
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metadynamics simulations of ligands binding to protein surfaces: a novel tool for rational drug design†
Ke
Zuo
 abcd,
Agata
Kranjc
a,
Riccardo
Capelli
e,
Giulia
Rossetti
afg,
Rachel
Nechushtai
c and
Paolo
Carloni
*abh
abcd,
Agata
Kranjc
a,
Riccardo
Capelli
e,
Giulia
Rossetti
afg,
Rachel
Nechushtai
c and
Paolo
Carloni
*abh
aComputational Biomedicine, Institute of Advanced Simulation IAS-5 and Institute of Neuroscience and Medicine INM-9, Forschungszentrum Jülich GmbH, Jülich 52425, Germany. E-mail: p.carloni@fz-juelich.de
bDepartment of Physics, RWTH Aachen University, Aachen 52074, Germany
cThe Alexander Silberman Institute of Life Science, The Hebrew University of Jerusalem, Edmond J. Safra Campus at Givat Ram, Jerusalem 91904, Israel
dDepartment of Physics, Università degli Studi di Ferrara, Ferrara 44121, Italy
eDepartment of Biosciences, Università degli Studi di Milano, Via Celoria 26, Milan 20133, Italy
fJülich Supercomputing Center (JSC), Forschungszentrum Jülich GmbH, Jülich 52425, Germany
gDepartment of Neurology, Faculty of Medicine, RWTH Aachen University, Aachen 52074, Germany
hJARA Institute: Molecular Neuroscience and Imaging, Institute of Neuroscience and Medicine INM-11, Forschungszentrum Jülich GmbH, Jülich 52425, Germany
First published on 5th May 2023
Abstract
Structure-based drug design protocols may encounter difficulties to investigate poses when the biomolecular targets do not exhibit typical binding pockets. In this study, by providing two concrete examples from our labs, we suggest that the combination of metadynamics free energy methods (validated against affinity measurements), along with experimental structural information (by X-ray crystallography and NMR), can help to identify the poses of ligands on protein surfaces. The simulation workflow proposed here was implemented in a widely used code, namely GROMACS, and it could straightforwardly be applied to various drug-design campaigns targeting ligands’ binding to protein surfaces.
1. Introduction
Ligands targeting protein surfaces (LTPS) may exert their beneficial pharmacological action in several ways. For instance, they may act by directly affecting protein/protein interactions1–5 and/or as allosteric ligands, i.e. by altering the structures of receptors’ binding sites, these ligands may increase drug or substrate affinity.6–9 Also, they may affect protein reactivity, preventing, for instance, the release of prosthetic groups.10 Unfortunately, LTPS often bind in the micromolar range, which is considered too low for classical pharmaceutical applications. This is calling for the medicinal chemists to adapt other experimental and/or computational approaches to the identification of high-affinity LTPS. This issue is however far from trivial. On the one hand, the power of drug-screening approaches based on machine learning (ML) to improve the potency of ligands with micromolar affinity might be limited due to the rather small datasets. An analysis of the Protein Data Bank (PDB) (https://www.rcsb.org/) showed that only ∼0.3% of the 190![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 protein hits featured complexes with LTPS whose binding affinity data have been measured. On the other hand, rational drug design is also not void of problems. The overwhelming majority of these LTPS/protein structures are determined by X-ray crystallography. In the solid state, the poses from LTPS might differ significantly from those in solution as high-energy intermolecular contacts between LTPS and the protein can be artificially stabilized by a protein's crystal packing.11–13 This situation differs significantly from ligands’ poses in receptors’ and enzymes’ cavities, which are often very similar to those in solution.14–16
000 protein hits featured complexes with LTPS whose binding affinity data have been measured. On the other hand, rational drug design is also not void of problems. The overwhelming majority of these LTPS/protein structures are determined by X-ray crystallography. In the solid state, the poses from LTPS might differ significantly from those in solution as high-energy intermolecular contacts between LTPS and the protein can be artificially stabilized by a protein's crystal packing.11–13 This situation differs significantly from ligands’ poses in receptors’ and enzymes’ cavities, which are often very similar to those in solution.14–16
The NMR structures of LTPS/protein complexes in the PDB are only a fraction of those solved by X-ray (less than 0.01% of the PDB). However, NMR may also provide useful information at the qualitative level in case it is not able to determine the structure; furthermore, the data coming from NMR experiments intrinsically contain the fluctuations of the relevant movements of the system. This information can be rationalized, for instance, by monitoring the changes in chemical shifts of a protein upon ligand binding in aqueous solution.17–19 Finally, computer-aided drug design also presents difficulties when addressing LTPS/protein complexes: the lack of deep binding pockets makes it difficult to efficiently use molecular docking protocols and high-throughput screening - the first steps of the “in silico” design of new potential therapeutic ligands.20
Here we underline a general way to address the unique binding mode of LTPS. We propose a combination of advanced molecular simulation methods, such as metadynamics,21–23 as a possible route to accurately identify LTPS poses on the target proteins in an aqueous solution, if the X-ray structures or NMR information is available. Metadynamics is an excellent method to be used here as it allows efficiently predicting the complex free energy landscape associated with ligand binding as a function of several collective variables (CVs) (for instance, Section 2 uses three CVs). There are several other excellent methods able to calculate free energy profiles (such as umbrella sampling), but metadynamics is more cost-effective than umbrella sampling, and as a purely explorative approach (especially in ligand binding), it is one of the few viable techniques.
