
Layered-perovskite oxides with in situ exsolved Co–Fe alloy nanoparticles as highly efficient electrodes for high-temperature carbon dioxide electrolysis†
Xiang
Sun
a,
Yongjian
Ye
a,
Mengzhen
Zhou
a,
Huijun
Chen
a,
Ying
Li
 a,
Peirong
Chen
a,
Dehua
Dong
b,
Yihan
Ling
c,
Majid
Khan
d and
Yan
Chen
*a
a,
Peirong
Chen
a,
Dehua
Dong
b,
Yihan
Ling
c,
Majid
Khan
d and
Yan
Chen
*a
aSchool of Environment and Energy, State Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou 510006, P. R. China. E-mail: escheny@scut.edu.cn
bSchool of Material Science and Engineering, University of Jinan, Jinan 250022, P. R. China
cSchool of Materials Science and Physics, China University of Mining and Technology, Xuzhou, 221116, P. R. China
dDepartment of Physics, Abdul Wali Khan University Mardan, Mardan, 23200, Pakistan
First published on 29th November 2021
Abstract
Carbon dioxide (CO2) reduction using solid oxide electrolysis cells (SOECs) has attracted great attention because of the high efficiency and fast kinetics enabled by high operating temperatures. Electrode materials with high catalytic activity for CO2 reduction and good stability over long-term operation, nevertheless, are still to be developed. In this work, layered-perovskite oxide electrodes with in situ exsolved Co–Fe alloy nanoparticles are developed for efficient CO2 electrolysis to produce carbon monoxide (CO). Using double perovskite oxide Sr2Ti0.8Co0.2FeO6−δ as a solid precursor, a Ruddlesden–Popper phase oxide matrix with exsolved Co–Fe alloy nanoparticles uniformly distributed on the surface (Co–Fe–STCF) is synthesized by thermal reduction. The cell with a mixture of Co–Fe–STCF and Sm0.2Ce0.8O2−δ (SDC) as the fuel electrode exhibits outstanding performance for CO2 electrolysis, with a polarization resistance (Rp) as low as 0.22 Ω cm2 at 800 °C. A current density of 1.26 A cm−2 is acquired at a bias of 1.6 V at 800 °C, and the CO production rate reached 8.75 mL min−1 cm−2 with a high value of Faraday efficiency (∼100%). Moreover, the Co–Fe–STCF–SDC electrode shows good stability for long-term (100 h) operation with no carbon deposition on the surface. Such high performance of the Co–Fe–STCF–SDC electrode is attributed to abundant oxygen vacancies in the oxide matrix and the high catalytic activity of the exsolved metal nanoparticles. The result of this work can guide the design of highly active and stable (electro)catalysts for high-temperature energy and environmental devices.
Introduction
The use of fossil fuels has released large amounts of carbon dioxide (CO2) over the past few decades, which poses great threat to the environment. Great efforts have been devoted to developing an efficient approach to reduce CO2 emissions.1,2 Electrochemical conversion of CO2 into valuable products using electricity from renewable energy has attracted much attention because it not only provides an effective way for energy storage, but also reduces the environmental effects of CO2. Among various electrochemical devices, solid oxide electrolysis cells (SOECs) which function at high temperature (600–1000 °C) exhibit great advantages such as outstanding conversion efficiency, high Faraday efficiency and fast kinetics.3–6 The performance of SOECs for CO2 electrolysis, nevertheless, is still strongly limited by the lack of high-performance fuel electrode materials. Ni-based composites are the most widely investigated fuel electrodes for SOECs.7,8 Despite its high catalytic activity, metal Ni gets easily oxidized to insulating NiO and faces serious carbon deposition issues when exposed to an atmosphere with a high concentration of CO2.9,10 Developing new electrode materials with high activity toward CO2 reduction and good stability over long-term operation at high temperature is of great significance for the practical application of SOECs for CO2 electrolysis.Perovskite-based oxides, such as SrFeO3−δ, doped-SrTiO3−δ and La0.75Sr0.25Cr0.5Mn0.5O3−δ, have been widely studied as alternative fuel electrode materials for solid oxide fuel/electrolysis cells (SOFCs/SOECs) because of their good stability.11–16 Their catalytic activity, nevertheless, is not sufficient for practical applications. It was reported that active B-site cations, such as Ni, Co, and Fe, could exsolve from the perovskite-based oxide matrix to form metal nanoparticles on the surface upon thermal reduction or applying negative bias.17–26 Because the metal nanoparticles were exsolved from the inside of the oxide matrix to the surface, the metal particles were normally embedded in oxide matrices with robust heterointerfaces.25,26 As a result, the exsolved metal nanoparticles and oxide matrix normally exhibited strong interfacial interaction, which facilitated electron-ionic transport across the metal–oxide interface and restricted the aggregation of metal nanoparticles.25–27 Exsolution materials were reported to exhibit comparable performance as Ni-based fuel electrodes for high-temperature CO2 electrolysis.18 However, auxiliary gases, such as CO and H2, were normally required to be added to the fuel electrode surface to prevent the electrode from being oxidized and improve the electrolysis performance.28–31 The addition of auxiliary gas diluted the reaction gas and made the research system more complex. Only very limited studies showed that pure CO2 can be directly reduced to CO by SOECs with appropriate exsolution materials as the fuel electrode.32–35 Nevertheless, the production rate of CO was still quite low, and the stability of the electrode materials required further improvement.
In this work, we demonstrated layered-perovskite oxides with in situ exsolved Co–Fe alloy nanoparticles as a highly efficient and stable fuel electrode for high-temperature electrolysis of pure CO2 to produce CO. Ruddlesden–Popper (R–P) phase oxides with exsolved Co–Fe alloy nanoparticles uniformly distributed on the surface (Co–Fe–STCF) were prepared by reducing stoichiometric double perovskite oxide Sr2Ti0.8Co0.2FeO6−δ in a H2 atmosphere at high temperature. The cell with the obtained exsolution materials as the fuel electrode (Co–Fe–STCF–SDC) exhibited outstanding activity toward CO2 reduction, with an electrolysis current density of 1.26 A cm−2, CO production rate of 8.75 mL min−1 cm−2 and a Faraday efficiency close to 100% at 1.6 V at 800 °C. The Co–Fe–STCF–SDC electrode also showed good stability for operation at 800 °C under 1.2 V for 100 h in pure CO2 environment. Such high performance was attributed to oxygen vacancies in the oxide matrix and the exsolved metal nanoparticles. This work demonstrates exsolution materials as promising high-performance catalysts for high-temperature energy and environmental devices.
