
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insight into the evaluation of complex mixture biodegradability: an UHPLC-qToF “all-ion MS/MS” acquisition technique for the untargeted and targeted analysis of pharmaceutical formulation biodegradation†
Luisa
Mattoli‡
 a,
Giacomo
Proietti‡
a,
Claudio Marzio
Quintiero
a,
Giada
Fodaroni
a,
Michela
Burico
a,
Mattia
Gianni
a,
Emiliano
Giovagnoni
a,
Valentino
Mercati
a and
Claudio
Santi
*bc
a,
Giacomo
Proietti‡
a,
Claudio Marzio
Quintiero
a,
Giada
Fodaroni
a,
Michela
Burico
a,
Mattia
Gianni
a,
Emiliano
Giovagnoni
a,
Valentino
Mercati
a and
Claudio
Santi
*bc
aAboca SpA Società Agricola, Innovation & Medical Science Division, Loc. Aboca 20, Sansepolcro 52037, Italy. E-mail: LMattoli@aboca.it; GProietti@aboca.it; CMQuintiero@aboca.it; GFodaroni@aboca.it; MBurico@aboca.it; EGiovagnoni@aboca.it; MGianni@aboca.it; VoMercati@aboca.it
bGroup of Catalysis, Synthesis and Organic Green Chemistry – Department of Pharmaceutical Sciences University of Perugia, Via del Liceo 1, Perugia, 06123, Italy. E-mail: claudio.santi@unipg.it
cCentro di Eccellenza Materiali Innovativi Nanostrutturati (CEMIN) University of Perugia, Via Elce di Sotto 8, Perugia, 06123, Italy
First published on 11th August 2022
Abstract
UHPLC-qToF analysis is reported here as a modern and reliable spectrometric technique, for a more accurate evaluation of the biodegradation of pharmaceutical formulations that should be considered as complex mixtures of organic compounds interacting with naturally occurring microfauna. The untargeted approach evidences that natural and non-natural compounds are not equally biodegraded even when both passed the requirements of the respirometry test. In particular, we demonstrated that for mixtures containing synthetic non-naturally occurring compounds, a new cluster of derivatives can be detected and in part interpreted by a target analysis. We also demonstrated that the fate of the biodegradation of an active ingredient pure or inserted in a pharmaceutical formulation is not the same, confirming that complex chemical systems should be better analyzed using a holistic approach rather than a reductionistic one. The biodegradation of a pharmaceutical formulation could present a different fate compared to that predicted by considering the biodegradability of each component (the global result is not the sum of the parts). Considering these results, an update of the protocols currently used for the evaluation of the environmental impact of chemicals and chemical mixtures used in our daily life can be proposed, through the use of modern technologies nowadays widely diffused in analytical laboratories.
Environmental significanceA correct assessment of pharmaceutical formulation biodegradability is a relevant issue in the right forecast of their potential environmental impact. In our opinion, the currently used methods present some limitations in understanding the real fate of a mixture of chemicals. When organic molecules are partially degraded, a pool of new unknown compounds can be produced leading to an unpredictable effect on the biosphere; furthermore, a mixture cannot be considered as simply the sum of each single component. We demonstrated that UHPLC-qToF analysis could be a modern alternative leading to a more accurate profiling of the biodegradation output for a pharmaceutical product by coupling targeted and untargeted methods in order to holistically consider the formulations as a complex system. |
Introduction
Biodegradation is the process by which organic substances are decomposed by microorganisms into the simplest natural building blocks (e.g. CO2, H2O, and NH3) that can be integrated into natural biogeochemical cycles. Anthropic and industrial activities have led to the emergence of a series of new polluting compounds and their release into the environment is responsible for a number of adverse effects, stimulating the development of a number of protocols to attempt their removal.1–4 The scientific interest in biodegradation is clearly demonstrated by more than 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 papers that are referenced in PubMed starting from the seminal one reported by Kagawa in 1960.5
000 papers that are referenced in PubMed starting from the seminal one reported by Kagawa in 1960.5
The evaluation of the biodegradability of chemicals is one of the main issues in environmental risk assessment. Biodegradability tests are designed to evaluate, under batch conditions, a chemical substance as the sole carbon source for the survival of microfauna.5 The ready biodegradation tests (RBT) are the basis of the integrated testing strategy on pure substance biodegradation. They are a series of tests (from no. 301A to 301F and no. 310) proposed by the Organization for Economic Co-operation and Development (OECD). Microorganisms and the tested substance are usually incubated in a buffered pH 7 medium containing N, P, and a trace element (named the “mineral medium”). The kinetics of biodegradation is monitored during at least 28 days by the evaluation of metabolic parameters such as oxygen consumption, carbon dioxide production, or dissolved organic carbon consumption. The RBT measures ultimate biodegradability, or complete biodegradation and a chemical can be classified as readily biodegradable if has passed one of the RBTs.6 Specific chemical analysis can be used to assess primary biodegradation of the tested substance and to determine the concentration of any newly formed intermediate. This additional evaluation is mandatory only in the MITI method (301 C), but it is optional for all other RBTs.6
The term primary biodegradation indicates the structural modification of a substance caused by a biological event, which results in the loss of a specific property of that substance. It can be calculated from supplemental chemical analysis for parent compounds made at the beginning and end of the tests (OECD 301, 310).6–9
A not-readily biodegradable substance is considered persistent unless its environmental degradability is proven in more expensive and complex simulation tests, (tests method no. 303, 306, 307, 308, and 309, OECD, 1992c, d, f, g, and h). The type of simulation test to be performed depends on the potential receptor environments that are causing concern (wastewater treatment plant, surface water, sediment, and soil). Simulation tests are designed to evaluate the long-term chemical behavior in the environment. However, these tests are limited to a very small number of pure substances because they are expensive, technically sophisticated, and time-consuming (e.g. need the use of several different tests and the use of 14C-labeled chemicals).10–12
In silico models have been developed13 and are available under REACH regulations.14 However, the prediction using these methods remains limited to existing data and may be not adapted to assess environmental persistence as a function of both chemical intrinsic properties and environmental conditions.12,15–17
Even though the currently adopted tests for biodegradability are cheap and easy to be performed, they present some limitations. As an example, they are conducted under standardized conditions, which do not reflect highly variable environmental situations like seasonality. Furthermore, even if they allow risk assessment by conservative models, tests most chemicals at concentrations that are unlikely occurring in the environment.12,18–21 Other criticisms are related to the preparation of the microbial inoculum used for the biodegradation tests. Before being used, inoculum sources need to be either washed (to limit carbon contamination other than that from the tested substance) or acclimated (e.g. for the 301C test).22 In addition, the final output may be influenced by the total cell density, the diversity of species, the origin and history of the inoculum sample, the ratio between food and biomass, and the duration of the evaluation period that, in the standard experiment, has been defined at 28 days.21,22
A further limitation of these tests is that usually they do not take into consideration the outcome of biodegradation, supposing that the investigated molecule is fully degraded into elemental bricks. Nowadays, the development of modern and reliable analytical techniques offers the possibility to investigate the biodegradability of a substance (or a mix of substances like in a pharmaceutical nutraceutical or cosmetic formulation) in a more accurate manner both at qualitative and quantitative levels, offering an interesting and unprecedented complementary analysis in the currently used primary biodegradation studies. The use of novel technologies enables, in our opinion, a more accurate and realistic evaluation of the environmental risks and impacts, looking toward a more sustainable development.
Furthermore, ready biodegradability tests and simulation tests are usually referred to pure chemicals, and only a few cases of mixtures composed of structurally similar chemicals are reported. One example is that of petroleum and surfactants. It was reported that the kinetics observed for the biodegradation of the mixture and those of the isolated chemical entities are considerably different, with these latter in general faster than that of the mixture.23 Clearly, moving from a reductionistic analytical investigation of single molecule biodegradation to a holistic one that will take into consideration the biodegradability of a pool of chemical ingredients could represent a strong innovation and, potentially, a real improvement in the design of a new reliable sustainable development.
In this paper, two commercially available cough syrups (one containing synthetic ingredients [A] and one containing only natural ingredients[B]) were chosen as real examples of largely distributed formulations of pharmaceutical ingredients. Their biodegradation has been evaluated and compared with that of the pure non-naturally occurring API (Active Pharmaceutical Ingredient) bromhexine, using an UHPLC-qToF “all-ion MS/MS” acquisition technique. The chemical compositions of mixtures A and B declared in the package insert of the analyzed pharmaceutical mixtures are summarized in Table 1.
| Mixture A | Mixture B |
|---|---|
| a As reported on the package leaflet. | |
| Bromhexine, maltitol, sucralose, preservative and flavouring | Honey “Polyresin” composed of resins, polysaccharides and flavonoids from grindelia, plantago and helichrysum |
| Sugar cane essential oil: lemon, sweet orange, and myrtle; lemon natural flavouring; arabic gum; xanthan gum | |
| Water | Water |
An ultra-high performance liquid chromatography-quadrupole time-of-flight mass spectrometer (UHPLC-qToF) is a recent type of spectrometer characterized by an ultra-high performance chromatographic separation of the molecular species that has been successfully employed for its fast, high-resolution separations, high-resolution mass spectra and high sensitivity.
Even if high resolution mass spectrometry as well as other spectroscopic methods were previously reported for the investigation of the environmental impact produced by pharmaceuticals and cosmetic formulations,24–28 to the best of our knowledge, this is the first UHPLC-qToF “all-ion MS/MS” approach to obtain metabolite fingerprints by untargeted data treatment of pharmaceutical formulation submitted to a ready biodegradability test and targeted analyses for deeper investigation of the readily biodegradation output.