In our proposed protocol, the simulation predicts a ligand's pose and potency, while the experiment validates the model. To illustrate how this works in practice, we consider two examples from our research. First, we compare our predictions with accurate X-ray crystallography data, to provide an insight into the drug binding modes on passing from the solid state to solution. We focus here on the human nutrient deprivation autophagy factor-1 (NAF-1) protein target for a variety of diseases, including cancer (work performed here). The second exploits the qualitative NMR information to predict the poses of a ligand on the prion protein, as described in ref. 24. The results are validated by comparing the calculated free energy of binding with the corresponding experimental values. In both cases, our prediction suggested multiple poses for the association of the ligands with the proteins in an aqueous solution. The latter resulted in a quite complex free energy landscape of ligand binding, with multiple minima separated by small barriers. The protocol can be straightforwardly extended to other ligand/proteins complexes having similar characteristics of ligand binding and available experimental structural information.
2. NAF-1 case study
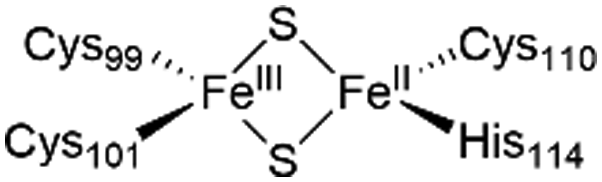
The NAF-1 NEET protein contains two [2Fe–2S] clusters, which are ligated by a unique 3Cys:1His coordination through the two iron atoms (Fig. 1a).10 The clusters exist in an oxidized state, with two ferric iron ions (Fe(III)), and in a reduced state, in which the His-bound iron is reduced (Fe(II)).25,26 The His residue confers the cluster lability, and the clusters can be transferred to a variety of acceptor proteins.10 An abnormal expression of NAF-1 was found to be involved in various neurological disorders, e.g. Alzheimer's disease,27 and to be the cause of Wolfram Syndrome type 2.28,29 Thus, ligands binding NAF-1 may disrupt cluster release; hence, their development can be a powerful therapy against various diseases.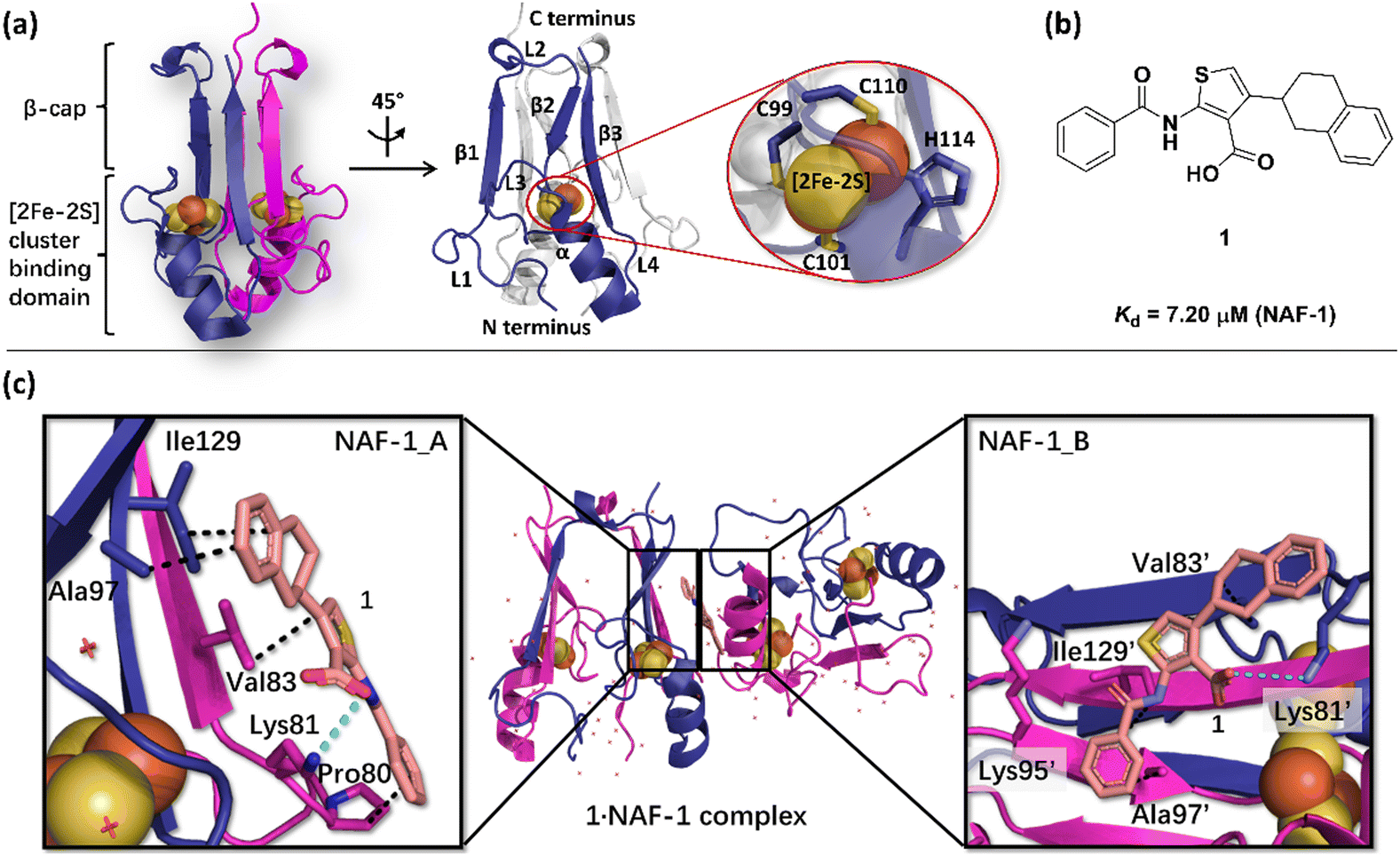 | ||
| Fig. 1 X-Ray structure of NAF-1, and its complex with ligand 1. (a) The protein (PDB ID: 4OO732) is a homodimer. Each monomer (shown in deep blue and magenta, respectively) contains a [2Fe–2S] cluster with a 3Cys:1His coordination (Cys99, Cys101, Cys110, and His114) and a beta cap. (b) Chemical structure of ligand 1. (c) 1·NAF-1 complex X-ray structure (PDB ID: 7P0P33). The ligand, shown as a wheat stick model, is sandwiched between two adjacent proteins in the crystal unit cell, coloured as in (a). | ||
The NAF-1 protein is anchored by two α-helices to the outer membrane of mitochondria (OMM), the endoplasmic reticulum (ER) membrane, and the ER-mitochondria-associated membrane (MAM).30–32 The protein is a homodimer, with each monomer composed of a β-cap and a [2Fe–2S] cluster binding domain. The topology of the second structural domain is in the order of L1-β1-L2-β2-L3-α-L4-β3 (L: loop, β: β sheet). Each monomer also contains a [2Fe–2S] cluster, of which one Fe is coordinated by two cysteines (Cys99 and Cys101) buried inside the protein and the other one is coordinated by one cysteine (Cys110) and one histidine (His114) on the surface of each monomer (Fig. 1a).32 The two monomers also form hydrophobic interactions with each other.32 The iron-bound histidine His114 can exist in a protonated or deprotonated form according to the protonation state of the Nε of the imidazole ring. Thus, NAF-1 can exist in four different states at physiological pH (states I–IV described in Table 1). For state I, the experimental ligand binding free energy has been measured by isothermal titration calorimetry at pH 8.0.33