Experimental procedures
Materials preparation
Double perovskite oxide Sr2Ti0.8Co0.2FeO6−δ (STCF) powders were synthesized by solid-state reactions. SrCO3, TiO2, Co3O4, and Fe2O3 powders (Macklin, China) with a stoichiometric ratio and appropriate amount of ethanol were mixed and ball milled for 24 h. The obtained mixtures were dried in a muffle furnace and then calcined at 1200 °C for 8 h in air to form stoichiometric STCF powders. The as-prepared STCF powders were heated in a tubular furnace at different temperatures (800 °C and 900 °C) and times (1, 2, 4, and 8 h) in a H2 atmosphere. After hydrogen reduction, Co–Fe alloy nanoparticles with different population densities and diameters exsolved from the oxide matrix to the surface.Oxygen electrode materials, La0.7Sr0.3CoO3−δ (LSC), were synthesized using a similar method to the one used for STCF, with La2O3, SrCO3, and Co3O4 powders (Macklin, China) as the precursors and ethanol as the solvent. The mixtures were calcined at 1000 °C for 6 h to form the stoichiometric LSC phase. Electrolyte materials, La0.8Sr0.2Ga0.83Mg0.17O3−δ (LSGM), were prepared by solid-state reactions with the detailed procedure described in the Experimental section of the ESI.†
Electrolysis cell fabrication
CO2 electrolysis was carried out using an electrolyte-supported single cell with STCF–SDC as the fuel electrode, LSGM as the electrolyte and LSC–SDC–PrOx as the oxygen electrode (STCF–SDC/LSGM/LSC–SDC–PrOx). Sm0.2Ce0.8O2−δ (SDC) (Ningbo SOFCMAN Energy Technology Co.) was mixed with STCF to enhance the adhesion between the fuel electrode and the electrolyte (LSGM). When the pure STCF electrode material was prepared at high temperature, the electrode suffered from sintering effects, which resulted in the reduction of the specific surface area (Fig. S1†). Adding high temperature resistant cerium-based oxide to the fuel electrode can also effectively prevent such sintering of the electrode structure. Moreover, the presence of cerium-based oxide with high oxygen ion conductivity in the medium temperature region36,37 can improve the ionic conductivity of the electrode. The fuel electrode slurry was prepared by mixing STCF, SDC and graphite. The oxygen electrode slurry composed of LSC and SDC was prepared using a similar approach to that of the fuel electrode, and a PrOx decoration layer was impregnated on the LSC–SDC oxygen electrode by using Pr(NO3)3·6H2O as the precursor. A Ag grid was painted on both sides of the electrode surface to serve as current collectors. The effective area of the cell was 0.2 cm−2.Electrochemical measurement
The obtained cell was tested in both SOFC and SOEC modes using an electrochemical workstation (IM6, Zahner, Germany). In SOFC mode, dry H2 was fed to the fuel electrode side with a flow rate of 50 mL min−1, while the oxygen electrode was directly exposed to air. In SOEC mode, the fuel electrode side was fed with pure CO2 at a flow rate of 50 mL min−1, and pure Ar was fed to the oxygen electrode side with a flow rate of 50 mL min−1. Before the electrochemical test, the STCF–SDC fuel electrode was reduced in pure H2 gas at 900 °C for 1 hour to obtain Co–Fe alloy nanoparticles on the surface of the oxide matrix (denoted as Co–Fe–STCF–SDC). The production of CO was analysed quantitatively using gas chromatography (GC9790II, Fuli, Zhejiang, China).Materials characterization
The crystal structure of the materials was analysed by X-ray diffraction (XRD, Bruker D8 Advance, Germany) with Cu-Kα radiation. The topography was investigated by scanning electron microscopy (SEM, SU8010, Japan). The microstructure and local composition were probed by transmission electron microscopy (TEM) which was coupled with energy dispersive X-ray spectroscopy (EDS) analysis (FEI Tecnai G2 F20) operating at 200 kV. X-ray photoelectron spectroscopy (XPS) (ESCALAB250Xi, Thermo Scientific) with an Al Kα X-ray source was used to analyse the surface composition and chemical environment of different elements. The binding energies of all the XPS spectra were calibrated by adventitious carbon (C 1s) at 284.6 eV. The XPS spectra were fitted using a Shirley-type background and peaks with 80% Gaussian and 20% Lorentzian functions.38,39 Raman spectra (LabRAM HR 800) were collected to characterize the possible carbon deposition on the fuel electrode after the stability test. Thermogravimetric analysis (TGA SDT Q600) was performed from 50 to 800 °C in air for STCF before and after reduction. Temperature-programmed desorption of CO2 (CO2-TPD) was conducted to probe the CO2 adsorption characteristics on the sample surface. The detailed experimental procedure of CO2-TPD is shown in the ESI.†Results and discussion
To identify the optimal reduction conditions for the exsolution of metal nanoparticles, we first investigated the dependence of the material microstructure on the hydrogen reduction conditions. The morphology of STCF before and after reduction at different temperatures (800 to 900 °C) and for various times (1 to 8 h) was observed by SEM, and the results are shown in Fig. 1a–h. The as-prepared STCF exhibited a smooth surface with a particle size of about ∼1 μm (Fig. 1a). After reduction at 800 °C for 1 hour, the morphology remained unchanged and no exsolved nanoparticles were observed on the surface (Fig. S2†). By elongating the reduction time to 2 hours at 800 °C, nanoparticles with a size of ∼15 nm and a population density of 220 μm−2 were observed on the surface of the oxide matrix (Fig. 1b and i). Further increasing the reduction time to 4 h and 8 h led to an increase of the particles size to ∼30 and ∼43 nm, respectively, and a decrease of the particle population density to 156 and 112 μm−2, respectively (Fig. 1c, d and i). By increasing the annealing temperature from 800 °C to 900 °C, the appearance of exsolved nanoparticles was clearly observed after 1 h reduction (Fig. 1e). After reduction at 900 °C for 1, 2, 4 and 8 h, the sizes of the nanoparticles were ∼23, ∼35, ∼48 and ∼58 nm, and the particle population densities were 222, 188, 152 and 143 μm−2, respectively (Fig. 1e–h and i).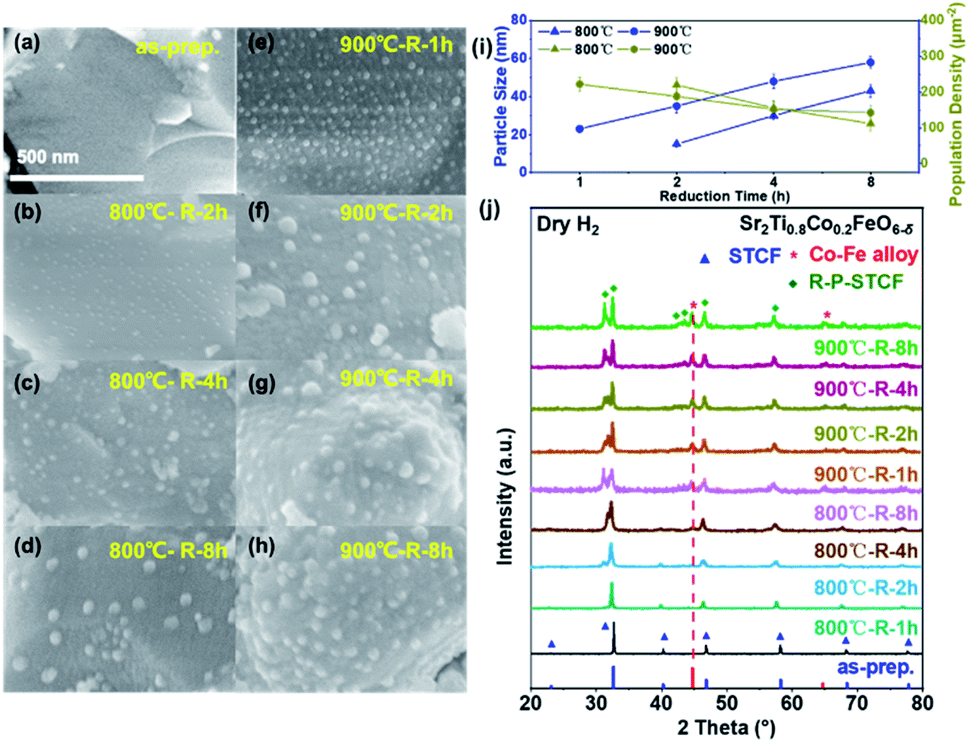 | ||
| Fig. 1 (a–h) SEM patterns of STCF before and after reduction treatment at different temperatures (800 °C and 900 °C) and reduction times (1, 2, 4, and 8 h), respectively, in pure H2. (i) Particle size and population density of STCF after reduction treatment obtained from SEM images. (j) XRD patterns of STCF before and after reduction related to SEM images. | ||
The SEM results showed that the exsolved nanoparticles grew slightly larger when the annealing time increased. Importantly, STCF subjected to hydrogen annealing for 1 hour at 900 °C exhibited the optimal morphology with a high particle population density and small particle size. We also observed that exsolution occurred more easily at higher reduction temperature. Increasing the thermal reduction temperature is likely to facilitate cation migration from the bulk to the surface during exsolution.18,40,41 More importantly, the thermodynamic driving force for exsolution was also impacted by the reduction temperature. Table 1 shows the Gibbs free energy change (ΔG) of each element from metal oxides (M+) to metal states (M°) upon reduction at two different temperatures.25,42 The comparison of ΔG values provides critical information about the thermodynamic feasibility of different cations to exsolve as metals upon reduction. The Gibbs free energy change (ΔG) for the conversion of Fe2O3 to metal Fe is −4.09 kJ mol−1 and −16.54 kJ mol−1 at 600 °C and 900 °C, respectively (Table 1). The ΔG at 900 °C is much more negative than that at 600 °C, suggesting that Fe exsolution is more conducive at higher reduction temperature. The same trend of ΔG is also observed for Co2O3. Moreover, the Gibbs free energy change for the conversion of titanium dioxide to metal Ti is positive at both 900 and 600 °C, indicating that it cannot be reduced at these temperatures. Consistent with these calculation results, we observed experimentally that Co and Fe exsolved to the surface after reduction, and Ti remained stable in the lattice.
