
A reverse intersystem crossing managing assistant dopant for high external quantum efficiency red organic light-emitting diodes†
Dong Jin
Shin
,
Seung Chan
Kim
and
Jun Yeob
Lee
 *
*
School of Chemical Engineering, Sungkyunkwan University, 2066, Seobu-ro, Jangan-gu, Suwon-si, Gyeonggi-do 16419, Korea. E-mail: leej17@skku.edu
First published on 16th December 2021
Abstract
Thermally activated delayed fluorescence (TADF) materials that can serve as assistant dopants with reduced Dexter energy transfer rates were designed by replacing the donor moiety of 2,3,5,6-tetra(9H-carbazol-9-yl)terephthalonitrile (4CzTPN) with 5H-benzo[4,5]thieno[3,2-c]carbazole (BTCz). The employment of the BTCz donor strengthened the charge transfer character, inducing high reverse intersystem crossing (RISC) rate. Careful management of the number of BTCz donor moieties optimized the RISC rates and photoluminescence quantum yield value for high efficiency in TADF-assisted fluorescent devices. A TADF-assisted fluorescent device with the BTCz-modified TADF assistant dopant exhibited a high external quantum efficiency of 13.0% compared with 11.3% for that with the 4CzTPN assistant dopant due to suppressed Dexter energy transfer.
Introduction
Recently, thermally activated delayed fluorescence (TADF) emitters have undergone active research due to their capability of achieving 100% internal quantum efficiency (IQE) in electroluminescent (EL) devices. TADF molecules utilize 75% of electrogenerated triplet excitons for radiative transition through the reverse intersystem crossing (RISC) process. In general, they are designed to have donor and acceptor moieties for efficient RISC through spatial orbital separation between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), which induces a small energy difference between singlet (S1) and triplet (T1) excited states (ΔEST). The small ΔEST makes it possible to convert triplet excitons into singlet excitons at room temperature through the RISC process.1–14However, the use of TADF materials as dopant molecules is limited by their broad emission spectra due to the high degree of geometry changes in the excited state in spite of a high IQE close to 100%. Instead, the triplet exciton harvesting function of TADF materials allowed them to be used as assistant dopants to harvest the singlet excitons of a fluorescent emitter for an IQE of 100% (hyperfluorescence).12,15 In general, only 25% of electrogenerated excitons are utilized by fluorescent emitters, but 100% electrogenerated excitons can be completely used by the fluorescent emitter in the hyperfluorescent device employing TADF molecules as the assistant dopant to harvest triplet excitons for Förster resonance energy transfer (FRET) to the fluorescent emitter. Furthermore, high color purity can also be achieved by the hyperfluorescence process because the main emission originates from the fluorescent emitter. Recently, several articles demonstrated outstanding performance in organic light emitting diodes (OLEDs) via the TADF-assisted fluorescence process by managing either the TADF assistant dopant or the fluorescent emitter. However, the design of the TADF assistant dopant to effectively harvest the singlet excitons of a fluorescent emitter is still limited and further exploration of TADF materials is demanded.16–25
In this work, we synthesized two compounds, 2-(5H-benzo[4,5]thieno[3,2-c]carbazol-5-yl)-3,5,6-tri(9H-carbazol-9-yl)terephthalonitrile (BTCz3CzTPN) and 2,5-bis(5H-benzo[4,5]thieno[3,2-c]carbazol-5-yl)-3,6-di(9H-carbazol-9-yl)terephthalonitrile (2BTCz2CzTPN), adopting terephthalonitrile (TPN) as a strong acceptor moiety combined with 9H-carbazole (Cz) and 5H-benzo[4,5]thieno[3,2-c]carbazole (BTCz) donor moieties to develop orange-red TADF assistant dopants. The number of BTCz moieties was controlled to manage the RISC rate of the TADF molecules. It was demonstrated that the introduction of BTCz instead of Cz accelerated the RISC process and red-shifted the emission spectrum. In particular, BTCz3CzTPN proved its potential as an assistant dopant by achieving a high external quantum efficiency (EQE) of 13.0% compared to 11.3% for the 2,3,5,6-tetra(9H-carbazol-9-yl)terephthalonitrile (4CzTPN) reference material.26
Results and discussion
