
Regulating solvation and interface chemistry to inhibit corrosion of the aluminum anode in aluminum–air batteries†
Chaonan
Lv
a,
Yuxin
Zhang
a,
Jianjun
Ma
 b,
Yuanxin
Zhu
a,
Dan
Huang
c,
Yixin
Li
*a,
Haiyan
Wang
*a and
Yougen
Tang
*a
b,
Yuanxin
Zhu
a,
Dan
Huang
c,
Yixin
Li
*a,
Haiyan
Wang
*a and
Yougen
Tang
*a
aHunan Provincial Key Laboratory of Chemical Power Sources, College of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, P. R. China. E-mail: wanghy419@csu.edu.cn; ygtang@csu.edu.cn; yixinli@csu.edu.cn
bMaterial Corrosion and Protection Key Laboratory of Sichuan Province, Sichuan University of Science & Engineering, Zigong, 643000, P. R. China
cGuangxi Novel Battery Materials Research Center of Engineering Technology, School of Physical Science and Technology, Guangxi University, Nanning 530004, P. R. China
First published on 16th March 2022
Abstract
As a promising energy storage technology, aluminum–air batteries possess the advantages of high energy density, safety and low-cost. However, the severe self-corrosion of the aluminum anode greatly limits their practical applications. To address such issues, solvation chemistry and interface chemistry are combined together to reduce the aluminum anode corrosion by adding a concentrated potassium acetate electrolyte component and sodium stannate electrolyte additive, which can greatly decrease the number of free H2O molecules and improve the potential of hydrogen evolution. Thus, the aluminum anode exhibits significantly reduced corrosion rate behavior in the as-prepared electrolyte (0.085 mg cm−2 min−1 for the blank electrolyte, and 0.011 mg cm−2 min−1 for the hybrid high concentration electrolyte). The full cell with the optimized electrolyte shows a remarkably increased discharge capacity of 2439 mA h g−1 at a current density of 25 mA cm−2 and the cathode electrocatalyst is not damaged after discharging.
1. Introduction
Lithium-ion batteries are ubiquitous in portable electronics because of their high energy density and excellent cycling stability.1–12 However, they inevitably encounter the potential safety and environmental hazards caused by toxic nonaqueous electrolytes. In this regard, aqueous batteries are drawing increased interest owing to the utilization of aqueous electrolytes which have the advantages of safety, environmental friendliness, higher ionic conductivity and lower cost compared with battery systems based on nonaqueous electrolytes. With inexhaustible oxygen in air as the active material of cathodes, aqueous metal–air batteries have received great attention because of their high theoretical energy density (2–10 times that of lithium-ion batteries), constant discharge voltage and light weight. Among the reported aqueous metal–air batteries (such as Li, Na, Zn, Mg, Al, and Fe), aluminum–air batteries stand out due to high theoretical specific capacity (2.98 A h g−1, 8035 mA h cm−3) and low cost of the aluminum metal anode. Generally, aluminum–air batteries can exhibit a high theoretical energy density of 8.1 kW h kg−1, which far surpasses that of state-of-the-art lithium-ion batteries.13–19As a rule, the strong alkaline solution is utilized as an electrolyte to remove the passivation layer on the aluminum surface and reduce the battery polarization.15 However, the aluminum metal anode suffered from serious self-corrosion and the parasitic hydrogen evolution reaction (HER) in both open-circuit state and working mode under alkaline conditions, leading to inferior battery performance and sharply curtails the safety level.16,17 Numerous efforts have been made to address this issue, including aluminum anode alloying,18 electrolyte additive,19 gel electrolyte,13 nonaqueous electrolyte,20 and oil displacement and draining the electrolyte after shutdown,10,21,22 but the efficiency is still unsatisfactory. Recently, a “water-in-salt” (WIS) electrolyte has been designed for aqueous batteries to reduce the water electroactivity and broaden the electrochemical windows by decreasing the content of free H2O molecules.23–25 Inspired by this, our group once developed a hybrid high-concentration potassium acetate–potassium hydroxide electrolyte (16 M potassium acetate (KOAc) in 4 M KOH, HCPA–KOH) for aluminum–air batteries, which obviously inhibited the self-corrosion of the aluminum anode and increased the discharge capacity (2324 mA h g−1vs. 745 mA h g−1 of the blank one).12 However, the utilization of such 16 M KOAc results in low ionic conductivity and high viscosity of the HCPA–KOH electrolytes, impeding their practical applications in aluminum–air batteries. Besides, the high dosage of salt in WIS electrolytes tends to precipitate at low temperatures, confining the low temperature performance of aluminum–air batteries.26 The formation of the protective layer on the surface of the aluminum anode would hinder the direct contact between the anode and electrolyte and effectively inhibit the corrosion of the aluminum anode in alkaline solutions.19,27–29 Therefore, rational design formulations of the electrolyte to ensure the reduction of free H2O molecules and continuous generation of the protective layer is necessary for high performance aluminum–air batteries.
Herein, a novel strategy from the structure of solvation and interface is designed for the first time to mitigate the self-corrosion of the aluminum anode for alkaline aluminum–air batteries. It was found out that by introducing 0.02 mol L−1 sodium stannate (Na2SnO3) and 8 mol kg−1 potassium acetate (KOAc) in 4 mol L−1 KOH electrolyte as the hybrid high concentration electrolyte (HHCE), the corrosion potential of the aluminum anode is significantly increased from −1.39 V to −1.53 V vs. Hg/HgO. Both molecular dynamics (MD) simulations and spectral characterization experiments indicate that the number of free H2O molecules are reduced and the acetate anions (OAc−) partially substitute coordinated H2O molecules as part of the cation solvent sheath, which significantly reduces the H2O activity in the electrolyte. In addition, Na2SnO3 in the electrolyte will be spontaneously reduced to the Sn layer on the surface of the aluminum anode, which can protect the aluminum anode by covering hydrogen adsorption sites and reducing the contact between the aluminum anode and electrolyte. Benefitting from the regulation strategy of solvation and interface chemistry, the corrosion rate of the aluminum anode is significantly decreased from 0.085 mg cm−2 min−1 (4 mol L−1 KOH) to 0.011 mg cm−2 min−1 (HHCE). As a result, a superior high discharge capacity of 2439 mA h g−1 is achieved at 25 mA cm−2 in HHCE. We believe that the encouraging performance will provide a new direction for development of practical aluminum–air batteries.