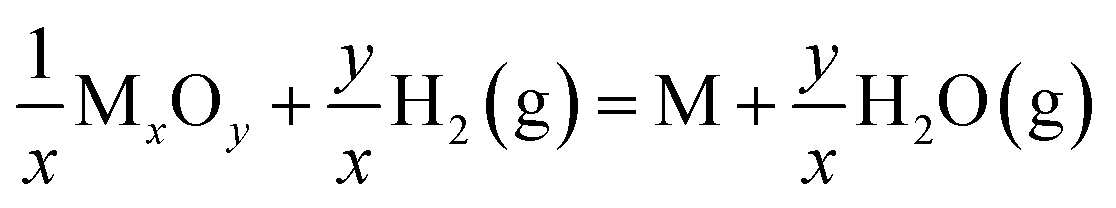 . The data are generated using the HSC chemistry software43
. The data are generated using the HSC chemistry software43
| Elements | TiO2 | Fe2O3 | Co3O4 |
|---|---|---|---|
| 600 °C | 385.82 | −4.09 | −74.96 |
| 900 °C | 365.89 | −16.54 | −90.78 |
The crystal structure of the as-prepared and thermally reduced STCF samples was probed by XRD measurement.17,44 As shown in Fig. 1j, the as-prepared STCF and the one subjected to thermal reduction treatment at 800 °C for 1 h had a double perovskite structure and showed no other impurity, which was consistent with the absence of exsolved nanoparticles in the SEM images (Fig. 1a and S2†). All the other STCF samples that were reduced at higher temperature or longer time exhibited characteristic XRD peaks of the Co–Fe alloy. As shown in Fig. 1j, the two peaks marked with “*” were assigned to the (110) and (200) peaks of the Co3Fe7 alloy (PDF no. 48-1817).45 With the increase of the annealing time and temperature, the intensity of Co–Fe alloy peaks showed noticeable enhancement, suggesting the increase of Co–Fe alloy contents. The peak position of the alloy did not show a noticeable shift, suggesting a similar composition of the alloy nanoparticles.33 This result is consistent with the increased amount of exsolved nanoparticles on the surface, as observed in SEM measurement. Accompanied by the presence of a Co–Fe alloy peak, a new diffraction peak, which was assigned to R–P phases, appeared in the XRD pattern. This result implied that the double perovskite structure partially converted into an R–P phase structure (Srm+1(TiCoFe)mO3m+1) during the exsolution process of metal nanoparticles. The phase transformation became more pronounced with the prolongation of the reduction time and the increase of the reduction temperature. For the sample subjected to hydrogen reduction at 900 °C for longer than one hour, the double perovskite completely converted into the R–P phase structure. Importantly, the R–P phase perovskite was reported to exhibit good stability at elevated temperature in a CO2 atmosphere in previous research studies.46,47 The combination of SEM and XRD results suggests that after thermal reduction, Co–Fe alloy nanoparticles successfully exsolved to the surface. STCF subjected to 1 h annealing at 900 °C (denoted as Co–Fe–STCF) exhibited the optimal distribution of nanoparticles, and thereby will be the focus of the following investigation.
To further confirm that we have obtained the oxide matrix with exsolved Co–Fe alloy nanoparticles on the surface, we further carried out TEM and EDS analysis of the Co–Fe–STCF sample. As shown in the TEM image and EDS elemental distribution (Fig. 2a and S3†), the nanoparticles on the surface exhibited significantly higher concentrations of Co and Fe than the matrix. The lattice parameter of the surface nanoparticles (Fig. 2b and c) was quantified to be 0.202 nm, which corresponds to the (110) plane at an angle of 44.7° for the Co–Fe alloy. All these TEM results are consistent with the SEM and XRD results above, implying the exsolution of Co–Fe alloy nanoparticles on the surface. The structural changes during the exsolution process are described in Fig. 2d.
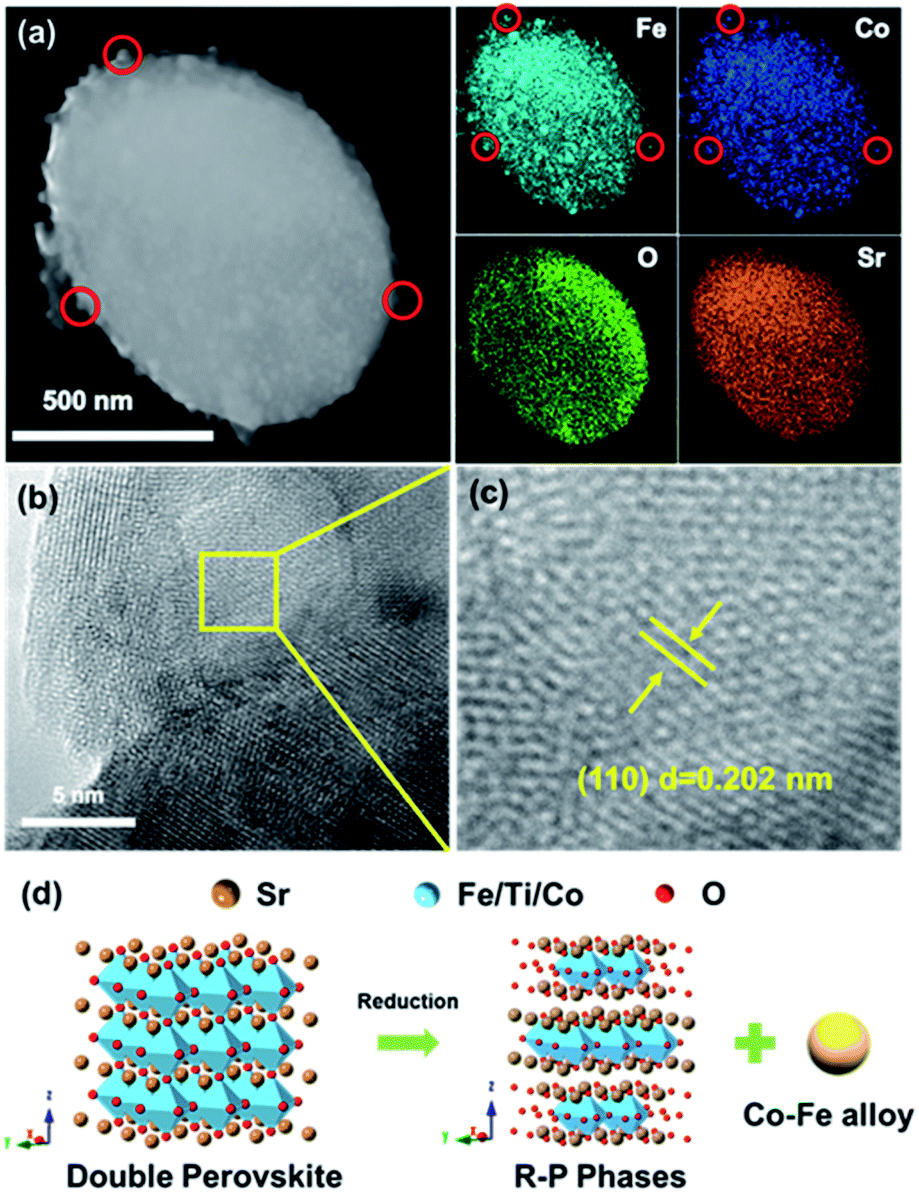 | ||
| Fig. 2 (a) STEM image and elemental maps of STCF subjected to 1 h hydrogen annealing at 900 °C (Co–Fe–STCF). (b and c) HRTEM images of the exsolved nanoparticles. (d) Illustration of structural transformation of STCF during the exsolution process. | ||
It is important to note that parts of the exsolved nanoparticles were embedded in the oxide matrix (Fig. 2b), suggesting a robust heterointerface between the oxide matrix and exsolved metal nanoparticles. Such a robust heterointerface can potentially facilitate electron-ionic transport between the metal particles and oxide matrices,25,26 and prevent the agglomeration of the metal nanoparticles under the operating conditions of SOECs.35,44,48,49