Materials and methods
Biodegradation method
Glucose purum, magnesium sulphate heptahydrate for analysis ACS Reag. Ph. Eur and Reag. USP, sodium acetate anhydrous USP, phosphoric acid 99%, potassium phosphate dibasic 99+% anhydrous for analysis, potassium phosphate monobasic 99% anhydrous for analysis, iron(III) chloride hexahydrate pure, sodium phosphate dibasic dihydrate 98+%, and calcium chloride anhydrous 99% pure, were purchased from Carlo Erba Reagents srl (Cornaredo, Milano, Italy). Ammonium chloride 99% puriss and potassium hydroxide 85% puriss were purchased from Italchimica SPA (Pontecchio Polesine, Rovigo, Italy). Buffered peptone water (ISO) was purchased from ThermoFisher. Purified water was prepared by using an Arium Mini water purification system (from Sartorius Goettingen, Germany).Sample preparation
The sample was treated according to the procedure reported in the respirometric–manometric method no. 301F (OECD). To be able to have the correct registration by using the BOD sensor and enough material to perform the planned instrumental analysis, few adjustments were made, as shown in Table S4.†Sodium acetate was used as the reference substance. Tests were done at a constant temperature of 22 °C.
Inoculum preparation
The inoculum was prepared by collecting 5 samples of activated sludge and 5 samples of river water from different places and mixing them in equivalent volume. The inoculum is oxygenated, stirred and fed with glucose, peptone, dibasic potassium phosphate, and the values of redox potential, oxygen consumption and total dry matter are monitored daily. The dry substance was determined at 100 °C to measure the same quantity (30 mg mL−1) in the vessels containing the test substance.Inoculum composition
The inoculum was obtained by mixing active sludge from 8 different sectors and river water taken from two different rivers in equal parts. The assembled inoculum was oxygenated, stirred and fed with glucose, peptone and monopotassium orthophosphate. Oxygen, redox, and total suspended solid values were monitored daily.Before the use of inoculum, the total dry matter is determined.
The composition of the microfauna was determined by optical microscope analysis and was composed of the following species: Aspidisca cicada 20%, Zoothamnium 20%, Rotiferi 40%, and Euglyphia 20%.
Instrumental parameters
The biological oxygen demand was constantly monitored by means of a BOD EVO Sensor System 6, being made up of six BOD EVO sensors, a six-position stirring base, dark glass bottles, and sub-caps for the absorption of carbon dioxide and magnetic stir bars. The BOD EVO sensor transmits wireless data to the dedicated BODSoft™ software (VELP Scientifica, Usmate, Monza–Brianza, Italy). All the experiments were conducted at a controlled temperature in a refrigerated thermostat provided with an auto-tuning thermoregulation system and forced air circulation in order to have stability and homogeneity of the internal temperature (VELP Scientifica, Usmate, Monza-Brianza, Italy). The temperature of the thermostat was monitored by means of a datalogger with an external probe with a range of temperature from −40 to +80 °C (XS Instruments, Carpi, Modena, Italy).UHPLC ESI-qToF methods
All solvents were of high purity analytical grade and were used without further purification. ULC/MS grade absolute methanol was purchased from Biosolve (Dieuze, France). Ultrahigh purified water was prepared in a PURELAB® Ultra water purification system (ELGA, UK). formic acid 98–100% for LC-MS LiChropur® and dimethyl sulfoxide ≥99% were purchased from Sigma-Aldrich (St. Louis, MO). Mixture A was purchased (at a local pharmacy, batch no. 181092) and mixture B was produced by Aboca (batch no. 19C2076); bromhexine hydrochloride, sucralose, maltitol and ambroxol were purchased from Sigma-Aldrich (St. Louis, MO).The high-purity reference standards, used both for the construction of the in-house database and for the construction of the calibration curves, were purchased from Extrasynthese (Genay, France), Sigma-Aldrich (St. Louis, MO), PhytoLab GmbH & Co. KG (Vestenbergsgreuth, Germany) and ChromaDex (Irvine, CA). High-purity reference standard stock solutions were prepared in methanol, water/methanol (80:20, v/v) or methanol/dimethyl sulfoxide (80:20, v/v) at 500 ppm. The working solutions were prepared by diluting appropriate volumes of the stock solutions with water/methanol (50:50, v/v). Internal standard sulfadimethoxine-d6 was purchased from Sigma-Aldrich (St. Louis, MO). Internal standard stock solution was prepared in methanol at 5 ppm. All the stock solutions were stored in glass vials at −80 °C.
Targeted method
The UHPLC system was coupled to a qToF mass spectrometer endowed with a Dual AJS ESI source operating in negative and positive ionization modes with a scan range from 50 m/z to 1700 m/z. The optimized instrument parameters are reported in Table S6.†
The construction of the in-house database was performed by acquiring each analytical standard reference compound in data dependent mode (Auto MS/MS, Agilent Technologies) using three different collision energy values (20, 30 and 40 eV) by N2, while the acquisition of the sample metabolite fingerprint under investigation was performed in All-Ions mode22,25 using a collision energy of 30 eV. In both the acquisition methods, a reference mass solution containing purine and hexakis(1H, 1H, 3H-tetrafluoropropoxy)phosphazine was directly injected into the ESI source by using an isocratic pump and ionized together with the sample solution for mass correction allowing accurate mass time-of-flight data to be obtained. MassHunter software version B.07 (Agilent Technologies, Santa Clara, CA) was used for data acquisition and processing.
The exact masses of the various molecular species were calculated by means of an isotope distribution calculator, a tool of MassHunter Data Analysis core, version 8.0.8208.0 (Agilent Technologies, Santa Clara, CA).
Untargeted method
Multivariate statistical analysis
Instrumental parameters were the same as those used for the targeted UHPLC-qToF method, positive ions for mixture A and negative ions for mixture B.
Variables with a frequency of missing value and coefficient of variation of more than 20% have been eliminated and data imputation was performed. Additionally, a filter was applied to eliminate those variables present in the analytical blank samples.
The median of the replicates was applied to remove the effect of the random noise, while data normalization was performed with PQN (Probabilistic Quotient Normalization) on the reference pooled samples.
The processed data were saved in CSV format and the data matrix in CSV format has been reworked by the program SIMCA (Version 16.0.2.10561, Jan 22 2020). Later on, the data matrix was mean centered and Pareto scaling was applied prior to performing data analysis.
Statistical model performance evaluation was carried out and a confidence level of 95% was used.
The goodness of fit and the goodness of prediction were evaluated by means of R2X and Q2, respectively (Table S7†).
The diagnostic tools Hotelling T2 and DModX were calculated to be sure that the model is not deformed. The models were robust, as the observations were below the calculated critical values and there were no outliers (Table S8†).
Relative ratio (RR) calculation. The relative ratio (RR) is calculated by using the ratio between the area counts of the compound and the area count of the internal standard (sulfadimethoxine-d6).
Calculation of the percentage of reduction. The percentage of reduction of a certain compound is the ratio between the RR at T28 and the RR at T0 expressed as a percentage.
Results and discussion
The UHPLC chromatographic conditions were optimized to obtain the best ionization along with the elution of all the species of interest within 21 minutes. Pure water and methanol, both acidified with formic acid, were used as the mobile phase, composed according to a linear gradient suitable for eluting hydrophilic and lipophilic compounds in a time frame compatible with a UHPLC analysis.Through the UHPLC-qToF “all-ion MS/MS”29 acquisition technique high resolution mass spectrometry (HRMS) data can be acquired using different conditions: (1) with a low value of collision energy and (2) with a high energy value. The low energy spectra predominantly show just the molecular (or precursor) ions for the compounds and the high-energy spectra provide the precursors plus their fragment ions.
With All ions MS/MS data, a targeted qualitative screening analysis with structure elucidation and identification of fragmentation patterns of the compounds presents in complex products can also be confidently carried out during the biodegradability studies.
Without resorting to structural identification, the large number of signals obtained from UHPLC-qToF analyses can be used to perform untargeted studies. Untargeted analysis gives information about the physical-chemical changes in samples during the RBT, by observing the route of their grouping in statistical models by principal component analysis (PCA) and cluster analysis (CA).
The respirometric–manometric test no. 301F (according to OECD guidelines)6 was chosen as the method for the biodegradability evaluation.
It was performed at two concentrations required by the OECD 301F method (50 mg L−1 and 100 mg L−1) and, in addition, we tested an out-of-range concentration (1000 mg L−1) aiming for better identification of all the metabolites formed during the test. The samples were analysed at the beginning of the ready biodegradation test and after 28 days by means of the untargeted and targeted approaches.
Performing the RBT at a concentration of 50 mg L−1,mixture A passed the ten-day window criteria and after 28 days of incubation reached 81% of biodegradation, while mixture B passed the ten-day window criteria and reached 74% of biodegradation on the 28th day. The curves of biodegradation are shown in Fig. 1.