| State of NAF-1 | Redox state of [2Fe–2S] | Protonation of Nε@His114 | Lowest binding free energy (kcal mol−1) |
|---|---|---|---|
| I |
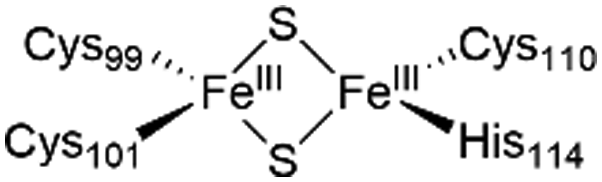
|
Deprotonated | −7.4 ± 0.8 |
| II |
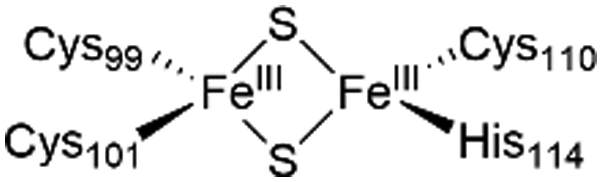
|
Protonated | −6.1 ± 0.8 |
| III |
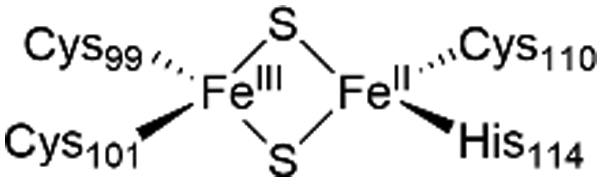
|
Deprotonated | −6.7 ± 0.8 |
| IV |
|
Protonated | −5.8 ± 0.8 |
Ligand 1 (2-benzamido-4-(1,2,3,4-tetrahydronaphthalen-2-yl)-thiophene-3-carboxylate), binds to the NAF-1 surface with a relatively low binding affinity (Kd = 7.20 μM, Fig. 1b)‡. The X-ray structure of the 1·NAF-1 complex33 shows that (i) the ligand's tetralin ring forms hydrophobic interactions with Ala97, Ile129 of one protein, and with Val83′ of another, adjacent protein – its residues are indicated with a prime (′) symbol; (ii) the ligand's phenyl forms hydrophobic interactions with Pro80, Ala97′, and Ile129; (iii) the carboxylate group forms salt bridges with Lys81 and Lys81′ (Fig. 1c).
We used volume-based well-tempered metadynamics,21 an exact method to reconstruct the free energy landscape associated with 1 bound to human NAF-1 in an aqueous solution at physiological temperature (310 K). The free energy was calculated as a function of three collective variables, using the same computational protocol as in ref. 11 (see ESI† for computational details). Our calculations were based on an educated guess of the 1·NAF-1 complex structure and then compared with the experimental structure of the complex.33
The calculated lowest free energy of binding in the state I, where the Fe ions are in their oxidized states (Table 1), was in good agreement with the experimental value (−6.6 kcal mol−133), considering that the latter was obtained at a lower temperature. In the global minimum 1 (Fig. 2), ligand 1's phenyl moiety forms hydrophobic interactions with Ile86 and Leu91, and polar van der Waals (vdW) interactions with Asp90, Asn87, and Glu85. The carbonyl oxygen forms an H-bond with Tyr98, whereas the thiophene ring and tetralin ring form hydrophobic interactions with Ala109, Pro108, and Phe107, Thr106, respectively. In minimum 2 (−4.7 ± 0.8 kcal mol−1, Fig. 2), higher by 2.7 kcal mol−1 from the minimum 1, the tetralin group forms a π-cation interaction with Lys116, the thiophene ring forms hydrophobic interactions with Leu120, and the carboxylate oxygens interacts with Glu119 through a water molecule. In minimum 3 (−3.5 ± 0.8 kcal mol−1, Fig. 2), higher than minimum 1 by 3.9 kcal mol−1, ligand 1 binds to the L2 loop. Its phenyl group forms metastable interactions with Ser92, the thiophene ring with Leu93, Thr94, and the tetralin ring forms metastable interactions with Lys95 (see Fig. 2). The binding of the other three states is reported in the ESI.† Multiple binding poses could be identified in all cases, with a free energy of binding slightly lower (in absolute values) than that of state I (see ESI†).