The valence states of the cations in STCF and Co–Fe–STCF were further compared by using XPS measurements. Ti 2p, Co 2p, Fe 2p, O 1s and survey XPS spectra were acquired, as shown in Fig. 3 and S4.† The XPS survey spectra of STCF and Co–Fe–STCF powders suggested that the samples contained Sr, Ti, Co, Fe and O elements, and no other impurity elements were observed (Fig. S4a†). The valence states of Co and Fe are determined on the basis of Co 2p and Fe 2p spectra, as shown in Fig. 3a and b. The Co 2p and Fe 2p spectra were fitted to three sets of spin–orbit splitting doublets. The doublet located at 777.8 eV and 792.6 eV can be clearly observed for the Co–Fe–STCF sample, which is attributed to Co0.49 Similarly, the Fe0 2p3/2 and 2p1/2 doublet located at 706.6 eV and 722.1 eV, respectively, was observed for the Co–Fe–STCF sample.50 In contrast, Co and Fe in the as-prepared STCF were dominated by 2+ and 3+.51–53 These XPS results further confirmed the exsolution of Co and Fe alloy metal nanoparticles on the surface after hydrogen reduction. Furthermore, the atomic ratio of Co0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe0 in Co–Fe–STCF annealed at 900 °C for 1 hour is quantified to be 0.37:1 based on the Co0 and Fe0 peaks in the XPS spectra (Fig. 3, Table S1†), which is in agreement with the XRD results. The Ti 2p spectra (Fig. S4b†) exhibited negligible changes upon reduction, implying no noticeable changes in the valence state of Ti. This result is consistent with what is reported in the literature.25,54–57
:Fe0 in Co–Fe–STCF annealed at 900 °C for 1 hour is quantified to be 0.37:1 based on the Co0 and Fe0 peaks in the XPS spectra (Fig. 3, Table S1†), which is in agreement with the XRD results. The Ti 2p spectra (Fig. S4b†) exhibited negligible changes upon reduction, implying no noticeable changes in the valence state of Ti. This result is consistent with what is reported in the literature.25,54–57
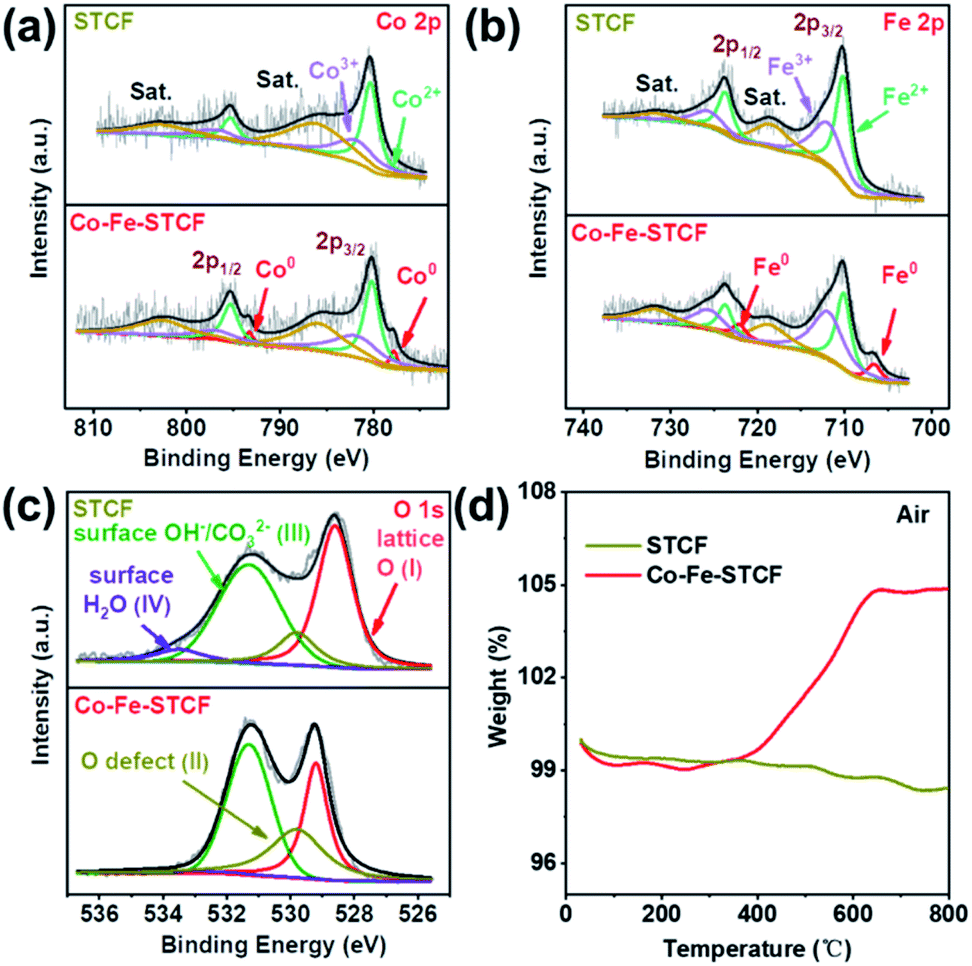 | ||
| Fig. 3 XPS spectra of (a) Co 2p, (b) Fe 2p, and (c) O 1s for STCF and Co–Fe–STCF. (d) Thermogravimetric analyses of weight increase of STCF powders before and after reduction treatment in air flow. | ||
The decrease of Co and Fe valence states implied that large amounts of oxygen vacancies were generated in the oxide matrix after thermal reduction. The appearance of oxygen vacancies was further confirmed by the analysis of O 1s spectra, as shown in Fig. 3c. The O 1s spectra were fitted to four components located at 528.6, 529.8, 531.3 and 533.5 eV. These peaks corresponded to oxygen species of lattice oxygen (Olat, peak I), defective oxygen (peak II), hydroxyl groups or carbonate groups (peak III), and adsorbed molecular water (peak IV) on the surface of STCF.49,58–62 As shown in Fig. 3c, there was an apparent decrease in lattice oxygen (peak I) on the Co–Fe–STCF surface with respect to the one on the STCF surface.48 Moreover, the peak of defective O (peak II) showed a significant increase on the Co–Fe–STCF surface. Both results implied the formation of excessive oxygen vacancies in the oxide matrix during the exsolution of the Co–Fe alloy upon thermal reduction.58
To further reveal the oxygen vacancy characteristics, TGA measurement was carried out for the STCF and Co–Fe–STCF samples in air from 50 to 800 °C. As shown in Fig. 3d, the weight change before 100 °C was caused by the desorption of water molecules on the surface. The overall weight change of Co–Fe–STCF was 4.864% after the temperature reached 800 °C. Such a change was attributed to oxygen uptake from air during the re-oxidation processes of metal to the oxide phase and/or the oxygen vacancy filling.49 In contrast, STCF powders exhibit quite small changes in weight. These TGA results suggest that the oxide matrix of Co–Fe–STCF contained large amounts of oxygen vacancies, which may be beneficial for the CO2 reduction reaction to occur.
Having confirmed the formation of Co–Fe alloy nanoparticles on the Co–Fe–STCF surface, we fabricated an electrolyte supported single cell with the configuration of STCF–SDC/LSGM/LSC–SDC–PrOx. The fuel electrode with the Co–Fe alloy on the surface was prepared by exposing the STCF–SDC fuel electrode to hydrogen gas at 900 °C for 1 hour. The microstructure of the single cell before in situ reduction is shown in Fig. 4a. The thicknesses of the fuel electrode (STCF–SDC) and electrolyte are ∼29 μm and ∼230 μm, respectively. After in situ thermal reduction, the porous structure of the fuel electrode was well-maintained (Fig. 4b). As shown in the inset figure of Fig. 4b, the exsolved Co–Fe alloy nanoparticles are uniformly embedded in the oxide matrix of the Co–Fe–STCF phase, while no particles are found on the surface of SDC. We further carried out chemical compatibility tests of STCF with LSGM and SDC. The mixture of STCF, LSGM and SDC in pairs was ball-milled for 3 h and annealed at 1150 °C for 10 hours. No extra peaks appeared in the XRD pattern of the obtained mixture (Fig. S5†), indicating the good compatibility of STCF with LSGM and SDC.
