DOI:
10.1039/D2SC04727F
(Edge Article)
Chem. Sci., 2022,
13, 13403-13408
Total synthesis of the antibacterial polyketide natural product thailandamide lactone†
Received
24th August 2022
, Accepted 20th October 2022
First published on 21st October 2022
Abstract
Stereoselective total synthesis of the structurally intriguing polyketide natural product thailandamide lactone was accomplished, and done so using a convergent approach for the first time to the best of our knowledge. The key features of this synthesis included use of a Crimmins acetate aldol reaction, Evans methylation, Urpi acetal aldol reaction, Sharpless asymmetric epoxidation and subsequent γ-lactonization for the installation of six asymmetric centers and the use of the Negishi reaction, Julia-Kocienski olefination, cross metathesis, HWE olefination and intermolecular Heck coupling for construction of a variety of unsaturated linkages. Pd(I)-based Heck coupling was introduced, for the first time to the best of our knowledge, quite efficiently to couple the major eastern and sensitive western segments of the molecule. The antibacterial activity of thailandamide lactone was also evaluated.
Introduction
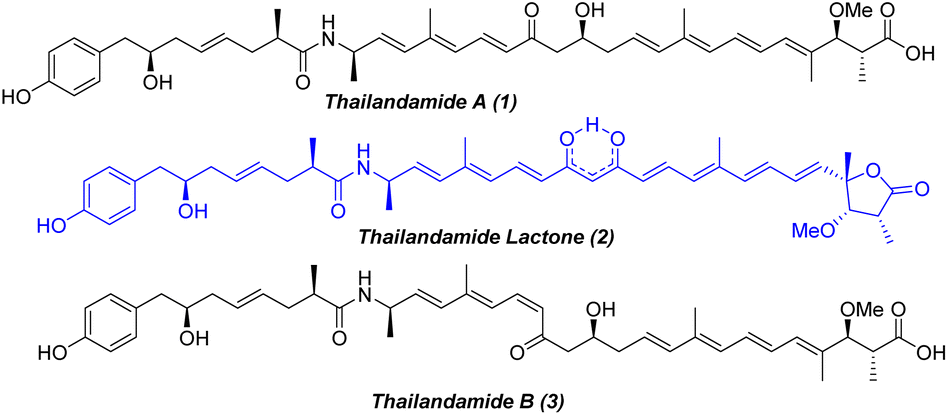
During the mining of the genome of Burkholderia thailandensis, a bacterium isolated from rice paddies in central and northeastern Thailand, Hertweck and co-workers1 in 2008 first observed the labile polyene polyketide thailandamide A (1, Fig. 1), albeit in minute quantities. To better understand thailandamide biosynthesis, the silent tha PKS-NRPS gene cluster of B. thailandensis was activated by a research group in 2010 through manipulation of a quorum sensing (QS) regulatory system that produced a mutant with a dramatically altered metabolic profile.2 This process resulted in the isolation of the structurally challenging new polyketide thailandamide lactone (2, Fig. 1), which was not detected in wild type broth initially. Moreover, the production of thailandamide A (1) was significantly greater here than when the wild type was used.2 Later, the same group developed an elegant biosynthetic route yielding the first total synthesis of thailandamide A; this development enabled them to find another unstable metabolite, namely thailandamide B (3, Fig. 1), a geometrical isomer of thailandamide A.3 Broad biological screening of thailandamide A revealed its selective and potential inhibitory activity against various pathogenic Gram-positive and Gram-negative bacteria with a specific mode of action.4 However, the antibacterial activity of thailandamide lactone and thailandamide B remained undisclosed. The highly challenging architectural features and natural scarcity of thailandamide lactone and lack of a synthetic route to this lactone—together with our continual interest in natural products chemistry5—encouraged us to seek out its total synthesis. Structurally, thailandamide lactone2 is a linear polyene polyketide where a tetraene conjugated with a γ-butyrolactone is fused with a conjugated triene through an enolized dione moiety. It consists of six asymmetric centers including a quaternary center at one terminus of the molecule and a phenolic moiety at the other terminus. Herein, we report a convergent and highly modular route for the first total synthesis of thailandamide lactone and report its antibacterial activity against various pathogenic and non-pathogenic bacterial strains.
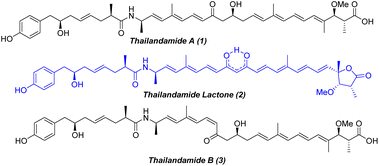 |
| | Fig. 1 Chemical structures of thailandamide family natural products. | |
Results and discussion
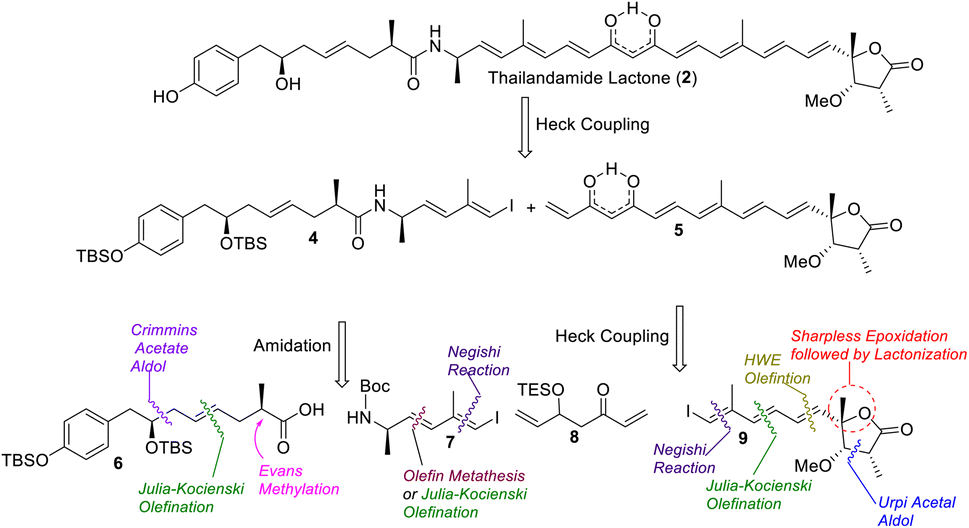
A retrosynthetic analysis of thailandamide lactone (2) is shown in Scheme 1. We envisioned that the target molecule could be constructed from vinyl iodide 4 and 1,3-dione-containing polyene 5 adopting intermolecular Heck coupling as the key step. Vinyl iodide 4 could further be made from compounds 6 and 7 by performing amide coupling. Compound 6 could be assembled using the Crimmins acetate aldol reaction, Julia-Kocienski olefination and Evans methylation as the key steps, whereas compound 7 could be accessed utilizing cross olefin metathesis or Julia-Kocienski olefination and the Negishi reaction as the pivotal steps. On the other hand, keto-alkene 5 could be prepared from intermediates 8 and 9 using Heck coupling, whereas compound 9 could be synthesized using the Negishi reaction, Julia-Kocienski olefination, HWE olefination, Urpi acetal aldol reaction, Sharpless asymmetric epoxidation and subsequent γ-lactonization as the salient steps.
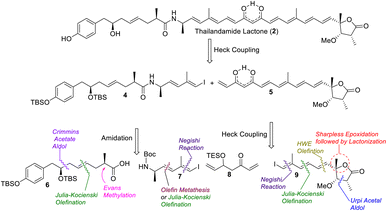 |
| | Scheme 1 Retrosynthetic analysis of thailandamide lactone (2). | |
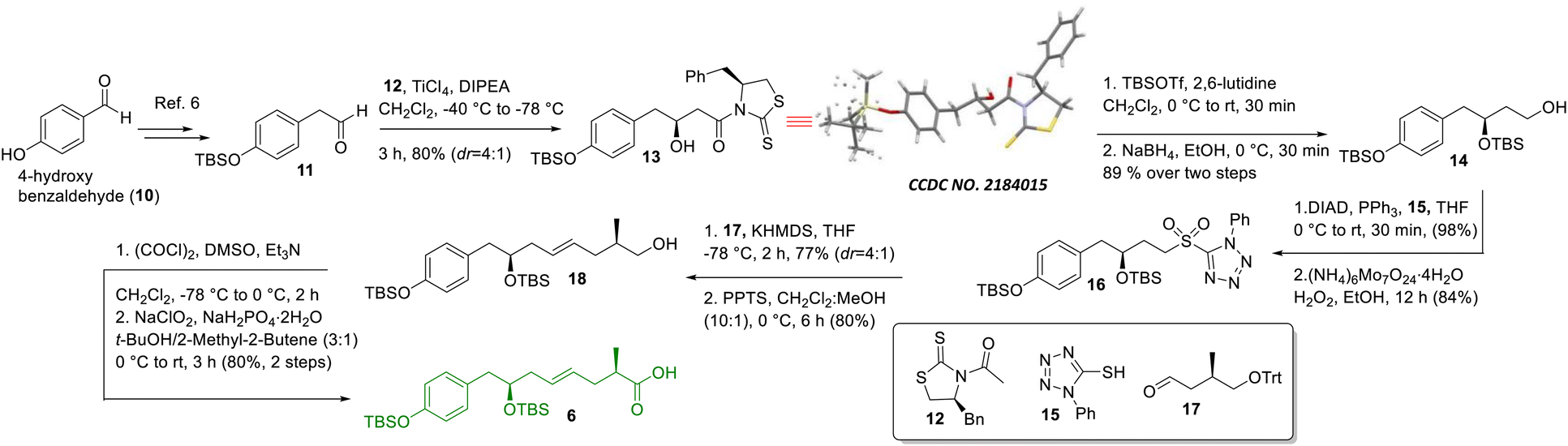
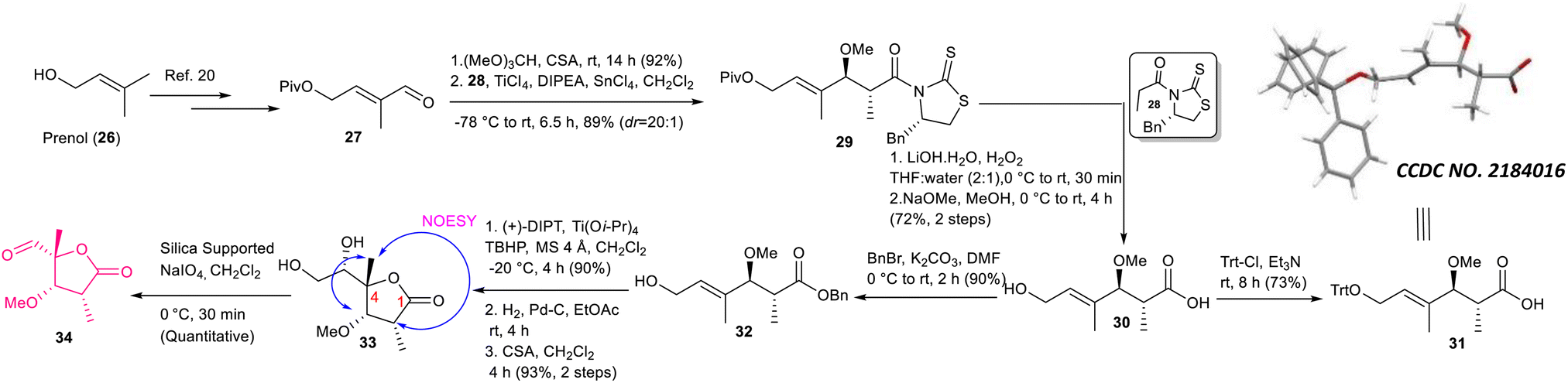
The synthesis of intermediate 6 was commenced with the known compound 11 (Scheme 2) prepared from commercially available 4-hydroxy benzaldehyde (10) following a literature procedure;6 compound 11 was subjected to a Crimmins acetate aldol reaction7 using the known auxiliary 127b in the presence of TiCl4/DIPEA to obtain compound 13 as the major product in 80% yield along with its minor counterpart (dr = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The major aldol product was separated from other components using silica gel column chromatography and its structure was confirmed unambiguously using X-ray crystallographic analysis. Next, compound 13 was treated with TBSOTf/2,6-lutidine followed by NaBH4 to access compound 14, which was subjected to the Mitsunobu reaction8 using 1-phenyl-5-thiotetrazole (15) in the presence of DIAD/PPh3 and oxidized further using (NH4)6Mo7O24·4H2O/H2O2 in ethanol5c to achieve sulfone 16 in very good overall yield. Next, sulfone 16 was subjected to Julia-Kocienski olefination5c,9 with the known aldehyde 1710using KHMDS to obtain the corresponding E-coupled isomer as the major product along with its minor Z-isomer (dr = 4:1). The purified major isomer was subsequently treated with PPTS to obtain alcohol 18, which finally was oxidized to acid 6 using Swern oxidation followed by Pinnick oxidation.11
:1). The major aldol product was separated from other components using silica gel column chromatography and its structure was confirmed unambiguously using X-ray crystallographic analysis. Next, compound 13 was treated with TBSOTf/2,6-lutidine followed by NaBH4 to access compound 14, which was subjected to the Mitsunobu reaction8 using 1-phenyl-5-thiotetrazole (15) in the presence of DIAD/PPh3 and oxidized further using (NH4)6Mo7O24·4H2O/H2O2 in ethanol5c to achieve sulfone 16 in very good overall yield. Next, sulfone 16 was subjected to Julia-Kocienski olefination5c,9 with the known aldehyde 1710using KHMDS to obtain the corresponding E-coupled isomer as the major product along with its minor Z-isomer (dr = 4:1). The purified major isomer was subsequently treated with PPTS to obtain alcohol 18, which finally was oxidized to acid 6 using Swern oxidation followed by Pinnick oxidation.11
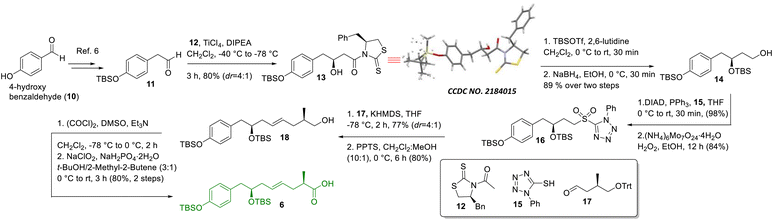 |
| | Scheme 2 Synthesis of intermediate 6. | |
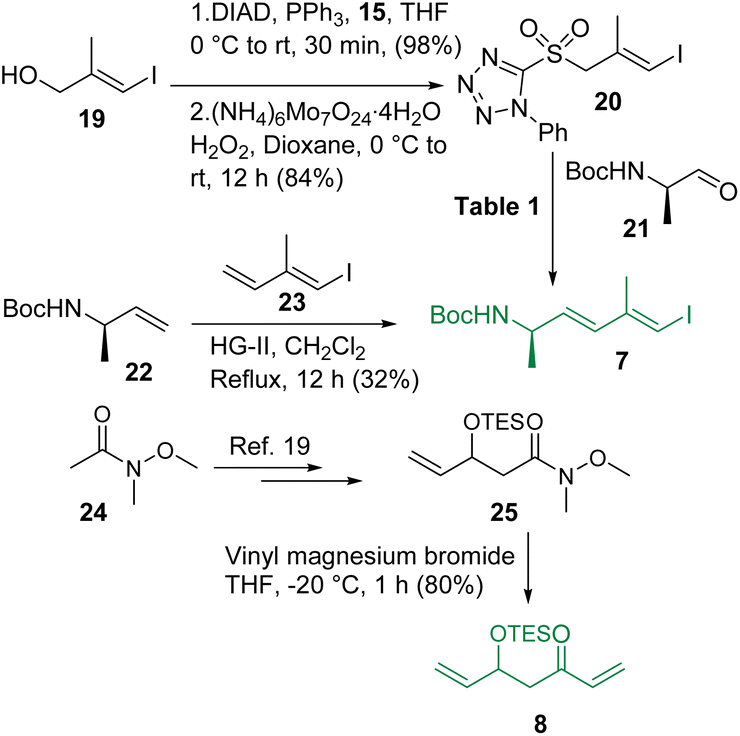
The synthesis of intermediates 7 and 8 is depicted in Scheme 3. The known vinyl iodide 19,12 prepared from propargylic alcohol using the Negishi reaction as the key step, was subjected to the Mitsunobu reaction using 1-phenyl-5-thiotetrazole (15) and the resultant sulfide was oxidized using (NH4)6Mo7O24·4H2O/H2O2 in dioxane13 to access sulfone 20. Notably, the production of sulfone 20 from its corresponding sulfide was found to be much more efficient in dioxane than in the commonly used ethanol. Sulfone 20 was then reacted with the known aldehyde 2114 following the Julia-Kocienski olefination protocol.9,15 Several conditions were screened for synthesizing compound 7 (Table 1) and the use of KHMDS in DME (entry-4) was found to be the best (E/Z = 3:1). In parallel, the cross metathesis16 between the known alkenes 2217 and 2318 was also investigated and it was observed that HG-II produced compound 7 in 32% yield with much better selectivity (E:Z = 10:1) compared to Julia-Kocienski olefination whereas G-II and HG-I functioned ineffectively, leaving a trace amount of the desired product. However, the geometrical isomers remained inseparable at this stage. On the other hand, commercially available Weinreb amide 24 was transformed to the known compound 25 following a literature procedure19 and subjected further to a reaction with vinyl magnesium bromide to access intermediate 8 in very good overall yield.
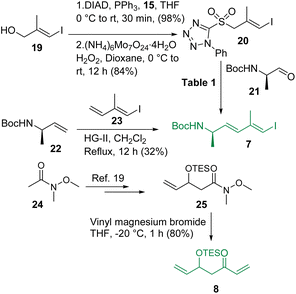 |
| | Scheme 3 Synthesis of intermediates 7 and 8. | |
Table 1 Efforts to optimize Julia-Kocienki olefination
| Entry |
Conditions |
Yield (E:Z) |
| 1 |
NaHMDS, THF, −78 °C |
66% (1:1.2) |
| 2 |
KHMDS, THF, −78 °C |
70% (1:1) |
| 3 |
LiHMDS, DME, −60 °C |
62% (1.5:1) |
| 4 |
KHMDS, DME, −60 °C |
68% (3:1) |
The synthesis of aldehyde 34 is described in Scheme 4. The known aldehyde 27 prepared from prenol (26) following a literature method20 was converted to the corresponding acetal using (MeO)3CH/CSA, which was subjected further to the Urpi acetal aldol reaction5b,21 in the presence of TiCl4/DIPEA/SnCl4 to access compound 29 with excellent selectivity (dr = 20:1). The purified compound was then treated with LiOH·H2O/H2O2 followed by NaOMe/MeOH to obtain compound 30 in 72% yield. The stereochemistry of asymmetric centers newly generated using Urpi acetal aldol reaction was confirmed further from an X-ray crystallographic analysis of compound 31, which was synthesized from compound 30 by performing tritylation. Next, compound 30 was reacted with BnBr/K2CO3 to obtain benzyl ester 32, which was subjected to Sharpless asymmetric epoxidation22 followed by hydrogenation to produce the corresponding epoxy acid. The stage was set for γ-lactonization.23 The corresponding epoxy acid was treated with CSA/CH2Cl2 to access the 5-exo cyclized product 33 exclusively. The characteristic NOESY correlation of C4–Me with C2–H and C3–H confirmed its structure unambiguously. We did not observe the formation of any other possible γ-lactone originating via 6-endo cyclization followed by concomitant acyl migration.23 Our exhaustive efforts for achieving oxidative cleavage of the diol moiety of compound 33 using either NaIO4 or NaIO4/NaHCO3 did not produce aldehyde 34 in an isolable yield due to its rapid decomposition. Delightfully, silica-supported NaIO424 provided considerable relief here, where the required aldehyde was obtained quantitatively.
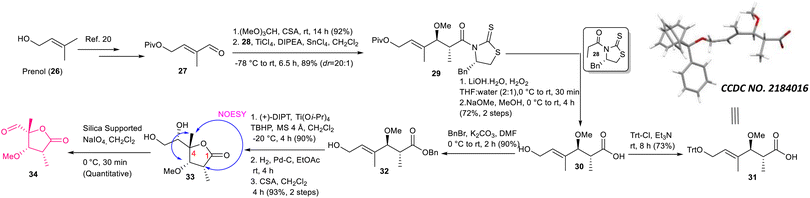 |
| | Scheme 4 Synthesis of aldehyde 34. | |
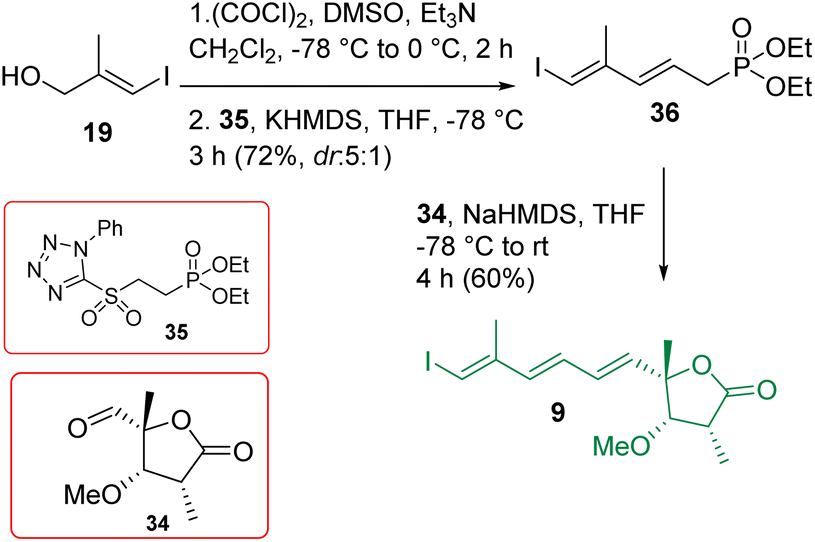
The construction of compound 9 is shown in Scheme 5. Alcohol 19 was oxidized using the Swern condition and subjected to Julia-Kocienski olefination9 with the known sulfone 3525 to access compound 36 as a major product (dr = 5:1). The purified major isomer was reacted further with aldehyde 34 in the presence of NaHMDS/THF following the HWE olefination protocol25 to achieve intermediate 9 exclusively. The initially encountered isomerization problem with the α-methyl center was overcome by performing a controlled addition of NaHMDS and also by reducing the reaction time (see the optimization in Table S1 in ESI†).
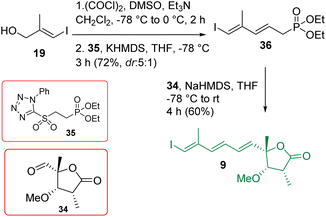 |
| | Scheme 5 Synthesis of intermediate 9. | |
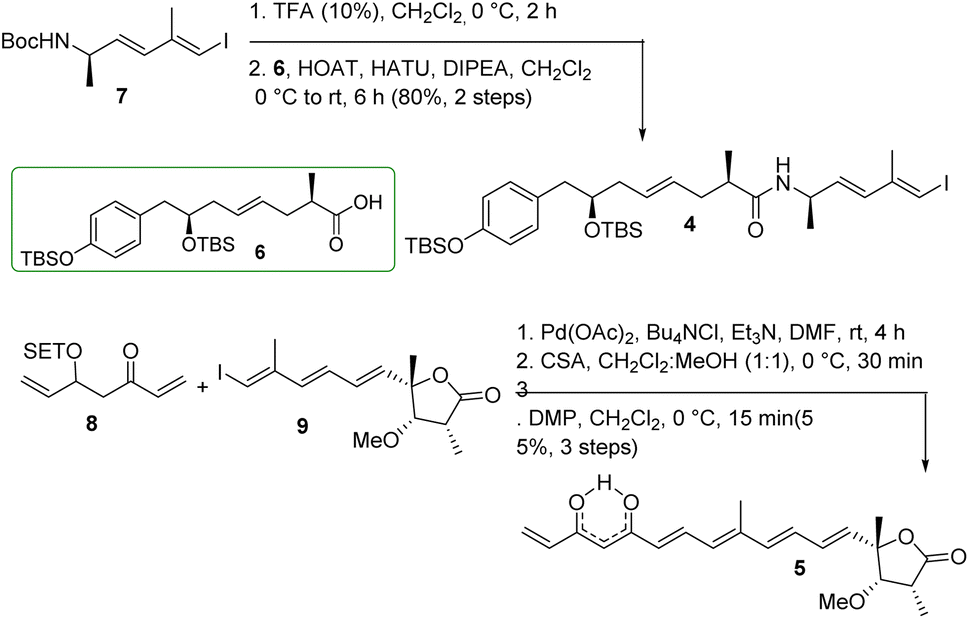
The synthesis of major coupling partners (4 and 5) of thailandamide lactone is described in Scheme 6. Compound 7 was treated with 10% TFA/CH2Cl2 and the resultant Boc-deprotected amine was coupled with acid 6 to access the western segment 4 in 80% yield (over two steps). On the other hand, vinyl ketone 8 was subjected to intermolecular Heck coupling with compound 9 in the presence of Pd(OAc)2/Bu4NCl/Et3N in DMF26 to obtain the corresponding coupled product in complete regioselectivity, and this coupled species was further treated with CSA to obtain the corresponding β-hydroxy ketone in 77% yield after two steps. Substantial trials have been conducted to optimize its conversion to the eastern segment 5. Most of the oxidizing agents including DMP/NaHCO3 did not function properly as their use ended up with complete decomposition of the product. However, DMP without NaHCO3 produced the required product 5 in 73% yield. The appearance of a signal at a δ of 15.6 ppm in the 1H NMR spectrum of compound 5 and two carbonyl carbons at δ 183.0, 183.9 ppm in its 13C NMR spectrum clearly ascertained its existence as keto–enol tautomeric mixtures.
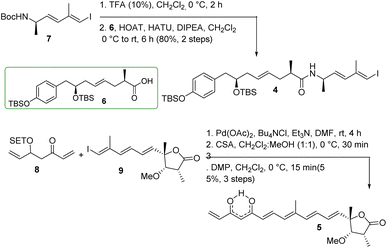 |
| | Scheme 6 Synthesis of coupling partners 4 and 5. | |
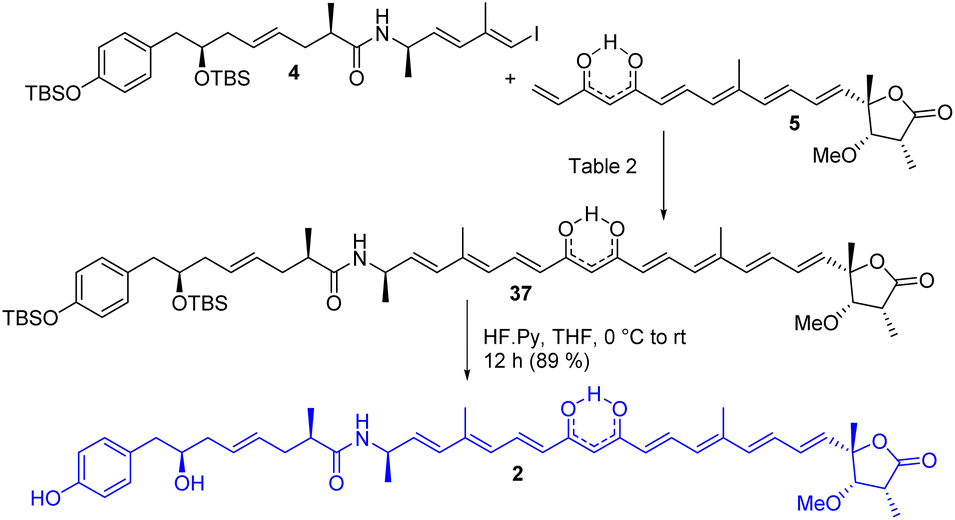
The completion of the total synthesis of thailandamide lactone is depicted in Scheme 7 where the stage was set for the crucial coupling between the western (4) and eastern (5) segments. Extensive efforts were made to optimize the Heck coupling (Table 2). Trials with PdCl2(MeCN)2/Et3N/HCO2H in MeCN (entry-1)27 ended up with complete decomposition of staring materials, whereas those with Pd(PPh3)4/Et3N/Bu4NCl in DMF (entry-2)27 provided the coupled product 37 in a trace amount. Use of Pd(PPh3)2Cl2/K2CO3/Bu4NCl in DMF (entry-3), Pd(OAc)2/Et3N/Bu4NCl in DMF (entry-4)27 and Pd(OAc)2/K3PO4 in DMF (entry-5)27c resulted in the required product in 10%, 45% and 40% yields, respectively. A mixture of some unidentified compounds was formed along with the required compound 37 in most of the cases. Having moderate success in the transformation of compound 37 using either Pd(0) or Pd(II), we then turned our attention towards Pd(I)-catalyzed Heck coupling as it has provided excellent results in some cases.28 Thus, [Pd(μ-I)(Pt-Bu3)]2 (entry-6),28b prepared from PdI2/PtBu3 following a literature report,28a was then screened in the presence of DIPEA/toluene to furnish the coupled product in an improved yield (58%). Later, an equimolar mixture of Pd(OAc)2 and Pd(PPh3)4 in the presence of K3PO4/DMF (entry-7)28e was tested. Delightfully, this reaction was found to proceed in a considerably cleaner manner than those with all the other tested conditions, and the coupled product 37 was obtained in 77% yield. All the reactions were performed at room temperature to reduce the rate of decomposition. A detailed NMR study unambiguously confirmed the identity of compound 37 (see the 2D spectra in ESI†). Notably the attempted synthesis of the corresponding compound requisite for an alternative Heck coupling with compound 9 was not successful—because the corresponding β-hydroxy ketone obtained from the Heck coupling between compounds 4 and 8 followed by subsequent TES ether deprotection was found to be very sensitive to various oxidizing agents including DMP. Next, compound 37 was subjected to global deprotection using HF Py to access compound 229 in 89% yield. 1H and 13C NMR data (see comparison Table S2 in ESI†), optical rotation results {observed [α]D28 = −43.20 (c 0.24, methanol); reported [α]D = −45.76}, and HRMS, FT-IR and UV-visible spectra (see ESI†) of synthesized compound 2 were found to be in good agreement with reported data of the isolated thailandamide lactone, which unambiguously confirmed its first total synthesis.
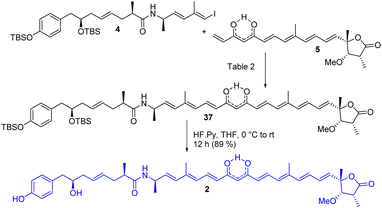 |
| | Scheme 7 Completion of the synthesis of thailandamide lactone (2). | |
Table 2 Optimization of final Heck coupling
| Entry |
[Pd] (mol%) |
Condition |
Yield (%) |
| 1 |
PdCl2(MeCN)2 (10) |
Et3N, HCO2H, MeCN, rt, 3 h |
Decomposition |
| 2 |
Pd(PPh3)4 (5) |
Et3N, Bu4NCl, DMF, rt, 6 h |
Trace |
| 3 |
PdCl2(PPh3)2 (10) |
K2CO3, Bu4NCl, DMF, rt, 12 h |
10 |
| 4 |
Pd(Oac)2 (5) |
Et3N, Bu4NCl, DMF, rt, 4 h |
45′ |
| 5 |
Pd(Oac)2 (5) |
K3PO4, DMF, rt, 12 h |
40 |
| 6 |
[Pd(μ-I)(PtBu3)]2 (7.5) |
DIPEA, toluene, rt, 9 h |
58 |
| 7 |
Pd(Oac)2, (10) Pd(PPh3)4 (10) |
K3PO4, DMF, rt, 18 h |
77 |
Having thailandamide lactone in hand, we then screened its antibacterial activity against different non-pathogenic and pathogenic Gram-positive bacteria such as Bacillus subtilis (PY79), Bacillus megaterium (2G), Staphylococcus aureus as well as Gram-negative bacteria such as Vibrio cholerae (N16961), Enteropathogenic Escherichia coli (EPEC e2348/69), and Escherichia coli (MC1061)]. This screening revealed its moderate to potent antibacterial activity (Table 3). The efficacies of thailandamide lactone even against Gram-negative strains were found to be promising.
Table 3 Antibacterial activities of thailandamide lactone
| Staining type |
Strains |
MIC (μg ml−1) |
| Gram negative |
V. cholerae (N16961) (pathogenic) |
71.3 |
| EPEC (e2348/69) (pathogenic) |
71.3 |
|
E. coli (MC1061) |
53.5 |
| Gram positive |
B. subtilis (PY79) |
57.0 |
|
B. megaterium (2G) |
53.5 |
Conclusions
In summary, we have developed a convergent route for the first total synthesis of a structurally challenging and labile polyketide natural product, namely thailandamide lactone, starting from the known compound 26 in 17 LLS with 8.5% overall yield. Our synthesis includes several coupling operations including two intermolecular Heck reactions. Notably, Pd(I)-based Heck coupling has been introduced for the first time, to the best of our knowledge, in the total synthesis of natural product. The possible site to couple the highly sensitive eastern and western polyene segments of thailandamide lactone was determined. The antibacterial activities of thailandamide lactone against different bacterial strains have been disclosed. Importantly, our modular strategy is expected to be amenable to thailandamide A, another member of this family as well as to structurally simplified designed analogues for further antibacterial study.
Data availability
Please see the ESI† for the data related to the manuscript.
Author contributions
R. K. G. conceived the idea, designed the hypothesis and managed overall manuscript preparation. R. R. P. planned and supervised the biological testing and assisted with manuscript preparation. H. S. and J. M. completed the total synthesis, A. K. G. was responsible for biological testing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
H. S., J. M. and A. K. G. thank the Council of Scientific and Industrial Research (CSIR), New Delhi and the Indian Association for the Cultivation of Science, respectively, for their research fellowship. The financial support from the Science and Engineering Research Board (Project no-CRG/2019/001664) of India to carry out this work is gratefully acknowledged.
Notes and references
- T. Nguyen, K. Ishida, H. Jenke-Kodama, E. Dittmann, C. Gurgui, T. Hochmuth, S. Taudien, M. Platzer, C. Hertweck and J. Piel, Nat. Biotechnol., 2008, 26, 225–233 CrossRef CAS PubMed.
- K. Ishida, T. Lincke, S. Behnken and C. Hertweck, J. Am. Chem. Soc., 2010, 132, 13966–13968 CrossRef CAS PubMed.
- K. Ishida, T. Lincke and C. Hertweck, Angew. Chem., Int. Ed., 2012, 51, 5470–5474 CrossRef CAS PubMed.
- Y. Wu and M. R. Seyedsayamdost, Biochemistry, 2018, 57, 4247–4251 CrossRef CAS PubMed.
-
(a) S. Saha, D. Paul and R. K. Goswami, Chem. Sci., 2020, 11, 11259–11265 RSC;
(b) T. K. Kuilya and R. K. Goswami, Org. Lett., 2017, 19, 2366–2369 CrossRef CAS PubMed;
(c) S. Das, D. Paul and R. K. Goswami, Org. Lett., 2016, 18, 1908–1911 CrossRef CAS PubMed;
(d) S. Guchhait, S. Chatterjee, R. S. Ampapathi and R. K. Goswami, J. Org. Chem., 2017, 82, 2414–2435 CrossRef CAS PubMed;
(e) D. Saha, G. H. Mandal and R. K. Goswami, J. Org. Chem., 2021, 86, 10006–10022 CrossRef CAS PubMed.
- A. Ruiz-Olalla, M. A. Wurdemann, M. J. Wanner, S. Ingemann, J. H. van Maarseveen and H. Hiemstra, J. Org. Chem., 2015, 80, 5125–5132 CrossRef CAS PubMed.
-
(a) M. T. Crimmins, B. W. King, E. A. Tabet and K. Chaudhary, J. Org. Chem., 2001, 66, 894–902 CrossRef CAS PubMed;
(b) M. T. Crimmins, Org. Lett., 2007, 9, 149–152 CrossRef CAS PubMed.
- O. Mitsunobu, Synthesis, 1981, 1, 1–28 CrossRef.
- P. Liu and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 10772–10773 CrossRef CAS PubMed.
- A.-F. Salit, C. Meyer and J. Cossy, Synlett, 2007, 6, 934–938 Search PubMed.
- P. Li, J. Li, F. Arikan, W. Ahlbrecht, M. Dieckmann and D. Menche, J. Am. Chem. Soc., 2009, 131, 11678–11679 CrossRef CAS PubMed.
- C. L. Rand, D. E. Van Horn, M. W. Moore and E. Negishi, J. Org. Chem., 1981, 46, 4093–4096 CrossRef CAS.
- T. P. Ahern, H. O. Fong, R. F. Langler and P. M. Mason, Can. J. Chem., 1980, 58, 878–883 CrossRef CAS.
- H. Yao and K. Yamamoto, Chem.–Asian J., 2012, 7, 1542–1545 CrossRef CAS PubMed.
- T. J. Harrison, S. Ho and J. L. Leighton, J. Am. Chem. Soc., 2011, 133, 7308–7311 CrossRef CAS PubMed.
-
(a) D. Saha, S. Guchhait and R. K. Goswami, Org. Lett., 2020, 22, 745–749 CrossRef CAS PubMed;
(b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed;
(c) S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS;
(d) Y. Zhang, M. Dlugosch, M. Jubermann, M. G. Banwell and S. J. Ward, J. Org. Chem., 2015, 80, 4828–4833 CrossRef CAS PubMed.
- J. Ryan, M. Saiuciulis, A. Gomm, B. Maciá, E. O'Reilly and V. Caprio, J. Am. Chem. Soc., 2016, 138, 15798–15800 CrossRef CAS PubMed.
- S. Ma and E.-i. Negishi, J. Org. Chem., 1997, 62, 784–785 CrossRef CAS.
- J. L. Tyler, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2021, 60, 11824–11829 CrossRef CAS PubMed.
- J. A. González-Delgado, J. F. Arteaga, M. M. Herrador and A. F. Barrero, Org. Biomol. Chem., 2013, 11, 5404–5408 RSC.
- A. Cosp, P. Romea, P. Talavera, F. Urpí, J. Vilarrasa, M. Font-Bardia and X. Solans, Org. Lett., 2001, 3, 615–617 CrossRef CAS PubMed.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS.
- J. S. Clark, C. A. Baxter, A. G. Dossetter, S. Poigny, J. L. Castro and W. G. Whittingham, J. Org. Chem., 2008, 73, 1040–1055 CrossRef CAS PubMed.
- Y. L. Zhong and T. K. M. Shing, J. Org. Chem., 1997, 62, 2622–2624 CrossRef CAS PubMed.
- N. Cichowicz and P. Nagorny, Org. Lett., 2012, 14, 1058–1061 CrossRef CAS PubMed.
- R. F. Heck, Org. React., 1982, 27, 345–390 CrossRef CAS.
-
(a) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed;
(b) A. de Meijere and F. E. Meyer, Angew. Chem., Int. Ed. Engl., 1994, 33, 2379–2411 CrossRef;
(c) Q. Yao, E. P. Kinney and Z. Yang, J. Org. Chem., 2003, 68, 7528–7531 CrossRef CAS PubMed.
-
(a) R. Vilar, D. M. P. Mingos and C. J. Cardin, J. Chem. Soc., Dalton Trans., 1996, 23, 4313–4314 RSC;
(b) F. Schoenebeck, T. Sperger and C. Stirner, Synthesis, 2016, 49, 115–120 CrossRef;
(c) M. Aufiero, T. Sperger, A. S. Tsang and F. Schoenebeck, Angew. Chem., Int. Ed., 2015, 54, 10322–10326 CrossRef CAS PubMed;
(d) F. Proutiere, M. Aufiero and F. Schoenebeck, J. Am. Chem. Soc., 2012, 134, 606–612 CrossRef CAS PubMed;
(e) G. Magnin, J. Clifton and F. Schoenebeck, Angew. Chem., Int. Ed., 2019, 58, 10179–10183 CrossRef CAS PubMed;
(f) C. Fricke, T. Sperger, M. Mendel and F. Schoenebeck, Angew. Chem., Int. Ed., 2021, 60, 3355–3366 CrossRef CAS PubMed.
- Compound 2 was found highly sensitive to light as well as quite unstable. It decomposed to a mixture of unidentified compounds. The colour changed from yellow to dark red. It was observed that use of Silica gel for purification and glass vessel for storing was not suitable as it underwent faster decomposition. Notably, the major part of compound stuck to reverse phase C18 column (Xbridge RP18) during purification..
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Rajib Kumar
Goswami
b and
Rajib Kumar
Goswami