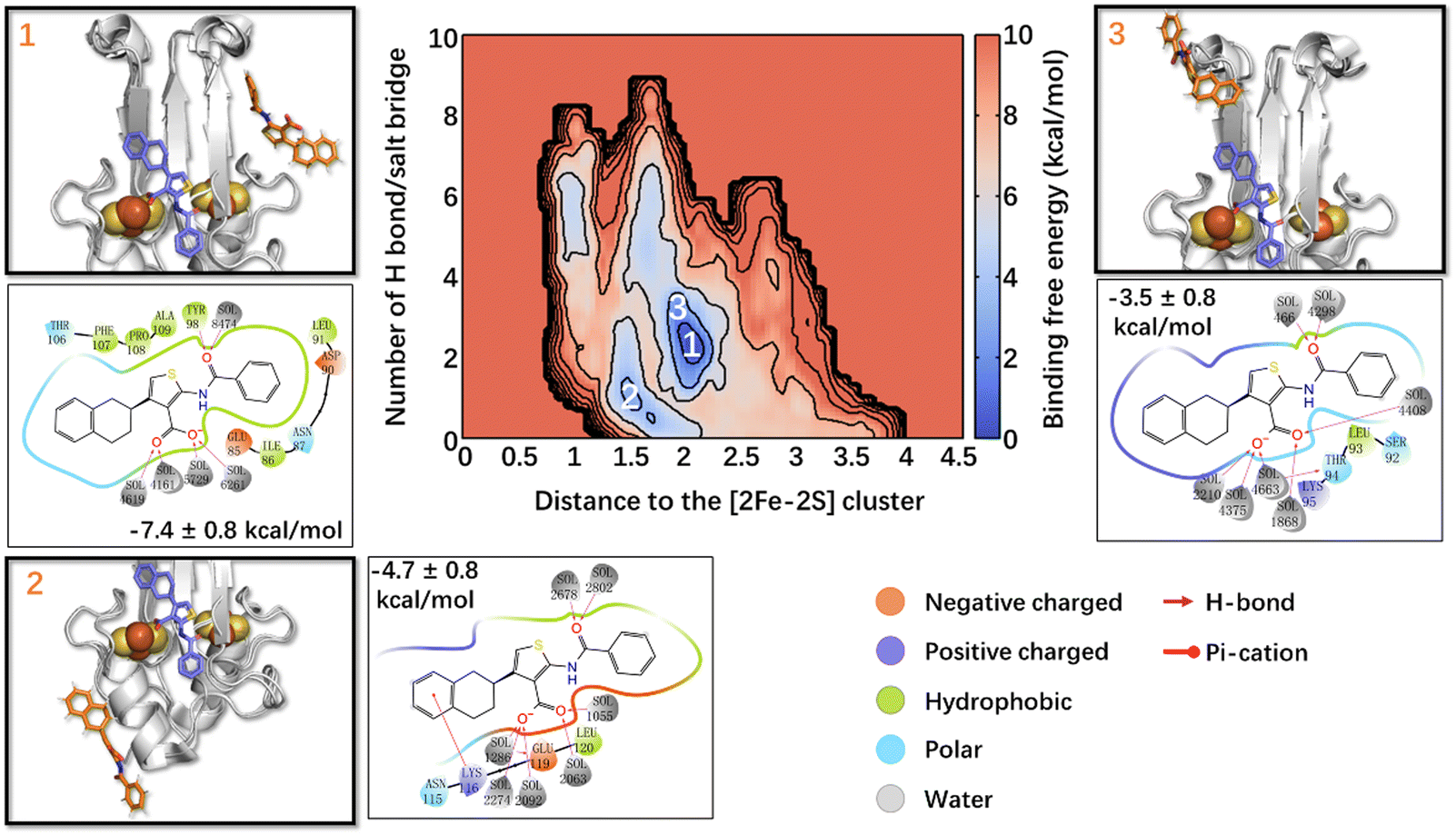 | ||
| Fig. 2 Free energy landscape associated with ligand 1 binding to NAF-1(I). The crystal binding pose is shown in the violet sticks as a reference, while the calculated/simulated representative binding poses of the three deepest free energy local minima (panels 1–3) are shown in orange. The ligand/protein contacts are also shown. | ||
In summary, our simulations could fairly reproduce the binding affinity of ligand 1 to human NAF-1 in aqueous solutions. Ligand 1 turns out to bind to the [2Fe–2S] cluster region differently from the X-ray structure. It also binds to the L1/L3 and L2 loops. We attribute this difference, at least in part, to the different binding properties of the drug on passing from the solid state to the solution, where roughly half of the drug's contacts are replaced by the solvent.§ Finally, the redox state of the [2Fe–2S] clusters and the protonation states of the Fe-bound histidine residues have a significant impact on the ligand binding free energies and poses (See ESI†).