4CzTPN is a well-known TADF molecule having two para positioned CN units and four Cz donors around a benzene core.26,27 The two para positioned CN units induce a deep LUMO energy level and strong acceptor character for red-shifted emission, whereas the four donors rendered strong donor character and a sterically crowded environment to suppress Dexter energy transfer (DET) from 4CzTPN to a fluorescent emitter in TADF-assisted fluorescent devices. However, the RISC rate of 4CzTPN is not high enough to manage FRET and DET to the fluorescent emitter. Therefore, the donor of 4CzTPN was modified with the BTCz donor with stronger donor properties than Cz. The introduction of the BTCz donor may intensify the charge transfer (CT) character, inducing a small ΔEST and a high RISC rate.28,29 One or two BTCz donors were employed in the design of TADF emitters because further replacement of Cz with the BTCz donor may shift the emission wavelength to the red emission range, which is not suitable for sensitization of the red fluorescent emitter.The synthetic schemes of BTCz3CzTPN and 2BTCz2CzTPN are shown in Scheme 1 and detailed synthetic procedures are described in the ESI.† In all synthetic procedures, cesium carbonate was used as the base material and dimethylformamide (DMF) as the solvent for nucleophilic substitution. 2BTCz2CzTPN and BTCz3CzTPN were synthesized in good yields of 90.3% and 87.2%, respectively, without any noticeable byproducts, thanks to the electron deficiency of the TPN moiety. Selective substitution of two and three Cz units was carried out and then BTCz was reacted with Cz-modified intermediates to obtain the target compounds. The final products were purified by sublimation and column chromatography.
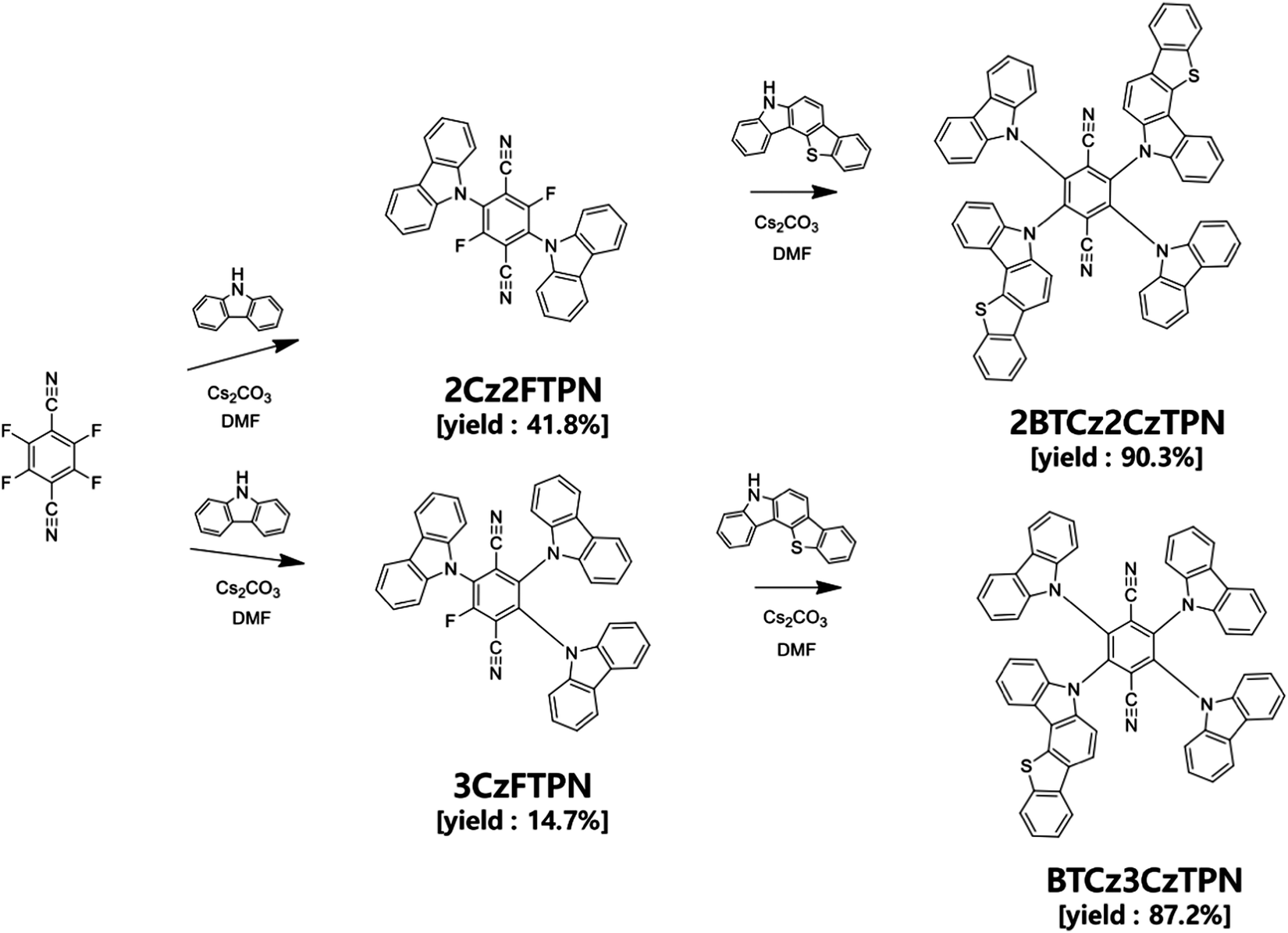 | ||
| Scheme 1 Synthetic schemes of BTCz3CzTPN and 2BTCz2CzTPN. | ||
The geometry optimization and electronic orbital calculation in the ground state were studied by Gaussian simulation using the B3LYP 6-31G basis set. The optimized geometrical structures and energy details of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN are shown in Fig. 1(a), (b) and (c), respectively. 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN showed a similar LUMO distribution positioned on the TPN moiety, resulting in LUMO energy levels of −2.80, −2.83, and −2.87 eV, respectively, whereas the HOMO distribution of the TADF molecules was dissimilar. The HOMO of 4CzTPN was evenly dispersed over the four Cz donors, but those of BTCz3CzTPN and 2BTCz2CzTPN were localized on the BTCz donor due to the strong donor character. As a result, BTCz3CzTPN and 2BTCz2CzTPN had the same shallow HOMO energy level of −5.58 eV compared to −5.65 eV for 4CzTPN. The simulated HOMO–LUMO gaps of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were 2.85, 2.75 and 2.71 eV, respectively, which can be interpreted that the BTCz donor decreased the HOMO–LUMO gap.30
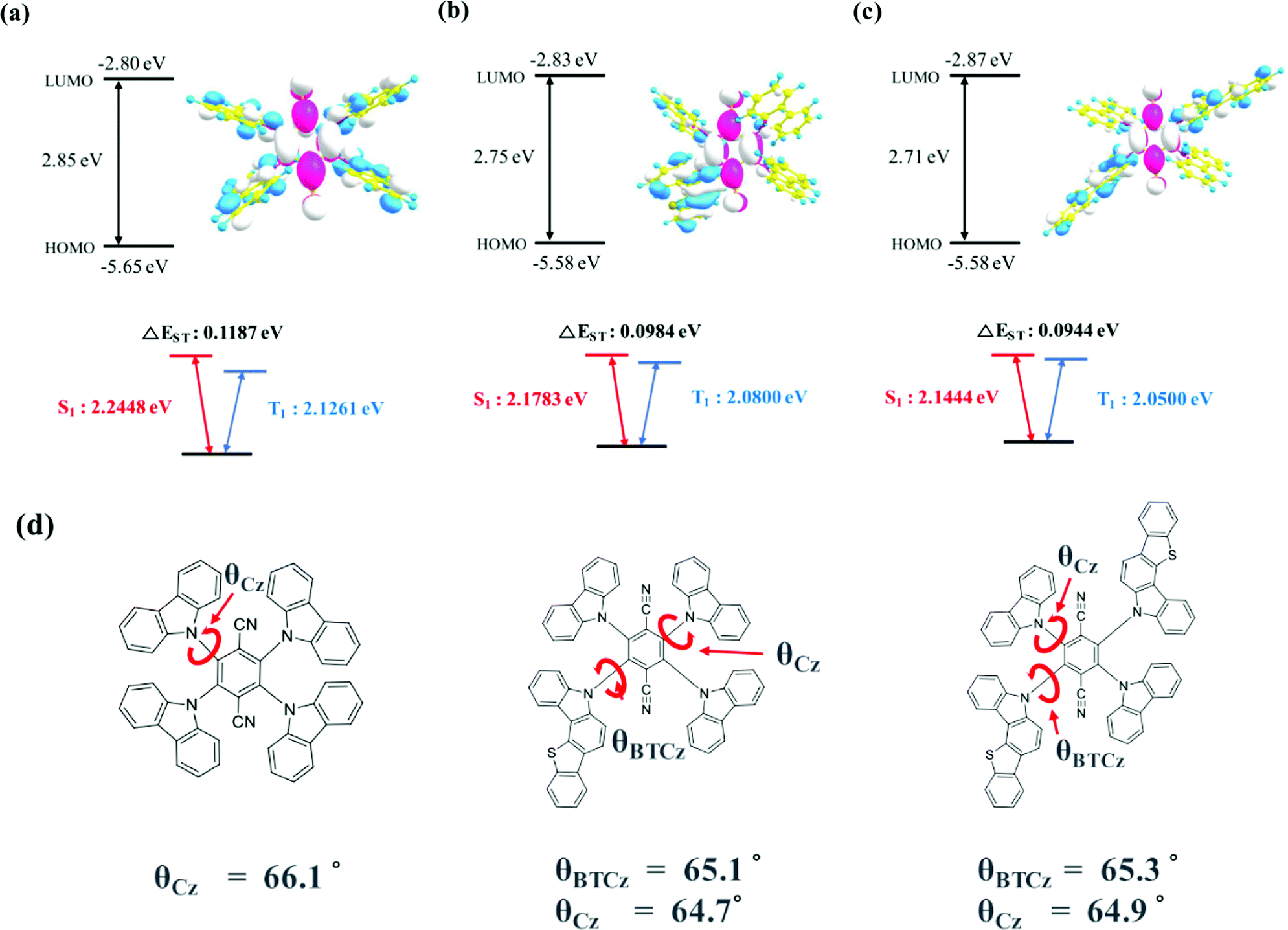 | ||
| Fig. 1 Optimized ground state HOMO (blue) and LUMO (red) distribution and calculated energy details of (a) 4CzTPN, (b) BTCz3CzTPN and (c) 2BTCz2CzTPN. (d) Dihedral angles of the Cz and BTCz moieties in 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN. | ||
The singlet and triplet energies of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were governed by the BTCz donor. As shown in Fig. 1(d), the dihedral angles of BTCz and Cz were similar, that is, about 65°, indicating that the BTCz donor does not affect the dihedral angle in the optimized ground state geometry. Therefore, it is expected that CT strength would be significantly dominated by donor strength. The adopted BTCz donor induced strengthened CT properties, resulting in decreased singlet and triplet energies. Singlet and triplet energies were lowered as the BTCz number increased and the calculated singlet/triplet energies of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were 2.2448/2.1261 eV, 2.1783/2.0800 eV and 2.1444/2.0500 eV, respectively. The calculated ΔEST values of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were 0.1187, 0.0984 and 0.0944 eV, respectively, which demonstrates the potential of a high RISC rate in BTCz3CzTPN and 2BTCz2CzTPN.
The ultraviolet-visible (UV-vis) absorption, fluorescence and phosphorescence spectra of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were analyzed in tetrahydrofuran and toluene solutions (1 × 10−5 M). As shown in Fig. 2, strong absorption peaks below 400 nm originating from the π–π* transition of the donor and acceptor structures were observed in the UV-vis absorption spectra of the three TADF molecules. Additionally, they showed broad absorption spectra above 400 nm which can be assigned to CT absorption by the main skeleton. As depicted in Figure S1, the degree of red shift of the CT absorption spectra was in the order of 4CzTPN < BTCz3CzTPN < 2BTCz2CzTPN, confirming that the BTCz donor strengthened the CT properties.