2. Experimental
2.1 Electrode preparation
The aluminum alloy used in this work was purchased from Changzhou Yoteco, and its element composition is shown in Table S1.† The aluminum alloy was mechanically polished with sandpaper, then washed with deionized water and absolute ethanol in an ultrasonic cleaner for 5 min, respectively, and then dried in a vacuum at 70 °C for 8 h to obtain the anode material. The air cathode is commercially available which is composed of a MnxOy@Ag catalyst, gas diffusion layer and Cu mesh current collector. More details can be seen in our previous work.122.2 Electrolyte preparation
Potassium hydroxide, potassium acetate and sodium stannate were all of the analytical grade from Shanghai Chemical Reagent Company of China. Hybrid electrolytes were prepared by dissolving 8 mol kg−1 KOAc and 0.02 mol L−1 Na2SnO3 in prepared 4 mol L−1 KOH, respectively.2.3 Characterization
The morphology and composition of the aluminum surface were systematically investigated by using a scanning electron microscope (SEM, Nova Nano-SEM 230), in situ optical microscope (Nikon, SMZ25), X-ray diffraction spectrometer (XRD, Shimadzu XRD-6000). Raman spectra were collected on a Renishaw inVia spectrometer. Fourier transform infrared (FT-IR) spectra were obtained using a Bruker Vertex 70 FT-IR spectrophotometer. 1H nuclear magnetic resonance (NMR) spectra were recorded on a 300.5 mm glass NMR tube. Samples were analysed at 25 ± 0.1 °C. Data were processed in MestReNova 11.0.2.2.4 Electrochemical measurements
Electrochemical measurement was carried out in a conventional three-electrode cell by using a CHI760 electrochemical workstation, which used an aluminum alloy (10 mm × 10 mm × 3 mm) as the working electrode (WE), a Hg/HgO electrode as the reference electrode (RE) and platinum as the counter electrode (CE). The potentiodynamic polarization curves were obtained from −0.5 V to 1.5 V vs. the open circuit potential (OCP). The electrochemical impedance spectroscopy (EIS) experiments were performed at the OCP in the frequency from 100 kHz to 0.01 Hz with 5 mV amplitude. The electrochemical windows of the different electrolytes were obtained by linear sweep voltammetry (LSV) at 1 mV s−1 in the three-electrode system.2.5 Aluminum–air full battery tests
The aluminum–air batteries were composed of an aluminum plate anode, electrolyte and two cathode films with a MnxOy@Ag catalyst. To obtain discharge curves, the electrolytes (100 mL) were put in sealed bottles and kept flowing when the battery was operated at 25 mA cm−2. The mass-specific capacity of the full battery was calculated by dividing the mass difference of the aluminum anode before and after galvanostatic discharge.2.6 Computational details
The simulation box consisted of a cube box containing K+, OH−, and OAc− ions and H2O molecules. Initial structures for molecular dynamics simulations were constructed by using the Forcite package in Material Studio. The ions and H2O molecules were randomly added in the simulated box for providing homogeneous state. Before performing the dynamics simulations, the geometrical optimization was carried out to pre-equalize the system. After that, NPT (constant temperature, constant pressure) run was performed at 298 K for 10 ns to ensure the system equilibrium. Then, NVT (constant temperature, constant volume) run was performed at room condition for 10 ns to accelerate the ion aggregation.3. Results and discussion
Taking into account the battery polarization and corrosion rate, we choose 8 M KOAc and 0.02 M Na2SnO3 introduced in 4 M KOH as the optimal electrolyte (Fig. S1 and Table S2†). First, we performed a series of experimental studies and calculations to understand the solvation configuration of the HHCE. Hydrogen proton nuclear magnetic resonance (1H NMR) was used to explore the chemical states of hydrogen nuclei in different electrolytes (Fig. 1a). Two peaks can be clearly observed, which can be assigned to hydrogen nuclei of OAc− (∼1.50 ppm) and H2O (∼4.80 ppm), respectively.30,31 As we can see, the 1H signal of H2O shifts to a high field with addition of 8 M KOAc, which can be attributed to that the formation of the solvation sheath containing H2O molecules and OAc− anions destroys shielding of protons from the H2O molecules.32 Note that the chemical shift in the HHCE is almost identical to that in the solution with 8 M KOAc, indicating that the addition of 0.02 M Na2SnO3 does not affect the solvation configuration of the electrolyte. FT-IR curves shown in Fig. 1b demonstrate that the H–O–H bending vibration of the bare KOH electrolyte exhibits a blue shift from 1636 to 1647 cm−1 after the addition of 8 M KOAc, suggesting the reduction of the free H2O content.12 Raman spectroscopy was also performed to explore the structure of different electrolytes. Fig. S2† shows the representative Raman spectra of different electrolytes, crystalline KOAc and pure water as the reference. The broad band located at 3150–3500 cm−1 is assigned to the stretching vibration (O–H) of