3. Prion protein case study
The prion protein is present ubiquitously in the animal kingdom.34 In prion diseases (Creutzfeldt–Jakob disease, bovine spongiform encephalopathy, chronic wasting disease, and scrapie),35,36 it transforms from its native state PrPC (the alpha-helix protein shown in Fig. 3a) to a beta-rich pathogenic form PrPSc (scrapie prion protein). The latter oligomerizes, leading to large fibrillar aggregates, leading to pathological changes of the brain.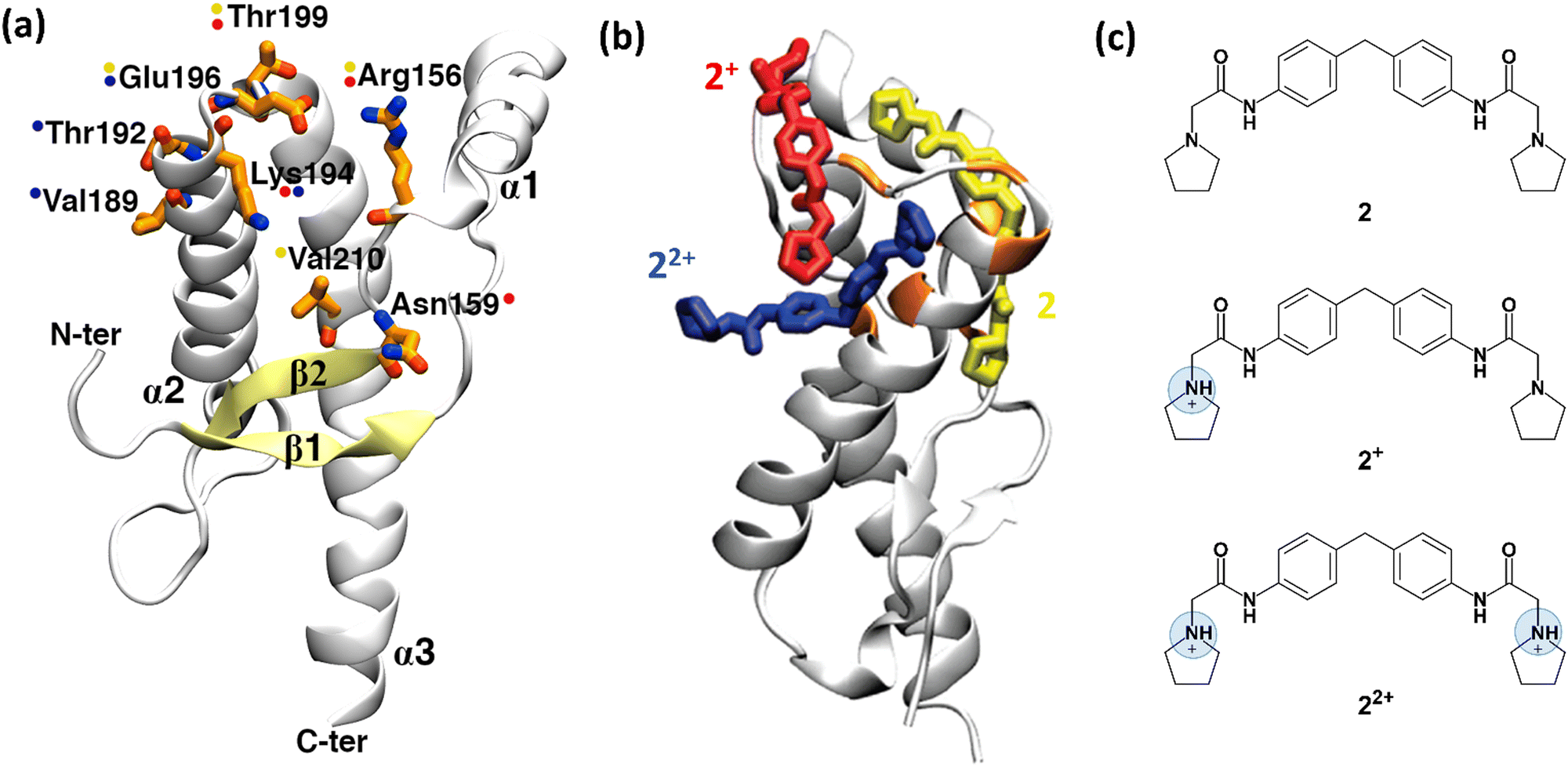 | ||
| Fig. 3 Structure of human prion protein and ligand 2. (a) The structured part of the human PrPC protein (Leu125-Arg228) is composed of three α-helices and a β-sheet (PDB ID: 1HJM41). Residues, shown by NMR experiments as being involved in binding are depicted in orange sticks. Coloured spots next to the residues show which representative 2 protomer (shown in (b)) interacts with them after the metadynamics simulations. (b) The representative binding poses of the three 2 protomers with human PrPC protein resulting from metadynamics. Uncharged protomer 2 is shown as yellow sticks; 2+ and 22+ are shown in red and blue colours, respectively. (c) Chemical structure of ligand 2. At acidic pH, at which the NMR experiments were performed,37 except uncharged 2, the ligand can also be present in two other ionic forms, 2+ and 22+. Fig. 3a and b were adapted and reprinted, respectively, from ref. 24. Copyright 2009, American Chemical Society (ACS). | ||
Ligands binding to the protein may interfere with the interconversion to the scrapie form. Changes in 1H-15N HSQC (heteronuclear single quantum coherence) NMR spectra of the protein have established that ligands such as ligand 2 (2-pyrrolidin-1-yl-N-[4-[4-(2-pyrrolidin-1-yl-acetylamino)-benzyl]-phenyl]-acetamide, Fig. 3c)¶ bind to the surface of the mouse prion protein (mPrPC, which has 98% sequence similarity and 86% sequence identity with the human protein).37 Ligand 2 can exist in three protomers under the conditions of the NMR experiments: uncharged 2, and positive 2+ and 22+ (Fig. 3c). Although one could suggest that the ligand binds to different PrP regions, based on the NMR data, only one binding site could be identified.37
Bias-exchange metadynamics simulations based on the AMBER99 force field38,39 provided a ligand binding/unbinding free energy (7.8 ± 0.9 kcal mol−1) that agreed well with the experimental value37 (7.5 kcal mol−1). The calculations identified different poses for the three protomers (Fig. 3b)||24, including the one suggested in ref. 37. Taken together, the poses were consistent with all the ligand/protein contacts deduced by the NMR data (Fig. 3a and b). Approaches combining NMR and enhanced sampling, such as that presented here, could be applied to other proteins undergoing fibril formation in neurodegenerative diseases, e.g. amyloid-beta in Alzheimer's disease, α-synuclein in Parkinson's disease, and huntingtin in Huntington's disease.40 All of them feature no binding cavities.
4. Conclusions