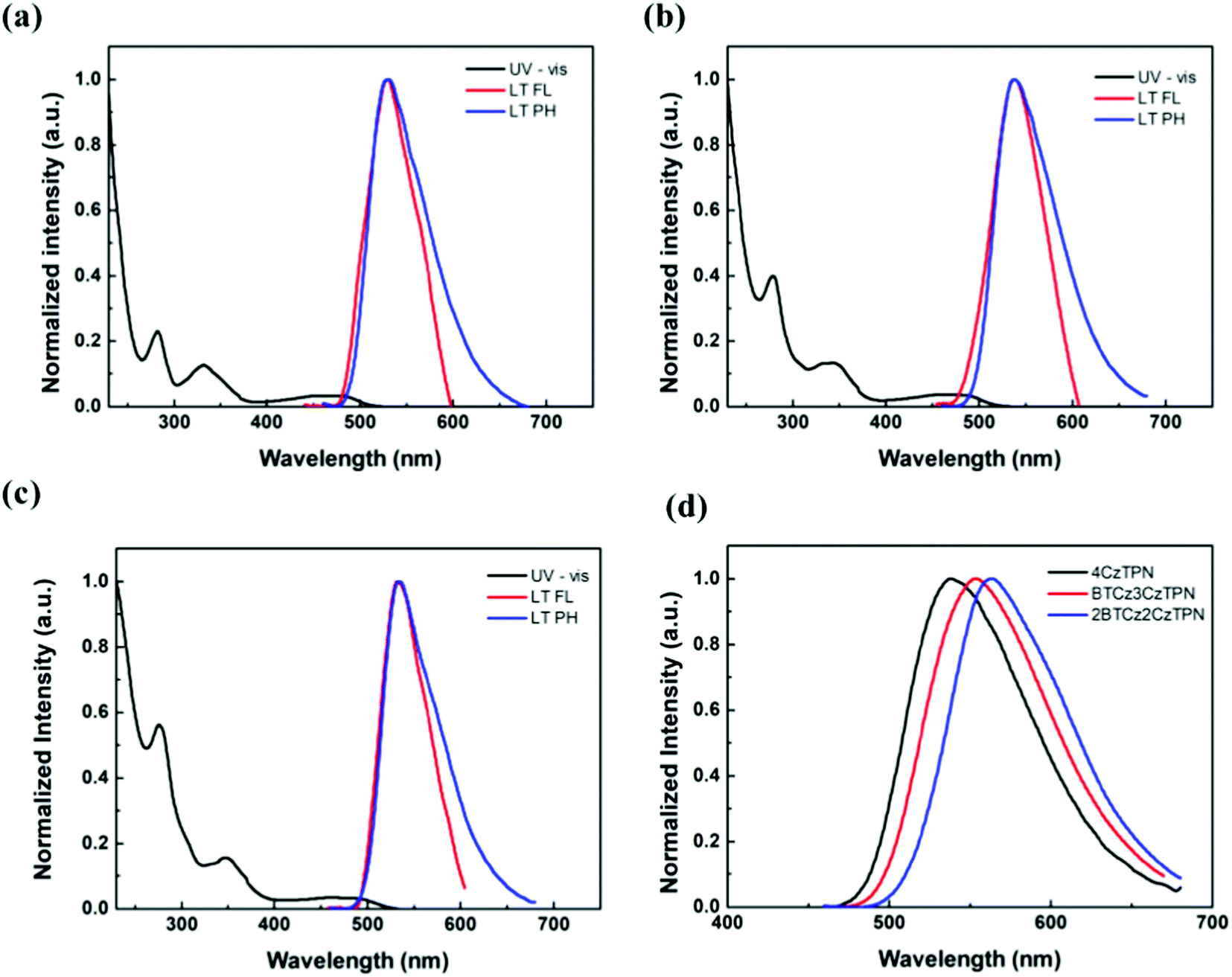 | ||
| Fig. 2 Ultraviolet-visible and photoluminescence (PL) spectra of (a) 4CzTPN, (b) BTCz3CzTPN and (c) 2BTCz2CzTPN. (d) Room temperature fluorescence (RTFL) of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN. | ||
The singlet and triplet energy values of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were analyzed from the low temperature fluorescence (LT FL) and low temperature phosphorescence (LT PH) spectra. The LT PH spectra were measured after a delay time of 1 ms to remove the prompt emission component. The singlet and triplet energy values of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN from the onset wavelengths of LT FL and LT PH were 2.561/2.519 eV, 2.524/2.488 eV and 2.508/2.479 eV, respectively. The calculated ΔEST values of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were 0.042 eV, 0.036 eV and 0.029 eV, respectively. The singlet and triplet energy values decreased in the order of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN, which was in good agreement with the calculated data. Since the BTCz donor possesses stronger donor strength than the Cz donor, the attached BTCz donor rendered intensified CT properties, resulting in reduced singlet and triplet energy values.31,32 Moreover, as shown in Fig. 2(d), clearly red-shifted spectra in the order of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were observed. Furthermore, the strengthened CT properties triggered a large HOMO and LUMO separation, decreasing ΔEST in the BTCz-derived TADF compounds.
Photoluminescence quantum yield (PLQY) and transient PL decay measurements were performed to analyze the TADF performance of the three molecules using a 1 wt% emitter-doped bis[2-(diphenylphosphino)phenyl]ether oxide (DPEPO) film. The PLQYs of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were 34.1, 27.9 and 25.3% under nitrogen, respectively. The PLQY was reduced as the number of attached BTCz moieties increased. This might be caused by the red-shifted emission spectrum activating non-radiative transition according to the energy gap law.33,34 The transient PL decay curves of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN are shown in Fig. 3. The prompt lifetimes (τp) and delayed fluorescence lifetimes (τd) of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were 14.5 ns/5.51 μs, 16.4 ns/3.55 μs, and 17.6 ns/2.47 μs, respectively.35 The order of τd of the three TADF molecules matched well with that of the ΔEST value. Therefore, this can be interpreted that the small ΔEST initiated facile triplet to singlet crossover for up-conversion, resulting in a short τd. Based on the measured τp, τd, and PLQY, PL-related rate constants were calculated and are summarized in Table 1.36–38 The rate constant for RISC (kRISC) increased in the order of 4CzTPN (8.03 × 105 s−1) < BTCz3CzTPN (9.08 × 105 s−1) < 2BTCz2CzTPN (10.22 × 105 s−1) due to the shortened τd. However, the red-shifted emission spectra of BTCz3CzTPN and 2BTCz2CzTPN resulted in increased non-radiative rate constants (knr) of 2.18 × 105 s−1 and 3.27 × 105 s−1, respectively, by the energy gap law. The prompt (kp), delayed (kd), intersystem crossing (kISC), and radiative transition (kr) rate constants are given in Table S1 (ESI†).
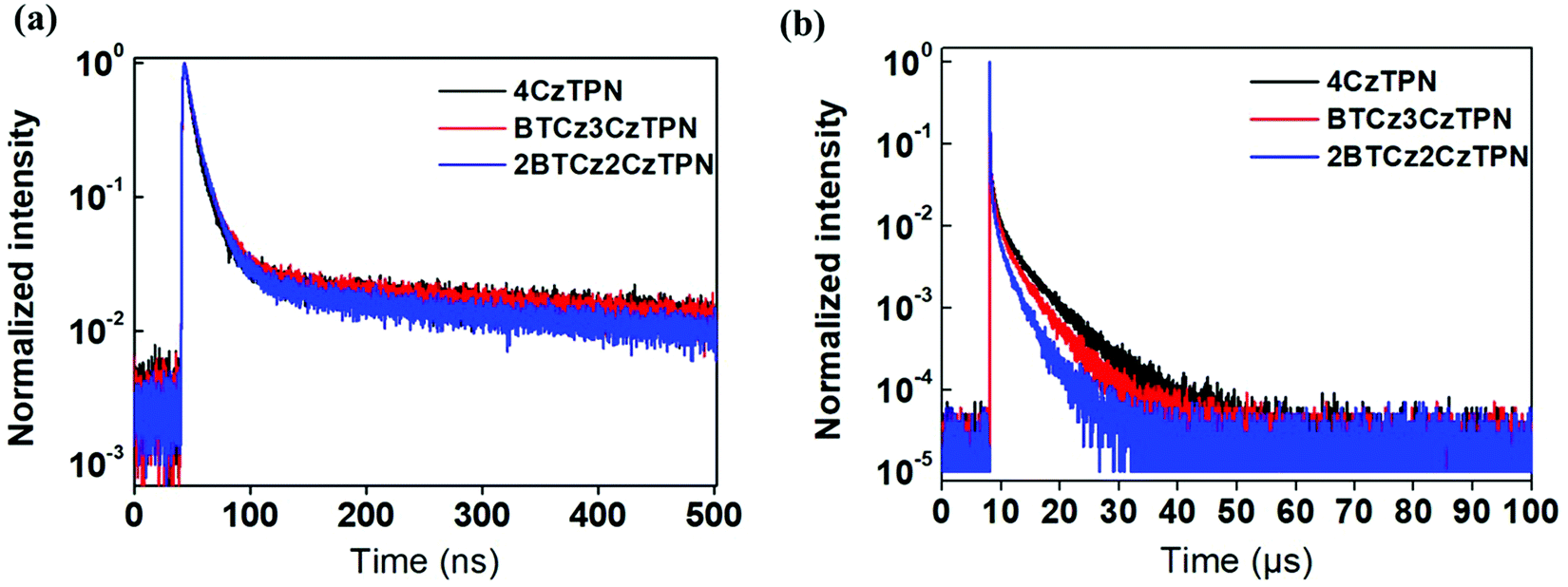 | ||
| Fig. 3 Transient PL decay curves of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN. | ||
| HOMO/LUMO [eV] | Singlet energya/triplet energya [eV] | ΔEST [eV] | PLQYb [%] | τ p [ns] | τ d [μs] | k RISC [×105 s−1] | k nr [×105 s−1] | |
|---|---|---|---|---|---|---|---|---|
| a Measured in 10−5 M toluene solution. Onset energy parameter of the PL spectrum. b Measured using a 1 wt% doped DPEPO film with TADF under nitrogen. c Measured using a 1 wt% doped DPEPO film with TADF. | ||||||||
| 4CzTPN | −6.36/−3.95 | 2.561/2.519 | 0.042 | 34.1b | 14.5 | 5.51 | 8.03 | 1.28 |
| Btcz3CzTPN | −6.30/−3.91 | 2.524/2.488 | 0.036 | 27.9b | 16.4 | 3.55 | 9.08 | 2.18 |
| 2Btcz2CzTPN | −6.25/−3.90 | 2.508/2.479 | 0.029 | 25.3b | 17.6 | 2.47 | 10.22 | 3.27 |
The electrochemical properties were examined by cyclic voltammetry measurements. The scanned data of oxidation and reduction are shown in Fig. S2 (ESI†). The LUMO energy levels of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were −3.95 eV, −3.91 eV and −3.90 eV, respectively. The similar LUMO energy levels of the three TADF molecules were derived from the same electron acceptor moiety. However, the HOMO energy levels of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN were −6.36 eV, −6.30 eV and −6.25 eV, respectively. The HOMO energy levels became shallow sequentially as the strong BTCz donor was introduced.