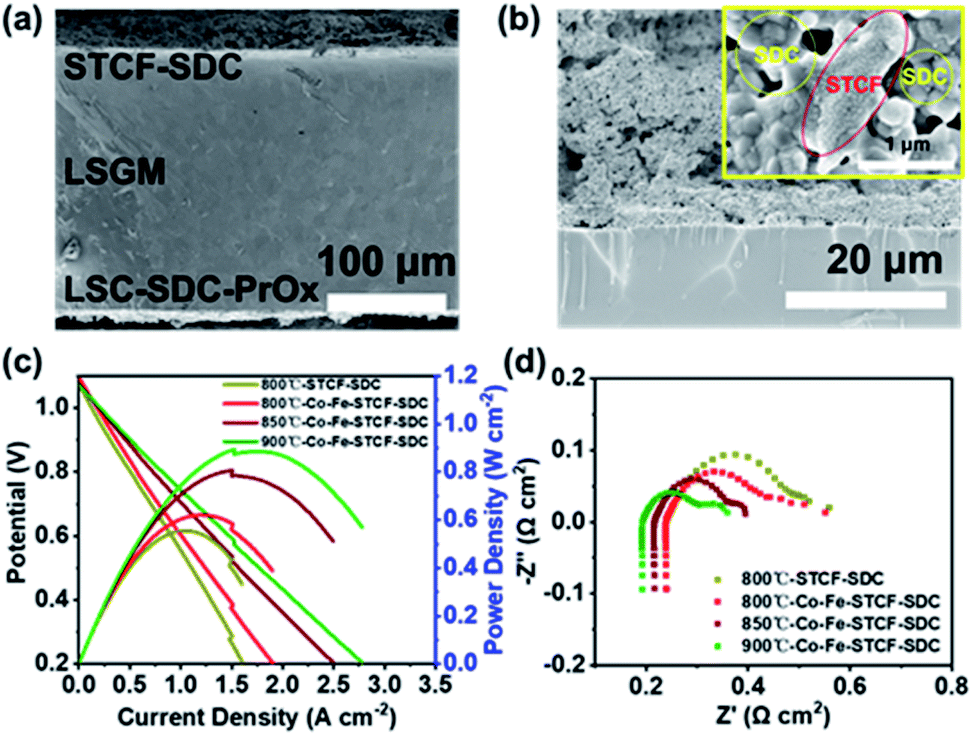 | ||
| Fig. 4 (a and b) SEM images of the cell with STCF–SDC (a) and Co–Fe–STCF–SDC (b) fuel electrodes. (c) Current–voltage (I–V) and current–power (I–P) curves and (d) electrochemical impedance spectroscopy (EIS) results of the cell with the STCF–SDC fuel electrode at 800 °C and the cell with the Co–Fe–STCF–SDC fuel electrode at 800, 850 and 900 °C. | ||
To check the quality of the fabricated cells, we first evaluate the cell performance in the SOFC mode with pure H2 fed onto the fuel electrode surface. Fig. 4c shows the current–voltage (I–V) and current–power (I–P) curves of the SOFCs with Co–Fe–STCF–SDC as the fuel electrode at 800, 850 and 900 °C. The OCV of the cell is around 1.05–1.10 V and was found to decrease with the rise of temperature. The Pmax of the cell with the Co–Fe–STCF–SDC fuel electrode reached 621, 806 and 885 mW cm−2 at 800, 850, and 900 °C, respectively. The power density of the cell with the STCF–SDC fuel electrode is about 555 mW cm−2 at 800 °C, which is lower than that of the cell with the Co–Fe–STCF–SDC fuel electrode under the same conditions (621 mW cm−2). The electrochemical impedance spectroscopy (EIS) results of all the cells are shown in Fig. 4d. The intercept of the semicircle in high and low frequency regions was attributed to the ohmic resistances (Ro) of the single cell and total resistances of the cell, respectively, while the electrode polarization resistance was the difference between the two intercepts.39,63 The ohmic resistances of the cell with the STCF–SDC electrode and the one with the Co–Fe–STCF–SDC electrode were similar under the same conditions. In contrast, the Rp value of the cell with the Co–Fe–STCF–SDC electrode is smaller than that of the cell with the STCF–SDC electrode. Considering the fact that both cells had the same oxygen electrode, such a decrease in the Rp value indicated an improved activity of the fuel electrode toward H2 oxidation after exsolution.
The SOFC results above suggest that we have successfully assembled the cells with STCF–SDC or Co–Fe–STCF–SDC fuel electrodes. These cells were then operated in SOEC mode to convert CO2 to CO and the performance was systematically compared. The schematic diagram of high-temperature electrolysis of carbon dioxide in SOEC mode is shown in Fig. 5a. Fig. 5b shows the I–V curves for high-temperature electrolysis of CO2 at applied voltage of 0 to 1.7 V at 800 °C. The current density began to increase rapidly near 1.1 V for both cells with STCF–SDC and Co–Fe–STCF–SDC fuel electrodes (Fig. 5b). These results indicated that the electrochemical reduction of CO2 to CO only occurred at an applied potential higher than 1.1 V, which was consistent with the reported theoretical value under the same conditions.64 The current density of the cell with the Co–Fe–STCF–SDC (red line) electrode was much higher than that of the cell with the STCF–SDC electrode, implying the strongly enhanced CO2 reduction performance after the exsolution of the Co–Fe alloy. The potentiostatic measurements of the electrochemical CO2 reduction reaction at various voltages are shown in Fig. 5c. The cell with the Co–Fe–STCF–SDC electrode exhibited noticeable higher current density at all voltages than the cell with the STCF–SDC electrode, implying the better CO2 reduction performance of the Co–Fe–STCF–SDC electrode. The CO production rate and faradaic efficiency are the most critical parameters to represent the CO2 reduction performance of the cells. As shown in Fig. 5d, the CO production rate of the cell with the Co–Fe–STCF–SDC electrode reached 8.75 mL min−1 cm−2 at 1.6 V and 800 °C, which is noticeably higher than 5.90 mL min−1 cm−2 for the cell with the STCF–SDC electrode. The cells with the Co–Fe–STCF–SDC and STCF–SDC fuel electrodes both exhibited faradaic efficiencies of ∼100% at 1.6 V. This result implies that the main product is CO and no other species, such as carbon deposition, form during the electrochemical CO2 reduction process.
 | ||
| Fig. 5 (a) Illustration of high-temperature CO2 electrolysis using SOECs, (b) I–V curves, (c) potentiostatic tests at different voltages, (d) CO production rate and faradaic efficiency, and (e) electrode polarization resistance (Rp) of SOECs at different potentiostatic voltages for the cells with STCF–SDC or Co–Fe–STCF–SDC as the fuel electrodes measured at 800 °C. (f) CO2-TPD curves of STCF–SDC and Co–Fe–STCF–SDC powder samples. | ||
We also carried out EIS measurement for both cells tested at 1.2, 1.4 and 1.6 V at 800 °C. Similar to the results for the SOFC mode, both cells exhibited similar ohmic resistance at various applied voltages. The polarization resistance of the cell with the Co–Fe–STCF–SDC electrode was much lower than that of the cell with the STCF–SDC electrode at applied voltages (Fig. 5e, S6 and S7†). In particular, the Rp value of the cell with Co–Fe–STCF–SDC is 0.22 Ω cm2 at 1.6 V, which is noticeably lower than 0.33 Ω cm2 for the cell with the STCF–SDC electrode. These results indicated that exsolution greatly enhanced the electrocatalytic activity of the STCF phase for CO2 reduction.
Due to the lack of polarity of linear molecules, the adsorption and activation of CO2 on the fuel electrode surface are normally quite poor, which led to the starvation of CO2 during operation and low electrochemical performance.58 Therefore, the adsorption of CO2 on the surface plays a critical role in determining the activity of the fuel electrode toward CO2 reduction.65 To investigate the CO2 adsorption characteristics, we carried out CO2-TPD measurement of the STCF–SDC and Co–Fe–STCF–SDC electrode materials. As shown in the CO2-TPD curves of STCF–SDC and Co–Fe–STCF–SDC electrode materials (Fig. 5f), the first CO2 desorption peak (50–200 °C) was the desorption of physically adsorbed CO2 in a low-temperature range.58,66 The desorption peaks located in the temperature range from 300 °C to 800 °C were attributed to the chemically adsorbed CO2.32 In comparison to the STCF–SDC powder, the Co–Fe–STCF–SDC powder exhibited a stronger desorption peak in the high temperature range (500 °C to 800 °C), suggesting a larger amount of adsorbed CO2 on the surface of Co–Fe–STCF–SDC. As shown in the previous section, large amounts of oxygen vacancies existed in the oxide matrix of the Co–Fe–STCF powder. These oxygen vacancies were reported to be the active sites for accommodating nonpolar CO2, leading to the larger amount of adsorbed CO2 on the Co–Fe–STCF surface.58,67 The adsorbed CO2 on the defect sites can be further electrochemically reduced to CO. The defective oxide matrix is very likely to be one reason for the high CO2 reduction performance of the Co–Fe–STCF samples. In addition to the oxide matrix, the exsolved Co–Fe alloy was reported to be a good catalyst for CO2 conversion.11,68 For instance, Chen et al. prepared three distinct Co–Fe alloy catalysts at different temperatures by H2-reduction of a layered double hydroxide nanosheet precursor, and the obtained alloy showed outstanding activity towards CO2 hydrogenation.11 Therefore, we believe both the highly defective oxide matrix and the exsolved metal nanoparticles contribute to the high activity of Co–Fe–STCF–SDC for CO2 reduction.