Our two case studies reported in Sections 2 and 3 suggest that an enhanced sampling method, such as metadynamics, may be used as a practical tool for the rational design of LTPS,** complementing experimental structural information from X-ray or NMR. In our examples, the calculations, validated against experimental affinity measurements, (i) pointed to differences in ligand poses on passing from X-ray to solution, possibly because of the increased hydration of the ligands and/or change in conformations of the residues at the surface (1·NAF-1 complex); (ii) matched, at the qualitative levels, the poses found by NMR data in solution. One can anticipate that massively parallel enhanced sampling approaches, such as that developed by Mandelli et al.,48 which may meet the challenges of using current exascale machines, may further enlarge the scope of these computer-aided approaches to unique LTPS.Our proposed combined experimental/simulation protocol is by no means the only way to foresee how to overcome the current limitations. During the last decade, a plethora of methods devoted to the study of protein–ligand binding have been developed, such as funnel metadynamics,49 lambda-dependent umbrella sampling,50 and ligand Gaussian accelerated molecular dynamics (LiGaMD).51 One of them has been presented in this perspective (Section 2). Here we presented two possible flavours of metadynamics to tackle this problem, while, as already said, a large number of techniques (also not metadynamics-related) can be effectively applied. In addition, and most importantly, ML-based techniques are expected to significantly increase the predictive power with the expansion/growth of experimental structural information (along with affinity data). Finally, the increasing number of Cryo-EM protein/LTPS complex structures’ (currently ∼0.08% of the PDB) is expected to boost a variety of rational LTPS design campaigns,52 possibly assisted by molecular simulation and/or ML.
Author contributions
P. C. conceived the conceptualization of the perspective; K. Z. performed the calculations and data analysis of the NAF-1 part; A. K. summarized the PrP part. K. Z., A. K., R. C., G. R., R. N., and P. C. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K.Z. is supported by the Marie Skłodowska-Curie grant agreement No. 765048. We acknowledge the computing time granted by RWTH Compute Cluster (No. 3497 and No. 20134).References
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS PubMed.
- S. A. Serapian and G. Colombo, Chem. – Eur. J., 2020, 26, 4656–4670 CrossRef CAS PubMed.
- D. Ni, N. Liu and C. Q. Sheng, Adv. Exp. Med. Biol., 2019, 1163, 313–334 CrossRef CAS PubMed.
- D. Ni, S. Y. Lu and J. Zhang, Med. Res. Rev., 2019, 39, 2314–2342 CrossRef PubMed.
- W. H. Shin, K. Kumazawa, K. Imai, T. Hirokawa and D. Kihara, Adv. Appl. Bioinform. Chem., 2020, 13, 11–25 Search PubMed.
- Z. Cong, L.-N. Chen, H. Ma, Q. Zhou, X. Zou, C. Ye, A. Dai, Q. Liu, W. Huang, X. Sun, X. Wang, P. Xu, L. Zhao, T. Xia, W. Zhong, D. Yang, H. E. Xu, Y. Zhang and M.-W. Wang, Nat. Commun., 2021, 12, 3763 CrossRef CAS PubMed.
- P. Xiao, W. Yan, L. Gou, Y.-N. Zhong, L. Kong, C. Wu, X. Wen, Y. Yuan, S. Cao, C. Qu, X. Yang, C.-C. Yang, A. Xia, Z. Hu, Q. Zhang, Y.-H. He, D.-L. Zhang, C. Zhang, G.-H. Hou, H. Liu, L. Zhu, P. Fu, S. Yang, D. M. Rosenbaum, J.-P. Sun, Y. Du, L. Zhang, X. Yu and Z. Shao, Cell, 2021, 184, 943–956 CrossRef CAS PubMed.
