh, N. Bhargava and W. Hillesheim, Ullmann’s Encycl. Ind. Chem., 2000 DOI:10.1002/14356007.a20_181.
- A. Bardow and M. Wessling, Nat. Energy, 2019, 4, 440–441 CrossRef.
- L. Du, Y. Sun and B. You, Mater. Rep.: Energy, 2021, 1, 100004 CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrog. Energy, 2013, 38, 4901–4934 CrossRef CAS.
- M. Yang, C. H. Zhang, N. W. Li, D. Luan, L. Yu and X. W. Lou, Adv. Sci., 2022, 9, 2105135 CrossRef CAS PubMed.
- M. S. Yusubov and V. V. Zhdankin, Resour.-Effic. Technol., 2015, 1, 49–67 Search PubMed.
- T. C. T. Pham, S. Docao, I. C. Hwang, M. K. Song, D. Moon, P. Oleynikov and K. B. Yoon, Energy Environ. Sci., 2016, 9, 1050–1062 RSC.
- D. Nacapricha, K. Uraisin, N. Ratanawimarnwong and K. Grudpan, Anal. Bioanal. Chem. A., 2004, 378, 816–821 CrossRef CAS PubMed.
- M. Zimmermann and F. Delange, Eur. J. Clin. Nutr., 2004, 58, 979–984 CrossRef CAS PubMed.
- X. Li, X. Cao, J. Li, J. Xu, W. Ma, H. Wang, J. Wang and Y. Zhang, J. Trace Elem. Med. Biol., 2020, 62, 126575 CrossRef CAS PubMed.
- H. De Santana and M. Temperini, J. Electroanal. Chem. Interfacial Electrochem., 1991, 316, 93–105 CrossRef CAS.
- D. B. Adam, M.-C. Tsai, Y. A. Awoke, W.-H. Huang, C.-H. Lin, T. Alamirew, A. A. Ayele, Y.-W. Yang, C.-W. Pao and W.-N. Su, Appl. Catal., B, 2022, 316, 121608 CrossRef CAS.
- T. A. Dessie, W.-H. Huang, D. B. Adam, Y. A. Awoke, C.-H. Wang, J.-L. Chen, C.-W. Pao, N. G. Habtu, M.-C. Tsai and W.-N. Su, Nano Lett., 2022, 22, 7311–7317 CrossRef CAS PubMed.
- S.-M. Peng, S. B. Patil, C.-C. Chang, S.-T. Chang, Y.-C. Chen, K.-C. Wu, W.-N. Su, B. J. Hwang and D.-Y. Wang, J. Mater. Chem., 2022, 10, 23982–23989 RSC.
- E. Hu, Y. Yao, Y. Chen, Y. Cui, Z. Wang and G. Qian, Nanoscale Adv., 2021, 3, 604–610 RSC.
- G. Toyooka and K. I. Fujita, ChemSusChem, 2020, 13, 3820–3824 CrossRef CAS PubMed.
- R. Mittal and S. K. Awasthi, ACS Sustainable Chem. Eng., 2022, 10, 1702–1713 CrossRef CAS.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS PubMed.
- D. L. Sanchez and D. M. Kammen, Nat. Energy, 2016, 1, 1–4 Search PubMed.
- Y. Chen, A. Lavacchi, H. Miller, M. Bevilacqua, J. Filippi, M. Innocenti, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 CrossRef CAS.
- X. Han, H. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. Qiu and S. Jin, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- M. Sutter, E. D. Silva, N. Duguet, Y. Raoul, E. Metay and M. Lemaire, Chem. Rev., 2015, 115, 8609–8651 CrossRef CAS PubMed.
- Z. Zhang, G. Liu, X. Cui, Y. Gong, D. Yi, Q. Zhang, C. Zhu, F. Saleem, B. Chen and Z. Lai, Sci. Adv., 2021, 7, eabd6647 CrossRef CAS PubMed.
- Y.-C. Qin, F.-Q. Wang, X.-M. Wang, M.-W. Wang, W.-L. Zhang, W.-K. An, X.-P. Wang, Y.-L. Ren, X. Zheng and D.-C. Lv, Rare Met., 2021, 40(9), 1–15 CrossRef.
- A. Marchionni, M. Bevilacqua, C. Bianchini, Y. X. Chen, J. Filippi, P. Fornasiero, A. Lavacchi, H. Miller, L. Wang and F. Vizza, ChemSusChem, 2013, 6, 518–528 CrossRef CAS PubMed.