H2O molecules, which is corresponding to the hydrogen-bonded clusters of free H2O.26 Generally, the peak of O–H can be fitted as three peaks located at ∼3230, ∼3450 and ∼3620 cm−1, corresponding to the H2O molecules with strong, weak and non H-bonds, respectively.33 With the addition of 8 M KOAc, the peaks are shifted to high frequency, which results in reduction of strong H-bonded H2O and increasing non H-bonded H2O (Fig. 1c). This difference means that there are fewer free H2O molecules in the HHCE, which is beneficial to reduce H2O activity and enhance the stability window of the electrolyte. In addition, the wettability of the HHCE is significantly changed on the aluminum surface (Fig. S3†). The contact angle of HHCE is 80.4°, which is much higher than that of 4 M KOH (48.4°). The larger contact angle of the HHCE is critical to suppress corrosion inhibition.12,27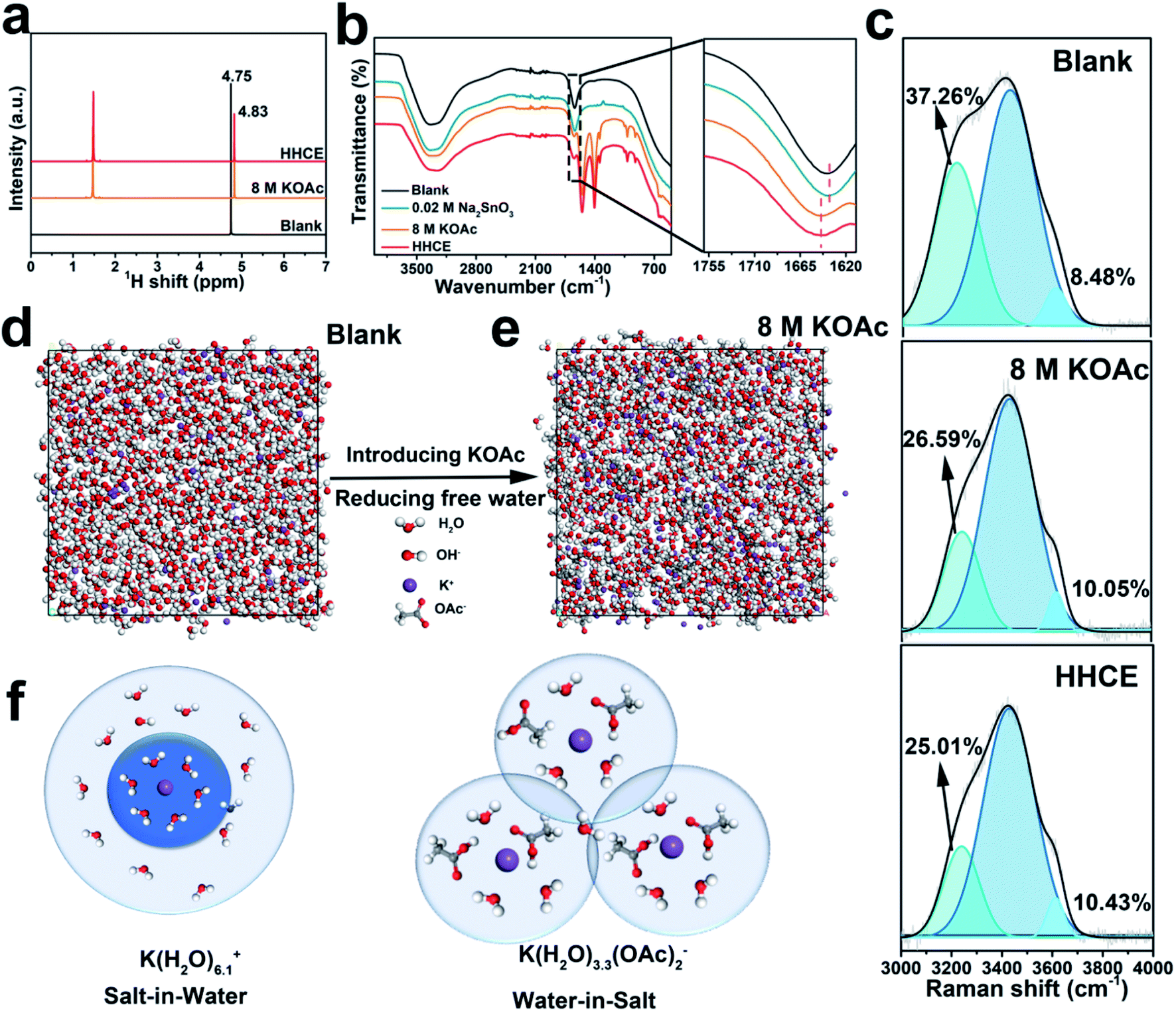 | ||
| Fig. 1 Physicochemical properties of different electrolytes. (a) 1H NMR spectra, (b) FT-IR spectra and (c) Raman spectra of different electrolytes. The snapshot of the MD simulation of (d) KOH electrolyte and (e) 8 M KOAc electrolyte. (f) Illustration of the evolution of the K+ primary solvation sheath in KOH and 8 M KOAc electrolytes. (blank: 4 mol L−1 KOH electrolyte; 8 M KOAc: 8 mol kg−1 KOAc in 4 mol L−1 KOH electrolyte; 0.02 M Na2SnO3: 0.02 mol L−1 Na2SnO3 in 4 mol L−1 KOH electrolyte; HHCE: 8 mol kg−1 KOAc and 0.02 mol L−1 Na2SnO3 in 4 mol L−1 KOH electrolyte) | ||
In a concentrated electrolyte, unlike the conventional aqueous electrolyte, the interionic interaction becomes dominant over the solvent–ion interaction, resulting in the chemical environment change of H2O molecules.32 K+ and OH− exhibit the hydration state in the solvation sheath in 4 M KOH electrolyte. As for the high concentration electrolyte, OAc− can cooperate with the sheath of K+, resulting in the reduced amount of free H2O molecules, so that the primary coordination sphere of K+ contains H2O molecules and OAc− anions.23 To unveil the solvation configurations in the electrolyte after introducing 8 M KOAc, the interactions among K+ ions and H2O were studied by MD (Fig. 1d and e). The radial distribution function (RDF) was also used to further investigate the solvation structure of K+ as shown in Fig. S4.† In 4 M KOH electrolyte, the RDF between K+ and the O atom in H2O molecules shows a coordination peak at 3.6 Å with coordination number of 6.1 (Fig. S4a†). As a comparison, the RDF between K+ and the O atom of H2O molecules in 8 M KOAc shows a coordination peak at 3.4 Å with coordination number of 3.3 (Fig. S4b†), which demonstrates that OAc− has participated in the solvation structures of K+ and the surrounding H2O shell is decreased (Fig. 1f). This result is in good agreement with the Raman and 1H NMR results.
The potentiodynamic polarization curves were measured to evaluate the corrosion of aluminum anodes in different electrolytes (Fig. 2a). The aluminum anode electrode potential gradually shifts toward negative with the addition of KOAc and Na2SnO3. Since the presence of 8 M KOAc reduces activity of H2O molecules and 0.02 M Na2SnO3 promotes the formation of the protective layer, the decomposition of H2O molecules on the surface and corrosion reaction of the aluminum anode are suppressed successfully. Especially, the HHCE results in much more negative shift of the corrosion potential to −1.53 V vs. Hg/HgO. Meanwhile, the cathodic current density of the aluminum anode in electrolytes with Na2SnO3 or KOAc is lower than that of the blank one. Considering the significant decrease in the cathodic process, it can be deduced that Na2SnO3 and KOAc hybrid high concentration electrolytes greatly promote the inhibition effect. The electrochemical stability windows of different electrolytes were evaluated by linear sweep voltammetry (LSV). As shown in Fig. 2b, the HHCE exhibits the widest electrochemical stability window compared with other electrolytes. The current density at −1.75 V vs. Hg/HgO is reduced from 2.7 mA cm−2 (KOH) to 1.2 mA cm−2 (HHCE), which is beneficial for suppressing hydrogen evolution and self-corrosion of the aluminum anode.12 The EIS results reveal that the resistance of the electrolyte and charge transfer resistance increase in the HHCE, causing by the fewer free H2O molecules in the electrolyte and the formation of the Sn layer on the aluminum anode surface (Fig. 2c, S5 and Table S3†). This result is in agreement with the Tafel curve shown in Fig. 2a. The ion conductivity and viscosity of different electrolytes were further recorded (Fig. 2d and Table S4†). As seen clearly, the ion conductivity decreases from 0.24 S cm−1 (blank) to 0.13 S cm−1 (HHCE) with the addition of 8 M KOAc and 0.02 M Na2SnO3, while the viscosity increases from 1.74 to 5.49 mPa s−1. The higher viscosity and lower ionic conductivity indicate that excessive KOAc is detrimental to the ion transmission, thus reducing the voltage of the full cell in galvanostatic discharge tests.