- C. J. Wenthur, P. R. Gentry, T. P. Mathews and C. W. Lindsley, Annu. Rev. Pharmacol., 2014, 54, 165–184 CrossRef CAS PubMed.
- L. J. Olson, S. K. Misra, M. Ishihara, K. P. Battaile, O. C. Grant, A. Sood, R. J. Woods, J. J. P. Kim, M. Tiemeyer, G. Ren, J. S. Sharp and N. M. Dahms, Commun. Biol., 2020, 3, 498 CrossRef CAS PubMed.
- R. Nechushtai, O. Karmi, K. Zuo, H.-B. Marjault, M. Darash-Yahana, Y. S. Sohn, S. D. King, S. I. Zandalinas, P. Carloni and R. Mittler, BBA, Mol Cell Res., 2020, 1867, 118805 CAS.
- K. Zuo, R. Capelli, G. Rossetti, R. Nechushtai and P. Carloni, J. Chem. Inf. Model., 2023, 63, 643–654 CrossRef CAS PubMed.
- L. G. Hoang, J. Gossen, R. Capelli, T. T. Nguyen, Z. X. Sun, K. Zuo, J. B. Schulz, G. Rossetti and P. Carloni, Front. Cell Dev. Biol., 2022, 10, 886568 CrossRef PubMed.
- D. D. Leonidas, G. B. Chavali, N. G. Oikonomakos, E. D. Chrysina, M. N. Kosmopoulou, M. Vlassi, C. Frankling and K. R. Acharya, Protein Sci., 2003, 12, 2559–2574 CrossRef CAS PubMed.
- J. Lameira, C. N. Alves, V. Moliner, S. Marti, R. Castillo and I. Tunon, J. Phys. Chem. B, 2010, 114, 7029–7036 CrossRef CAS PubMed.
- S. Piana, P. Carloni and U. Rothlisberger, Protein Sci., 2002, 11, 2393–2402 CrossRef CAS PubMed.
- A. Shiriaeva, D. Park, G. Kim, Y. J. Lee, X. Y. Hou, D. B. Jarhad, G. Kim, J. H. Yu, Y. E. Hyun, W. Kim, Z. G. Gao, K. A. Jacobson, G. W. Han, R. C. Stevens, L. S. Jeong, S. Choi and V. Cherezov, J. Med. Chem., 2022, 65, 11648–11657 CrossRef CAS PubMed.
- T. Zhou, J. Z. Lin, Y. G. Feng and J. F. Wang, Biochemistry, 2010, 49, 9604–9612 CrossRef CAS PubMed.
- G. Riviere, S. Oueslati, M. Gayral, J. B. Crechet, N. Nhiri, E. Jacquet, J. C. Cintrat, F. Giraud, C. van Heijenoort, E. Lescop, S. Pethe, B. I. Iorga, T. Naas, E. Guittet and N. Morellet, ACS Omega, 2020, 5, 10466–10480 CrossRef CAS PubMed.
- X. Wang, A. A. Gorfe and J. A. Putkey, J. Biomol. NMR, 2021, 75, 233–244 CrossRef CAS PubMed.
- F. Ballante, A. J. Kooistra, S. Kampen, C. Graaf and J. Carlsson, Pharmacol. Rev., 2021, 73, 1698–1736 CrossRef PubMed.
- R. Capelli, P. Carloni and M. Parrinello, J. Phys. Chem. Lett., 2019, 10, 3495–3499 CrossRef CAS PubMed.
- F. L. Gervasio, A. Laio and M. Parrinello, J. Am. Chem. Soc., 2005, 127, 2600–2607 CrossRef CAS PubMed.
- S. Piana and A. Laio, J. Phys. Chem. B, 2007, 111, 4553–4559 CrossRef CAS PubMed.
- A. Kranjc, S. Bongarzone, G. Rossetti, X. Biarnes, A. Cavalli, M. L. Bolognesi, M. Roberti, G. Legname and P. Carloni, J. Chem. Theory Comput., 2009, 5, 2565–2573 CrossRef CAS PubMed.
- M. M. Dicus, A. Conlan, R. Nechushtai, P. A. Jennings, M. L. Paddock, R. D. Britt and S. Stoll, J. Am. Chem. Soc., 2010, 132, 2037–2049 CrossRef CAS PubMed.
- K. Zuo, H. B. Marjault, K. L. Bren, G. Rossetti, R. Nechushtai and P. Carloni, J. Biol. Inorg. Chem., 2021, 26, 763–774 CrossRef CAS PubMed.
- Y. F. Chen, T. Y. Chou, I. H. Lin, C. G. Chen, C. H. Kao, G. J. Huang, L. K. Chen, P. N. Wang, C. P. Lin and T. F. Tsai, J. Pathol., 2020, 250, 299–311 CrossRef CAS PubMed.
- S. Tamir, M. L. Paddock, M. Darash-Yahana-Baram, S. H. Holt, Y. S. Sohn, L. Agranat, D. Michaeli, J. T. Stofleth, C. H. Lipper, F. Morcos, I. Z. Cabantchik, J. N. Onuchic, P. A. Jennings, R. Mittler and R. Nechushtai, BBA, Mol. Cell Res., 2015, 1853, 1294–1315 CAS.
- Y. F. Chen, C. H. Kao, Y. T. Chen, C. H. Wang, C. Y. Wu, C. Y. Tsai, F. C. Liu, C. W. Yang, Y. H. Wei, M. T. Hsu, S. F. Tsai and T. F. Tsai, Gene Dev., 2009, 23, 1183–1194 CrossRef CAS PubMed.