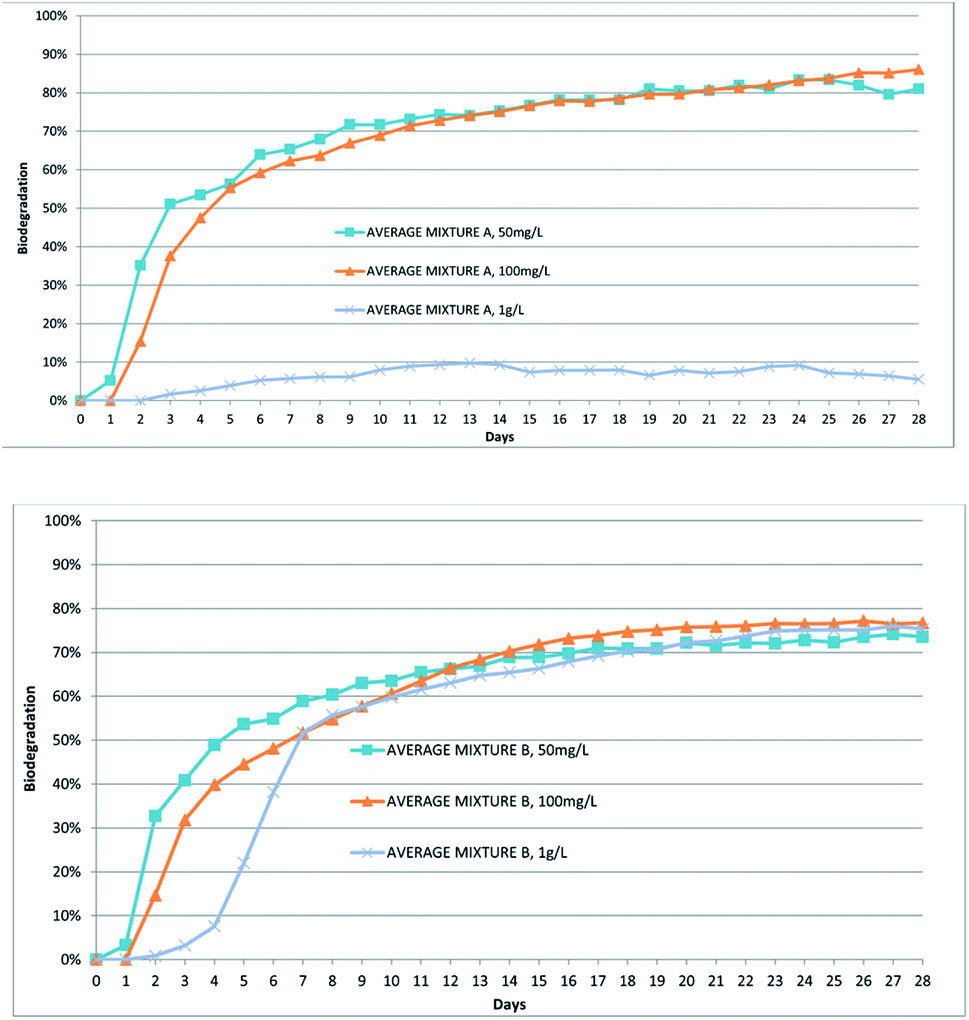 | ||
| Fig. 1 Biodegradation of the mixtures A and B at different concentrations. All the data are reported as an average of two or three replicates. | ||
The test was repeated at the concentrations of 100 mg L−1 and 1000 mg L−1. The results evidenced that mixture A (100 mg L−1 BD28 days 86%) was again readily biodegradable at 100 mg L−1 but not at 1000 mg L−1 (1000 mg L−1 BD28 days 6%), while mixture B was readily biodegradable under both conditions (100 mg L−1: BD28 days 77%, 1000 mg L−1 BD28: days 75%). These results are summarized in Table 2 as well as in Fig. 1.
| Mixture | Concentration | 10 day criteriaa | BD at the 28th day |
|---|---|---|---|
| a The pass levels for ready biodegradability are 60% of ThOD removal for respirometric methods. These pass values have to be reached in a 10 d window within the 28 d period of the test. The 10 d window begins when the degree of biodegradation has reached 10% ThOD and must end before day 28 of the test. Chemicals which reach the pass levels after the 28 d period are not deemed to be readily biodegradable. | |||
| A | 50 mg L−1 | Passed | 81% |
| 100 mg L−1 | Passed | 86% | |
| 1000 mg L−1 | Not passed | 6% | |
| B | 50 mg L−1 | Passed | 74% |
| 100 mg L−1 | Passed | 77% | |
| 1000 mg L−1 | Passed | 75% | |
Even if 1000 mg L−1 is not a concentration considered in the validated method, it is interesting to observe the different behaviours of the analysed samples. In the case of mixture A, the microfauna of the inoculum seem to be unable to adequately activate biodegradation, probably because it is negatively affected by the experimental conditions. In our opinion, this can be ascribed to the high concentration of both the substrate and/or some initially formed metabolites. A similar effect was not observed in the case of mixture B, indicating that different mixtures can exhibit a different dose-dependent impact on the microenvironment of the experiment.
In order to better investigate this phenomenon, after 28 days under the RBT conditions mixtures A at 50 mg L−1, 100 mg L−1 and 1000 mg L−1, respectively, were subjected to a spectrometric semiquantitative evaluation of the residual main components: sucralose 1, maltitol 2 and bromhexine 3 (see below and Table S1†). The sucralose was found to be fully non-degradable30 and in all the samples it was detected at the original concentration (see Table S1†). Differently, degradation of bromhexine 3 was scarcely affected by the sample concentration and only 6%, 12% and 12% of the parent compound was still present at the end of the experiment in the 50 mg sample and both 100 mg L−1 and 1000 mg L−1 samples, respectively. Maltitol 2, that was fully biodegraded in the samples at 50 mg L−1 and 100 mg L−1, was detected at 62% of the original concentration in the 1000 mg L−1 sample, largely contributing to the failure of the RBT at this concentration.
Furthermore, the composition of both the mixtures (A and B) at 100 mg L−1 and 1000 mg L−1 before and after 28 days under RBT conditions were studied by UHPLC-qToF analysis following the untargeted and targeted approaches.
The UHPLC-qToF method used to acquire data was characterized by a chromatographic separation in a reverse phase UHPLC column followed by qToF mass spectrometric detection. All the species eluted were selectively characterized by their retention time and high-resolution mass. Therefore, after the acquisition of the chemical fingerprint, the data were aligned. Variables with a frequency of missing value and coefficient of variation of more than 20% have been eliminated, noise was subtracted, and the corresponding raw data matrix was subjected to normalization and scaling.
The resulting set of data was used to build the statistical model, after passing the classical test as R2X, Q2 (model diagnostic tests), Hotelling T2, DModX (observation diagnostic tests). The covariance matrix and standardized principal components were selected for unsupervised principal component analysis (PCA) computation.
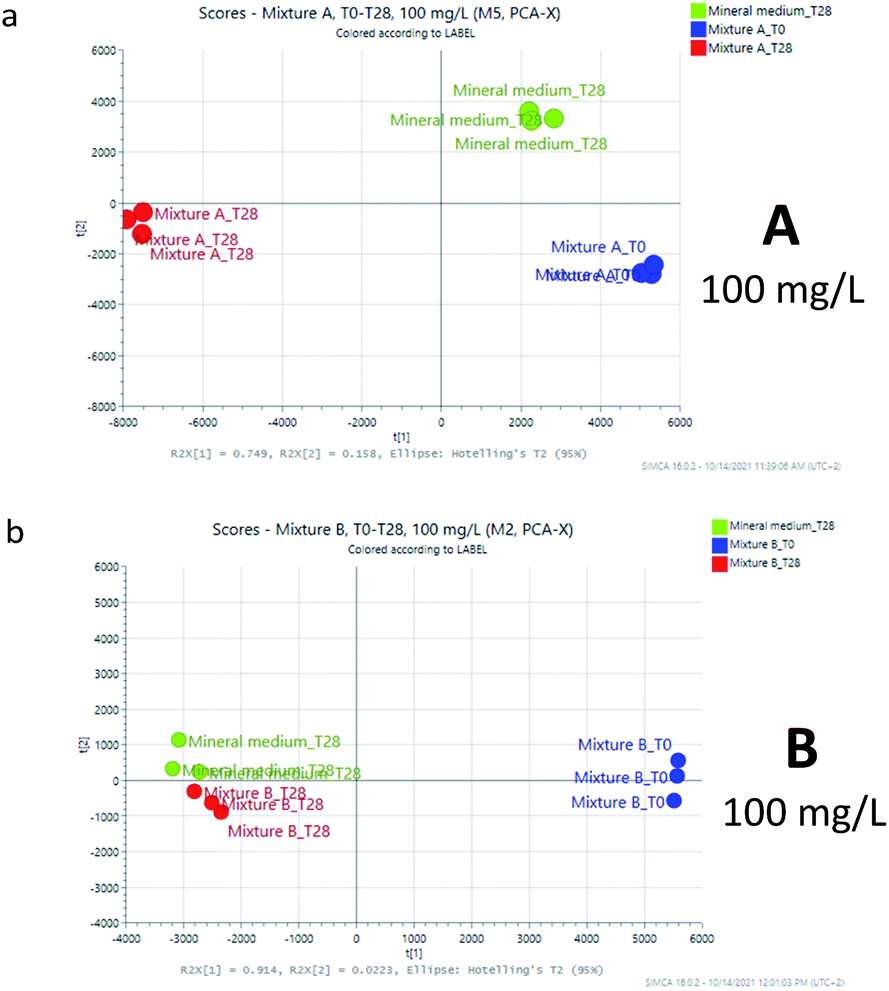 | ||
| Fig. 2 PCA models for samples A and B at 100 mg L−1. | ||
The first two components of the 2D-PCA were found to include more than 90% for mixture A (PC1 74.9%, PC2 15.8% 100 mg L; PC1 57.5%, and PC2 33.2% 1000 mg L−1) and for mixture B (PC1 91.4%, PC2 2.2% 100 mg L; PC1 96.1%, and PC2 2.1% 1000 mg L−1) of the total variance of the model, in both the experiments at 100 mg L−1 (Fig. 2) and 1000 mg L−1 (Fig. 3). The graphics evidenced that for mixture A the observations (score) are far from that of the mineral medium at 28 days while in the case of mixture B the biodegradation appear to be more advanced, but still not fully coincident with that of the mineral medium (compare Fig. 2a/2b and 3a/3b).