Device fabrication and electroluminescence properties
Based on the photophysical properties and energy levels of the three TADF molecules, EL properties were evaluated by fabricating OLEDs. The energy level details of the devices are shown in Fig. S3 (ESI†). TADF molecules were doped in 2-phenyl-4,6-bis(12-phenylindolo[2,3-a]carbazole-11-yl)-1,3,5-triazine (PBICT) and 4-(3-(triphenylen-2-yl)phenyl)dibenzo[b,d]thiophene (DBTTP1) hosts at 1 wt% and 10 wt% doping concentrations.39–41 The specific chemical structures of each layer are also depicted in Fig. S3 (ESI†). The EQE–luminance curves and EL spectra of the 1 wt% and 10 wt% emitter-doped devices are shown in Fig. 4. The current density–voltage–luminance data are shown in Fig. S4 (ESI†) and the summarized device data in Table 2.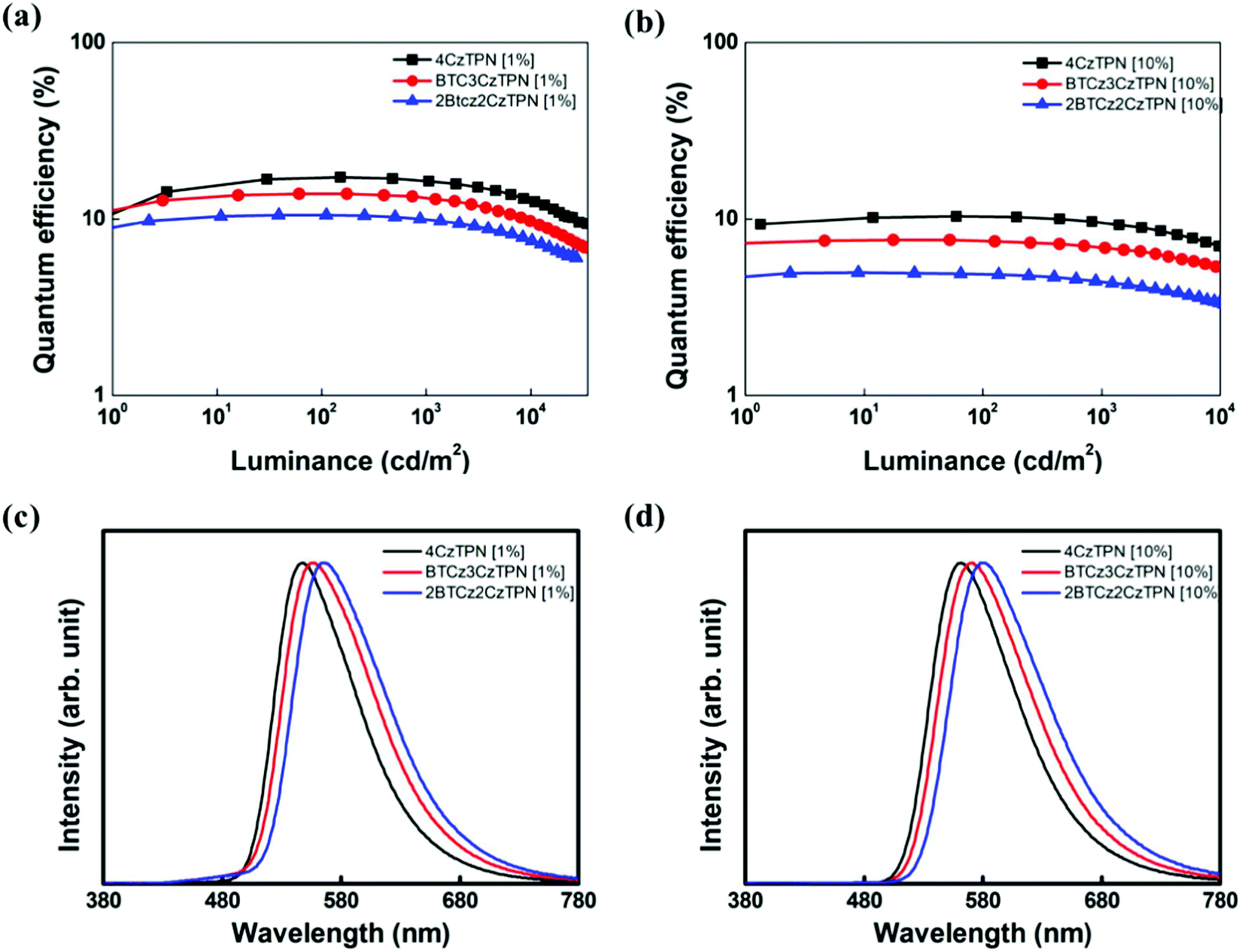 | ||
| Fig. 4 Quantum efficiency–luminance plots of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN at (a) 1 wt% and (b) 10 wt%. EL spectra of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN devices at (c) 1 wt% and (d) 10 wt%. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DBP [10:0.5 wt%] devices
:DBP [10:0.5 wt%] devices
| λ EL [nm] | CEmax [cd A−1] | PEmax [lm W−1] | QEab [%] | J 90% [mA cm−2] | CIE (x, y) | |
|---|---|---|---|---|---|---|
| a Measured at 5000 cd m−2. b Maximum value. | ||||||
| 4CzTPN [1 wt%] | 548 | 60.2 | 41.2 | 14.3a/17.2b | 4.5 | (0.42, 0.56) |
| 4CzTPN [10 wt%] | 561 | 32.5 | 22.7 | 8.0a/10.4b | 4.4 | (0.47, 0.52) |
| 4CzTPN:DBP [10:0.5 wt%] |
614 | 14.1 | 9.8 | 4.1a/11.3b | 0.1 | (0.60, 0.40) |
| BTCz3CzTPN [1 wt%] | 557 | 44.7 | 28.5 | 11.1a/13.9b | 5.2 | (0.45, 0.53) |
| BTCz3CzTPN [10 wt%] | 571 | 21.4 | 14.2 | 5.9a/7.6b | 6.8 | (0.50, 0.49) |
| BTCz3CzTPN:DBP [10:0.5 wt%] |
614 | 17.0 | 10.0 | 4.6a/13.0b | 0.1 | (0.60, 0.40) |
| 2BTCz2CzTPN [1 wt%] | 567 | 31.1 | 20.1 | 8.5a/10.5b | 7.2 | (0.48, 0.50) |
| 2BTCz2CzTPN [10 wt%] | 581 | 12.1 | 7.7 | 3.7a/5.0b | 8.2 | (0.53, 0.46) |
| 2BTCz2CzTPN:DBP [10:0.5 wt%] |
614 | 12.7 | 8.0 | 3.8a/10.2b | 0.1 | (0.62, 0.38) |
The maximum EQEs of the 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN devices at a 1/10 wt% doping concentration were 17.2/10.4, 13.9/7.6 and 10.5/5.0%, respectively, which were in good agreement with the PLQY values. The maximum EQEs at a 10 wt% doping concentration were decreased compared to those at a 1 wt% doping concentration due to a serious concentration quenching effect by the donor–acceptor structure.42 The replacement of the Cz donor with the BTCz donor reduced the EQE following the trend of PLQY measurement (PLQY: 4CzTPN > BTCz3CzTPN > 2BTCz2CzTPN). To estimate the efficiency roll-off behavior of the devices, the current density up to 90% of maximum EQE (J90%) was measured. The J90% values of the devices were in the order of 2BTCz2CzTPN > BTCz3CzTPN > 4CzTPN. Among the three TADF molecules, 2BTCz2CzTPN showed the highest J90% value, indicating that the BTCz donor suppresses exciton quenching at a high current density due to its highest RISC rate.43 The EL spectra of the TADF materials were red-shifted in the order of 2BTCz2CzTPN > BTCz3CzTPN > 4CzTPN by the strong BTCz donor strengthening the CT character. The peak wavelengths of the 2BTCz2CzTPN, BTCz3CzTPN, and 4CzTPN devices at a 1 wt% doping concentration were 567, 557, and 548 nm, respectively, offering color coordinates of (0.48, 0.50), (0.45, 0.53) and (0.42, 0.56). The EL spectra of BTCz3CzTPN and 2BTCz2CzTPN displayed shoulder peaks at around 480 nm at a 1 wt% doping concentration due to incomplete energy transfer from PBICT to the TADF materials as confirmed by the PBICT emission spectrum in Fig. S5 (ESI†). The PBICT emission was absent at a 10 wt% doping concentration owing to the complete energy transfer from the PBICT host material. At a 10 wt% doping concentration, the three TADF materials showed broad CT emission spectra. Full width at half maximum (FWHM) values slightly increased as the strength of CT increased (2BTCz2CzTPN (93 nm) > BTCz3CzTPN (90 nm) > 4CzTPN (84 nm)) due to strong intermolecular interactions.