To evaluate the stability of the Co–Fe–STCF–SDC electrode for practical applications, we tested the CO2 electrolysis performance of the cell with the Co–Fe–STCF–SDC electrode at 800 °C and 1.2 V in pure CO2 for 100 h. As shown in Fig. 6b, the current density decreased slightly (∼8.5%) after the long-term stability test. For comparison, Dong et al.10,28 reported that the cell with the Ni-based electrode (Ni/YSZ) showed rapid degradation with a sharp increase of the voltage up to above 2 V (from 1.2 V) during the electrolysis period of 12 h at a constant current density of 0.50 A cm−2 in pure CO2. After the stability test, the authors believed that oxidation of Ni particles led to an almost complete deterioration of the Ni/YSZ electrode. For the case of the Co–Fe–STCF–SDC electrode, the exsolved Co–Fe alloy nanoparticles could well-maintain their initial structure without agglomeration, as shown in the SEM image of Fig. 6a. The Raman spectrum of the Co–Fe–STCF–SDC electrode after the stability test is presented in Fig. 6c. No carbon peaks were observed in the range of 1000–2000 cm−1, especially at 1100, 1338 and 1568 cm−1, which were assigned to SrCO3, the carbon D-band and carbon G-band, respectively. This result indicated that there was no carbon deposition and SrCO3 formation after long-term operation. Since the microstructure remained unchanged after the test (Fig. 6a and S8†) and no carbon deposition was observed, we believe that the slight decline of SOEC performance is likely due to changes in the microstructure at the electrode/electrolyte interface during the high-temperature test in a pure carbon dioxide atmosphere (Fig. S8†). The above results show that the exsolution materials have excellent stability as the fuel electrode of SOECs for the electrolysis of pure CO2.
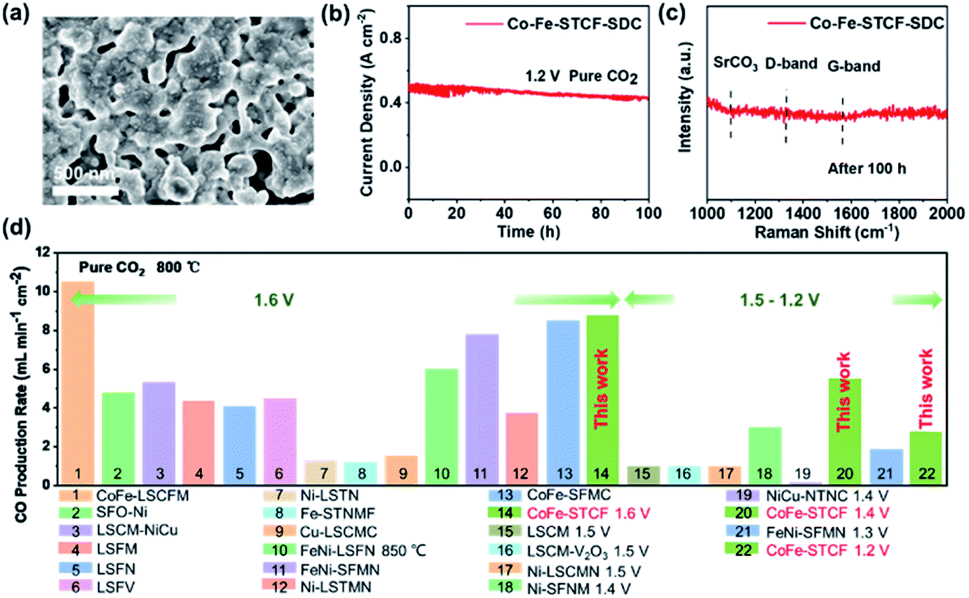 | ||
| Fig. 6 (a) The SEM image of the Co–Fe–STCF–SDC electrode after the stability test and (b) stability test of the SOECs at 1.2 V and 800 °C. (c) Raman spectrum of the Co–Fe–STCF–SDC electrode after the stability test. (d) Comparison of the CO production rate of SOECs with different fuel electrode materials reported in this work and in the literature. | ||
Finally, we compare the performance of the cell with the Co–Fe–STCF–SDC fuel electrode towards CO2 reduction to generate CO with other electrode materials reported in the literature, including SrFeO3−δ–Ni (SFO–Ni),11 La0.75Sr0.25Cr0.5Mn0.5O3−δ (LSCM),12 La0.75Sr0.25Cr0.5Mn0.5O3−δ–V2O5 (LSCM–V2O5),69 La0.75Sr0.25Cr0.5Mn0.5O3−δ–NiCu (LSCM–NiCu),70 La0.6Sr0.4Fe0.9Mn0.1O3−δ (LSFM),71 La0.6Sr0.4Fe0.8Ni0.2O3−δ (LSFN),72 La0.5Sr0.5Fe0.95V0.05O3−δ (LSFV),73 (La0.2Sr0.8)0.95Ti0.85Mn0.1Ni0.05O3−δ (Ni–LSTMN),32 (La0.75Sr0.25)0.9(Cr0.5Mn0.5)0.9Ni0.1O3−δ (Ni–LSCMN),74 Sr2Fe1.45Ni0.05Mo0.5O6−δ (Ni–SFNM),75 (La0.3Sr0.7)0.9Ti0.95Ni0.05O3−δ (Ni–LSTN),76 (Sr0.95)0.9(Ti0.8Nb0.1Mn0.1)0.9–Fe0.1O3−δ (Fe–STNMF),77 (La0.75Sr0.25)0.9(Cr0.5Mn0.5)0.9Cu0.1O3−δ (Cu–LSCMC),65 Sr2Fe1.35Mo0.45Co0.2O6−δ (CoFe–SFMC),33 Sr1.9Fe1.5Mo0.4Ni0.1O6−δ (FeNi–SFMN),34 La0.6Sr0.4Fe0.8Ni0.2O3−δ (FeNi–LSFN),78 Sr2Fe1.35Mo0.45Ni0.2O6−δ (FeNi–SFMN),58 La0.4Sr0.6Co0.2Fe0.7Mo0.1O3−δ (CoFe–LSCFM)35 and NbTi0.5(Ni0.75Cu0.25)0.5O4 (NiCu–NTNC).38 As shown in Fig. 6d and Table S2,† the cells with the Co–Fe–STCF–SDC electrode delivered outstanding performance for CO2 reduction to produce CO gas.
Conclusions
In this work, we developed highly active and stable fuel electrode materials, which consist of a porous R–P phase oxide matrix and exsolved Co–Fe alloy nanoparticles, for electrochemical reduction of CO2 to produce CO at high temperature. Stoichiometric double perovskite oxide Sr2Ti0.8Co0.2FeO6−δ (STCF) was thermally reduced in a H2 atmosphere at high temperature. The size and population density of the exsolved Co–Fe alloy nanoparticles were found to depend closely on the reduction temperature and time. The sample subjected to annealing at 900 °C for 1 h in H2 (Co–Fe–STCF) exhibited the optimal microstructure with a small particle size and high particle population density. The combination of SEM, XRD and TEM characterization suggested that the Co–Fe alloy metal particles were embedded in the oxide matrix with a robust interface. Moreover, XPS and TG analysis revealed that abundant oxygen vacancies were generated in the oxide matrix after the exsolution of metal particles. The cell with Co–Fe–STCF–SDC as the fuel electrode exhibited outstanding CO2 electrolysis performance, with the current density, CO production rate and faradaic efficiency as high as 1.26 A cm−2, 8.75 mL min−1 cm−2 and ∼100%, respectively, at 1.6 V and 800 °C. Such high activity was attributed to the promoted adsorption of CO2 molecules on the surface of Co–Fe–STCF–SDC as revealed by CO2-TPD measurement. Furthermore, the Co–Fe–STCF–SDC electrode exhibited excellent stability with no carbon deposition after the 100 h test of CO2 electrolysis at 1.2 V at 800 °C. These results demonstrated cation exsolution as an effective approach to construct high performance electrode materials for high-temperature CO2 electrolysis. The methodology and finding in this work can be applied to other high-temperature energy and environmental devices, such as solar thermal water splitting and volatile organic compound (VOC) removal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFA1501900); the National Natural Science Foundation of China (91745203); the State Key Laboratory of Pulp and Paper Engineering (2020C01); the Guangdong Pearl River Talent Program (2017GC010281). The authors acknowledge Dongdong Chen's assistance for the CO2-TPD measurement.References
- S. Vasireddy, B. Morreale, A. Cugini, C. Song and J. J. Spivey, Energy Environ. Sci., 2011, 4, 311–345 RSC.