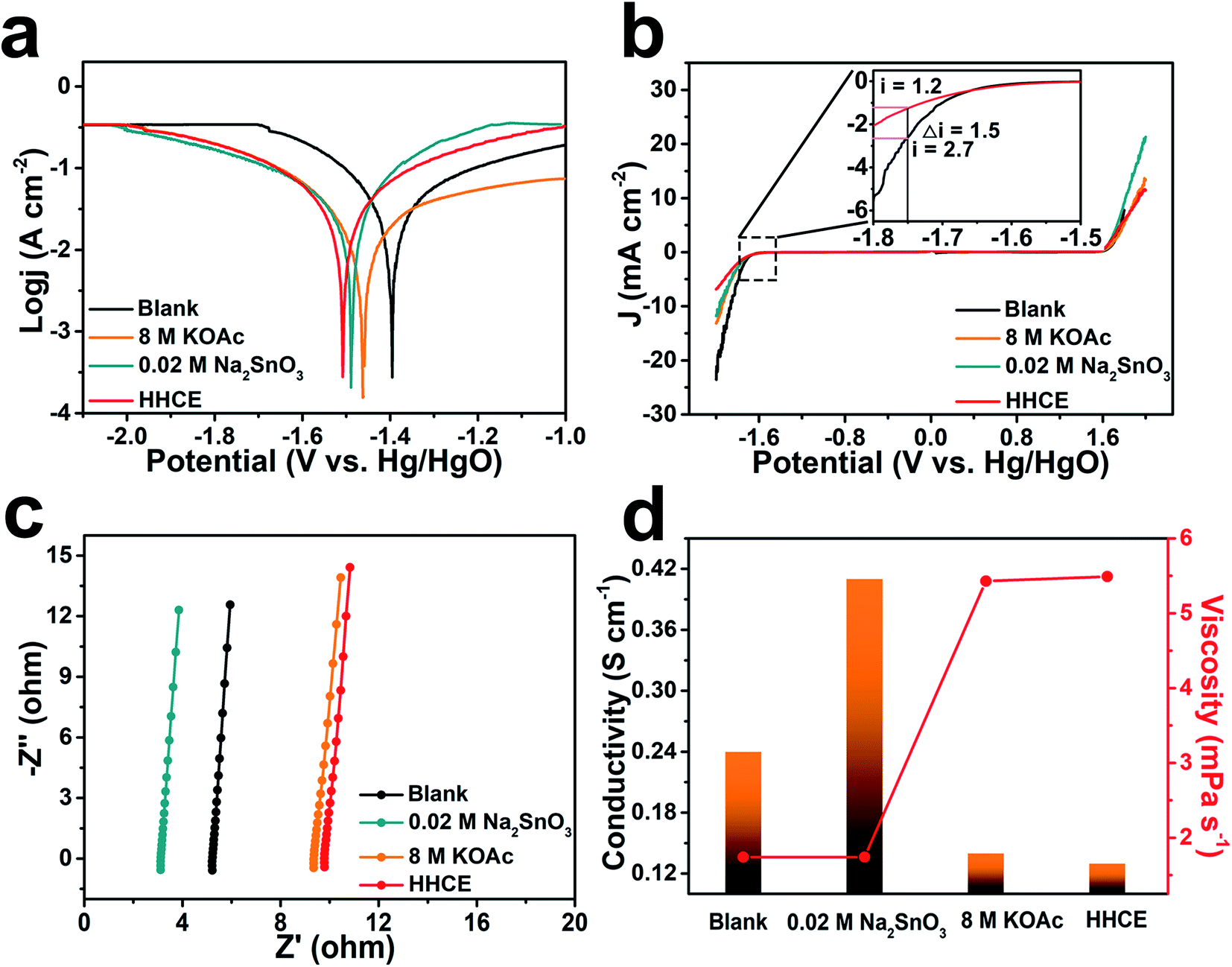 | ||
| Fig. 2 (a) Polarization curves of aluminum anodes in different electrolytes. (b) The electrochemical stability windows of different electrolytes. (c) The magnified curves of EIS in the high-frequency region. (d) The viscosity and ionic conductivity of different electrolytes. | ||
The corrosion measurement of aluminum anodes in different electrolytes is shown in Fig. S6.† Both the rate and amount of hydrogen gas generation are obviously decreased with addition of KOAc and Na2SO3 (Fig. S6a–e†). After immersing in different electrolytes for 3 h, we adopted the mass loss method to evaluate the hydrogen gas evolution performance of aluminum anodes. As shown in Fig. S6f,† the corrosion rate is 0.085 mg cm−2 min−1 in the blank electrolyte, which drops to 0.054 mg cm−2 min−1, 0.025 mg cm−2 min−1 and 0.011 mg cm−2 min−1 in 8 M KOAc, 0.02 M Na2SO3 and HHCE, respectively. And the highest inhibition efficiency (η%) can reach to 87.06% (Table S2,† HHCE). These results prove that the self-corrosion of aluminum anode is significantly suppressed. The pristine aluminum anode surface is smooth and contains slight Sn element (Fig. S7†). However, the surface of the aluminum anode immersed in 4 M KOH electrolyte exhibits a rough and cracked morphology, resulting in capacity loss and voltage instability during galvanostatic discharge (Fig. 3a). The surface morphologies of the aluminum anode immersed in 8 M KOAc solution are also loose and porous (Fig. 3b). In Fig. 3c, a protective layer is clearly observed on the surface of the aluminum anode immersed in the presence of 0.02 M Na2SnO3, which is proved to be the Sn layer by XRD (Fig. S8†). For the aluminum anode in the HHCE, a flat and smooth morphology can be seen as shown in Fig. 3d, which is attributed to even stripping of the aluminum anode and inhibition of self-corrosion caused by synergistic regulation of solvation and interface chemistry. An in situ optical microscope was employed to further investigate the surface corrosion of aluminum anodes in different electrolytes. As can be seen from Fig. S9,† the corrosion depth is nonuniform and the maximum corrosion depth is 55 μm in 4 M KOH electrolyte (Fig. S9a–c†). However, the corrosion depth of aluminum decreases to 45 μm (for 8 M KOAc), 24 μm (for 0.02 M Na2SnO3) and 16 μm (for HHCE), respectively. And the surface is more even than that in 4 M KOH electrolyte (Fig. S9d, g and j†), suggesting that the addition of KOAc and Na2SnO3 is beneficial for the uniform stripping of aluminum anodes, which is in agreement with SEM images.