The three TADF compounds were evaluated as assistant dopants of the TADF-assisted fluorescent devices doped with a 5,10,15,20-tetraphenylbisbenz[5,6]indeno[1,2,3-cd:1′,2′,3′-lm]perylene (DBP) fluorescent emitter.12,44 The energy level diagram of the devices is shown in Fig. 5 and the current density–voltage–luminance data are shown in Fig. S6 (ESI†).
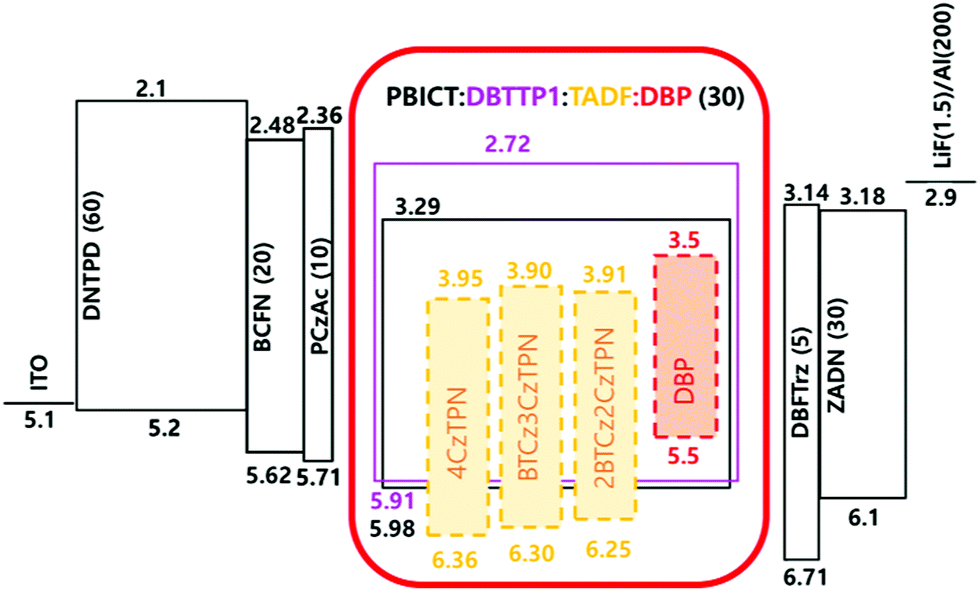 | ||
| Fig. 5 Energy level diagram of the TADF-assisted hyperfluorescent devices. | ||
The DBP fluorescent emitter was doped at a concentration of 0.5 wt% to minimize direct charge trapping and the TADF assistant dopants were added at a doping concentration of 10 wt%. The TADF-assisted DBP device based on BTCz3CzTPN showed the highest maximum EQE of 13.0%. Although the EQE of the BTCz3CzTPN TADF device was lower than that of the 4CzTPN device, that of the TADF-assisted fluorescent device was high in the BTCz3CzTPN device. It seems that the improved EQE of the BTCz3CzTPN device was achieved by efficient FRET and suppressed DET from the assistant dopant to DBP.45,46 4CzTPN may show efficient FRET due to its high PLQY, but the serious DET can be attributed to the low kRISC, whereas 2BTCz2CzTPN may exhibit poor FRET and suppressed DET due to a low PLQY and a high kRISC. In the case of BTCz3CzTPN, it allows efficient FRET and suppressed DET at the same time. However, among the three hyperfluorescent devices, the BTCz3CzTPN-based hyperfluorescent device showed serious efficiency roll-off. This might be due to the carrier imbalance at a high current density. The bulkiness of the BTCz donor moiety induced a high carrier hopping barrier for electrons, triggering a poor charge balance at a high current density. However, 2BTCz2CzTPN showed low efficiency roll-off possibly due to compensation for the charge balance with a high RISC rate. The EL spectra of the devices are shown in Fig. 6(b). The main emission of the devices originated from DBP, but weak TADF emission was also detected. The intensity of the TADF emission was relatively weak in 2BTCz2CzTPN. The color coordinates of the DBP devices with 2BTCz2CzTPN, BTCz3CzTPN, and 4CzTPN assistant dopants were (0.62, 0.38), (0.60, 0.40) and (0.60, 0.40), respectively.
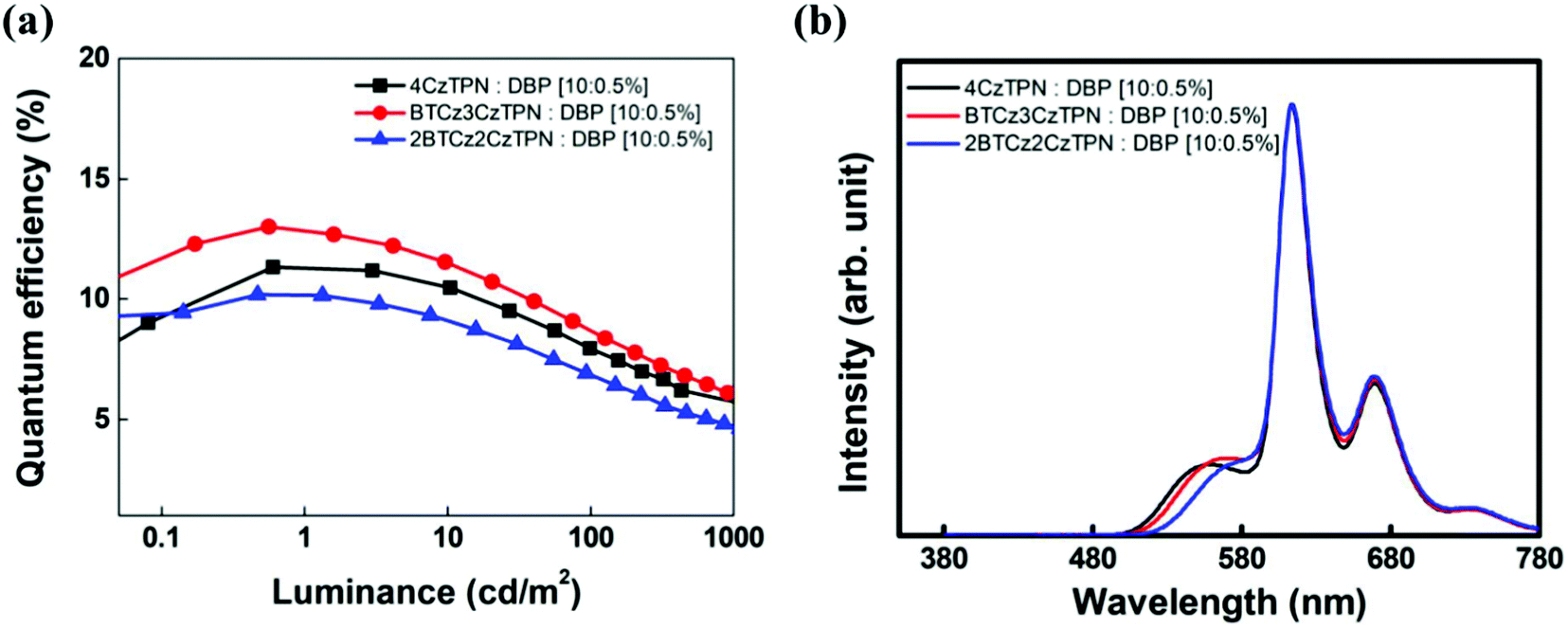 | ||
| Fig. 6 (a) Quantum efficiency–luminance plots and (b) EL spectra of TADF-assisted DBP devices. | ||
Time-resolved electroluminescence (TREL) of the BTCz3CzTPN-based TADF device and hyperfluorescent device was measured and is depicted in Fig. S7 (ESI†). In the hyperfluorescent device, a short exciton lifetime was observed compared to the TADF device. The EL-generated excitons in TADF were transferred to the DBP fluorescent emitter via FRET and DET in the hyperfluorescent device, resulting in faster decay of excitons.