- K. Xie, Y. Q. Zhang, G. Y. Meng and J. T. S. Irvine, Energy Environ. Sci., 2011, 4, 2218–2222 RSC.
- Y. F. Song, X. M. Zhang, K. Xie, G. X. Wang and X. H. Bao, Adv. Mater., 2019, 31, 1902033 CrossRef CAS PubMed.
- Y. Zheng, J. C. Wang, B. Yu, W. Q. Zhang, J. Chen, J. L. Qiao and J. J. Zhang, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- S. W. Kim, M. Park, H. Kim, K. J. Yoon, J. W. Son, J. H. Lee, B. K. Kim, J. H. Lee and J. Hong, Appl. Catal., B, 2017, 200, 265–273 CrossRef CAS.
- X. L. Yue and J. T. S. Irvine, J. Electrochem. Soc., 2012, 159, F442–F448 CrossRef CAS.
- Z. L. Zhan and L. Zhao, J. Power Sources, 2010, 195, 7250–7254 CrossRef CAS.
- R. Knibbe, M. L. Traulsen, A. Hauch, S. D. Ebbesen and M. Mogensen, J. Electrochem. Soc., 2010, 157, B1209 CrossRef CAS.
- S. McIntosh and R. J. Gorte, Chem. Rev., 2004, 104, 4845–4866 CrossRef CAS PubMed.
- D. H. Dong, S. S. Xu, X. Shao, L. Hucker, J. Marin, T. Pham, K. Xie, Z. M. Ye, P. Yang, L. B. Yu, G. Parkinson and C. Z. Li, J. Mater. Chem. A, 2017, 5, 24098–24102 RSC.
- G. B. Chen, R. Gao, Y. F. Zhao, Z. H. Li, G. I. N. Waterhouse, R. Shi, J. Q. Zhao, M. T. Zhang, L. Shang, G. y. Sheng, X. P. Zhang, X. D. Wen, L. Z. Wu, C. H. Tung and T. R. Zhang, Adv. Mater., 2018, 30, 1704663 CrossRef PubMed.
- S. S. Xu, S. S. Li, W. T. Yao, D. H. Dong and K. Xie, J. Power Sources, 2013, 230, 115–121 CrossRef CAS.
- N. Mahato, A. Banerjee, A. Gupta, S. Omar and K. Balani, Prog. Mater. Sci., 2015, 72, 141–337 CrossRef CAS.
- E. Rosa Silva, M. Curi, J. G. Furtado, H. C. Ferraz and A. R. Secchi, Ceram. Int., 2019, 45, 9761–9770 CrossRef CAS.
- B. Bochentyn, J. Karczewski, T. Miruszewski, A. Krupa, M. Gazda, P. Jasinski and B. Kusz, Solid State Ionics, 2012, 225, 118–123 CrossRef CAS.
- M. Curi, E. D. Silva, J. G. D. Furtado, H. C. Ferraz and A. R. Secchi, Quim. Nova, 2021, 44, 86–97 CAS.
- T. L. Zhu, H. E. Troiani, L. V. Mogni, M. F. Han and S. A. Barnett, Joule, 2018, 2, 478–496 CrossRef CAS.
- X. Sun, H. J. Chen, Y. M. Yin, M. T. Curnan, J. W. Han, Y. Chen and Z. F. Ma, Small, 2021, 17, 2005383 CrossRef CAS PubMed.
- Y. F. Li, W. Q. Zhang, Y. Zheng, J. Chen, B. Yu, Y. Chen and M. L. Liu, Chem. Soc. Rev., 2017, 46, 6345–6378 RSC.
- J. T. S. Irvine, D. Neagu, M. C. Verbraeken, C. Chatzichristodoulou, C. Graves and M. B. Mogensen, Nat. Energy, 2016, 1, 15014 CrossRef CAS.
- C. H. Zhao, Y. F. Li, W. Q. Zhang, Y. Zheng, X. M. Lou, B. Yu, J. Chen, Y. Chen, M. L. Liu and J. C. Wang, Energy Environ. Sci., 2020, 13, 53–85 RSC.
- O. Kwon, S. Joo, S. Choi, S. Sengodan and G. Kim, J. Phys.: Energy, 2020, 2, 032001 CAS.
- J. W. Zhang, M. R. Gao and J. L. Luo, Chem. Mater., 2020, 32, 5424–5441 CrossRef CAS.
- J. H. Myung, D. Neagu, D. N. Miller and J. T. S. Irvine, Nature, 2016, 537, 528–531 CrossRef CAS PubMed.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Ménard and J. T. S. Irvine, Nat. Chem., 2013, 5, 916–923 CrossRef CAS PubMed.
- D. Neagu, T. S. Oh, D. N. Miller, H. Ménard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. S. Irvine, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- K. J. Kim, H. Han, T. Defferriere, D. Yoon, S. Na, S. J. Kim, A. M. Dayaghi, J. Son, T. S. Oh, H. M. Jang and G. M. Choi, J. Am. Chem. Soc., 2019, 141, 7509–7517 CrossRef CAS PubMed.
- V. Kyriakou, D. Neagu, E. I. Papaioannou, I. S. Metcalfe, M. C. M. van de Sanden and M. N. Tsampas, Appl. Catal., B, 2019, 258, 117950 CrossRef CAS.
- S. Park, Y. Kim, H. Han, Y. S. Chung, W. Yoon, J. Choi and W. B. Kim, Appl. Catal., B, 2019, 248, 147–156 CrossRef CAS.
- S. Park, Y. Kim, Y. Noh, T. Kim, H. Han, W. Yoon, J. Choi, S. H. Yi, W. J. Lee and W. B. Kim, J. Mater. Chem. A, 2020, 8, 138–148 RSC.
- S. S. Hou and K. Xie, Electrochim. Acta, 2019, 301, 63–68 CrossRef CAS.
- O. Kwon, S. Sengodan, K. Kim, G. Kim, H. Y. Jeong, J. Shin, Y. W. Ju, J. W. Han and G. Kim, Nat. Commun., 2017, 8, 15967 CrossRef CAS PubMed.
- H. F. Lv, L. Lin, X. M. Zhang, Y. F. Song, H. Matsumoto, C. B. Zeng, N. Ta, W. Liu, D. F. Gao, G. X. Wang and X. H. Bao, Adv. Mater., 2020, 32, 1906193 CrossRef CAS PubMed.
- Y. H. Li, B. B. Hu, C. R. Xia, W. Q. Xu, J. P. Lemmon and F. L. Chen, J. Mater. Chem. A, 2017, 5, 20833–20842 RSC.
- H. F. Lv, T. F. Liu, X. M. Zhang, Y. F. Song, H. Matsumoto, N. Ta, C. B. Zeng, G. X. Wang and X. H. Bao, Angew. Chem. Int. Ed., 2020, 59, 15968–15973 CrossRef CAS PubMed.
- L. Holzer, B. Iwanschitz, T. Hocker, B. Münch, M. Prestat, D. Wiedenmann, U. Vogt, P. Holtappels, J. Sfeir, A. Mai and T. Graule, J. Power Sources, 2011, 196, 1279–1294 CrossRef CAS.
- T. Klemensø, K. Thydén, M. Chen and H. J. Wang, J. Power Sources, 2010, 195, 7295–7301 CrossRef.
- H. S. Wei, K. Xie, J. Zhang, Y. Zhang, Y. Wang, Y. Q. Qin, J. W. Cui, J. Yan and Y. C. Wu, Sci. Rep., 2014, 4, 5156 CrossRef CAS PubMed.