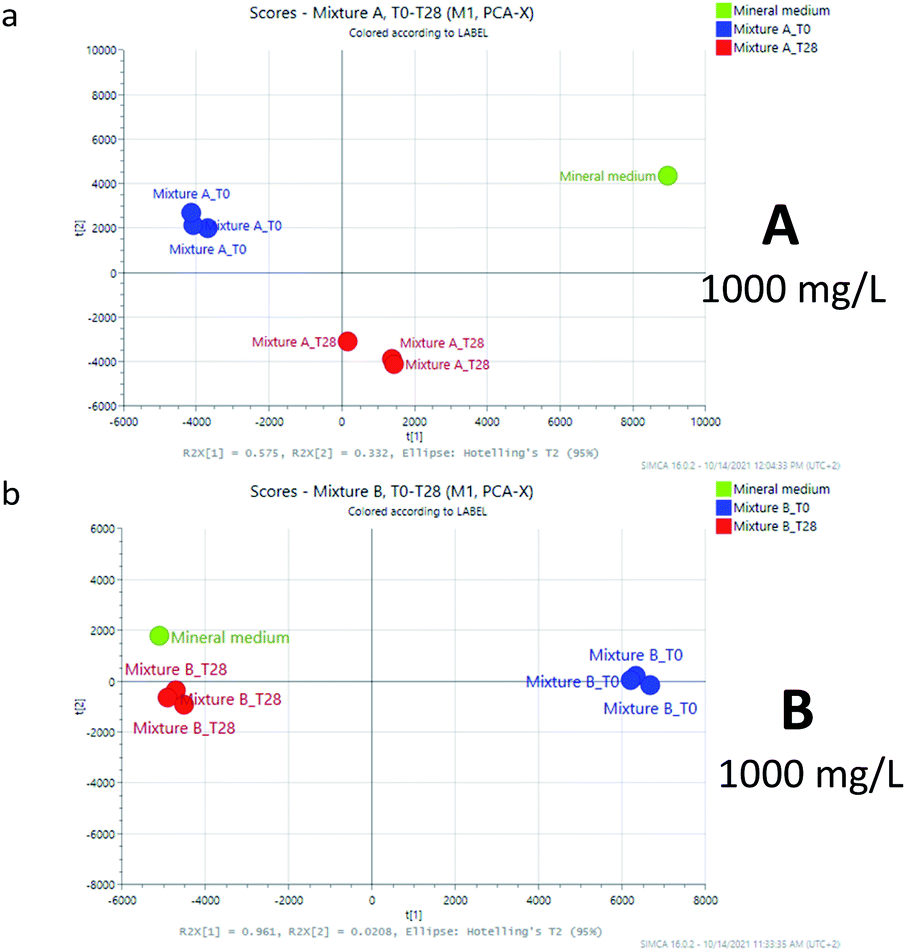 | ||
| Fig. 3 PCA models for samples A and B at 1000 mg L−1. | ||
Another way for a qualitative evaluation of metabolite fingerprinting obtained by UHPLC-qToF is statistical elaboration using unsupervised “cluster analysis” (CA) in order to obtain the natural hierarchical groupings. By means of CA, after applying the linkage method of Ward31 as a standard method for the formation of a hierarchical cluster, the distance between the groups was determined using the Euclidean distance and the results of the analysis of the clusters are presented as dendrograms (Fig. 4 and 5).
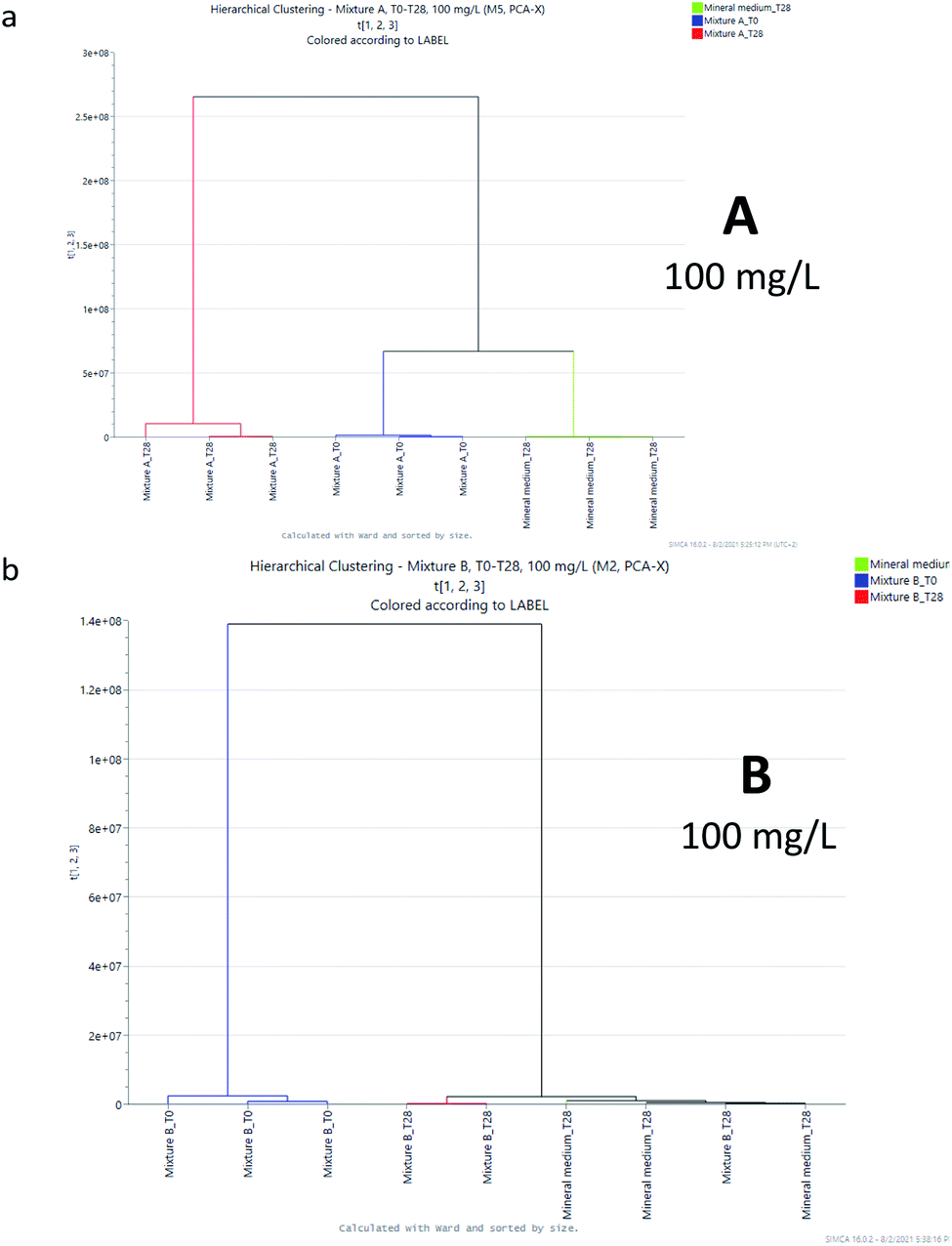 | ||
| Fig. 4 Cluster analysis for samples A and B at 100 mg L−1. | ||
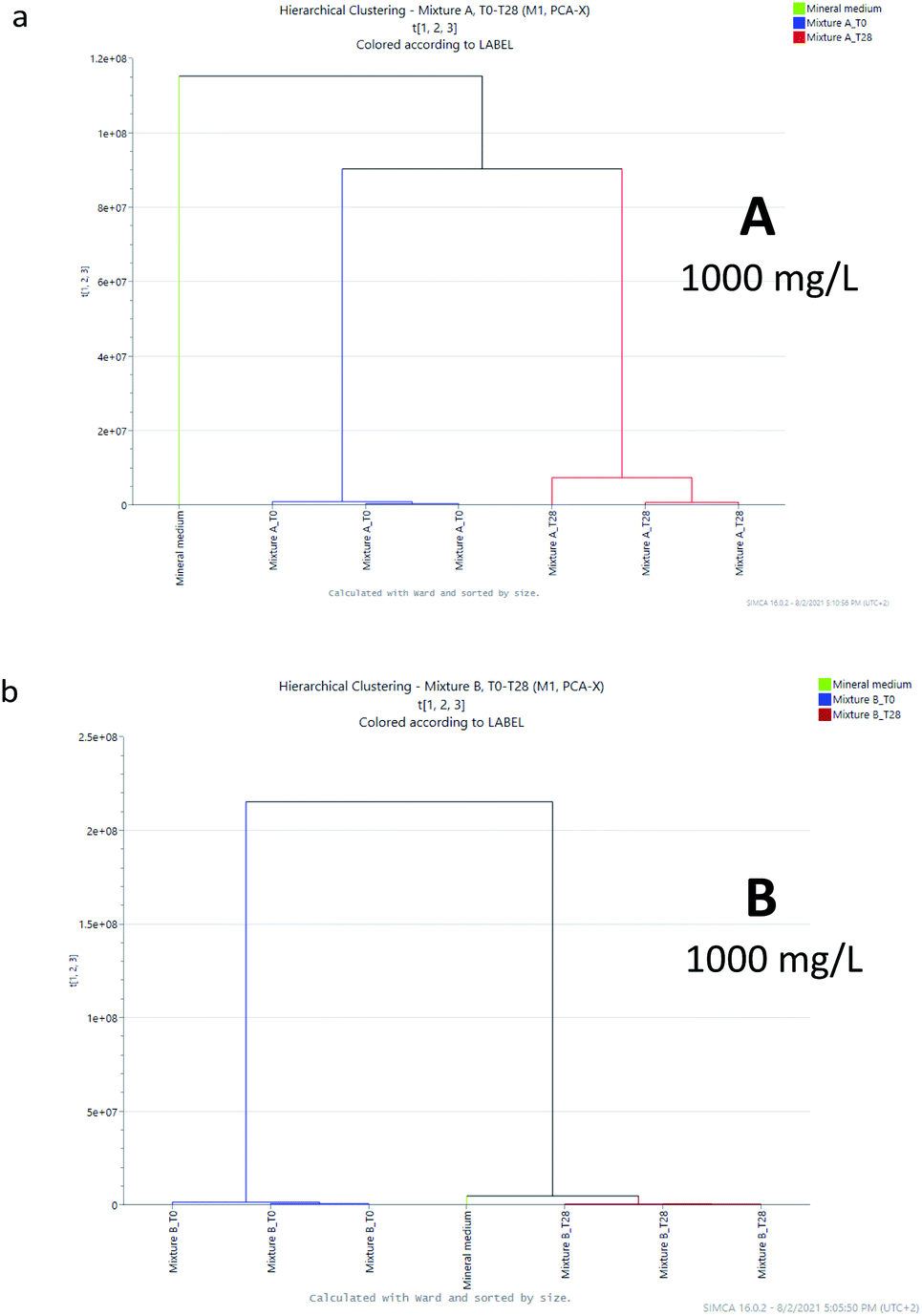 | ||
| Fig. 5 Cluster analysis for samples A and B at 1000 mg L−1. | ||
The clusters generated by mixture A (at 100 mg L−1) were essentially two: one formed by the samples collected after 28 days under the condition of the biodegradation test and the other by the samples collected at the beginning of the study and the mineral medium (Fig. 4a). For mixture B (at 100 mg L−1), similarly two clusters were identified: one is for the samples collected at the beginning of the test and the other for the samples collected after 28 days and the mineral medium (Fig. 4b), confirming the observation of PCA. Similarly, the untargeted analysis of the samples at a concentration of 1000 mg L−1 indicates that the degradation of mixture B produces a mixture of derivatives that are closer to those arising from the mineral medium, with respect to the molecular fingerprint of the mixture obtained by the biodegradation of A (compare Fig. 4a/4b and 5a/5b). Nevertheless, in both cases full mineralization seems to not be completely achieved.
These data clearly indicate that by considering a mixture of compounds the output of the biodegradation obtained after 28 days of the RBD test is strongly affected by the chemical composition of the starting material affording new mixtures of compounds that are not fully coincident with that of the mineral medium that is supposed to be fully degradable into basic bricks such as carbon dioxide, water, ammonia, and sulphide.