- Y. Qin, W. Zhang, F. Wang, J. Li, J. Ye, X. Sheng, C. Li, X. Liang, P. Liu and X. Wang, Angew. Chem., Int. Ed., 2022, 134, e202200899 Search PubMed.
- V. Bambagioni, M. Bevilacqua, C. Bianchini, J. Filippi, A. Marchionni, F. Vizza, L. Wang and P. Shen, Fuel Cells, 2010, 10, 582–590 CrossRef CAS.
- L. Demarconnay, S. Brimaud, C. Coutanceau and J.-M. Léger, J. Electroanal. Chem., 2007, 601, 169–180 CrossRef CAS.
- J. González-Cobos, S. Baranton and C. Coutanceau, ChemElectroChem, 2016, 3, 1694–1704 CrossRef.
- Y. Chen, A. Lavacchi, H. Miller, M. Bevilacqua, J. Filippi, M. Innocenti, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, Nat. Commun., 2014, 5, 4036 CrossRef CAS PubMed.
- D. Si, B. Xiong, L. Chen and J. Shi, Chem. Catal., 2021, 1, 941–955 CrossRef CAS.
- T. Yoshioka, T. Sato and A. Okuwaki, J. Appl. Polym. Sci., 1994, 52, 1353–1355 CrossRef CAS.
- H. Kurokawa, M.-a Ohshima, K. Sugiyama and H. Miura, Polym. Degrad. Stab., 2003, 79, 529–533 CrossRef CAS.
- L. Wang, G. A. Nelson, J. Toland and J. D. Holbrey, ACS Sustainable Chem. Eng., 2020, 8, 13362–13368 CrossRef CAS.
- S. Ügdüler, K. M. Van Geem, R. Denolf, M. Roosen, N. Mys, K. Ragaert and S. De Meester, Green Chem., 2020, 22, 5376–5394 RSC.
- V. Tournier, C. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M.-L. Desrousseaux, H. Texier and S. Gavalda, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- E. A. Moges, C. Y. Chang, W. H. Huang, K. Lakshmanan, Y. A. Awoke, C. W. Pao, M. C. Tsai, W. N. Su and B. J. Hwang, Adv. Funct. Mater., 2022, 32, 2206887 CrossRef CAS.
- A. A. Ayele, M.-C. Tsai, D. B. Adam, Y. A. Awoke, W.-H. Huang, C.-Y. Chang, S.-C. Liao, P.-Y. Huang, J.-L. Chen and C.-W. Pao, Appl. Catal., A, 2022, 646, 118861 CrossRef CAS.
- X. Liu, Z. Fang, D. Xiong, S. Gong, Y. Niu, W. Chen and Z. Chen, Nano Res., 2022, 1–9 Search PubMed.
- R. Shi, K.-S. Liu, F. Liu, X. Yang, C.-C. Hou and Y. Chen, Chem. Comm., 2021, 57, 12595–12598 RSC.
- J. H. Clark, V. Budarin, F. E. Deswarte, J. J. Hardy, F. M. Kerton, A. J. Hunt, R. Luque, D. J. Macquarrie, K. Milkowski and A. Rodriguez, Green Chem., 2006, 8, 853–860 RSC.
- Y. C. Lee, S. Dutta and K. C. W. Wu, ChemSusChem, 2014, 7, 3241–3246 CrossRef CAS PubMed.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed.
- F. M. Kerton, Y. Liu, K. W. Omari and K. Hawboldt, Green Chem., 2013, 15, 860–871 RSC.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- Y. D. Chiang, S. Dutta, C. T. Chen, Y. T. Huang, K. S. Lin, J. C. Wu, N. Suzuki, Y. Yamauchi and K. C. W. Wu, ChemSusChem, 2015, 8, 789–794 CrossRef CAS PubMed.
- S. Kaur and G. S. Dhillon, Crit. Rev. Biotechnol., 2015, 35, 44–61 CrossRef CAS PubMed.
- X. Gao, X. Chen, J. Zhang, W. Guo, F. Jin and N. Yan, ACS Sustainable Chem. Eng., 2016, 4, 3912–3920 CrossRef CAS.
- N. Yoneda, S. Kusano, M. Yasui, P. Pujado and S. Wilcher, Appl. Catal., 2001, 221, 253–265 CrossRef CAS.
- A. Jardine and S. Sayed, Curr. Opin. Green Sustain. Chem., 2016, 2, 34–39 CrossRef.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- B. K. Boggs, R. L. King and G. G. Botte, Chem. Commun., 2009, 4859–4861 RSC.
- G. Wang, J. Chen, Y. Li, J. Jia, P. Cai and Z. Wen, Chem. Commun., 2018, 54, 2603–2606 RSC.