- Y. X. Li, Y. Wang, W. Doherty, K. Xie and Y. C. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 8553–8562 CrossRef CAS PubMed.
- D. Neagu, V. Kyriakou, I. L. Roiban, M. Aouine, C. Tang, A. Caravaca, K. Kousi, I. Schreur Piet, I. S. Metcalfe, P. Vernoux, M. C. M. van de Sanden and M. N. Tsampas, ACS Nano, 2019, 13, 12996–13005 CrossRef CAS PubMed.
- S. K. Otto, K. Kousi, D. Neagu, L. Bekris, J. Janek and I. S. Metcalfe, ACS Appl. Energy Mater., 2019, 2, 7288–7298 CrossRef CAS.
- H. Tanaka, M. Uenishi, M. Taniguchi, I. Tan, K. Narita, M. Kimura, K. Kaneko, Y. Nishihata and J. i. Mizuki, Catal. Today, 2006, 117, 321–328 CrossRef CAS.
- Y. Gao, D. J. Chen, M. Saccoccio, Z. H. Lu and F. Ciucci, Nano Energy, 2016, 27, 499–508 CrossRef CAS.
- B. B. Niu, C. L. Lu, W. D. Yi, S. J. Luo, X. N. Li, X. W. Zhong, X. Z. Zhao and B. M. Xu, Appl. Catal., B, 2020, 270, 118842 CrossRef CAS.
- C. H. Yang, J. Li, Y. Lin, J. Liu, F. L. Chen and M. L. Liu, Nano Energy, 2015, 11, 704–710 CrossRef CAS.
- C. Chatzichristodoulou, B. C. Hauback and P. V. Hendriksen, J. Solid State Chem., 2013, 201, 164–171 CrossRef CAS.
- S. Park, H. Han, W. Yoon, J. Choi, Y. Kim, H. Kim and W. B. Kim, ACS Sustainable Chem. Eng., 2020, 8, 6564–6571 CrossRef CAS.
- S. B. Liu, Q. X. Liu and J. L. Luo, ACS Catal., 2016, 6, 6219–6228 CrossRef CAS.
- S. B. Liu, Q. X. Liu and J. L. Luo, J. Mater. Chem. A, 2016, 4, 17521–17528 RSC.
- D. J. Joyner, O. Johnson and D. M. Hercules, J. Am. Chem. Soc., 1980, 102, 1910–1917 CrossRef CAS.
- Y. M. Zhu, Z. Y. He, Y. M. Choi, H. J. Chen, X. B. Li, B. Zhao, Y. Yu, H. Zhang, K. A. Stoerzinger, Z. X. Feng, Y. Chen and M. L. Liu, Nat. Commun., 2020, 11, 4299 CrossRef PubMed.
- Y. M. Zhu, L. Zhang, B. Zhao, H. J. Chen, X. Liu, R. Zhao, X. W. Wang, J. Liu, Y. Chen and M. L. Liu, Adv. Funct. Mater., 2019, 29, 1901783 CrossRef.
- Y. Chen, D. D. Fong, F. W. Herbert, J. Rault, J. P. Rueff, N. Tsvetkov and B. Yildiz, Chem. Mater., 2018, 30, 3359–3371 CrossRef CAS.
- D. V. Ivanov, L. G. Pinaeva, L. A. Isupova, E. M. Sadovskaya, I. P. Prosvirin, E. Y. Gerasimov and I. S. Yakovleva, Appl. Catal., A, 2013, 457, 42–51 CrossRef CAS.
- W. Y. Hernández, M. N. Tsampas, C. Zhao, A. Boreave, F. Bosselet and P. Vernoux, Catal. Today, 2015, 258, 525–534 CrossRef.
- G. R. Yang, L. Wang, S. J. Peng, J. N. Wang, D. X. Ji, W. Yan and S. Ramakrishna, Small, 2017, 13, 1702357 CrossRef PubMed.
- Y. X. Li, K. Xie, S. G. Chen, H. X. Li, Y. Zhang and Y. C. Wu, Electrochim. Acta, 2015, 153, 325–333 CrossRef CAS.
- H. F. Lv, L. Lin, X. M. Zhang, D. F. Gao, Y. F. Song, Y. J. Zhou, Q. X. Liu, G. X. Wang and X. H. Bao, J. Mater. Chem. A, 2019, 7, 11967–11975 RSC.
- J. W. Yin, Y. M. Yin, J. Lu, C. M. Zhang, N. Q. Minh and Z. F. Ma, J. Phys. Chem. C, 2014, 118, 13357–13368 CrossRef CAS.
- Q. Xu, D. P. Huang, W. Chen, H. Wang, B. T. Wang and R. Z. Yuan, Appl. Surf. Sci., 2004, 228, 110–114 CrossRef CAS.
- N. A. Merino, B. P. Barbero, P. Eloy and L. E. Cadús, Appl. Surf. Sci., 2006, 253, 1489–1493 CrossRef CAS.
- M. J. Zhi, G. W. Zhou, Z. L. Hong, J. Wang, R. Gemmen, K. Gerdes, A. Manivannan, D. L. Ma and N. Q. Wu, Energy Environ. Sci., 2011, 4, 139–144 RSC.
- S. S. Xu, D. H. Dong, Y. Wang, W. Doherty, K. Xie and Y. C. Wu, J. Power Sources, 2014, 246, 346–355 CrossRef CAS.
- M. Ni, Chem. Eng. J., 2010, 164, 246–254 CrossRef CAS.
- W. T. Qi, Y. Gan, D. Yin, Z. Y. Li, G. J. Wu, K. Xie and Y. C. Wu, J. Mater. Chem. A, 2014, 2, 6904–6915 RSC.
- Z. Zhao, L. Liu, X. M. Zhang, W. M. Wu, B. F. Tu, D. A. Cui, D. R. Ou and M. J. Cheng, Int. J. Hydrogen Energy, 2013, 38, 15361–15370 CrossRef CAS.
- Y. Gao, Z. H. Lu, T. L. You, J. Wang, L. Xie, J. Q. He and F. Ciucci, J. Phys. Chem. C, 2018, 9, 3772–3778 CAS.
- A. S. Sandupatla, A. Banerjee and G. Deo, Appl. Surf. Sci., 2019, 485, 441–449 CrossRef CAS.
- X. Z. Zhang, L. T. Ye, J. P. Hu, J. Li, W. H. Jiang, C. J. Tseng and K. Xie, Electrochim. Acta, 2016, 212, 32–40 CrossRef CAS.
- C. L. Zhu, L. X. Hou, S. S. Li, L. Z. Gan and K. Xie, J. Power Sources, 2017, 363, 177–184 CrossRef CAS.
- X. Z. Peng, Y. F. Tian, Y. Liu, W. J. Wang, L. C. Jia, J. Pu, B. Chi and J. Li, J. CO2 Util., 2020, 36, 18–24 CrossRef CAS.
- Y. F. Tian, H. Y. Zheng, L. L. Zhang, B. Chi, J. Pu and J. Li, J. Electrochem. Soc., 2018, 165, F17–F23 CrossRef CAS.
- Y. J. Zhou, Z. W. Zhou, Y. F. Song, X. M. Zhang, F. Guan, H. F. Lv, Q. X. Liu, S. Miao, G. X. Wang and X. H. Bao, Nano Energy, 2018, 50, 43–51 CrossRef CAS.
- C. Ruan and K. Xie, Catal. Sci. Technol., 2015, 5, 1929–1940 RSC.
- S. Q. Hu, L. X. Zhang, H. Y. Liu, W. P. Li, Z. W. Cao, L. L. Cai, Y. Zhu, X. F. Zhu and W. S. Yang, J. Energy Chem., 2019, 36, 87–94 CrossRef.
- L. Z. Gan, L. T. Ye, S. W. Tao and K. Xie, Phys. Chem. Chem. Phys., 2016, 18, 3137–3143 RSC.
- J. Zhang, K. Xie, Y. Zhang, L. M. Yang, G. J. Wu, Q. Q. Qin, Y. X. Li and Y. C. Wu, RSC Adv., 2014, 4, 22697–22709 RSC.
- Y. F. Tian, L. L. Zhang, L. C. Jia, X. Wang, J. Yang, B. Chi, J. Pu and J. Li, J. CO2 Util., 2019, 31, 43–50 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta07251j |
| This journal is © The Royal Society of Chemistry 2022 |