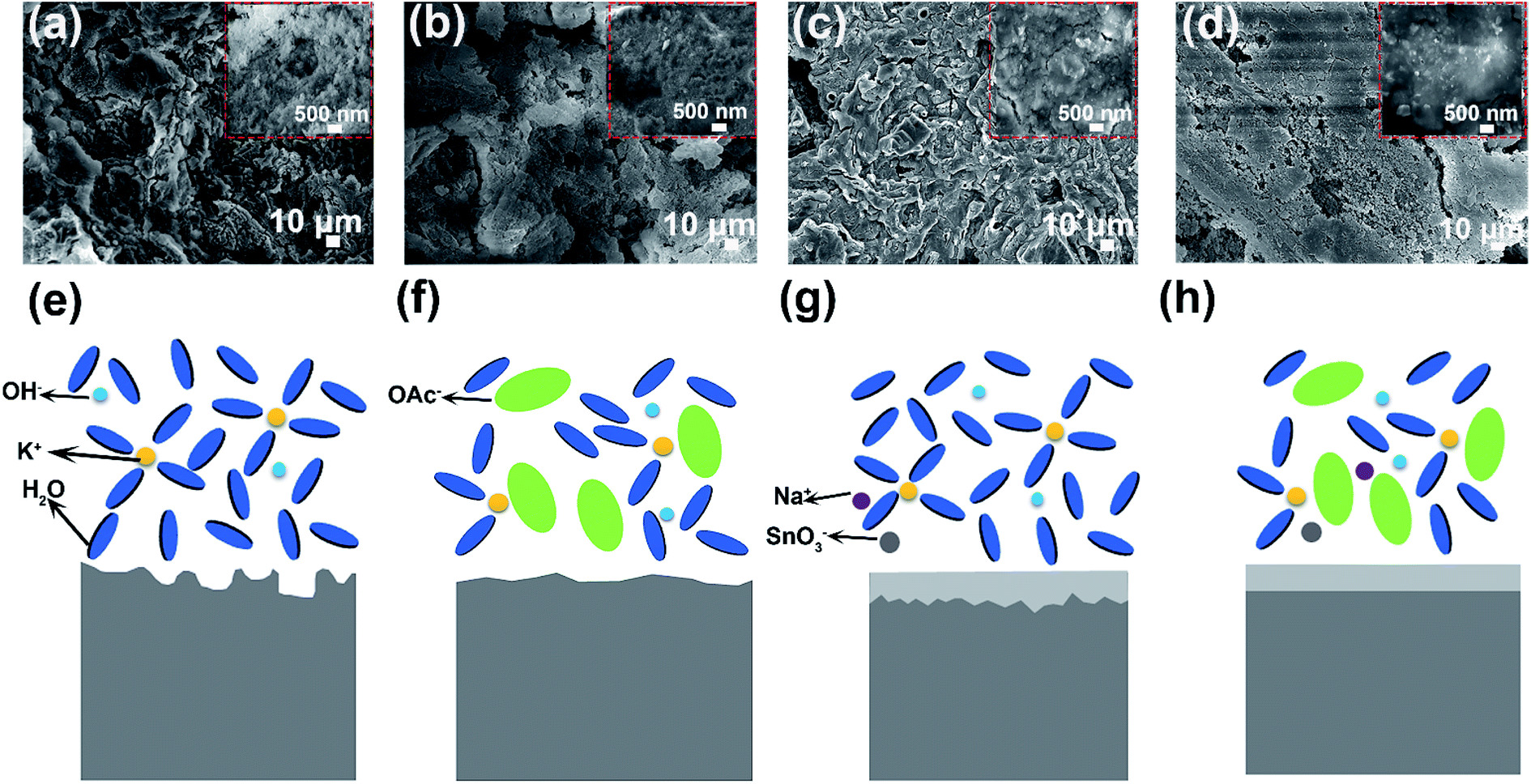 | ||
| Fig. 3 SEM images of aluminum anodes in (a) blank, (b) 8 M KOAc, (c) 0.02 M Na2SnO3 and (d) HHCE after 3 h of immersion. Schematic illustrations of the corrosion behavior of aluminum anodes in (e) blank, (f) 8 M KOAc, (g) 0.02 M Na2SnO3 and (h) HHCE. | ||
When the aluminum anode is immersed in alkaline solution, Al3+ will be formed due to the electrochemical force [eqn (1)].34 In bare KOH solution, Al3+ produced as shown in eqn (1) readily reacts with several OH− to form Al(OH)3 as shown in eqn (2). The generated Al(OH)3 can be easily dissolved [eqn (3)] and diffused from the surface of the aluminum anode to the electrolyte. The rapid transfer of produced Al3+ maintains a low Al3+ concentration at the interface, resulting in constant occurrence of the reactions shown in eqn (1) and (2). Consequently, the aluminum anode suffers from serious self-corrosion (Fig. 3e).
| Al → Al(ad)3+ + 3e− | (1) |
| Al(ad)3+ + 3OH− → Al(OH)3 | (2) |
| Al(OH)3 + OH− → Al(OH)4− | (3) |
However, the high concentration electrolyte with a large amount OAc− presents a different scenario. In this electrolyte, the generated Al(OH)3 shown in eqn (2) is sluggishly dissolved, leading to difficult diffusion. Therefore, the formation rate of Al3+ ions in 8 M KOAc electrolyte is slower than that in 4 M KOH electrolyte (Fig. 3f) [eqn (1)]. In addition, the high concentration of OAc− at the electrode/electrolyte interface hinders the access of solvent molecules to the aluminum anode and further suppresses the Al3+ dissolution from the electrode surface.35 As for the aluminum anode immersed in the alkaline solution containing 0.02 M Na2SnO3, the self-corrosion is remarkably suppressed. There is competition of two cathodic reactions between the reduction of SnO32− and water, as shown in eqn (4) and (5).36 So the deposition of the Sn layer covered in the surface of the aluminum anode improves the overpotential of parasitic hydrogen evolution and reduces direct contact between the aluminum anode and electrolyte, resulting in uniform stripping of Al3+ (Fig. 3g).
| SnO32− + 3H2O + 4e− → Sn + 6OH− | (4) |
| 4H2O + 4e− → 2H2↑ + 4OH− | (5) |
Benefiting from the synergistic effect of the interface and solvation regulation, the corrosion rate of the aluminum anode is significantly reduced and the aluminum strip is homogenized, which contribute to improving the capacity of aluminum–air batteries (Fig. 3h).