- L. E. Krausfeldt, A. T. Farmer, H. F. Castro Gonzalez, B. N. Zepernick, S. R. Campagna and S. W. Wilhelm, Front. Microbiol., 2019, 10, 1064 CrossRef PubMed.
- J. Du, D. Xiang, K. Zhou, L. Wang, J. Yu, H. Xia, L. Zhao, H. Liu and W. Zhou, Nano Energy, 2022, 107875 CrossRef CAS.
- R. P. Forslund, J. T. Mefford, W. G. Hardin, C. T. Alexander, K. P. Johnston and K. J. Stevenson, ACS Catal., 2016, 6, 5044–5051 CrossRef CAS.
- S.-K. Geng, Y. Zheng, S.-Q. Li, H. Su, X. Zhao, J. Hu, H.-B. Shu, M. Jaroniec, P. Chen and Q.-H. Liu, Nat. Energy, 2021, 6, 904–912 CrossRef CAS.
- L. Guo, J. Chi, J. Zhu, T. Cui, J. Lai and L. Wang, Appl. Catal., 2023, 320, 121977 CrossRef CAS.
- H. Jiang, M. Sun, S. Wu, B. Huang, C. S. Lee and W. Zhang, Adv. Funct. Mater., 2021, 31, 2104951 CrossRef CAS.
- Q. Zong, H. Yang, Q. Wang, Q. Zhang, J. Xu, Y. Zhu, H. Wang, H. Wang, F. Zhang and Q. Shen, Dalton Trans., 2018, 47, 16320–16328 RSC.
- F. Chen, F. Yang, C. Sheng, J. Li, H. Xu, Y. Qing, S. Chen, Y. Wu and X. Lu, J. Colloid Interface Sci., 2022, 626, 445–452 CrossRef CAS PubMed.
- L. Wang, Y. Zhu, Y. Wen, S. Li, C. Cui, F. Ni, Y. Liu, H. Lin, Y. Li and H. Peng, Angew. Chem., Int. Ed., 2021, 133, 10671–10676 CrossRef.
- F. Chen, Y. Wu, C. Sheng, H. Xu, Y. Qing, J. Li, S. Chen and Y. Wu, Colloids Surf., A, 2022, 651, 129695 CrossRef CAS.
- L. Wang, Y. Zhu, Y. Wen, S. Li, C. Cui, F. Ni, Y. Liu, H. Lin, Y. Li and H. Peng, Angew. Chem., 2021, 133, 10671–10676 CrossRef.
- F. Sun, J. Qin, Z. Wang, M. Yu, X. Wu, X. Sun and J. Qiu, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- Y. Zhu, J. Zhang, Q. Qian, Y. Li, Z. Li, Y. Liu, C. Xiao, G. Zhang and Y. Xie, Angew. Chem., Int. Ed., 2022, 61, e202113082 CAS.
- Y. Li, X. Yu, J. Gao and Y. Ma, J. Mater. Chem., 2023, 11, 2191–2202 RSC.
- S. Zhao, L. Wang, T. Wang, Q. Han and S. Xu, Appl. Surf. Sci., 2016, 369, 36–42 CrossRef CAS.
- W. Liu, T. Shi and Z. Feng, J. Colloid Interface Sci., 2023, 630, 888–899 CrossRef CAS PubMed.
- T. Wang, Q. Wang, Y. Wang, Y. Da, W. Zhou, Y. Shao, D. Li, S. Zhan, J. Yuan and H. Wang, Angew. Chem., 2019, 131, 13600–13605 CrossRef.
- L. Zhou, M. Shao, C. Zhang, J. Zhao, S. He, D. Rao, M. Wei, D. G. Evans and X. Duan, Adv. Mater., 2017, 29, 1604080 CrossRef PubMed.
- S. Ge, L. Zhang, J. Hou, S. Liu, Y. Qin, Q. Liu, X. Cai, Z. Sun, M. Yang and J. Luo, ACS Appl. Energy Mater., 2022, 5, 9487–9494 CrossRef CAS.
- Y. Li, X. Wei, L. Chen, J. Shi and M. He, Nat. Commun., 2019, 10, 1–12 CrossRef PubMed.
- Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li and G. Zhang, Adv. Funct. Mater., 2021, 31, 2103673 CrossRef CAS.
- L. Zhou, M. Shao, C. Zhang, J. Zhao, S. He, D. Rao, M. Wei, D. G. Evans and X. Duan, Adv. Mater., 2017, 29, 1604080 CrossRef PubMed.