Furthermore, these results demonstrate that untargeted analysis performed by UHPLC-qToF offers an interesting method to evaluate the actual biodegradation of chemical mixtures, overcoming some limitations on sensitivity shown by the currently used RBT.
These untargeted analyses provided a holistic overview of the mixture behaviour suggesting further investigations using a targeted approach, with the aim to understand the fate of each specific compound present in the complex mixture after 28 days of the RBT.
The same UHPLC-qToF technique used for the untargeted analysis was used to characterize some compounds present in mixtures A and B. By means of this method, pure reference samples of selected compounds such as sucralose, bromhexine and ambroxol (as its well-known metabolite), were analysed for their correct identification in mixture A. The MS and MS/MS experimental data of sucralose31,32 and bromhexine33,34 were in agreement with those reported in the literature (Table S2†). The tentative identification of compounds present in mixture B was achieved by using a personal compound database and library (PCDL) of natural compounds present in the laboratory.
In the starting mixtures A and B, based on the high resolution mass, retention time, MS/MS fragmentation patterns, isotopic profile and isotopic abundance the compounds 1–6 shown in Fig. 6 were unambiguously identified. (Table S2†).
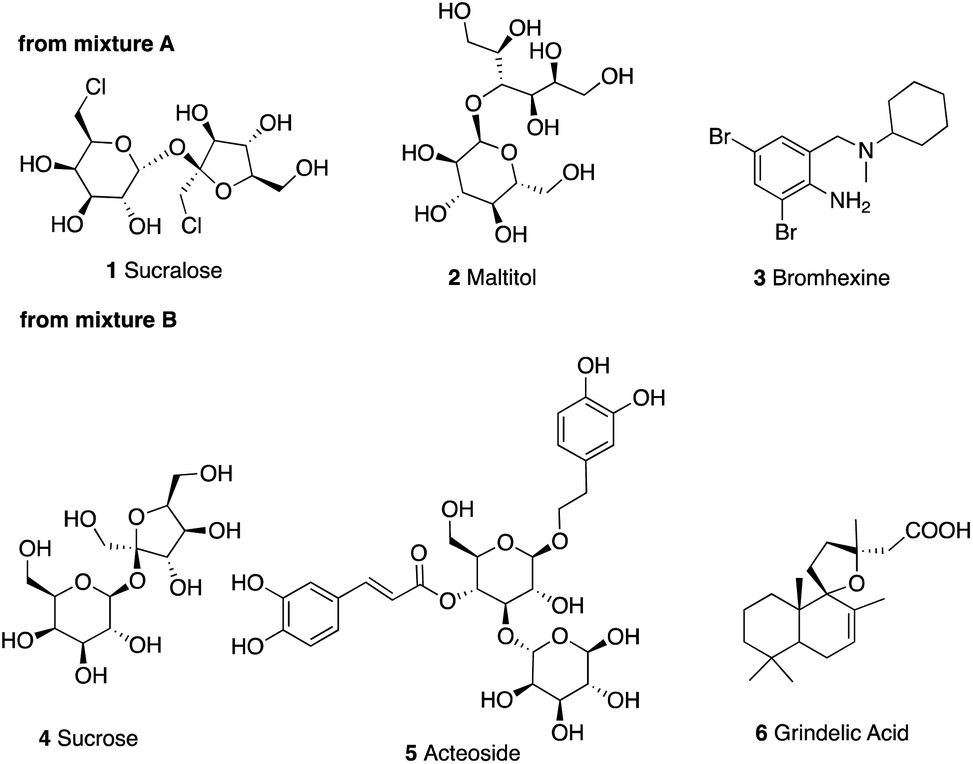 | ||
| Fig. 6 Main components of mixtures A and B identified by UHPLC-qToF. | ||
In mixture A 1000 mg L−1 after 28 days under the RBT conditions UHPLC-qToF analysis was used to detect bromhexine (3) and its metabolites. Bromhexine (3) has a typical MS/MS fragmentation characterized by the benzyl amino bond cleavage giving rise to two distinctive fragments, an aromatic one which brings the bromine atoms with a measured m/z at 263.8843, and a cyclohexane part bonded to the amino group with a measured at m/z 114.1277 (Fig. 7).
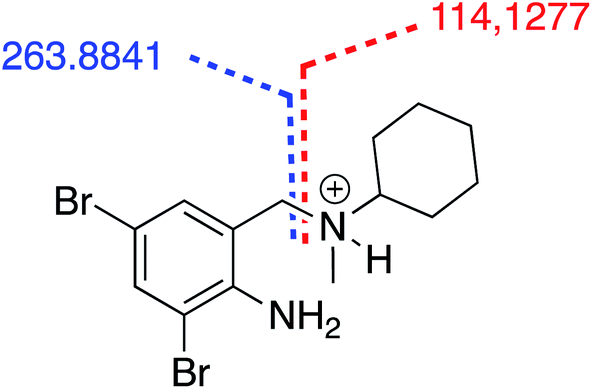 | ||
| Fig. 7 Bromhexine (3) typical ESI_MS/MS calculated fragment. | ||
Molecular ion m/z 377.0048 [(M + H+2)+] and of the corresponding fragment ions m/z 114.1280 and m/z 263.8843 (acceptable differences below 5 ppm with the calculated m/z are observed) clearly indicate that bromhexine and same related metabolites are still present after 28 days (Fig. 8).