Energy transfer analysis
In the light emission process of the TADF-assisted fluorescence, triplet exciton formation by DET or direct charge trapping in fluorescent emitters should be avoided for achieving high EQEs because the triplet excitons are wasted in the fluorescent emitters. Therefore, the FRET process should occur actively while suppressing DET. To understand the EQE differences of the 4CzTPN-, BTCz3CzTPN- and 2BTCz2CzTPN-assisted DBP devices, exciton dynamics in the emitting layer were studied by calculating the FRET radius (R0) and measuring the PL decay curves of the PBICT:DBTTP1:10 wt% assistant dopant:0.5 wt% DBP films and PBICT:DBTTP1:10 wt% assistant dopant films.47 The emission spectra and absorption spectra of the materials are shown in Fig. 7(a) to confirm the overlap between the emission spectra of the TADF assistant dopants and the absorption spectra of DBP. The PL spectra of the assistant dopant and DBP co-doped films are presented in Fig. 7(b).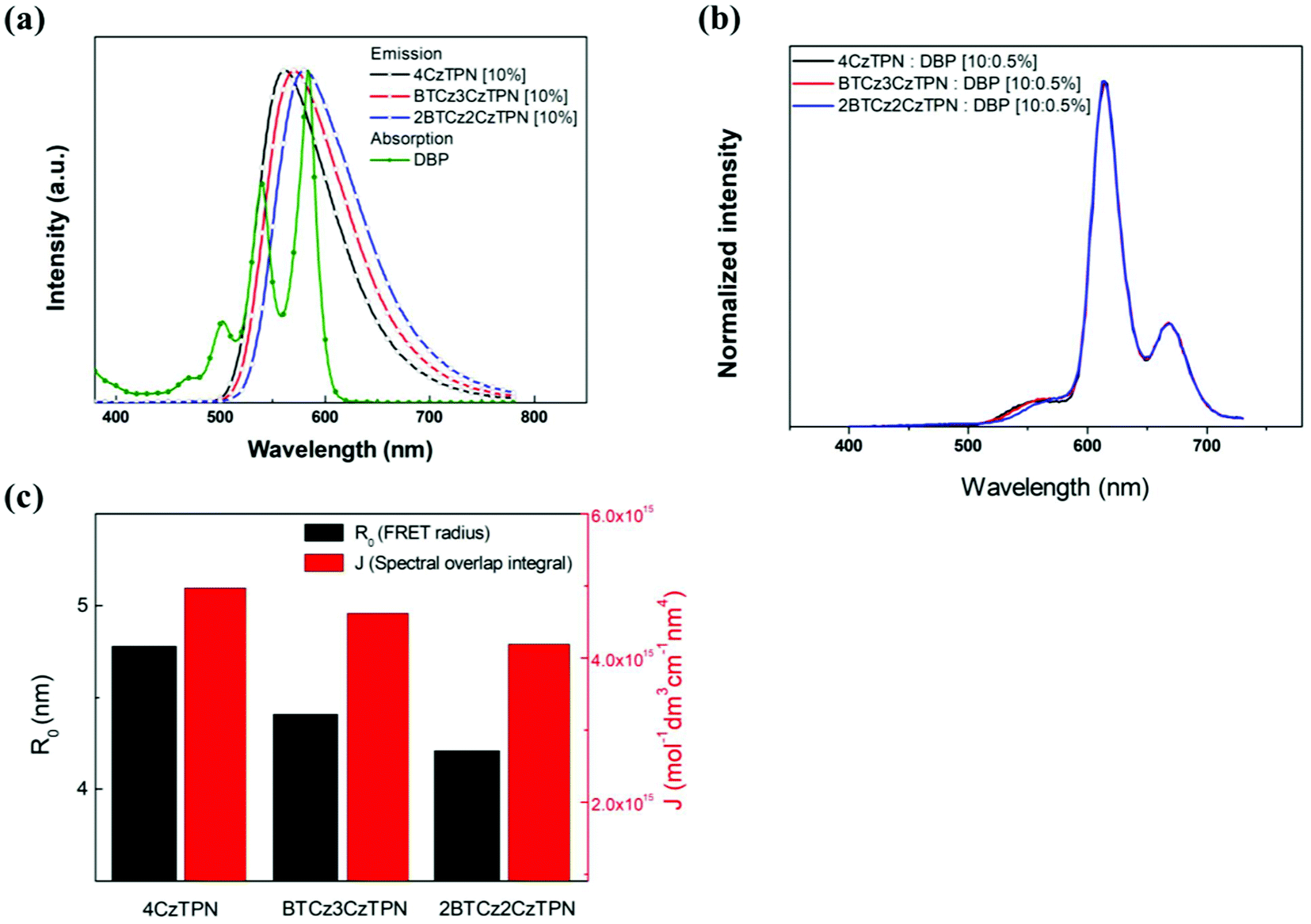 | ||
| Fig. 7 (a) EL emission spectra of the assistant dopant at 10 wt% and UV-vis absorption spectra of DBP. (b) PL emission spectra of 10 wt% assistant dopant and 0.5 wt% DBP co-doped films. (c) R0 and J values of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN. | ||
In general, the rate constant of FRET (kFRET) can be expressed by eqn (1):
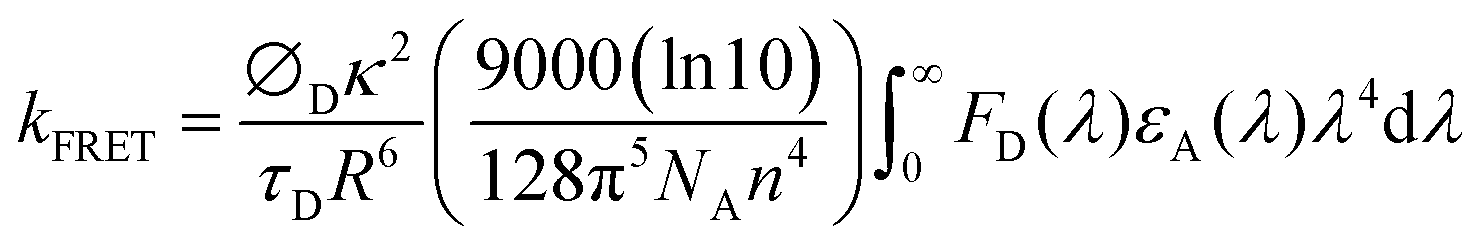 | (1) |
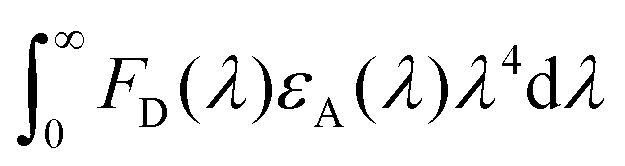 stands for the spectral overlap.
stands for the spectral overlap.
Considering the variables of eqn (1), eqn (1) clearly states that a high PLQY of the assistant dopant and a large spectral overlap between the assistant dopant and fluorescent emitter lead to a high kFRET. The spectral overlap integrals (J) of the DBP co-doped 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN emitting layer films were 4.97 × 1015, 4.62 × 1015, and 4.19 × 1015 mol−1 dm3 cm−1 nm4, respectively.47 The red shift of the emission spectra of BTCz3CzTPN and 2BTCz2CzTPN compared to that of 4CzTPN resulted in a slight decrease in the J value. Based on the J values, the R0 values were calculated using eqn (S1) and the calculated R0 values of BTCz3CzTPN (4.41 nm) and 2BTCz2CzTPN (4.21 nm) were smaller than that of 4CzTPN (4.78 nm) due to the low PLQY and J values. It can be predicted from this result that the BTCz substitution deteriorates the FRET between the assistant dopant and fluorescent emitter. The J and R0 values are summarized in Fig. 7(c).