To investigate the corrosion inhibition of different electrolytes during galvanostatic discharge, the aluminum–air full batteries were assembled by using commercial MnxOy/Ag and aluminum alloy as the cathode catalyst and anode, respectively. The surface morphology of the aluminum anode after discharging at 25 mA cm−2 for 3 h is shown in Fig. 4. There are numerous cracks and holes on the surface of the aluminum anode in 4 M KOH electrolyte (Fig. 4a). Especially, the corrosion of the aluminum surface is uneven and the corrosion depth is up to 45 μm (Fig. 4b–d). Uneven stripping of aluminum ions will lead to unstable discharge voltage. A similar structure is also observed on the surface of the aluminum anode in the electrolyte containing 8 M KOAc (Fig. 4e–h). The relatively flat aluminum anode surface is observed and the corrosion depth is 24 μm when the electrolyte contains 0.02 M Na2SnO3 (Fig. S10d–f†). In the case of HHCE, the surface is much smoother than that in the blank one and the EDS spectrum reveals the existence of abundant Sn element on the surface of the aluminum anode (Fig. 4i). In addition, the in situ optical microscope results also demonstrate that the surface of the aluminum anode in the HHCE is more uniform and the corrosion depth of the aluminum anode decreases to 11 μm (Fig. 4i–l), revealing the even stripping of Al3+, which is in good agreement with the results shown in Fig. 3. Fig. S11† shows the digital photographs of aluminum anodes in different electrolytes after galvanostatic discharge for 3 h. As can be seen, the surface of aluminum anodes is darker in the electrolytes containing Na2SnO3. XRD was used to determine the crystalline structures of the aluminum anode after galvanostatic discharge for 3 h in different electrolytes. As shown in Fig. S12a,† only aluminum peaks are observed in 4 M KOH electrolyte. However, the appearance of Sn peaks can be seen in the HHCE (Fig. S12b†),37 which can be ascribed to the reduction of Na2SnO3 [eqn (4)], that is, Sn is also deposited on the surface of the aluminum anode under working conditions. The anode utilization was further investigated under the same conditions according to the following equation:12
| Ua = 100IT/(ΔMF) | (6) |
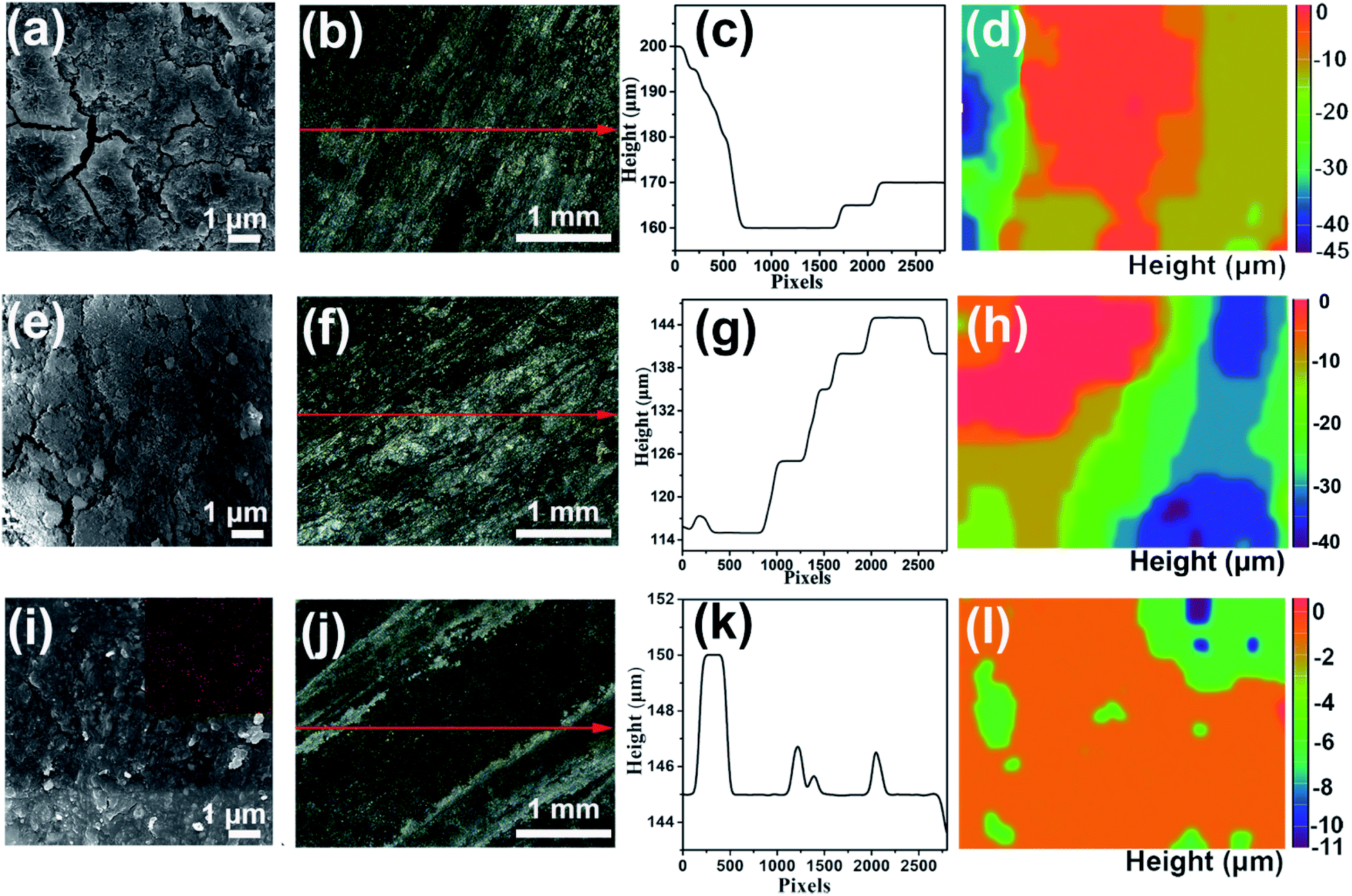 | ||
| Fig. 4 SEM-EDS and in situ optical microscope images of the aluminum anode surface in the (a–d) blank and (e–h) 8 M KOAc electrolytes, and (i–l) HHCE after 3 h galvanostatic discharge. | ||
As shown in Fig. S13,† the open circuit voltage (OCV) curve of the HHCE is more stable than the blank one. Due to the large amount of OAc− in the electrolyte and the Sn protective layer on the aluminum surface, the kinetics of the electrolytes are reduced, leading to increasing polarization of the battery and decreasing power density (Fig. S14 and S15†). The continuous discharge performance of full batteries employing the above-mentioned electrolytes was studied at different current densities. As shown in Fig. 5a, the mass specific capacity of the aluminum anode in 4 M KOH electrolyte is only 429 mA h g−1, which is far away from the theoretical capacity of aluminum (2980 mA h g−1), indicating the serious self-corrosion of the aluminum anode. The capacity increases with the addition of 8 M KOAc (621 mA h g−1) and 0.02 M Na2SnO3 (1072 mA h g−1) in the electrolyte. When HHCE was used, the battery exhibited the highest capacity of 2439 mA h g−1. It is worth noting that the working voltage of the battery using the HHCE is lower than that using 4 M KOH, which is caused by the reduced mass transfer kinetics in the HHCE. Fig. 5b shows the galvanostatic discharge of batteries at 5 mA cm−2. The mass specific capacity of the aluminum anode (100 mA h g−1 for KOH, 120 mA h g−1 for KOAc, 180 mA h g−1 for Na2SnO3 and 600 mA h g−1 for HHCE) is much lower than that at a current density of 25 mA cm−2. It can be found that the discharge curves show potential fluctuation, which may be attributed to the formation and peeling off of the discharge product layer (Al(OH)3) on the aluminum surface.38,39 The on/off cycling tests were carried out to simulate the operation of batteries under actual conditions. It can be clearly seen that the battery using the HHCE exhibits a much longer life (213 h) than that with 4 M KOH electrolyte (40 h) at 25 mA cm−2 (Fig. 5c). Besides, the long-term working time can be up to 388 h with the HHCE at 5 mA cm−2. Comparing the mass specific capacity of the aluminum anode at different current densities, we can conclude that the self-corrosion of the aluminum anode occupies the dominant reaction at a low current density. After the galvanostatic discharge test at a current density of 25 mA cm−2, it can be found that the aluminum surface is uneven and has numerous cracks and holes in the blank electrolyte (Fig. S16a†), which means that the aluminum anode suffers from serious self-corrosion. As for the HHCE, the surface is much smoother than that in the blank one (Fig. S16d†) and the XRD results reveal the existence of abundant Sn element on the surface of the aluminum anode (Fig. S17†). Moreover, the morphology of the MnxOy/Ag catalyst is maintained well in the HHCE but damaged in 4 M KOH electrolyte, implying the protective effect of the HHCE on the electrocatalyst in the cathode (Fig. S18†).