- Z. Wang, L. Xu, F. Huang, L. Qu, J. Li, K. A. Owusu, Z. Liu, Z. Lin, B. Xiang and X. Liu, Adv. Energy Mater., 2019, 9, 1900390 CrossRef.
- C. Zhu, A. L. Wang, W. Xiao, D. Chao, X. Zhang, N. H. Tiep, S. Chen, J. Kang, X. Wang and J. Ding, Adv. Energy Mater., 2018, 30, 1705516 CrossRef PubMed.
- L. Guo, Q. Yu, X. Zhai, J. Chi, T. Cui, Y. Zhang, J. Lai and L. Wang, Nano Res., 2022, 15, 8846–8856 CrossRef CAS.
- M. K. Birhanu, M.-C. Tsai, C.-T. Chen, A. W. Kahsay, T. S. Zeleke, K. B. Ibrahim, C.-J. Huang, Y.-F. Liao, W.-N. Su and B. J. Hwang, Electrochim. Acta, 2020, 356, 136756 CrossRef CAS.
- C. H. Chen, C. J. Pan, W. N. Su, L. S. Sarma, C. C. A. Andra, H. S. Sheu, D. G. Liu, J. F. Lee and B. J. Hwang, ChemNanoMat, 2016, 2, 117–124 CrossRef CAS.
- B.-J. Hwang, L. S. Sarma, J.-M. Chen, C.-H. Chen, S.-C. Shih, G.-R. Wang, D.-G. Liu, J.-F. Lee and M.-T. Tang, J. Am. Chem. Soc., 2005, 127, 11140–11145 CrossRef CAS PubMed.
- J. Fichtner, B. Garlyyev, S. Watzele, H. A. El-Sayed, J. N. Schwämmlein, W.-J. Li, F. D. R. M. Maillard, L. Dubau, J. Michalička and J. M. Macak, ACS Appl. Mater. Interfaces, 2019, 11, 5129–5135 CrossRef CAS PubMed.
- S. Anantharaj, S. Ede, K. Sakthikumar, K. Karthick, S. Mishra, S. Kundu and A. Catal, Adv. Mater., 2016, 28, 9266–9291 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- B. Garlyyev, K. Kratzl, M. Rück, J. Michalička, J. Fichtner, J. M. Macak, T. Kratky, S. Günther, M. Cokoja and A. S. Bandarenka, Angew. Chem., Int. Ed., 2019, 58, 9596–9600 CrossRef CAS PubMed.
- J. Shan, Y. Zheng, B. Shi, K. Davey and S.-Z. Qiao, ACS Energy Lett., 2019, 4, 2719–2730 CrossRef CAS.
- K.-i Otake, Y. Cui, C. T. Buru, Z. Li, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 8652–8656 CrossRef CAS PubMed.
- Q. Xue, Z. Ge, Z. Yuan, J. Huang, B. He and Y. Chen, Mater. Today Physics, 2023, 100980 CrossRef CAS.
- T. Sreekanth, R. Ramaraghavulu, S. P. Vattikuti, J. Shim and K. Yoo, Mater. Lett., 2019, 253, 450–453 CrossRef CAS.
- V. K. Singh, U. T. Nakate, P. Bhuyan, J. Chen, D. T. Tran and S. Park, J. Mater. Chem., 2022, 10, 9067–9079 RSC.
- K. Sivula, R. Zboril, F. Le Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Gratzel, J. Am. Chem. Soc., 2010, 132, 7436–7444 CrossRef CAS PubMed.
-
Y. Jia, T. Zhang, M. Liu, J. Zhang and Y. Han, J. Phys. Chem. C. 2022.
- L. Yu, L. Wu, B. McElhenny, S. Song, D. Luo, F. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2020, 13, 3439–3446 RSC.
- X. Zhao, L. Dai, Q. Qin, F. Pei, C. Hu and N. Zheng, Small, 2017, 13, 1602970 CrossRef PubMed.
- L. Huang, X. Zhang, Q. Wang, Y. Han, Y. Fang and S. Dong, J. Am. Chem. Soc., 2018, 140, 1142–1147 CrossRef CAS PubMed.
- Y. Hao, D. Yu, S. Zhu, C.-H. Kuo, Y.-M. Chang, L. Wang, H.-Y. Chen, M. Shao and S. Peng, Energy Environ. Sci., 2023, 16, 1100–1110 RSC.
- K. Xiang, D. Wu, X. Deng, M. Li, S. Chen, P. Hao, X. Guo, J. L. Luo and X. Z. Fu, Adv. Funct. Mater., 2020, 30, 1909610 CrossRef CAS.