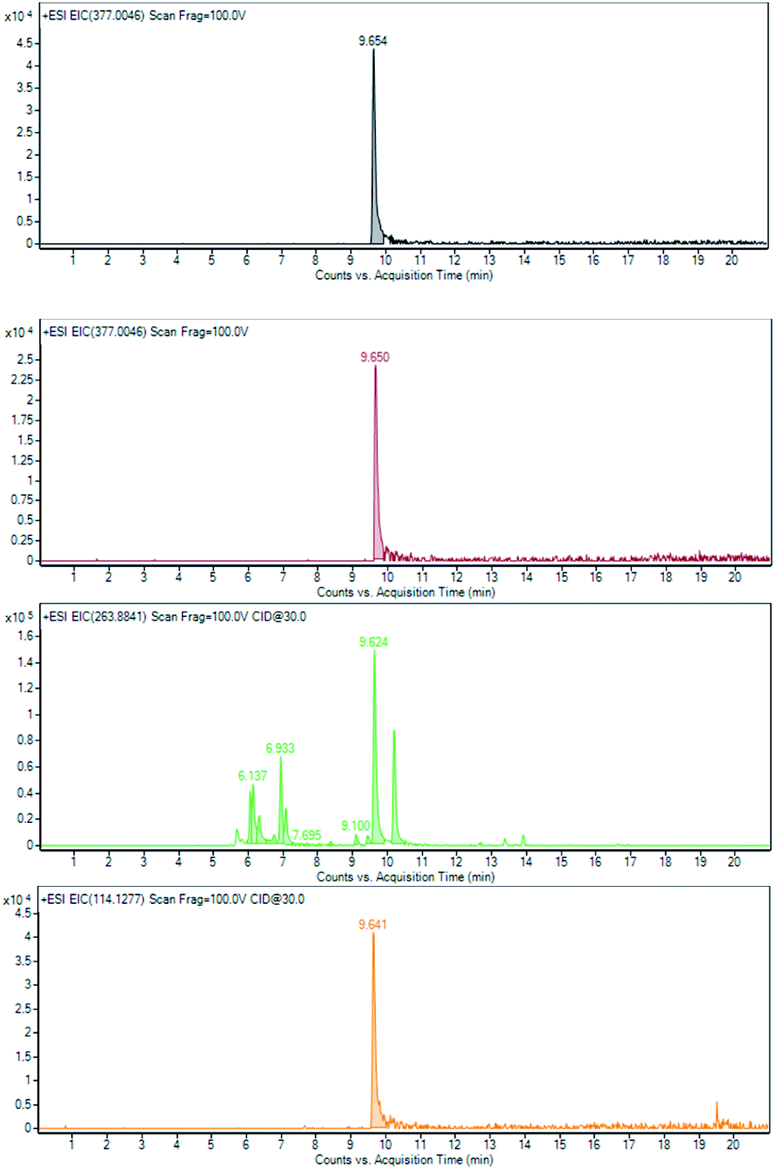 | ||
| Fig. 8 Mixture A: (i) bromhexine (5) at T0 EIC of m/z 377.0046 (black line) and at T28 EIC of m/z 377.0046 (brown line), 263.8841 (light green line) and 114.1227 (orange line). | ||
In particular, the extract ion at m/z of 263.8841 (Fig. 8, light green line) showed that there are several bromhexine metabolites having a bromo aryl fragment as a common characteristic. Some of them can be assigned (Table S3 and Fig. S1–5†) at different levels of confidence. As an example, it is possible to identify the structure of ambroxol 7 (level 1) with the experimental data fully superimposable to that of the pure reference standard identified by characterized fragmentation shown in Fig. 9.
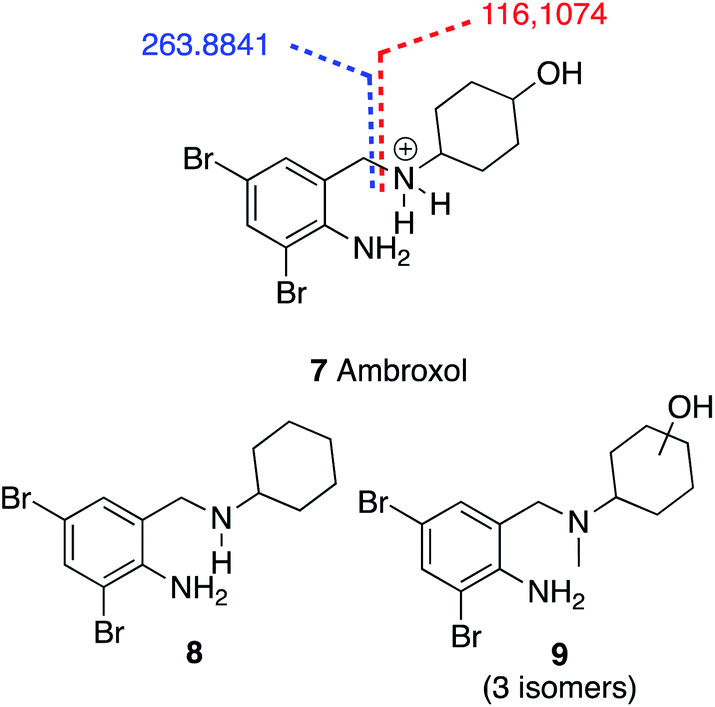 | ||
| Fig. 9 Ambroxol (6) typical ESI_MS/MS calculated fragment and most abundant metabolites 7 and 8. | ||
As described above, from the semiquantitative analysis of the spectroscopic data, it emerged that 12% of the initial bromhexine is not degraded after 28 days and that, among all, the metabolites 8 and 9 (Fig. 9) were the most abundant, probably because the N-demethylation of bromhexine and hydroxylation of the cyclohexyl ring were reactions kinetically favourable for the metabolism of RBT microfauna.
Similarly, the target analysis of sucralose was performed on mixture A. Searching for the molecular ion at an m/z of 419.0038 [(M + Na)+] after 28 days revealed its presence without any significant difference in concentration. This result was further confirmed by the EIC of its fragment at an m/z of 238.9848, that evidenced that in this case the presence of correlable metabolites having a common fragmentation was not detectable (Fig. 10).
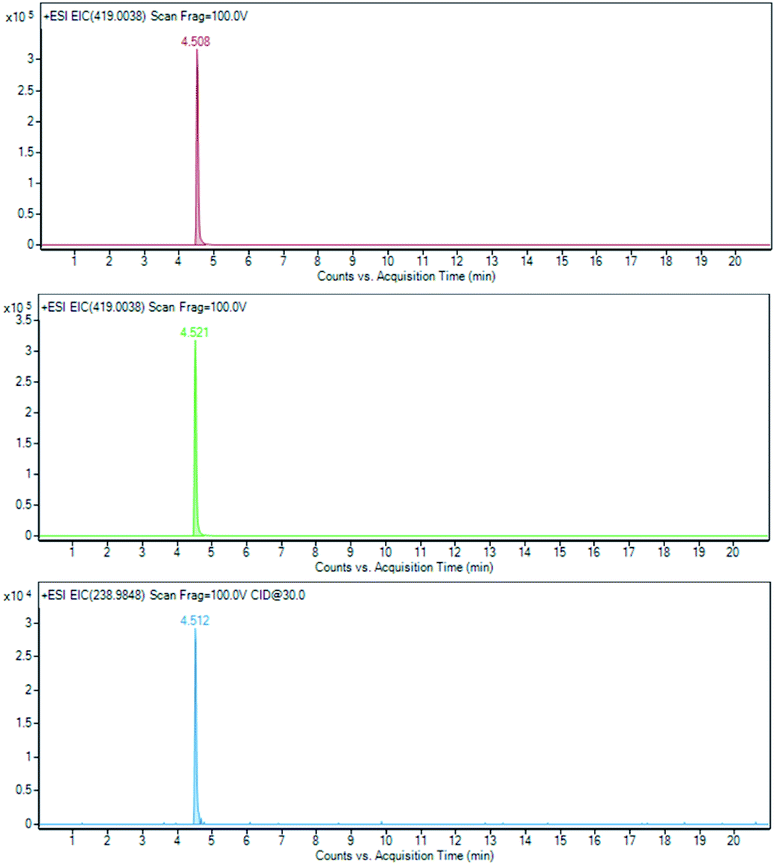 | ||
| Fig. 10 Sucralose (1) at T0 EIC of m/z 419.0038 (brown line); sucralose (1) at T28 EIC of m/z 419.0038 (green line) and its derivative 238.9848 (light blue line). | ||
The target study on biodegradability of mixture B was performed, focusing attention on the fate of sucrose (4), acteoside (5), and grindelic acid (6), selected as representative compounds of the mixture, identified at the T0 of the RBT.
The experimental MS/MS fragmentation patterns of acteoside (5)35,36 and grindelic acid (6) have been schematized in Fig. 14. Acteoside (5) showed the loss of the caffeoyl moiety corresponding to an m/z of 161.0244 [caffeic acid–H2O–H−], giving rise to the anion at an m/z of 461.1664 obtained by difference [M-H-caffeoyl−]. Grindelic acid (6) gave rise to the mass fragment 205.1596 m/z, assuming a spiro-tetrahydrofurane ring opening and further anion rearrangement not yet reported in literature. The elemental composition of the proposed fragment is C14H21O, with a calculated monoisotopic exact mass at m/z 205.1596, which corresponds to the m/z recorded from the fragmentation of grindelic acid pure standard as well as the sample.
As indicated by the extract ion chromatograms of ions shown in Fig. 11, 12 and 13 at m/z 341.1089 (M − H), 623.1981 (M − H)−, and 319.2278 (M − H)−, respectively, it was not possible to detect the presence of compounds 4, 5 and 6, or their direct derivatives after 28 days of the RBT.