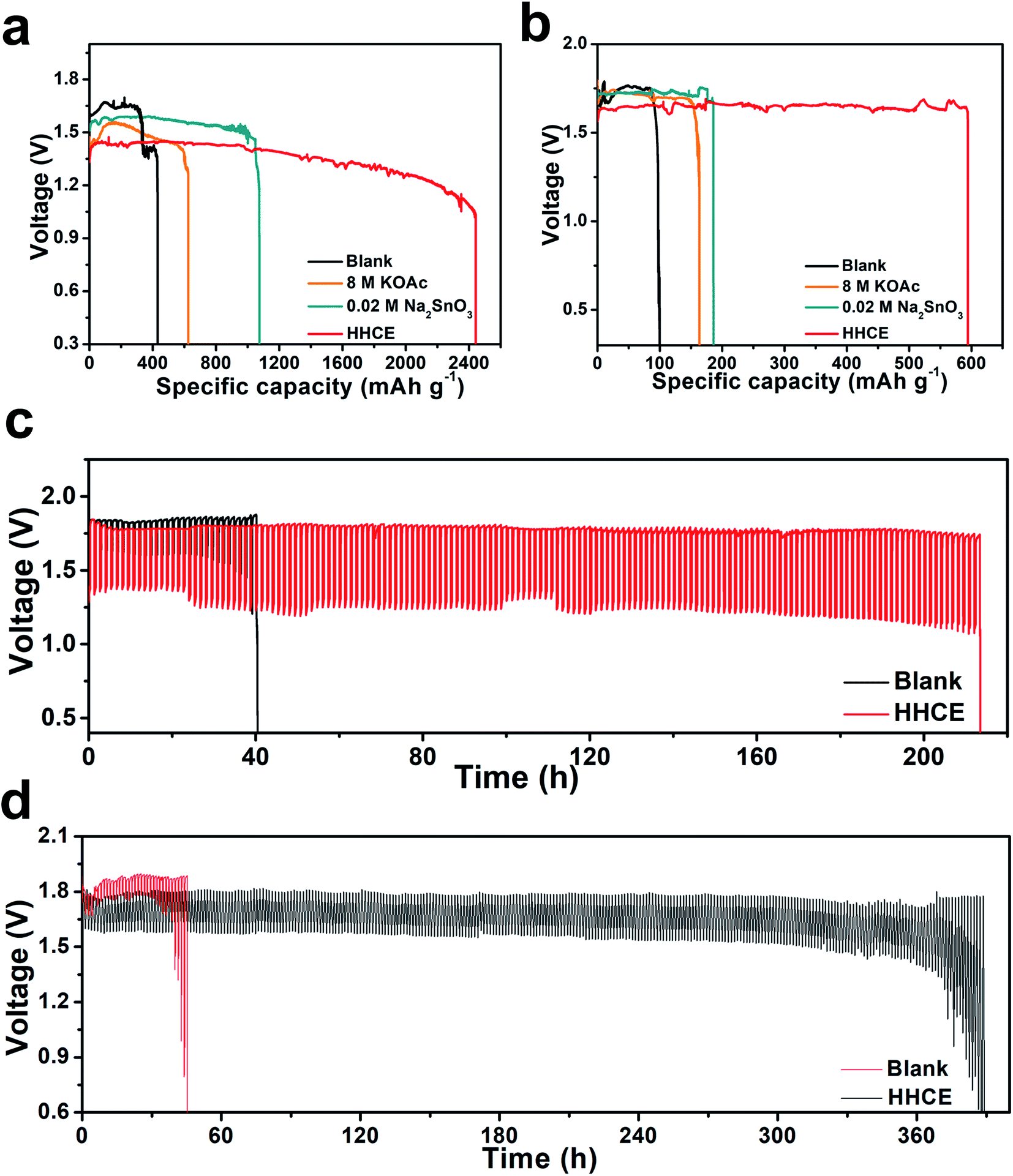 | ||
| Fig. 5 Galvanostatic discharge curves of full batteries with different electrolytes at a current density of (a) 25 mA cm−2, and (b) 5 mA cm−2. Voltage versus time plots for on/off cycling of the full batteries at a current density of (c) 25 mA cm−2, and (d) 5 mA cm−2. | ||
4. Conclusions
In summary, we for the first time presented a regulation of solvation and interface chemistry to inhibit the self-corrosion of the aluminum anode by synergistic regulation of a concentrated electrolyte and interface protective layer. The high concentration KOAc decreases the activity of H2O molecules and suppresses the decomposition of solvated H2O molecules. In addition, the in situ generated Sn protective layer on the surface of the aluminum anode blocks H2O molecule penetration, which further increases the overpotential of hydrogen evolution and significantly inhibits the self-corrosion of the aluminum anode. Benefiting from the regulation of solvation and interface chemistry, the aluminum–air battery with the HHCE exhibits a high capacity of 2439 mA h g−1 (vs. 429 mA h g−1 in 4 M KOH electrolyte) and almost 5.8 times longer on/off operating life than that with the traditional electrolyte at a current density of 25 mA cm−2. This work proposes an efficient and low-cost electrolyte for aluminum–air batteries and sheds light on the synergistic regulation mechanism of solvation and interface chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Hunan Provincial Science and Technology Plan Projects of China (2020JJ2042 and 2017TP1001), the National Nature Science Foundation of China (22179147), the Opening Project of Material Corrosion and Protection Key Laboratory of Sichuan Province of China (2020CL11).Notes and references
- J. Ryu, M. Park and J. Cho, Adv. Mater., 2019, 31, 180478 Search PubMed.
- Y. T. Sun, X. R. Liu, Y. M. Jiang, J. Li, J. Ding, W. B. Hu and C. Zhong, J. Mater. Chem. A, 2019, 7, 18183–18208 RSC.
- Y. J. Fang, Y. X. Zeng, Q. Jin, X. F. Lu, D. Y. Luan, X. T. Zhang and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 8515–8520 CrossRef CAS PubMed.
- H. Cheng, T. Wang, Z. Li, C. Guo, J. Q. Lai and Z. L. Tian, ACS Appl. Mater. Interfaces, 2021, 13, 51726–51735 CrossRef CAS PubMed.