- S. Feng, J. Luo, J. Li, Y. Yu, Z. Kang, W. Huang, Q. Chen, P. Deng, Y. Shen and X. Tian, Mater. Today Phys., 2022, 23, 100646 CrossRef CAS.
- T. Wang, X. Cao, H. Qin, X. Chen, J. Li and L. Jiao, J. Mater. Chem., 2021, 9, 21094–21100 RSC.
- C. Han, J. Zenner, J. Johny, N. Kaeffer, A. Bordet and W. Leitner, Nat. Catal., 2022, 5, 1110–1119 CrossRef CAS.
- Y. A. Awoke, M.-C. Tsai, D. B. Adam, A. A. Ayele, S.-C. Yang, W.-H. Huang, J.-L. Chen, C.-W. Pao, C. Y. Mou and W.-N. Su, Electrochim. Acta, 2022, 432, 141161 CrossRef CAS.
- Y. Li, X. Wei, S. Han, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 21464–21472 CrossRef CAS PubMed.
- B. Liu, G. Wang, X. Feng, L. Dai, Z. Wen and S. Ci, Nanoscale, 2022, 14, 12841–12848 RSC.
- K. Wang, W. Huang, Q. Cao, Y. Zhao, X. Sun, R. Ding, W. Lin, E. Liu and P. Gao, J. Chem. Eng., 2022, 427, 130865 CrossRef CAS.
- J. Wang, Z. Zhao, C. Shen, H. Liu, X. Pang, M. Gao, J. Mu, F. Cao and G. Li, Catal. Sci. Technol., 2021, 11, 2480–2490 RSC.
- T. Wang, X. Cao and L. Jiao, EScience, 2021, 1, 69–74 CrossRef.
- K. Li, B. Xie, D. Feng and Y. Tong, ChemSusChem, 2022, 15, e202201656 CAS.
- C. Li, Y. Liu, Z. Zhuo, H. Ju, D. Li, Y. Guo, X. Wu, H. Li and T. Zhai, Adv. Energy Mater., 2018, 8, 1801775 CrossRef.
- Y. Zhao, N. Jia, X.-R. Wu, F.-M. Li, P. Chen, P.-J. Jin, S. Yin and Y. Chen, Appl. Catal., 2020, 270, 118880 CrossRef CAS.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737–13783 RSC.
- P. Zhai, Y. Zhang, Y. Wu, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, L. Sun and J. Hou, Nat. Commun., 2020, 11, 5462 CrossRef CAS PubMed.
- K. B. Ibrahim, M. C. Tsai, S. A. Chala, M. K. Berihun, A. W. Kahsay, T. A. Berhe, W. N. Su and B. J. Hwang, JCCS, 2019, 66, 829–865 CAS.
- Y. Zhao, D. P. Adiyeri Saseendran, C. Huang, C. A. Triana, W. R. Marks, H. Chen, H. Zhao and G. R. Patzke, Chem. Rev., 2023, 123, 6257–6358 CrossRef CAS PubMed.
- M. J. Craig, G. Coulter, E. Dolan, J. Soriano-López, E. Mates-Torres, W. Schmitt and M. García-Melchor, Nat. Commun., 2019, 10, 4993 CrossRef PubMed.
- D. W. Shaffer, Y. Xie and J. J. Concepcion, Chem. Soc. Rev., 2017, 46, 6170–6193 RSC.
- D. García-Osorio, R. Jaimes, J. Vazquez-Arenas, R. Lara and J. Alvarez-Ramirez, J. Electrochem. Soc., 2017, 164, E3321 CrossRef.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Z. Li, Y. Yan, S.-M. Xu, H. Zhou, M. Xu, L. Ma, M. Shao, X. Kong, B. Wang and L. Zheng, Nat. Commun., 2022, 13, 147 CrossRef CAS PubMed.
- T. A. Dessie, W.-H. Huang, D. B. Adam, Y. A. Awoke, C.-H. Wang, J.-L. Chen, C.-W. Pao, N. G. Habtu, M.-C. Tsai and W.-N. Su, Nano Lett., 2022, 22, 7311–7317 CrossRef CAS PubMed.
- K. Drugkar, W. Rathod, T. Sharma, A. Sharma, J. Joshi, V. K. Pareek, L. Ledwani and U. Diwekar, Sep. Purif. Technol., 2022, 283, 120149 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Chia-Yu
Chang
a,
Meng-Che
Tsai
*ad,
Wei-Nien
Su
a,
Chia-Yu
Chang
a,
Meng-Che
Tsai
*ad,
Wei-Nien
Su