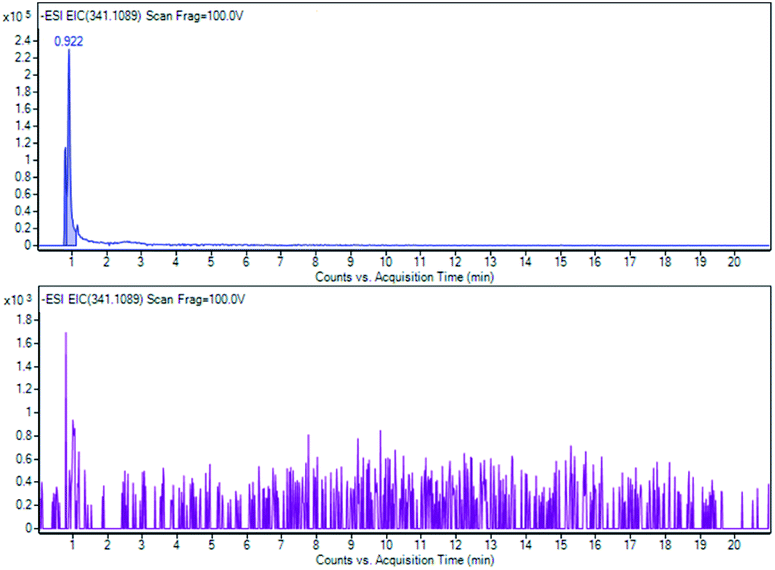 | ||
| Fig. 11 Sucrose (4) EIC of m/z 341.1089 at T0 and at T28. | ||
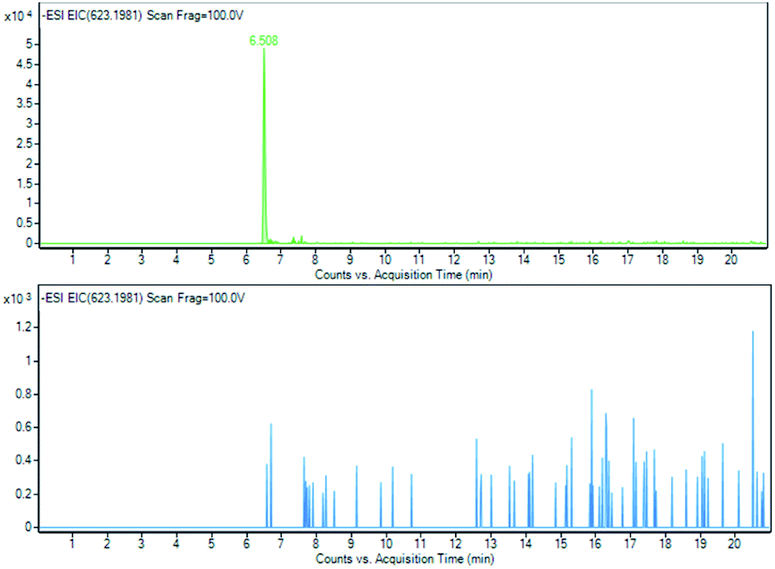 | ||
| Fig. 12 Acetoside (5) EIC of m/z 623.1981 at T0 and at T28. | ||
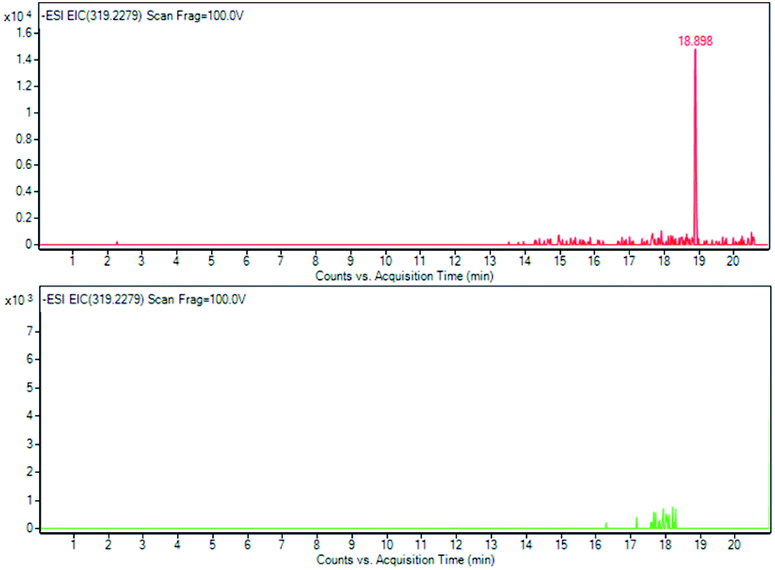 | ||
| Fig. 13 Grindelic acid (6) EIC of m/z 319, 2279 at T0 and at T28. | ||
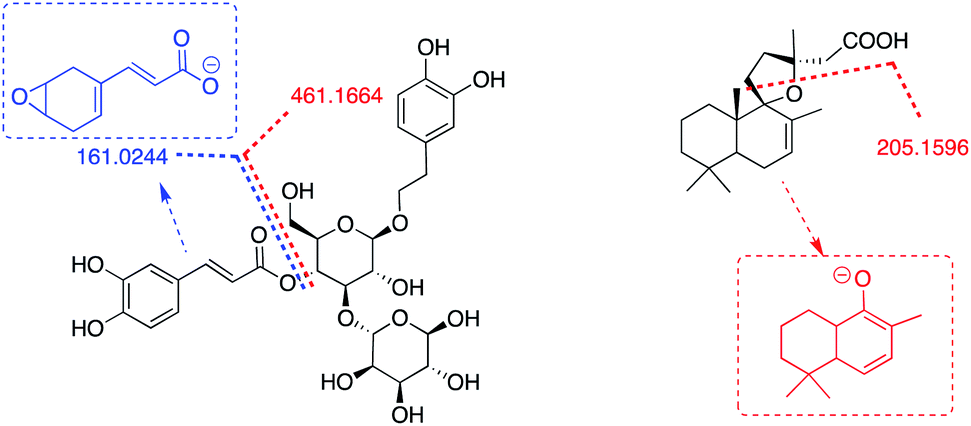 | ||
| Fig. 14 Acetoside (5) and grindelic acid (6) main ESI_ MS/MS fragments. | ||
Finally, in order to compare the fate of a not fully degradable API as a pure chemical entity or as a part of a pharmaceutical formulation the RTB test of pure bromhexine (3) was performed at 0.7 mg L−1 corresponding to its concentration in the mixture A 1000 mg L−1. After 28 days the evaluation of the relative area % (Table S1†) analysis evidenced that only 46% of the pure compound was degraded (vs. 88% observed in mixture A). This result is not easy to interpret and can be tentatively attributed to a higher activity of the microfauna in the presence of other readily available sources of energy. However, it clearly indicates that the degradation of a specific molecule can be considerably different when obtained in its pure form or as a part of a complex chemical mixture, suggesting that the mixture, similarly to a complex system, can exhibit some emerging and non-fully predictable properties.
Conclusions
In conclusion, based on the results collected in this manuscript we propose that the use of modern techniques, (such as UHPLC-qToF) should be considered for a more accurate and reliable evaluation of biodegradation and, consequently, the environmental impact of pharmaceutical and cosmetic formulations. We here demonstrated that using an untargeted approach, two different pharmaceutical formulations that, according to the currently used analytical protocol, can be considered readily biodegradable, are indeed not fully mineralized neither considering the synthetic components nor the natural ones even if, as expected, they showed a much more advanced degradation toward the mineral medium. The incomplete mineralization of a formulation affords a new mixture of derivatives that may deserve to be analyzed in detail using high performance methods like those based on mass spectrometry. Furthermore, we also observed that the biodegradability of a mixture of compounds cannot be considered as the simple sum of the biodegradability of each component. In this direction, the new technologies offer a holistic approach that is able to consider all at once the fate of the chemical system that has to be biodegraded and, at the same time, a very sensible target analysis that is able to elucidate most of the components of the final chemical fingerprint.For these reasons, in our opinion, the use of these modern technologies and the development of new protocols and rules based on them will improve the ability to predict, control and prevent the environmental impact of chemical mixtures as those present in pharmaceuticals, cosmetics, and personal care products.
The results reported here suggest a new approach in the evaluation of biodegradability, based more on systemic thinking, a paradigm that must always be considered when talking about the environment and biological systems.
Author contributions
Luisa Mattoli (L. M.), Giacomo Proietti (G. P.), Claudio Marzio Quintiero (C. M. Q.), Giada Fodaroni (G. F.), Michela Burico (M. B.), Mattia Gianni (M. G.) Emiliano Giovagnoni (E. G.) Valentino Mercati (V. M.) and Claudio Santi (C. S.); conceptualization: C. S., L. M., G. P., M. G., and V. M.; data curation: L. M., G. P., M. B., and C. M. Q.; formal analysis: G. P., M. B., and C. M. Q.; investigation: G. P., M. B., C. M. Q., and G. F.; methodology: G. P., L. M., and M. G.; software: M. B. and C. M. Q.; supervision: M. G. and V. M.; validation: G. P. and M. B.; visualization: G. P. and L. M.; writing – original draft: C. S., L. M., and G. P.; Writing – review & editing C. S., L. M., and E. G.Conflicts of interest
The authors listed below report the following details of affiliation or involvement in an organization or entity with a financial interest in the subject matter or materials discussed in this manuscript. L. M., G. P., C. M. Q., G. F., M. B., G. M., and E. G. are employed in Aboca who produce and commercialize the syrup used in the manuscript as mixture B. V. M. is the President of Aboca SPA. C. S. is the scientist responsible for a project related to the topic of the present manuscript and committed by Aboca to C.E.M.I.N (UniPG). The syrup used as mixture A is from a brand competitor of Aboca.Notes and references
- S. Zhu, M. A. Khan, F. Wang, Z. Bano and M. Xia, Exploration of adsorption mechanism of 2-phosphonobutane-1,2,4-tricarboxylic acid onto kaolinite and montmorillonite via batch experiment and theoretical studies, J. Hazard. Mater., 2021, 403, 123810 CrossRef CAS PubMed.