- J. R. Colca, W. G. McDonald, D. J. Waldon, J. W. Leone, J. M. Lull, C. A. Bannow, E. T. Lund and W. R. Mathews, Am. J. Physiol., 2004, 286, E252–E260 CAS.
- C. H. Lipper, O. Karmi, Y. S. Sohn, M. Darash-Yahana, H. Lammert, L. H. Song, A. Liu, R. Mittler, R. Nechushtai, J. N. Onuchic and P. A. Jennings, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 272–277 CrossRef CAS PubMed.
- A. R. Conlan, H. L. Axelrod, A. E. Cohen, E. C. Abresch, J. Zuris, D. Yee, R. Nechushtai, P. A. Jennings and M. L. Paddock, J. Mol. Biol., 2009, 392, 143–153 CrossRef CAS PubMed.
- H.-B. Marjault, O. Karmi, K. Zuo, D. Michaeli, Y. Eisenberg-Domovich, G. Rossetti, B. de Chassey, J. Vonderscher, I. Cabantchik, P. Carloni, R. Mittler, O. Livnah, E. Meldrum and R. Nechushtai, Commun. Biol., 2022, 5, 437 CrossRef CAS PubMed.
- M. A. Wulf, A. Senatore and A. Aguzzi, BMC Biol., 2017, 15, 34 CrossRef PubMed.
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10, S10–S17 CrossRef PubMed.
- C. Soto, Nat. Rev. Neurosci., 2003, 4, 49–60 CrossRef CAS PubMed.
- K. Kuwata, N. Nishida, T. Matsumoto, Y. O. Kamatari, J. Hosokawa-Muto, K. Kodama, H. K. Nakamura, K. Kimura, M. Kawasaki, Y. Takakura, S. Shirabe, J. Takata, Y. Kataoka and S. Katamine, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11921–11926 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- B. N. Dugger and D. W. Dickson, Csh Perspect Biol, 2017, 9, a028035 Search PubMed.
- L. Calzolai and R. Zahn, J. Biol. Chem., 2003, 278, 35592–35596 CrossRef CAS PubMed.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camilloni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci, R. A. Broglia and M. Parrinello, Comput. Phys. Commun., 2009, 180, 1961–1972 CrossRef CAS.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- M. Bonomi, G. Bussi, C. Camilloni, G. A. Tribello, P. Banas, A. Barducci, M. Bernetti, P. G. Bolhuis, S. Bottaro, D. Branduardi, R. Capelli, P. Carloni, M. Ceriotti, A. Cesari, H. C. Chen, W. Chen, F. Colizzi, S. De, M. De La Pierre, D. Donadio, V. Drobot, B. Ensing, A. L. Ferguson, M. Filizola, J. S. Fraser, H. H. Fu, P. Gasparotto, F. L. Gervasio, F. Giberti, A. Gil-Ley, T. Giorgino, G. T. Heller, G. M. Hocky, M. Iannuzzi, M. Invernizzi, K. E. Jelfs, A. Jussupow, E. Kirilin, A. Laio, V. Limongelli, K. Lindorff-Larsen, T. Lohr, F. Marinelli, L. Martin-Samos, M. Masetti, R. Meyer, A. Michaelides, C. Molteni, T. Morishita, M. Nava, C. Paissoni, E. Papaleo, M. Parrinello, J. Pfaendtner, P. Piaggi, G. Piccini, A. Pietropaolo, F. Pietrucci, S. Pipolo, D. Provasi, D. Quigley, P. Raiteri, S. Raniolo, J. Rydzewski, M. Salvalaglio, G. C. Sosso, V. Spiwok, J. Sponer, D. W. H. Swenson, P. Tiwary, O. Valsson, M. Vendruscolo, G. A. Voth and A. White, Nat. Methods, 2019, 16, 670–673 CrossRef PubMed.
- H. J. C. Berendsen, D. Vanderspoel and R. Vandrunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- T. M. M. J. Abraham, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham, III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao and P. A. Kollman, AMBER 2016, University of California, San Francisco, 2016 Search PubMed.
- D. Mandelli, B. Hirshberg and M. Parrinello, Phys. Rev. Lett., 2020, 125, 026001 CrossRef CAS PubMed.
- V. Limongelli, M. Bonomi and M. Parrinello, Proc. Nat. Acad. Sci., 2013, 110, 6358–6363 CrossRef CAS PubMed.
- S. T. Ngo, J. Chem. Comput., 2020, 42, 117–123 CrossRef PubMed.
- Y. Miao, A. Bhattarai and J. Wang, J. Chem. Theory Comput., 2020, 16, 5526–5547 CrossRef CAS PubMed.
- J. H. Van Drie and L. Tong, Bioorg. Med. Chem. Lett., 2020, 30, 127524 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp01388j |
| ‡ 1 accelerates the cluster release relative to the protein without ligands in vitro.33 |
| § Some of the residues at the protein surface might also change their conformations in solution as they may form contacts with protein images in the solid state. |
| ¶ 2 features three different protomers at pH 4.5, the pH at which the experiments were carried out37 (Fig. 3). |
| || This contrast with standard molecular docking protocol, which was not consistent with all the ligand/protein contacts identified by NMR. |
| ** The codes to be used are readily available in PLUMED42–44 (https://www.plumed.org//) which can be used in codes such as GROMACS45,46 AMBER,47etc. |
| This journal is © the Owner Societies 2023 |