Transient PL decay analysis of the assistant dopant and DBP co-doped films was carried out to analyze the energy transfer rate between the assistant dopant and DBP. The transient PL decay curves of prompt and delayed fluorescence are shown in Fig. 8(a and b), respectively. The rate constant for prompt fluorescence (kp) of the assistant dopant itself is a combination of the rate constants of radiative decay and intersystem crossing. However, implementation of the DBP fluorescent emitter induced additional FRET paths from the assistant dopant to the fluorescent emitter, resulting in a higher kp as proven by the fast decay in all films. Moreover, as shown in Fig. 8(b), it can be easily noticed that the rate constant for delayed fluorescence (kd) was also significantly increased by DBP due to reduced intersystem crossing probability and fast up-conversion by FRET. Based on the PL decay curves, the DET rate constant (kDET) and kFRET of the assistant dopant and DBP co-doped films were calculated using eqn (2) and (3) by assuming that kP ≫ kd and kr,S, kISC, kFRET ≫ knr,S, kr,T, kDET; here kr,S is the singlet radiative rate constant, knr,S is the singlet nonradiative rate constant and kr,T is the triplet radiative rate constant.48–50
| kFRET ≈ kp − kr,S − kISC | (2) |
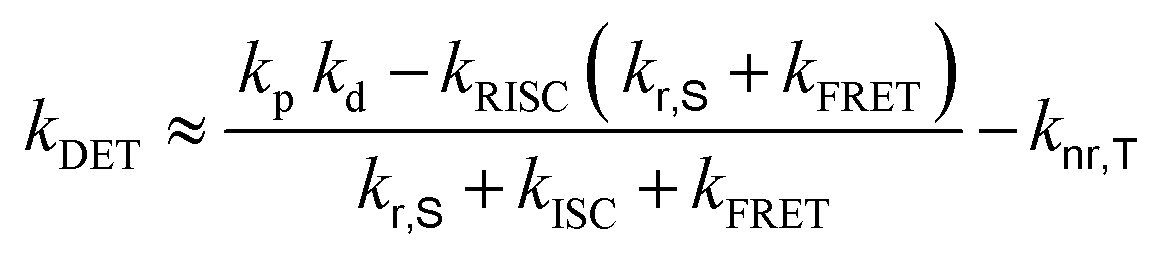 | (3) |
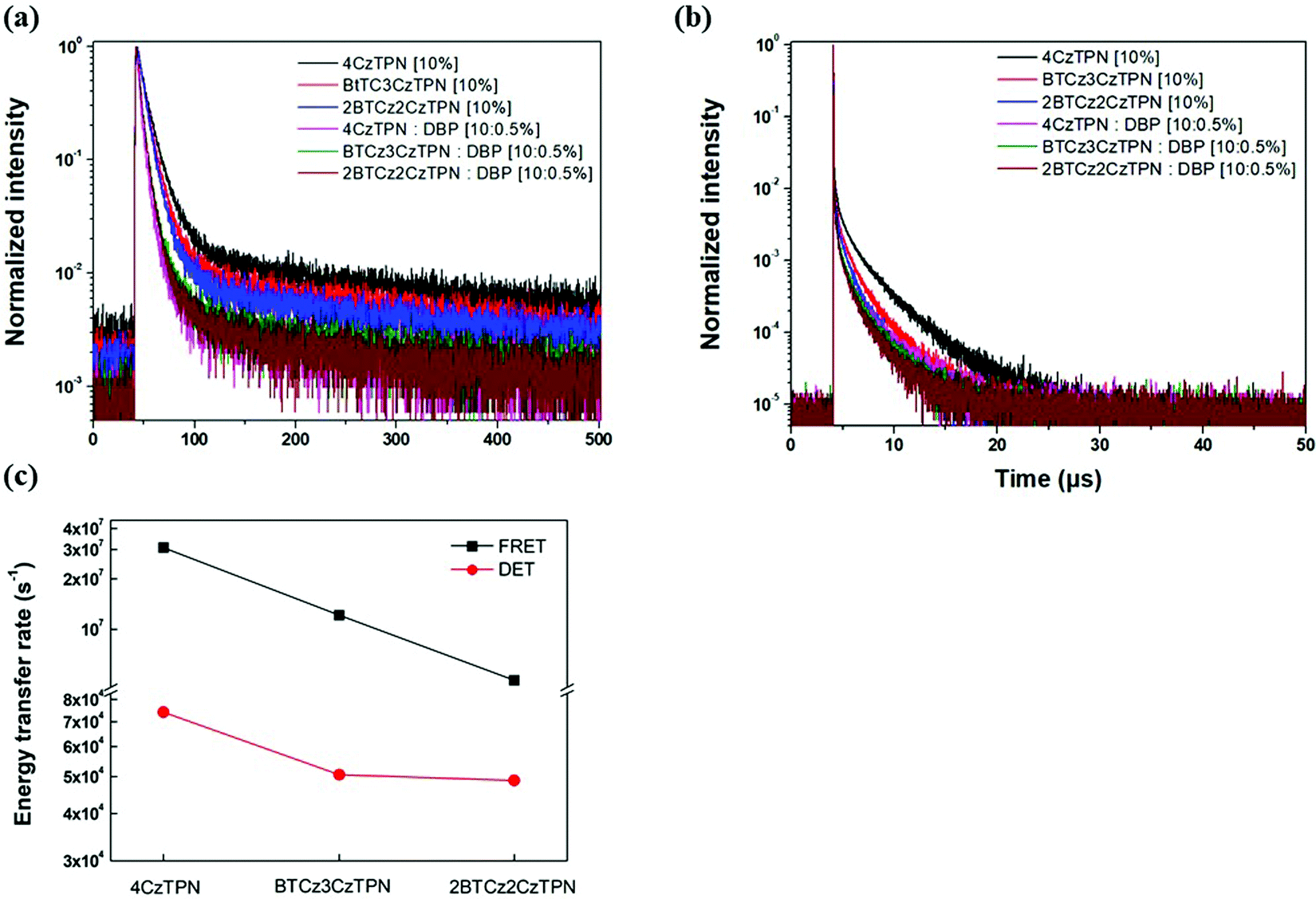 | ||
| Fig. 8 Transient PL decay curves of (a) prompt and (b) delayed fluorescence. (c) kFRET and kDET trends of 4CzTPN, BTCz3CzTPN and 2BTCz2CzTPN. | ||
In addition, to analyze the influence of donor bulkiness in DET, triplet spin density distribution (TSDD) was calculated using the Gaussian 16 and Schrödinger 2019-1 programs. As shown in Fig. 9, all three molecules had main triplet spin density in the TPN acceptor moieties. Therefore, it is expected that the bulkiness of the BTCz donor moiety increases the length between neighboring molecules decreasing the possibility of DET.