- Y. X. Zhu, L. Zhang, X. Zhang, Z. Y. Li, M. Zha, M. Li and G. Z. Hu, Chem. Eng. J., 2021, 405, 127002 CrossRef CAS.
- T. T. Liang, R. L. Hou, Q. Y. Dou, H. Z. Zhang and X. B. Yan, Adv. Funct. Mater., 2020, 31, 2006749 CrossRef.
- L. M. Suo, O. Borodin, Y. S. Wang, X. H. Rong, W. Sun, X. L. Fan, S. Y. Xu, M. A. Schroeder, A. V. Cresce, F. Wang, C. Y. Yang, Y.-S. Hu, K. Xu and C. S. Wang, Adv. Energy Mater., 2017, 7, 1701189 CrossRef.
- P. Li, H. Kim, J. Ming, H. G. Jung, I. Belharouak and Y. K. Sun, eScience, 2021, 1, 3–12 CrossRef.
- L. M. Suo, O. Borodin, W. Sun, X. L. Fan, C. Y. Yang, F. Wang, T. Gao, Z. H. Ma, M. Schroeder, A. V. Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. S. Wang, Angew. Chem., Int. Ed., 2016, 25, 7252–7257 CrossRef.
- B. J. Hopkins, Y. S. Horn and D. P. Hart, Science, 2018, 362, 658–661 CrossRef CAS PubMed.
- H. Zhang, Y. H. Chen, C. M. Li and M. Armand, SusMat, 2021, 1, 24–37 CrossRef.
- S. A. Wu, S. Y. Hu, Q. Zhang, D. Sun, P. F. Wu, Y. G. Tang and H. Y. Wang, Energy Storage Materials, 2020, 31, 310–317 CrossRef.
- X. B. Cheng, H. Liu, H. Yuan, H. J. Peng, C. Tang, J. Q. Huang and Q. Zhang, SusMat, 2021, 1, 38–50 CrossRef.
- Y. J. Liu, X. Y. Tao, Y. Wang, C. Jiang, C. Ma, O. W. Sheng, G. X. Lu and X. W. Lou, Science, 2022, 375, 739–745 CrossRef CAS PubMed.
- T. E. Hu, Y. D. Fang, L. Su and K. Li, Int. J. Energy Res., 2019, 43, 1839–1847 CrossRef CAS.
- M. A. Rahman, X. J. Wang and C. E. Wen, J. Electrochem. Soc., 2013, 160, A1759 CrossRef CAS.
- Y. G. Li and J. Lu, ACS Energy Lett., 2017, 2, 1370–1377 CrossRef CAS.
- I.-J. Park, S.-R. Choi and J.-G. Kim, J. Power Sources, 2017, 357, 47–55 CrossRef CAS.
- H. Jiang, S. Yu, W. Z. Li, Y. H. Yang, L. S. Yang and Z. J. Zhang, J. Power Sources, 2020, 448, 227460 CrossRef CAS.
- R. Revel, T. Audichon and S. Gonzalez, J. Power Sources, 2014, 272, 415–421 CrossRef CAS.
- T. A. Dougherty, A. P. Karpinski and S. P. Lapp, in Telecommunications Energy Conference, 1995. 17th International, 1995, pp. 821–827 Search PubMed.
- D. Tzidon and I. Yakupov, US Pat., US 9,627,726 B2, 2015 Search PubMed.
- L. M. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. L. Fan, C. Luo, C. S. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- C. Zhang, J. Holoubek, X. Y. Wu, A. Daniyar, L. D. Zhu, C. Chen, D. P. Leonard, I. A. Rodríguez-Perez, J.-X. Jiang, C. Fang and X. L. Ji, Chem. Commun., 2018, 54, 14097–14099 RSC.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. N. Bao, Energy Environ. Sci., 2018, 11, 2876–2883 RSC.
- Q. Y. Dou, S. L. Lei, D.-W. Wang, Q. N. Zhang, D. W. Xiao, H. W. Guo, A. P. Wang, H. Yang, Y. L. Li, S. Q. Shi and X. B. Yan, Energy Environ. Sci., 2018, 11, 3212–3219 RSC.
- S. A. Wu, Q. Zhang, D. Sun, J. Y. Luan, H. W. Shi, S. Y. Hu, Y. G. Tang and H. Y. Wang, Chem. Eng. J., 2020, 383, 123162 CrossRef CAS.
- M. A. Deyab, J. Power Sources, 2019, 412, 520–526 CrossRef CAS.
- Q. X. Kang, T. Y. Zhang, X. Wang, Y. Wang and X. Y. Zhang, J. Power Sources, 2019, 443, 227251 CrossRef CAS.
- G. F. Pauli, S.-N. Chen, D. C. Lankin, J. Bisson, R. J. Case, L. R. Chadwick, T. Gödecke, T. Inui, A. Krunic, B. U. Jaki, J. B. McAlpine, S. Y. Mo, J. G. Napolitano, J. Orjala, J. Lehtivarjo, S.-P. Korhonen and M. Niemitz, J. Nat. Prod., 2014, 77, 1473–1487 CrossRef CAS PubMed.
- H. S. Mobarakeh and M. H. Roudboneh, J. Polym. Res., 2006, 13, 421–426 CrossRef.
- N. Dubouis, P. Lemaire, B. Mirvaux, E. Salager, M. Deschampsbd and A. Grimaud, Energy Environ. Sci., 2018, 11, 3491–3499 RSC.
- Q. Zhang, Y. L. Ma, Y. Lu, L. Li, F. Wan, K. Zhang and J. Chen, Nat. Commun., 2020, 11, 4463 CrossRef CAS PubMed.
- Y. Yamada, C. H. Chiang, K. Sodeyama, J. Wang, Y. Tateyama and A. Yamada, ChemElectroChem, 2015, 2, 1687–1694 CrossRef CAS.
- J. Zheng, J. A. Lochala, A. Kwok, Z. D. Deng and J. Xiao, Adv. Sci., 2017, 4, 1700032 CrossRef PubMed.
- D. S. Stoychev, E. A. Stoyanova and S. Rashkov, Surf. Technol., 1984, 23, 127–141 CrossRef.
- Y. J. Nie, J. X. Gao, E. D. Wang, L. H. Jiang, L. An and X. Y. Wang, Electrochim. Acta, 2017, 248, 478–485 CrossRef CAS.
- C. Zhu, H. X. Yang, A. Q. Wu, D. Q. Zhang, L. X. Gao and T. Lin, J. Power Sources, 2019, 432, 55–64 CrossRef CAS.
- J. B. Wang, J. M. Wang, H. B. Shao, X. T. Chang, L. Wang, J. Q. Zhang and C. N. Cao, Corros. Mater., 2009, 60, 269–273 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ta01064j |
| This journal is © The Royal Society of Chemistry 2022 |