- S. Zhu, Y. Chen, M. A. Khan, H. Xu, F. Wang and M. Xia, In-Depth Study of Heavy Metal Removal by an Etidronic Acid- Functionalized Layered Double Hydroxide, ACS Appl. Mater. Interfaces, 2022, 14, 7450–7463 CrossRef CAS PubMed.
- S. Zhu, M. A. Khan, T. Kameda, H. Xu, F. Wang, M. Xia and T. Yoshioka, New insights into the capture performance and mechanism of hazardous metals Cr3+ and Cd2+ onto an effective layered double hydroxidebased material, J. Hazard. Mater., 2022, 426, 128062 CrossRef CAS PubMed.
- L. Ma, T. Hu, Y. Liu, J. Liu, Y. Wang, P. Wang, J. Zhou, M. Chen, B. Yang and L. Li, Combination of biochar and immobilized bacteria accelerates polyacrylamide biodegradation in soil by both bio-augmentation and bio-stimulation strategies, J. Hazard. Mater., 2021, 405, 124086 CrossRef CAS PubMed.
- G. Thouand, M.-J. Durand, A. Maul, C. Gancet and H. Blok, New concepts in the evaluation of biodegradation/persistence of chemical substances using a microbial inoculum, Front. Microbiol., 2011, 2, 164 Search PubMed.
- OECD, Revised Introduction to the OECD Guidelines for Testing of Chemicals, Section 3, OECD, 2006 and OECD, Test No. 301: Ready Biodegradability, 1992 Search PubMed.
- OECD, Test No. 311, Anaerobic Biodegradability of Organic Compounds in Digested Sludge: by Measurement of Gas Production, OECD, 2006 Search PubMed.
- REGULATION (EC), No. 440/2008 of 30 May, 2008.
- OECD, Test No. 310, Ready Biodegradability – CO2 in sealed vessels (Headspace Test), OECD, 2014 Search PubMed.
- N. Nyholm, A. Damborg and P. Lindgaard-Jørgensen, A comparative study of test methods for assessment of the biodegradability of chemicals in seawater—Screening tests and simulation tests, Ecotoxicol. Environ. Saf., 1992, 23, 173–190 CrossRef CAS PubMed.
- J. F. Ericson, Evaluation of the OECD 314B Activated Sludge Die-Away Test for Assessing the Biodegradation of Pharmaceuticals, Environ. Sci. Technol., 2010, 44, 375–381 CrossRef CAS PubMed.
- F. Brillet, A. Maul, M. J. Durand and T. Gérald, From laboratory to environmental conditions: a new approach for chemical's biodegradability assessment, Environ. Sci. Pollut. Res., 2016, 23, 18684–18693 CrossRef PubMed.
- J. W. Raymond, T. N. Rogers, D. R. Shonnard and A. A. Kline, A review of structure-based biodegradation estimation methods, J. Hazard. Mater., 2001, 84, 189–215 CrossRef CAS PubMed.
- F. Pizzo, A. Lombardo, A. Manganaro and E. Benfenati, In silico models for predicting ready biodegradability under REACH: a comparative study, Sci. Total Environ., 2013, 463–464, 161–168 CrossRef CAS PubMed.
- P. H. Howard, W. M. Stiteler, W. M. Meylan, A. E. Hueber, J. A. Beauman, M. E. Larosche and R. S. Boethling, Predictive model for aerobic biodegradability developed from a file of evaluated biodegradation data, Environ. Toxicol. Chem., 1992, 11, 593–603 CrossRef CAS.
- K. Fenner, M. Scheringer and K. Hungerbühler, Prediction of overall persistence and long-range transport potential with multimedia fate models: robustness and sensitivity of results, Environ. Pollut., 2004, 128, 189–204 CrossRef CAS PubMed.
- D. Aronson, R. Boethling, P. Howard and W. Stiteler, Estimating biodegradation half-lives for use in chemical screening, Chemosphere, 2006, 63, 1953–1960 CrossRef CAS PubMed.
- G. Thouand, P. Friant, F. Bois, A. Cartier, A. Maul and J. C. Block, Bacterial Inoculum Density and Probability of para-Nitrophenol Biodegradability Test Response, Ecotoxicol. Environ. Saf., 1995, 30, 274–282 CrossRef CAS PubMed.
- C. G. Van Ginkel, A. Haan, M. L. Luijten and C. A. Stroo, Influence of the size and source of the inoculum on biodegradation curves in closed-bottle tests, Ecotoxicol. Environ. Saf., 1995, 31, 218–223 CrossRef CAS PubMed.
- G. Vazquez-Rodriguez, G. Goma and J. L. Rols, Toward a standardization of the microbial inoculum for ready biodegradability testing of chemicals, Water Sci. Technol., 2000, 42, 43–46 CrossRef CAS.
- A. K. Goodhead, I. M. Head, J. R. Snape and R. J. Davenport, Standard inocula preparations reduce the bacterial diversity and reliability of regulatory biodegradation tests, Environ. Sci. Pollut. Res., 2014, 21, 9511–9521 CrossRef CAS PubMed.
- A. Kowalczyk, T. J. Martin, O. R. Price, J. R. Snape, R. A. Egmond, C. J. Finnegan, H. Schäfer, R. J. Davenport and G. D. Bending, Refinement of biodegradation tests methodologies and the proposed utility of new microbial ecology techniques, Ecotoxicol. Environ. Saf., 2015, 111, 9–22 CrossRef CAS PubMed.
- European Chemicals Agency, Guidance on Information Requirements and Chemical Safety Assessment Chapter R.7b: Endpoint specific guidance Draft Version 4.0, January, 2017 Search PubMed.
- M. Hannemann, B. Zonja, D. Barceló and S. Pérez, HRMS Approaches for Evaluating Transformations of Pharmaceuticals in the Aquatic Environment, in ACS Symposium Series, ed. J. E. Drewes and T. Letzel, American Chemical Society, Washington, DC, 2016, volume 1241, pp. 25–44 Search PubMed.
- C. Mejías, J. Martín, J. L. Santos, I. Aparicio and E. Alonso, Occurrence of pharmaceuticals and their metabolites in sewage sludge and soil: A review on their distribution and environmental risk assessment, Trends Environ. Anal. Chem., 2021, 30, e00125 CrossRef.
- U. Rozman and G. Kalčíková, The first comprehensive study evaluating the ecotoxicity and biodegradability of water-soluble polymers used in personal care products and cosmetics, Ecotoxicol. Environ. Saf., 2021, 228, 113016 CrossRef CAS PubMed.
- D. Rivas, B. Zonja, P. Eichhorn, A. Ginebreda, S. Pérez and D. Barceló, Using MALDI-TOF MS imaging and LC-HRMS for the investigation of the degradation of polycaprolactone diol exposed to different wastewater treatments, Anal. Bioanal. Chem., 2017, 409, 5401–5411 CrossRef CAS PubMed.
- 1D. Rivas, B. Zonja, P. Eichhorn, A. Ginebreda, S. Pérez and D. Barceló, Anal. Bioanal. Chem., 2017, 409, 5401–5411 CrossRef PubMed.
- S. Naz, H. Gallart-Ayala, S. N. Reinke, C. Mathon, R. Blankley, R. Chaleckis and C. E. Wheelock, Development of a Liquid Chromatography–High Resolution Mass Spectrometry Metabolomics Method with High Specificity for Metabolite Identification Using All Ion Fragmentation Acquisition, Anal. Chem., 2017, 89, 7933–7942 CrossRef CAS PubMed.
- G. Gatidou, N. Vazaiou, N. S. Thomaidis and A. S. Stadinakis, Biodegradability assessment of food additives using OECD 301F respirometric test, Chemosphere, 2020, 241, 125071 CrossRef CAS PubMed.
- J. H. Ward, Hierarchical Grouping to Optimize an Objective Function, J. Am. Stat. Assoc., 1963, 58, 236–244 CrossRef.
- Identification and Fragmentation of Sucralose Using Accurate-Mass Q-TOF LC/MS and Molecular Structure Correlator Software, Agilent Application Note 5991-4066EN, 2014 Search PubMed.
- H. M. El-Sayed and H. Hashem, Quality by Design Strategy for Simultaneous HPLC Determination of bromhexine HCl and Its Metabolite Ambroxol HCl in Dosage Forms and Plasma, Chromatographia, 2020, 83, 1075–1085 CrossRef CAS.
- J. Liu, X. Chen, Y. Hu, G. Cheng and D. Zhong, Quantification of the major metabolites of bromhexine in human plasma using RRLC-MS/MS and its application to pharmacokinetics, J. Pharm. Biomed. Anal., 2010, 51, 1134–1141 CrossRef CAS PubMed.
- Y. M. Attia, D. M. El-Kersh, H. A. Wagdy and M. M. Elmazar, Verbascoside: Identification, Quantification, and Potential Sensitization of Colorectal Cancer Cells to 5-FU by Targeting PI3K/AKT Pathway, Sci. Rep., 2018, 8, 16939 CrossRef PubMed.
- D. Su, W. Li, Q. Xu, Y. Liu, Y. Song and Y. Feng, New metabolites of acteoside identified by ultra-performance liquid chromatography/quadrupole-time-of-flight MS(E) in rat plasma, urine, and feces, Fitoterapia, 2016, 112, 45–55 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1va00038a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |