
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMapping paratopes of nanobodies using native mass spectrometry and ultraviolet photodissociation†
Luis A.
Macias
 a,
Xun
Wang
b,
Bryan W.
Davies
b and
Jennifer S.
Brodbelt
*a
a,
Xun
Wang
b,
Bryan W.
Davies
b and
Jennifer S.
Brodbelt
*a
aDepartment of Chemistry, University of Texas at Austin, Austin, TX 78712, USA. E-mail: jbrodbelt@cm.utexas.edu
bDepartment of Molecular Biosciences, University of Texas at Austin, Austin, TX 78712, USA
First published on 16th May 2022
Abstract
Following immense growth and maturity of the field in the past decade, native mass spectrometry has garnered widespread adoption for the structural characterization of macromolecular complexes. Routine analysis of biotherapeutics by this technique has become commonplace to assist in the development and quality control of immunoglobulin antibodies. Concurrently, 193 nm ultraviolet photodissociation (UVPD) has been developed as a structurally sensitive ion activation technique capable of interrogating protein conformational changes. Here, UVPD was applied to probe the paratopes of nanobodies, a class of single-domain antibodies with an expansive set of applications spanning affinity reagents, molecular imaging, and biotherapeutics. Comparing UVPD sequence fragments for the free nanobodies versus nanobody·antigen complexes empowered assignment of nanobody paratopes and intermolecular salt-bridges, elevating the capabilities of UVPD as a new strategy for characterization of nanobodies.
Antibodies must possess high specificity toward target antigens to enable recognition and activation of an immune response. Because of this specificity, antibodies or antibody fragments are being increasingly explored for deployment in diagnostic assays, vaccine design and other therapeutic applications.1 As such, unravelling the collection of noncovalent interactions and structural features that endow antibodies with specificity is a primary goal in biomedical research and critical for development of new biotherapeutics.2 Methods to decipher antibody–antigen interactions have advanced significantly in recent years, and well-established methods including X-ray crystallography, mutagenesis techniques, cryo-EM, and hydrogen–deuterium exchange mass spectrometry remain the most versatile methods.2–5 Another new strategy, native mass spectrometry (MS) has emerged as a powerful tool for structural biology, including analysis of macromolecular complexes and antibodies.6–10 In this technique, rapid but gentle ionization and transfer of protein complexes into the gas-phase via electrospray ionization (ESI) of aqueous solutions of near-physiological ionic strength preserves noncovalent interactions, allowing elucidation of ligand binding and stoichiometry of complexes.6–9 Coupled to advanced methodologies including ion mobility (IMS) or tandem mass spectrometry (MS/MS), native MS has proven an innovative strategy for interrogating protein structure, revealing topology, distinguishing conformations, identifying ligand binding sites, and determining folding thermodynamics.6–9,11,12 As one example of a premier application in biotherapeutic development, native MS has been utilized to rapidly measure stoichiometry, heterogeneity, and stability of antibody·antigen complexes.10,13–17
Detailed structural analyses by native MS often rely on controlled dissociation or disassembly of protein complexes via ion activation in MS/MS workflows.7,8,18–21 Collision-based dissociation methods comprise the most ubiquitous ion activation techniques and are capable of dismantling protein assemblies into subunits, detaching ligands, and facilitating determination of stoichiometries.7,8,18 However, the slow-heating process of collision induced dissociation (CID) causes protein unfolding and less effective fragmentation of the peptide backbone, limiting characterization of native protein complexes.7,8,18 Significantly more structural information can be acquired by employing alternative ion activation techniques.7,8,18 For example, surface induced dissociation (SID) is a collision-based method that promotes disassembly of protein complexes into subunits through a single high energy collision that minimizes protein unfolding and enables robust characterization of native protein assemblies.20,21 SID has proven especially versatile for the analysis of quaternary structure, as protein complexes disassemble to produce subcomplexes and subunits that reflect the native architecture.20,21 Most impressive, the outcome of SID correlates with the magnitude of protein interfacial area (cleavage of the weakest protein:protein interfaces) and has been integrated into computational modelling workflows to enhance the accuracy of protein assembly predictions, emphasizing the value of native MS/MS for structural interrogation.22–24 Alternatively, electron-based dissociation methods result in cleavage of the protein backbone to produce sequence fragments that are enhanced at surface exposed or flexible protein regions, informing topology (through the preservation of noncovalent interactions that prevent separation of reaction products) and degree of protein disorder.7,8,19,25
Also a method sensitive to protein structure, ultraviolet photodissociation (UVPD) is a photon-based ion activation method that has demonstrated exceptional use for structural biology. In particular, propensities of polypeptide backbone cleavages induced by 193 nm UVPD correlate with backbone flexibility and arrangement of non-covalent interactions, an outcome related to the likelihood of separation (and detection) of nascent product ions after a backbone cleavage event. Product ions enmeshed by stabilizing non-covalent interactions are less likely to separate (i.e. UVPnoD), causing an apparent suppression of backbone fragmentation. This correlation empowers characterization of conformational changes induced by point mutations, ligand binding, and protein complexation by UVPD.26–33 Additionally, the high sequence coverage and rapid timescale of photodissociation enable detailed analyses of protein gas-phase structure, even informing proton sequestration with single residue resolution.34 As native MS and UVPD increasingly gain broader utility for new protein applications, development and establishment of strategies to routinely study protein structure become imperative to cement these methodologies as cornerstones in the fields of structural biology and biotechnology that encompass development of new therapeutics, imaging agents, diagnostics and drug delivery agents.
One fascinating new class of biotherapeutics are nanobodies, single domain antibodies derived from the variable domain of functional heavy chain antibodies found in camelids and certain shark species.35,36 In contrast to conventional ∼150 kDa heterotetrameric immunoglobulin (IgG) antibodies, nanobodies feature a single peptide chain and overall reduced size of ∼15 kDa. Nanobodies offer high stability, solubility, affinity, and specificity, features that have propelled these single domain antibodies as a valuable alternative biotechnology.37–42 Concurrently, these same features facilitate native MS analysis, for which decreased size, increased solubility and antigen affinity are favorable for rapid and routine analysis of nanobody complexes, circumventing analytical challenges and tedious sample preparations, such as proteolysis and deglycosylation steps,14,17,43 often required for native MS of typical IgG·antigen complexes. One recent native MS study mapped the location of the epitope of influenza A hemagglutinin (HA1) bound to an antibody based on a decrease in backbone cleavages of HA1 when bound in the Ab·2HA1 complex relative to the free HA1 antigen during UVPD-MS analysis.31 This innovative approach motivated our interest in exploring an intriguing inverse strategy to map paratopes of nanobodies. In the present work, native MS and 193 nm UVPD are showcased as a valuable combination for determination of intersubunit salt-bridges and nanobody·antigen interfaces, ultimately localizing nanobody paratopes.
Three nanobodies44–46 with distinct proteinaceous antigens of green fluorescent protein (GFP), ribonuclease A (RNAseA), and porcine pancreatic amylase (PPA), referred to here as Gnb,44 Rnb,45 and Anb,46 respectively, were targeted to evaluate native MS and UVPD for characterization of nanobody·antigen complexes. Each of these nanobodies interact with the antigen via differing contributions of the complementarity-determining regions (CDR) 1–3 and framework residues to the protein interface, which vary in surface area from 554 Å2 to 683 Å2 to 1062 Å2 for Rnb, Gnb, and Anb, respectively. These three pairs of nanobodies and respective antigens were first ionized individually using native conditions (Fig. S1†), and as nanobody·antigen complexes (Fig. 1). In each case, native MS produced the expected 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex in the full mass spectrum (MS1), in accordance with known crystal structures, in a range of charge states. A single charge state of the antigen-bound (bound state) nanobody was isolated and subjected to 193 nm UVPD (Fig. 1B, D and F), resulting in disassembly to release the free nanobody and antigen as well as sequence fragments from backbone cleavage of each protein, the latter of which will be the focus of UVPD analysis. UVPD mass spectra of each free nanobody are shown in Fig. S2.† The corresponding backbone cleavage maps for the free nanobodies and corresponding nanobody·antigen complexes are shown in Fig. S3.†
:1 complex in the full mass spectrum (MS1), in accordance with known crystal structures, in a range of charge states. A single charge state of the antigen-bound (bound state) nanobody was isolated and subjected to 193 nm UVPD (Fig. 1B, D and F), resulting in disassembly to release the free nanobody and antigen as well as sequence fragments from backbone cleavage of each protein, the latter of which will be the focus of UVPD analysis. UVPD mass spectra of each free nanobody are shown in Fig. S2.† The corresponding backbone cleavage maps for the free nanobodies and corresponding nanobody·antigen complexes are shown in Fig. S3.†
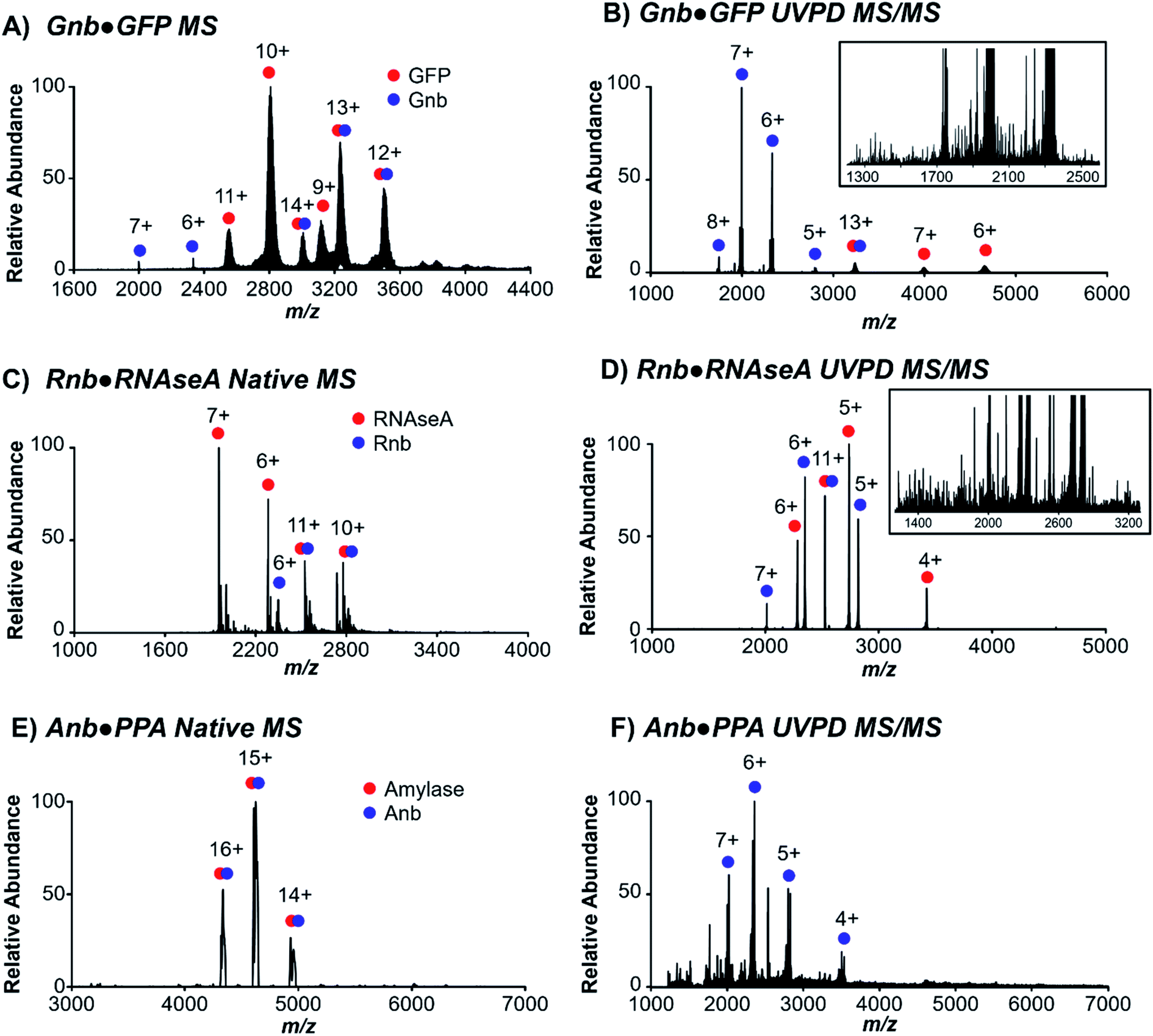 | ||
| Fig. 1 Native MS and UVPD of nanobody·antigen complexes. (A) MS1 of Gnb·GFP complex and (B) UVPD of the 13+ charge state. (C) MS1 of Rnb·RNAseA complex and (D) UVPD of the 11+ charge state. (E) MS1 of Anb·PPA complex and (F) UVPD of the 15+ charge state. Insets display expanded views of m/z regions populated by sequence fragments of low relative abundance. For UVPD, 1 laser pulse at 3 mJ was applied. | ||
Backbone cleavages induced by UVPD have been shown to be favored at protein regions with higher flexibility, typically ones less stabilized by networks of non-covalent interactions which might prevent separation and release of fragment ions.26–31,47 Accordingly, comparing abundances of fragment ions produced by free and bound nanobodies should reveal regions in which backbone fragmentation is suppressed or enhanced, thus uncovering those residues involved in stabilizing interactions with the antigen and effectively localizing the paratope. Ten types of fragment ions (a, a + 1, b, x, x + 1, y, y − 1, y − 2, and z, where +1, −1, and +2 indicate the gain or loss of hydrogen atoms) commonly generated by UVPD were monitored across the three nanobodies in both the free and bound states. Among the collection of ions detected, a- and x-type ions are the most prevalent and dominant for the nanobodies and complexes, thus providing the greatest sequence coverage. Those fragment ions displaying statistically significant differences in abundances (p < 0.05, n = 5) upon complexation of the nanobody to the antigen are shown in Fig. S4–S6† and mapped onto the backbone position cleaved to generate the fragment ions. The high diversity of fragment types characteristic of UVPD originates from competing pathways: direct dissociation from excited electronic states yields a/a + 1/x/x + 1 ions, and internal conversion to the ground state following intramolecular vibrational energy redistribution (IVR) produces b/y fragments.32,33 IVR processes may preferentially sever weak non-covalent interactions, whereas direct dissociation from excited states occurs on a faster time-scale minimizing disruption of non-covalent interactions.32,33 Consequently, the latter dissociation pathways and respective products, a/a + 1/x/x + 1 ions, are best suited to evaluate antigen-induced changes in nanobody topology. Variations in abundances of these four fragment types tended to be greater at the interface and CDRs, and inspection of the color-coded maps in Fig. S4–S6† reveals that in most cases the abundances of these a/x-type ions decreased for the nanobody·antigen complexes relative to the free nanobodies, signifying suppression of backbone fragmentation.
To underscore the impact of antigen binding on UVPD, significant changes in backbone fragmentation (ΔUVPD) based on differences in summed abundances of a/a + 1/x/x + 1 fragments for each free nanobody versus nanobody·antigen complex are shown in Fig. 2, along with color maps highlighting residues at the CDRs and protein interfaces. Generally, apparent suppression of backbone fragmentation (e.g., less efficient separation of nascent fragment ions owing to stabilizing non-covalent interactions) is the greatest at or adjacent to the interface residues, an outcome which is especially notable and consistent for patches of residues near CDR3 in Gnb (Fig. 2A), near CDR1 and CDR3 in Rnb (Fig. 2B), and near CDR2 and CDR3 in Anb (Fig. 2C). These patterns in suppression of backbone fragmentation upon antigen binding correlate with structural features of the respective crystal structures: main antigen contacts in Gnb are predominantly present on CDR3;44 only CDR1 and CDR3 participate in antigen binding for Rnb;45 CDR2 and CDR3 primarily mediate antigen binding for Anb.46 Although nanobodies characteristically feature a disulfide bond spanning C23 to a paired cysteine directly N-terminal to the CDR3, cleavage of the disulfide bond via UVPD was sufficient to unlock the nanobody and allow release of sequence ions from other concomitant backbone cleavages within this region, including CDR1 and CDR2. Moreover, any statistically significant enhancement of backbone cleavages induced by antigen binding was sparse and remote from interaction sites. These infrequent increases in UVPD fragmentation for nanobody·antigen complexes relative to the free nanobody possibly indicate disruption of non-covalent interactions and increased flexibility in those limited regions of the nanobody. However, this sporadic enhancement of backbone cleavages upon antigen binding occurs in stark contrast to more prevalent suppression of backbone cleavages spanning larger swaths of neighboring residues in the nanobody·antigen complexes. Overall, the observed suppression of fragmentation at the protein·protein interface serves as a strong basis for the development of native MS-UVPD strategies to discern nanobody paratopes.
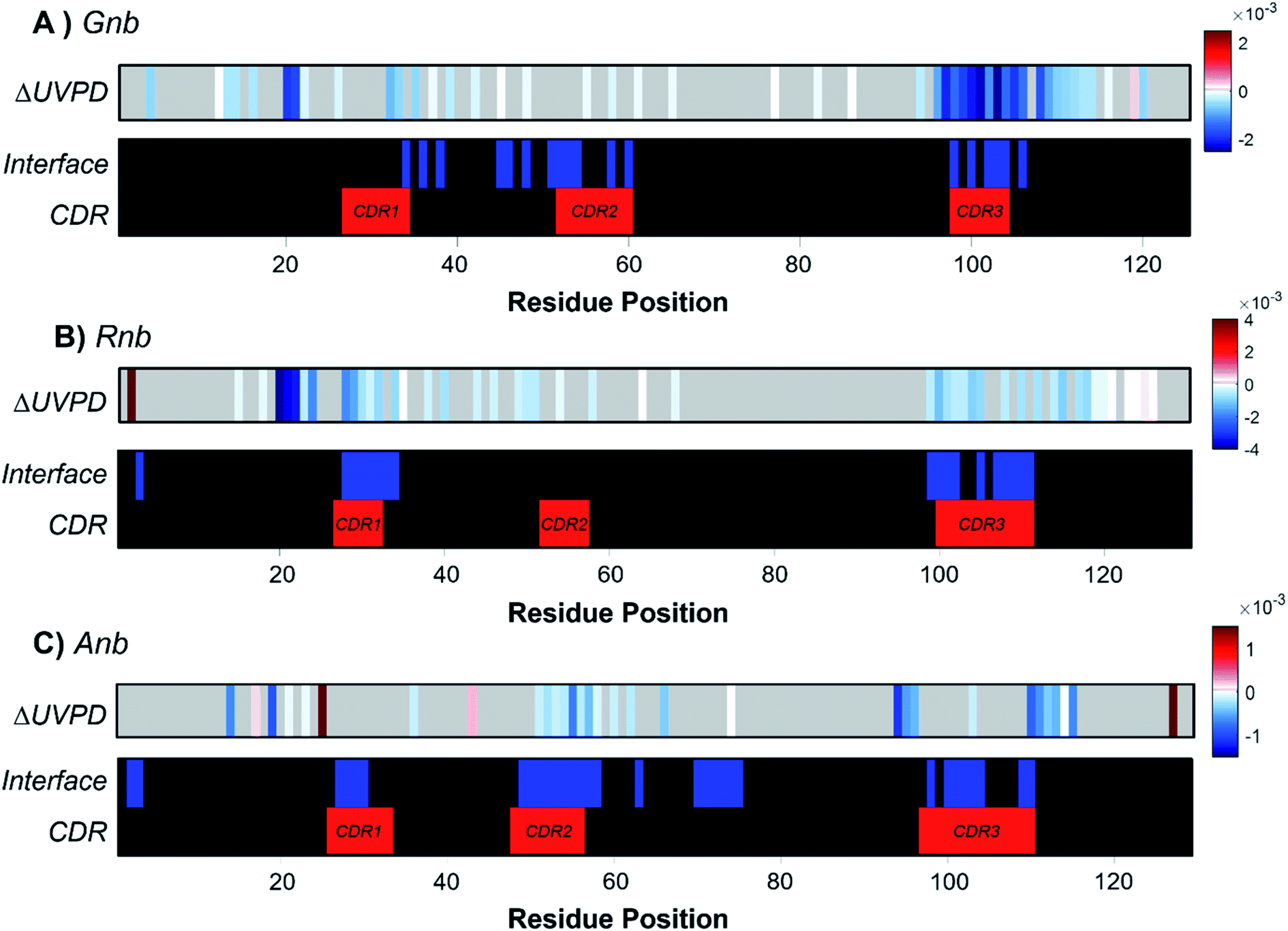 | ||
| Fig. 2 Suppression and enhancement of backbone cleavage sites based on abundances of UVPD fragment ions induced for each nanobody by antigen binding for (A) Gnb, (B) Rnb, and (C) Anb. ΔUVPD heat plots display significant differences (p < 0.05, n = 5) for the abundances of a- and x-type fragment ions between the free and bound nanobody. Blue and red indicate suppression and enhancement of fragment abundances, respectively, for the nanobody upon complexation. Positions that display no significant change are shown in grey, while white indicates small significant changes. Color maps highlighting interface residues and CDRs are also included for each nanobody. ΔUVPD values correspond to the fragment abundance per residue for the bound state minus the fragment abundance per residue for the free state. All product ions had a signal-to-noise ratio > 3. | ||
Contrary to classical IgGs where paratopes are primarily localized to CDRs,48 nanobody paratopes feature greater diversity in terms of residue identity and position, and also include the involvement of framework residues as well as the potential absence of interactions from certain CDRs, obfuscating assignment of paratopes.48 By leveraging the trends in the reduction of fragmentation observed in Fig. 2 for the three nanobody·antigen complexes relative to the free nanobodies, a strategy for the approximation of surface patches contributing to the paratope was developed. Because the most structurally significant changes of fragmentation related to antigen binding are demarcated by apparent suppression of backbone cleavages for stretches of neighboring residues, the UVPD data was analyzed by averaging abundances of fragment ions originating from backbone cleavages across every five residues (non-overlapping box-car average) prior to comparing fragment abundances for the free and bound states using Welch's t-test (n = 5). A stringent significance cutoff of p < 0.001 was subsequently applied to limit false assignments of protein sections involved at the interface. Protein sections (5 residues long) displaying significant suppression of backbone cleavages (i.e. reduction in abundances of fragment ions originating from backbone cleavages in each 5 residue segment) according to this method are plotted onto the crystal structures in Fig. 3. Impressively, only protein sections adjacent to the protein·protein interfaces displayed significant suppression for Gnb and Anb. Similarly, Rnb mostly featured suppression adjacent to the interface, while only two sections remote from the interface (spanning residues 15–24 and 45–49) were also suppressed. Furthermore, a significant enhancement of fragmentation was only observed for Rnb near the N-terminus and C-terminus (Fig. S7†), remote from the paratope. This Rnb complex featured the smallest interfacial area, which may lead to instability, compaction, or distortion during ion transmission, resulting in the unexpected suppression and enhancement at these regions. Regardless, UVPD suppression was predominant at the interfaces of each of the three nanobody complexes, presenting a new strategy for the approximation of nanobody residue patches that contribute to the paratope.
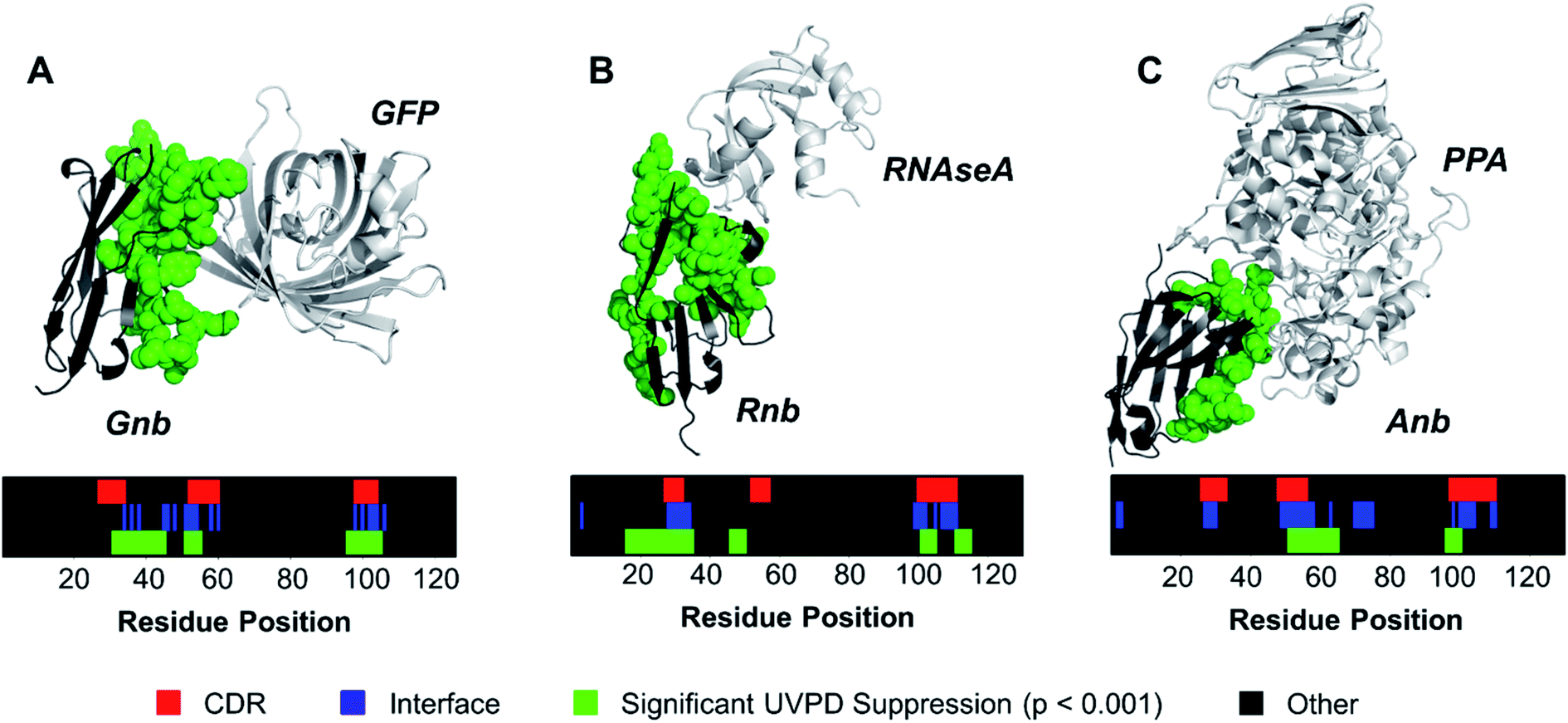 | ||
| Fig. 3 The a and x fragment ion abundances originating from backbone cleavages were averaged across every 5 residues for the free and bound forms of each nanobody. Sections displaying significant UVPD suppression upon complexation (p < 0.001, n = 5) were mapped onto the crystal structure as green spheres for (A) Gnb, (B) Rnb, and (C) Anb. Residue positions displaying significant suppression of backbone cleavages, interface residues, and CDR regions are shown for each nanobody as color maps. For comparative purposes, the CDR regions of each nanobody are demarcated on the crystal structures in Fig. S8.† | ||
Tracking charge states of specifically a- and x-type ions produced by 193 nm UVPD also informs proton sequestration along the protein sequence,34 a feature that is capitalized on here to identify the formation of inter-subunit salt-bridges. In this strategy, charge states for each detected a- or x-type ion are weighted based on intensity and plotted for each backbone cleavage position of the nanobody. For example, if both a42+ and a43+ have equal intensities, the weighted average charge state at backbone cleavage position 4 (between residues 4 and 5) would be 2.5. Because electrostatic interactions, such as salt-bridges, are highly stable in the gas-phase,49,50 applying this method to monitor changes in proton sequestration across free and bound states promises to reveal the locations of inter-subunit salt bridges introduced by complexation, if charge partitioning during subunit ejection is due to heterolytic scission of salt-bridges as previously proposed.26,51,52 Indeed, this is demonstrated in the analysis of the Rnb·RNAseA complex, for which one inter-chain salt bridge has been identified by X-ray crystallography between nanobody R107 and antigen E111. Charge site analysis of the a-ion series for free Rnb (6+ charge state) (Fig. 4A) displays discrete step-changes in charge states at residues R39, between residues 43–47 (suggesting protonation at R45), R68, Q111, and H128, indicating localized protons at these sites. Although coverage of the x-ion series is sparser, it nonetheless corroborates proton localization at Q111 as well as near the N-terminus in the span of residues Q2–L5. Protonation of side-chains is not unexpected for basic amino acids like R and K, but protonation of backbone heteroatoms is also possible for non-basic residues like Q at either the amide oxygen or the amide nitrogen of the peptide bond.53,54 The step analysis reported here localizes all 6 protons of the 6+ charge state of free Rnb.
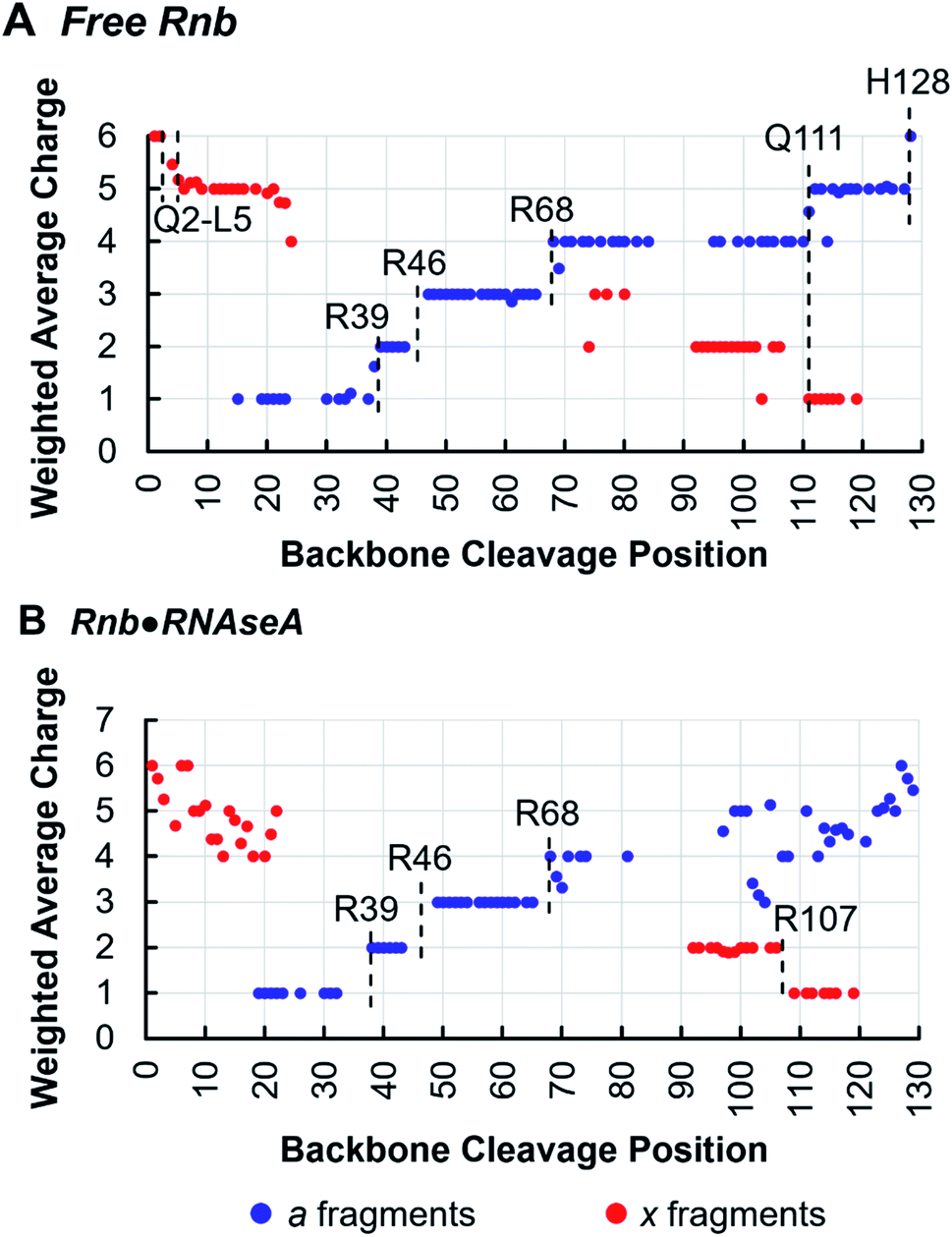 | ||
| Fig. 4 Weighted average charge of a-type and x-type fragment ions attributed to Rnb produced by UVPD of (A) free Rnb (6+ charge state) and (B) Rnb·RNAseA (11+ charge state), delineated based on the backbone cleavage site along the sequence of the nanobody. | ||
Charge site analysis of Rnb in the Rnb·RNAseA complex was not as comprehensive, but nonetheless many charge sites were elucidated (Fig. 4B). Specifically, R29, R46, and R68 remained protonated, while a shift occurred from protonation at Q111 in free RNAseA to protonation of the span of residues 108–106 in the Rnb·RNAseA complex, indicating proton sequestration at R107, a residue engaged in a salt-bridge with the antigen in solution. Charge migration observed upon antigen binding based on this charge-site analysis thus evinces formation of salt-bridges between binding partners. Similarly for Gnb·GFP, charge site analysis enabled localization of multiple charges in both the free and bound states (Fig. S9†) including protonation at R36 on Gnb, which is involved in electrostatic interactions with E142 on the GFP antigen. Additionally, a charge was located at R58 for bound Gnb that is absent for free Gnb. Although slightly higher than canonical distance cutoff of 4 Å for salt bridges, Gnb R58 is within 4.5–5.5 Å of the E172 and D173 side-chains of GFP according to the crystal structure. Salt-bridge formation spanning this distance may be possible in the gas-phase, according to charge site analysis derived from the UVPD data. The companion residues, E45 and E104, of the nanobody are also engaged in putative interchain salt-bridges when bound to GFP based on the X-ray structure, however, localization of deprotonation sites of acidic residues is not possible in the positive ion mode.
For Anb, coverage of a- and x-type fragments from the Anb·PPA complex was less comprehensive and precluded detailed charge site analyses (Fig. S10†). Regardless, data for Gnb and Rnb demonstrate that basic residues engaged in inter-subunit salt-bridges maintain a proton upon disassembly of the nanobody·antigen complexes, highlighting an exceptional use of MS/MS to localize gas-phase salt-bridges between protein subunits. However, it is also noted that these changes in protonation sites may be caused by reasons other than those postulated here. Specifically, it is possible that proton migration is caused by vibrational redistribution of deposited energy from photoabsorption, leading to proton mobilization,53,54 and is not due to heterolytic scission of salt bridges. These results are nonetheless intriguing and may guide future interpretations of charge partitioning during UVPD of protein complexes.
Following incredible advances in native MS, routine and rapid analysis of biotherapeutics for quality control and drug development has become commonplace.13–17 At the same time, UVPD has been propelled over the past decade as a premier ion activation method capable of uncovering structural features of proteins that are not revealed by other MS/MS methods.47 As shown in this study, we extended the combination of native MS and UVPD to characterize nanobody·antigen complexes, particularly aiming to showcase UVPD for mapping the binding interface. The pattern of fragment ions generated by UVPD for nanobody complexes resulted in the discernment of inter-chain salt-bridges. Additionally, tracking trends in suppression of fragmentation enabled the localization of nanobody paratopes using only micromolar quantities of nanobodies and circumventing some of the limitations of traditional structural biology methods such as NMR and X-ray crystallography. We anticipate that further improvements in data analysis/informatics methods will further extend the level of structural detail gleaned from the very dense UVPD mass spectra generated for large macromolecular assemblies akin to nanobody·antigen complexes.
Experimental
Reagents
Ammonium acetate, porcine pancreatic amylase, and bovine pancreas ribonuclease A were acquired from Sigma-Aldrich (St. Louis, MO). Ammonium acetate solutions were prepared in HPLC grade water (Millipore, Burlington, MA).Expression and purification of recombinant proteins
Nanobodies cloned with a C-terminal 6XHis tag and expressed and purified from E. coli strain SHuffle T7 express. C-Terminal 6XHis GFP was expressed and purified from BL21DE3. Following immobilized metal affinity chromatography (IMAC) purification, samples were dialyzed against dialysis buffer (50 mM phosphate, 150 mM NaCl). Purified nanobodies were stored at 4 °C before use. For simplicity, nanobody naming used here is as follows: GFP-nanobody44 = Gnb, cAb-RN05 (ref. 45) = Rnb, and AMD9 (ref. 46) = Anb.Mass spectrometry
All spectra were collected on a Thermo Scientific Q Exactive UHMR mass spectrometer (Bremen, Germany) modified with a Coherent Excistar excimer laser (Santa Cruz, CA) for 193 nm UVPD as previously described.55 All nanobody, antigen, and nanobody·antigen samples were desalted and buffer exchanged into 100 mM ammonium acetate using 10 kDa molecular weight cutoff filters (MilliporeSigma, Burlington, MA) and diluted to a 30 μM working concentration prior to MS analysis. Samples were loaded onto gold/palladium coated borosilicate emitters (prepared in house) for positive mode nano-ESI using a source voltage of 1.0–1.3 kV and source temperature of 200 °C. MS1 spectra were collected by averaging 25 microscans at 200 K resolution at m/z 200, except for the MS1 of free PPA and Anb·PPA for which 3 K resolution was utilized, as the high mass (>50 kDa) inhibited isotopic resolution and deteriorated mass analyses at high resolution. UVPD mass spectra were acquired by quadrupole isolation of the selected precursor ion, prior to transfer to the HCD cell where the ion cloud was irradiated by a single laser pulse set to 3 mJ, then subsequent mass analysis of resulting fragment ions in the Orbitrap analyzer. All UVPD mass spectra were collected at 200 K resolution at m/z 200 by averaging 500 microscans. The ion population was controlled by modulating the injection time (IT) between 50 and 100 ms for MS1 spectra and 500–800 ms for UVPD mass spectra.Data analysis
Low resolution free PPA and Anb·PPA MS1 spectra were analyzed with the aid of UniDec, an algorithm that allows deconvolution of lower resolution spectra lacking isotopic detail commonly acquired for higher mass ions.56 All other MS1 and all UVPD spectra were deconvoluted using Xtract algorithm with a signal-to-noise ratio threshold of 3, fit factor of 44%, remainder of 25%, and max charge set to the precursor charge state. UVPD spectra were searched for a, a + 1, b, c, x, x + 1, y, y − 1, y − 2, and z fragment ions with the aid of Prosight Lite57 and UVPOSIT.58 Charge site analysis was performed as previously described34 and used to calculate weighted average charge states per backbone cleavage position. Fragment matches and abundances determined by UVPOSIT were further analyzed using custom R scripts with the aid of the dplyr package. Abundances of fragment ions originating from backbone cleavages for each residue at position n for protein of length L correspond to the sum ion current of the an, bn, cn, xL−n+1, yL−n+1, and zL−n+1 fragments divided by the total ion current of the MS/MS spectrum. Statistical comparisons between UVPD fragment abundances of free and bound states per residue or per section (5 residue stretches) of each nanobody were performed using Welch's t-test, based on 5 replicate UVPD analyses of the free nanobody and 5 replicate UVPD analyses of the bound nanobody. Interface residues, interface surface areas, and salt-bridges were determined from crystal structures PDB 3OGO, PDB 1KXQ, and PDB 1BZQ using PDBePISA online tool.59Author contributions
LAM, BWD and JSB contributed to conceptualization of this project. XW and BWD provided all proteins, while JSB provided all other resources. LAM performed all MS experiments (data curation), formal analysis, investigation and methodology. JSB coordinated project administration. JSB, LAM and BWD are responsible for acquisition of funding. LAM, BWD, and JSB prepared figures (visualization) and the manuscript (writing – original draft). All authors contributed to the final manuscript (writing – review & editing).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Funding from the National Institutes of Health [R35GM139658] and the Robert A. Welch Foundation [F-1155] is acknowledged by JSB. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number F31CA257404 to LAM. Funding from the National Institutes of Health [R01AI148419, R01AI125337, R21AI159203] is acknowledged by BWD. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- M. L. Chiu, D. R. Goulet, A. Teplyakov and G. L. Gilliland, Antibody Structure and Function: The Basis for Engineering Therapeutics, Antibodies, 2019, 8(4), 55, DOI:10.3390/antib8040055.
- W. M. Abbott, M. M. Damschroder and D. C. Lowe, Current Approaches to Fine Mapping of Antigen–Antibody Interactions, Immunology, 2014, 142(4), 526–535, DOI:10.1111/imm.12284.
- A. Antanasijevic, C. A. Bowman, R. N. Kirchdoerfer, C. A. Cottrell, G. Ozorowski, A. A. Upadhyay, K. M. Cirelli, D. G. Carnathan, C. A. Enemuo, L. M. Sewall, B. Nogal, F. Zhao, B. Groschel, W. R. Schief, D. Sok, G. Silvestri, S. Crotty, S. E. Bosinger and A. B. Ward, From Structure to Sequence: Antibody Discovery Using CryoEM, Sci. Adv., 2022, 8(3), eabk2039, DOI:10.1126/sciadv.abk2039.
- H. Wei, J. Mo, L. Tao, R. J. Russell, A. A. Tymiak, G. Chen, R. E. Iacob and J. R. Engen, Hydrogen/Deuterium Exchange Mass Spectrometry for Probing Higher Order Structure of Protein Therapeutics: Methodology and Applications, Drug Discovery Today, 2014, 19(1), 95–102, DOI:10.1016/j.drudis.2013.07.019.
- G. R. Masson, M. L. Jenkins and J. E. Burke, An Overview of Hydrogen Deuterium Exchange Mass Spectrometry (HDX-MS) in Drug Discovery,, Expert Opin. Drug Discovery, 2017, 12(10), 981–994, DOI:10.1080/17460441.2017.1363734.
- K. R. Karch, D. T. Snyder, S. R. Harvey and V. H. Wysocki, Native Mass Spectrometry: Recent Progress and Remaining Challenges, Annu. Rev. Biophys., 2022, 51:1, 157–179, DOI:10.1146/annurev-biophys-092721-085421.
- M. Zhou, C. Lantz, K. A. Brown, Y. Ge, L. Paša-Tolić, J. A. Loo and F. Lermyte, Higher-Order Structural Characterisation of Native Proteins and Complexes by Top-down Mass Spectrometry, Chem. Sci., 2020, 11(48), 12918–12936, 10.1039/D0SC04392C.
- S. Tamara, M. A. den Boer and A. J. R. Heck, High-Resolution Native Mass Spectrometry, Chem. Rev., 2022, 122(8), 7269–7326, DOI:10.1021/acs.chemrev.1c00212.
- C. V. Robinson, Mass Spectrometry: From Plasma Proteins to Mitochondrial Membranes, PNAS, 2019, 116(8), 2814–2820, DOI:10.1073/pnas.1820450116.
- I. D. G. Campuzano and W. Sandoval, Denaturing and Native Mass Spectrometric Analytics for Biotherapeutic Drug Discovery Research: Historical, Current, and Future Personal Perspectives, J. Am. Soc. Mass Spectrom., 2021, 32(8), 1861–1885, DOI:10.1021/jasms.1c00036.
- J. W. McCabe, M. J. Hebert, M. Shirzadeh, C. S. Mallis, J. K. Denton, T. E. Walker and D. H. Russell, The Ims Paradox: A Perspective on Structural Ion Mobility-Mass Spectrometry, Mass Spectrom. Rev., 2021, 40(3), 280–305, DOI:10.1002/mas.21642.
- G. Ben-Nissan and M. Sharon, The Application of Ion-Mobility Mass Spectrometry for Structure/Function Investigation of Protein Complexes, Curr. Opin. Chem. Biol., 2018, 42, 25–33, DOI:10.1016/j.cbpa.2017.10.026.
- S. Vimer, G. Ben-Nissan, M. Marty, S. J. Fleishman and M. Sharon, Direct-MS Analysis of Antibody-Antigen Complexes, Proteomics, 2021, 21(21–22), 2000300, DOI:10.1002/pmic.202000300.
- G. Terral, A. Beck and S. Cianférani, Insights from Native Mass Spectrometry and Ion Mobility-Mass Spectrometry for Antibody and Antibody-Based Product Characterization, J. Chromatogr. B, 2016, 1032, 79–90, DOI:10.1016/j.jchromb.2016.03.044.
- Y. Tian and B. T. Ruotolo, The Growing Role of Structural Mass Spectrometry in the Discovery and Development of Therapeutic Antibodies, Analyst, 2018, 143(11), 2459–2468, 10.1039/C8AN00295A.
- D. Goswami, J. Zhang, P. V. Bondarenko and Z. Zhang, MS-Based Conformation Analysis of Recombinant Proteins in Design, Optimization and Development of Biopharmaceuticals, Methods, 2018, 144, 134–151, DOI:10.1016/j.ymeth.2018.04.011.
- S. Rosati, Y. Yang, A. Barendregt and A. J. R. Heck, Detailed Mass Analysis of Structural Heterogeneity in Monoclonal Antibodies Using Native Mass Spectrometry, Nat. Protoc., 2014, 9(4), 967–976, DOI:10.1038/nprot.2014.057.
- L. A. Macias, I. C. Santos and J. S. Brodbelt, Ion Activation Methods for Peptides and Proteins, Anal. Chem., 2020, 92(1), 227–251, DOI:10.1021/acs.analchem.9b04859.
- F. Lermyte, D. Valkenborg, J. A. Loo and F. Sobott, Radical Solutions: Principles and Application of Electron-Based Dissociation in Mass Spectrometry-Based Analysis of Protein Structure, Mass Spectrom. Rev., 2018, 37(6), 750–771, DOI:10.1002/mas.21560.
- A. Q. Stiving, Z. L. VanAernum, F. Busch, S. R. Harvey, S. H. Sarni and V. H. Wysocki, Surface-Induced Dissociation: An Effective Method for Characterization of Protein Quaternary Structure, Anal. Chem., 2019, 91(1), 190–209, DOI:10.1021/acs.analchem.8b05071.
- D. T. Snyder, S. R. Harvey and V. H. Wysocki, Surface-Induced Dissociation Mass Spectrometry as a Structural Biology Tool, Chem. Rev., 2022, 122(8), 7442–7487, DOI:10.1021/acs.chemrev.1c00309.
- S. R. Harvey, J. T. Seffernick, R. S. Quintyn, Y. Song, Y. Ju, J. Yan, A. N. Sahasrabuddhe, A. Norris, M. Zhou, E. J. Behrman, S. Lindert and V. H. Wysocki, Relative Interfacial Cleavage Energetics of Protein Complexes Revealed by Surface Collisions, PNAS, 2019, 116(17), 8143–8148, DOI:10.1073/pnas.1817632116.
- J. T. Seffernick, S. R. Harvey, V. H. Wysocki and S. Lindert, Predicting Protein Complex Structure from Surface-Induced Dissociation Mass Spectrometry Data, ACS Cent. Sci., 2019, 5(8), 1330–1341, DOI:10.1021/acscentsci.8b00912.
- J. T. Seffernick, S. M. Canfield, S. R. Harvey, V. H. Wysocki and S. Lindert, Prediction of Protein Complex Structure Using Surface-Induced Dissociation and Cryo-Electron Microscopy, Anal. Chem., 2021, 93(21), 7596–7605, DOI:10.1021/acs.analchem.0c05468.
- N. M. Riley and J. J. Coon, The Role of Electron Transfer Dissociation in Modern Proteomics, Anal. Chem., 2018, 90(1), 40–64, DOI:10.1021/acs.analchem.7b04810.
- L. J. Morrison and J. S. Brodbelt, 193 Nm Ultraviolet Photodissociation Mass Spectrometry of Tetrameric Protein Complexes Provides Insight into Quaternary and Secondary Protein Topology, J. Am. Chem. Soc., 2016, 138(34), 10849–10859, DOI:10.1021/jacs.6b03905.
- M. B. Cammarata and J. S. Brodbelt, Structural Characterization of Holo- and Apo-Myoglobin in the Gas Phase by Ultraviolet Photodissociation Mass Spectrometry, Chem. Sci., 2015, 6(2), 1324–1333, 10.1039/C4SC03200D.
- M. B. Cammarata, R. Thyer, J. Rosenberg, A. Ellington and J. S. Brodbelt, Structural Characterization of Dihydrofolate Reductase Complexes by Top-Down Ultraviolet Photodissociation Mass Spectrometry, J. Am. Chem. Soc., 2015, 137(28), 9128–9135, DOI:10.1021/jacs.5b04628.
- M. B. Cammarata, C. L. Schardon, M. R. Mehaffey, J. Rosenberg, J. Singleton, W. Fast and J. S. Brodbelt, Impact of G12 Mutations on the Structure of K-Ras Probed by Ultraviolet Photodissociation Mass Spectrometry, J. Am. Chem. Soc., 2016, 138(40), 13187–13196, DOI:10.1021/jacs.6b04474.
- M. R. Mehaffey, M. B. Cammarata and J. S. Brodbelt, Tracking the Catalytic Cycle of Adenylate Kinase by Ultraviolet Photodissociation Mass Spectrometry, Anal. Chem., 2018, 90(1), 839–846, DOI:10.1021/acs.analchem.7b03591.
- M. R. Mehaffey, J. Lee, J. Jung, M. B. Lanzillotti, E. E. Escobar, K. R. Morgenstern, G. Georgiou and J. S. Brodbelt, Mapping a Conformational Epitope of Hemagglutinin A Using Native Mass Spectrometry and Ultraviolet Photodissociation, Anal. Chem., 2020, 92(17), 11869–11878, DOI:10.1021/acs.analchem.0c02237.
- L. A. Macias, S. N. Sipe, I. C. Santos, A. Bashyal, M. R. Mehaffey and J. S. Brodbelt, Influence of Primary Structure on Fragmentation of Native-Like Proteins by Ultraviolet Photodissociation, J. Am. Soc. Mass Spectrom., 2021, 32(12), 2860–2873, DOI:10.1021/jasms.1c00269.
- R. Julian, The Mechanism behind Top-Down UVPD Experiments: Making Sense of Apparent Contradictions, J. Am. Soc. Mass Spectrom., 2017, 28(9), 1823, DOI:10.1007/s13361-017-1721-0.
- L. J. Morrison and J. S. Brodbelt, Charge Site Assignment in Native Proteins by Ultraviolet Photodissociation (UVPD) Mass Spectrometry, Analyst, 2016, 141(1), 166–176, 10.1039/C5AN01819F.
- S. Muyldermans, Nanobodies: Natural Single-Domain Antibodies, Annu. Rev. Biochem., 2013, 82, 775–797 CrossRef CAS PubMed.
- M. Arbabi-Ghahroudi, Camelid Single-Domain Antibodies: Historical Perspective and Future Outlook, Front. Immunol., 2017, 8 DOI:10.3389/fimmu.2017.01589.
- G. Bao, M. Tang, J. Zhao and X. Zhu, Nanobody: A Promising Toolkit for Molecular Imaging and Disease Therapy, EJNMMI Res., 2021, 11(1), 1–13, DOI:10.1186/s13550-021-00750-5.
- E. Beghein and J. Gettemans, Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein–Protein Interaction Analysis, and Protein Function Exploration, Front. Immunol., 2017, 8 DOI:10.3389/fimmu.2017.00771.
- G. Gonzalez-Sapienza, M. A. Rossotti and S. Tabares-da Rosa, Single-Domain Antibodies As Versatile Affinity Reagents for Analytical and Diagnostic Applications, Front. Immunol., 2017, 8 DOI:10.3389/fimmu.2017.00977.
- Y. Hu, C. Liu and S. Muyldermans, Nanobody-Based Delivery Systems for Diagnosis and Targeted Tumor Therapy, Front. Immunol., 2017, 8 DOI:10.3389/fimmu.2017.01442.
- Y. Wu, S. Jiang and T. Ying, Single-Domain Antibodies As Therapeutics against Human Viral Diseases, Front. Immunol., 2017, 8 DOI:10.3389/fimmu.2017.01802.
- I. Jovčevska and S. Muyldermans, The Therapeutic Potential of Nanobodies, BioDrugs, 2020, 34(1), 11–26, DOI:10.1007/s40259-019-00392-z.
- J.-F. Greisch, M. A. den Boer, F. Beurskens, J. Schuurman, S. Tamara, A. Bondt and A. J. R. Heck, Generating Informative Sequence Tags from Antigen-Binding Regions of Heavily Glycosylated IgA1 Antibodies by Native Top-Down Electron Capture Dissociation, J. Am. Soc. Mass Spectrom., 2021, 32(6), 1326–1335, DOI:10.1021/jasms.0c00461.
- M. H. Kubala, O. Kovtun, K. Alexandrov and B. M. Collins, Structural and Thermodynamic Analysis of the GFP:GFP-Nanobody Complex, Protein Sci., 2010, 19(12), 2389–2401, DOI:10.1002/pro.519.
- K. Decanniere, A. Desmyter, M. Lauwereys, M. A. Ghahroudi, S. Muyldermans and L. Wyns, A Single-Domain Antibody Fragment in Complex with RNase A: Non-Canonical Loop Structures and Nanomolar Affinity Using Two CDR Loops, Structure, 1999, 7(4), 361–370, DOI:10.1016/S0969-2126(99)80049-5.
- A. Desmyter, S. Spinelli, F. Payan, M. Lauwereys, L. Wyns, S. Muyldermans and C. Cambillau, Three Camelid VHH Domains in Complex with Porcine Pancreatic α-Amylase: Inhibition and Versatility of Binding Topology, J. Biol. Chem., 2002, 277(26), 23645–23650, DOI:10.1074/jbc.M202327200.
- J. S. Brodbelt, L. J. Morrison and I. Santos, Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules, Chem. Rev., 2020, 120(7), 3328–3380, DOI:10.1021/acs.chemrev.9b00440.
- L. S. Mitchell and L. J. Colwell, Analysis of Nanobody Paratopes Reveals Greater Diversity than Classical Antibodies, Protein Eng., Des. Sel., 2018, 31(7–8), 267–275, DOI:10.1093/protein/gzy017.
- S. Yin, Y. Xie and J. A. Loo, Mass Spectrometry of Protein-Ligand Complexes: Enhanced Gas Phase Stability of Ribonuclease-Nucleotide Complexes, J. Am. Soc. Mass Spectrom., 2008, 19(8), 1199–1208, DOI:10.1016/j.jasms.2008.05.012.
- A. S. Woods and S. Ferré, Amazing Stability of the Arginine–Phosphate Electrostatic Interaction, J. Proteome Res., 2005, 4(4), 1397–1402, DOI:10.1021/pr050077s.
- R. R. Ogorzalek Loo and J. A. Loo, Salt Bridge Rearrangement (SaBRe) Explains the Dissociation Behavior of Noncovalent Complexes, J. Am. Soc. Mass Spectrom., 2016, 27(6), 975–990, DOI:10.1007/s13361-016-1375-3.
- S. Tamara, A. Dyachenko, K. L. Fort, A. A. Makarov, R. A. Scheltema and A. J. R. Heck, Symmetry of Charge Partitioning in Collisional and UV Photon-Induced Dissociation of Protein Assemblies, J. Am. Chem. Soc., 2016, 138(34), 10860–10868, DOI:10.1021/jacs.6b05147.
- V. H. Wysocki, G. Tsaprailis, L. L. Smith and L. A. Breci, Mobile and Localized Protons: A Framework for Understanding Peptide Dissociation, J. Mass Spectrom., 2000, 35(12), 1399–1406, DOI:10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R.
- B. J. Bythell, S. Suhai, Á. Somogyi and B. Paizs, Proton-Driven Amide Bond-Cleavage Pathways of Gas-Phase Peptide Ions Lacking Mobile Protons, J. Am. Chem. Soc., 2009, 131(39), 14057–14065, DOI:10.1021/ja903883z.
- M. R. Mehaffey, J. D. Sanders, D. D. Holden, C. L. Nilsson and J. S. Brodbelt, Multistage Ultraviolet Photodissociation Mass Spectrometry To Characterize Single Amino Acid Variants of Human Mitochondrial BCAT2, Anal. Chem., 2018, 90(16), 9904–9911, DOI:10.1021/acs.analchem.8b02099.
- M. T. Marty, A. J. Baldwin, E. G. Marklund, G. K. A. Hochberg, J. L. P. Benesch and C. V. Robinson, Bayesian Deconvolution of Mass and Ion Mobility Spectra: From Binary Interactions to Polydisperse Ensembles, Anal. Chem., 2015, 87(8), 4370–4376, DOI:10.1021/acs.analchem.5b00140.
- R. T. Fellers, J. B. Greer, B. P. Early, X. Yu, R. D. LeDuc, N. L. Kelleher and P. M. Thomas, ProSight Lite: Graphical Software to Analyze Top-Down Mass Spectrometry Data, Proteomics, 2015, 15(7), 1235–1238, DOI:10.1002/pmic.201570050.
- J. Rosenberg, W. R. Parker, M. B. Cammarata and J. S. Brodbelt, UV-POSIT: Web-Based Tools for Rapid and Facile Structural Interpretation of Ultraviolet Photodissociation (UVPD) Mass Spectra, J. Am. Soc. Mass Spectrom., 2018, 29(6), 1323–1326, DOI:10.1007/s13361-018-1918-x.
- E. Krissinel and K. Henrick, Inference of Macromolecular Assemblies from Crystalline State, J. Mol. Biol., 2007, 372(3), 774–797, DOI:10.1016/j.jmb.2007.05.022.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary MS1 and UVPD spectra, fragment maps, charge site analyses, fragmentation heat plots, and crystal structure color maps. See https://doi.org/10.1039/d2sc01536f |
| This journal is © The Royal Society of Chemistry 2022 |