 | ||
| Fig. 9 Triplet spin density distribution (TSDD) simulation of (a) 4CzTPN, (b) BTCz3CzTPN and (c) 2BTCz2CzTPN. | ||
Conclusion
BTCz3CzTPN was designed to have a perfect-fit performance as an assistant dopant by introducing and managing the BTCz donor moiety. BTCz3CzTPN demonstrated a high RISC rate and a moderate PLQY as an assistant dopant in TADF assisted fluorescent devices, resulting in suppression of DET without significant reduction of FRET compared to the well-known 4CzTPN. BTCz3CzTPN showed enhanced device performance having an EQE of 13.0% compared to 11.3% for the 4CzTPN device in the TADF assisted fluorescent devices. Therefore, the design route employing the BTCz donor in the assistant dopant is effective in improving the EQE of the TADF-assisted fluorescent devices.Experimental
The specified device structure for analyzing the TADF device performance is indium tin oxide (ITO) (50 nm)/N4,N4′-bis[4-[bis(3-methylphenyl)amino]phenyl]-N4,N4′-diphenyl-[1,1′-biphenyl]-4,4′-diamine (DNTPD) (60 nm)/N-([1,1′-biphenyl]-4-yl)-9,9-dimethyl-N-(4-(9-phenyl-9H-carbazol-3-yl)-phenyl)-9H-fluoren-2-amine (BCFN) (20 nm)/9,9-dimethyl-10-(9-phenyl-9H-carbazol-2-yl)-9,10-dihydro-acridine (PCzAc) (10 nm)/2-phenyl-4,6-bis(12-phenylindolo[2,3-a]carbazole-11-yl)-1,3,5-triazine (PBICT):4-(3-(triphenylen-2-yl)phenyl)dibenzo[b,d]thiophene (DBTTP1):emitting materials (30 nm)/2,8-bis(4,6-diphenyl-1,3,5-triazin-2-yl)dibenzo-[b,d]furan (DBFTrz) (5 nm)/2-[4-(9,10-di-naphthalen-2-yl-anthracen-2-yl)-phenyl]-1-phenyl-1H-benzoimidazole (ZADN) (30 nm)/LiF (1.5 nm)/Al (200 nm).10,51 The DNTPD, BCFN, and PCzAc materials were deposited for hole injection, hole transport and electron blocking. Furthermore, ZADN and DBFTrz were deposited for electron transport and hole confinement. The emitting materials were the 1 or 10 wt% TADF emitter in the TADF devices and 10 wt% TADF assistant dopant:0.5 wt% DBP co-doped materials in the TADF-assisted fluorescent devices. The evaluation of the devices followed the method reported in previous work.Conflicts of interest
There are no conflicts to declare.References
- Y. Im, S. Y. Byun, J. H. Kim, D. R. Lee, C. S. Oh, K. S. Yook and J. Y. Lee, Adv. Funct. Mater., 2017, 27, 1603007 CrossRef.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- T. A. Lin, T. Chatterjee, W. L. Tsai, W. K. Lee, M. J. Wu, M. Jiao, K. C. Pan, C. L. Yi, C. L. Chung, K. T. Wong and C. C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Q. S. Zhang, D. Tsang, H. Kuwabara, Y. Hatae, B. Li, T. Takahashi, S. Y. Lee, T. Yasuda and C. Adachi, Adv. Mater., 2015, 27, 2096–2100 CrossRef CAS PubMed.
- J. H. Kim, J. H. Yun and J. Y. Lee, Adv. Opt. Mater., 2018, 6, 1800255 CrossRef.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- H. J. Jang, J. Y. Lee, J. Kim, J. Kwak and J. H. Park, J. Inf. Disp., 2020, 21, 1–9 CrossRef CAS.
- Q. S. Zhang, J. Li, K. Shizu, S. P. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476 CrossRef CAS PubMed.
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 CrossRef CAS PubMed.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. S. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- Q. S. Zhang, B. Li, S. P. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- D. D. Zhang, X. Z. Song, M. H. Cai and L. Duan, Adv. Mater., 2018, 30, 1705250 CrossRef PubMed.
- S. Y. Byeon, D. R. Lee, K. S. Yook and J. Y. Lee, Adv. Mater., 2019, 31, 1803714 CrossRef PubMed.
- S. K. Jeon, H. Lee, K. S. Yook and J. Y. Lee, Adv. Mater., 2019, 31, 1803524 CrossRef PubMed.
- D. R. Lee, M. Kim, S. K. Jeon, S. H. Hwang, C. W. Lee and J. Y. Lee, Adv. Mater., 2015, 27, 5861 CrossRef CAS PubMed.
- C. Y. Chan, M. Tanaka, Y. T. Lee, Y. W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Nat. Photonics, 2021, 15, 245 CrossRef CAS.
- S. O. Jeon, K. H. Lee, J. S. Kim, S. G. Ihn, Y. S. Chung, J. W. Kim, H. Lee, S. Kim, H. Choi and J. Y. Lee, Nat. Photonics, 2021, 15, 208–215 CrossRef CAS.
- D. D. Zhang and L. Duan, Nat. Photonics, 2021, 15, 173–174 CrossRef CAS.
- F. M. Xie, J. X. Zhou, Y. Q. Li and J. X. Tang, J. Mater. Chem. C, 2020, 8, 9476–9494 RSC.
- S. Y. Yang, Y. K. Qu, L. S. Liao, Z. Q. Jiang and S. T. Lee, Adv. Mater., 2021, 2104125 CrossRef PubMed.
- S. Y. Yang, Y. K. Wang, C. C. Peng, Z. G. Wu, S. Yuan, Y. J. Yu, H. Li, T. T. Wang, H. C. Li, Y. X. Zheng, Z. Q. Jiang and L. S. Liao, J. Am. Chem. Soc., 2020, 142, 17756–17765 CrossRef CAS PubMed.
- S. Y. Yang, Y. L. Zhang, A. Khan, Y. J. Yu, S. Kumar, Z. Q. Jiang and L. S. Liao, J. Mater. Chem. C, 2020, 8, 3079–3087 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- R. Ishimatsu, S. Matsunami, T. Kasahara, J. Mizuno, T. Edura, C. Adachi, K. Nakano and T. Imato, Angew. Chem., Int. Ed., 2014, 53, 6993–6996 CrossRef CAS PubMed.
- Y. Olivier, M. Moral, L. Muccioli and J. C. Sancho-Garcia, J. Mater. Chem. C, 2017, 5, 5718–5729 RSC.
- P. Stachelek, J. S. Ward, P. L. dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce and A. P. Monkman, ACS Appl. Mater. Interfaces, 2019, 11, 27125–27133 CrossRef CAS PubMed.
- W. L. Che, Y. J. Xie and Z. Li, Asian J. Org. Chem., 2020, 9, 1262–1276 CrossRef CAS.
- R. J. Huang, N. Kukhta, J. S. Ward, A. Danos, A. S. Batsanov, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2019, 7, 13224–13234 RSC.
- J. Hwang, H. Kang, J. E. Jeong, H. Y. Woo, M. J. Cho, S. Park and D. H. Choi, Chem. Eng. J., 2021, 416, 129185 CrossRef CAS.
- J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630–632 CrossRef CAS.
- R. Englman, Mol. Phys., 1988, 145–164 Search PubMed.
- J. Wlodarczyk and B. Kierdaszuk, Biophys. J., 2003, 85, 589–598 CrossRef CAS PubMed.
- K. Masui, H. Nakanotani and C. Adachi, Org. Electron., 2013, 14, 2721–2726 CrossRef CAS.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- X. Zhao, X. T. Tang, H. Q. Zhu, C. H. Ma, Y. Wang, S. N. Ye, L. Y. Tu and Z. H. Xiong, ACS Appl. Electron. Mater., 2021, 3, 3034–3043 CrossRef CAS.
- Y. Wang, J. H. Yun, L. Wang and J. Y. Lee, Adv. Funct. Mater., 2021, 31, 2008332 CrossRef CAS.
- Y. Im, W. Song and J. Y. Lee, J. Mater. Chem. C, 2015, 3, 8061–8065 RSC.
- D. D. Zhang, L. Duan, Y. L. Li, H. Y. Li, Z. Y. Bin, D. Q. Zhang, J. Qiao, G. F. Dong, L. D. Wang and Y. Qiu, Adv. Funct. Mater., 2014, 24, 3551–3561 CrossRef CAS.
- H. S. Kim, S. R. Park and M. C. Suh, J. Phys. Chem. C, 2017, 121, 13986–13997 CrossRef CAS.
- C. Murawski, K. Leo and M. C. Gather, Adv. Mater., 2013, 25, 6801–6827 CrossRef CAS PubMed.
- K. Okumoto, H. Kanno, Y. Hamada, H. Takahashi and K. Shibata, Appl. Phys. Lett., 2006, 89, 013502 CrossRef.
- N. Haase, A. Danos, C. Pflumm, P. Stachelek, W. Brutting and A. P. Monkman, Mater. Horiz., 2021, 8, 1805–1815 RSC.
- L. Paterson, A. Mondal, P. Heimel, R. Lovrincic, F. May, C. Lennartz and D. Andrienko, Adv. Electron. Mater., 2019, 5, 160901 Search PubMed.
- C. F. Wu, Y. L. Zheng, C. Szymanski and J. McNeill, J. Phys. Chem. C, 2008, 112, 1772–1781 CrossRef CAS PubMed.
- C. Zhang, Y. Lu, Z. Y. Liu, Y. W. Zhang, X. W. Wang, D. D. Zhang and L. Duan, Adv. Mater., 2020, 32, 2004040 CrossRef CAS PubMed.
- N. Aizawa, S. Shikita and T. Yasuda, Chem. Mater., 2017, 29, 7014–7022 CrossRef CAS.
- X. Z. Song, D. D. Zhang, Y. W. Zhang, Y. Lu and L. Duan, Adv. Opt. Mater., 2020, 8, 2000483 CrossRef CAS.
- M. Kim, S. K. Jeon, S. H. Hwang and J. Y. Lee, Adv. Mater., 2015, 27, 2515–2520 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc04875a |
| This journal is © The Royal Society of Chemistry 2022 |