
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuzuki-type cross-coupling of alkyl trifluoroborates with acid fluoride enabled by NHC/photoredox dual catalysis†
Hua
Huang
ab,
Qing-Song
Dai
a,
Hai-Jun
Leng
a,
Qing-Zhu
Li
a,
Si-Lin
Yang
a,
Ying-Mao
Tao
a,
Xiang
Zhang
a,
Ting
Qi
a and
Jun-Long
Li
 *a
*a
aAntibiotics Research and Re-evaluation Key Laboratory of Sichuan Province, Sichuan Industrial Institute of Antibiotics, School of Pharmacy, Chengdu University, Chengdu 610106, P. R. China. E-mail: lijunlong709@hotmail.com
bState Key Laboratory of Southwestern Chinese Medicine Resources, School of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China
First published on 21st January 2022
Abstract
The Suzuki–Miyaura cross-coupling of C(sp3)-hybridised boronic compounds still remains a challenging task, thereby hindering the broad application of alkyl boron substrates in carbon–carbon bond-forming reactions. Herein, we developed an NHC/photoredox dual catalytic cross-coupling of alkyl trifluoroborates with acid fluorides, providing an alternative solution to the classical acylative Suzuki coupling chemistry. With this protocol, various ketones could be rapidly synthesised from readily available materials under mild conditions. Preliminary mechanistic studies shed light on the unique radical reaction mechanism.
Introduction
The Suzuki–Miyaura cross-coupling of boron compounds with electrophiles, such as organic halides, has played a central role in modern organic synthesis, as it provides a robust protocol to forge carbon–carbon chemical bonds.1 As one of the most popular synthetic strategies in pharmaceutical science, the Suzuki–Miyaura reaction has been well received by medicinal chemists, with the accomplishment of more than half of the C–C bond-forming processes for drug synthesis.2 Despite its significant contributions, the cross-coupling method is still imperfect in many aspects and further improving the reaction outcome remains an important research subject. Typically, the transition-metal-catalysed cross-coupling proceeds through a three-stage catalytic cycle, including oxidative addition, transmetalation and reductive elimination. In particular, the rate-determining step of most Suzuki cross-coupling reactions was found to be the transmetalation process, and the transmetalation rate of C(sp3)-hybridised boronic reagents is often sluggish and problematic, and generally exhibits a much slower rate than that of the C(sp)- or C(sp2)-hybridised boron substrates.3 In this scenario, several slow alkyl transmetalation processes were alternatively realised by using more reactive but air-/moisture-sensitive organometallic species, such as alkyl Grignard reagents and alkylzincs, to replace the bench-stable boron reagents for cross-coupling.4 Thus, the broad application of alkyl boronic compounds in Suzuki–Miyaura reactions still represents a challenging task. Despite a few examples that used the means of heating, strong bases or ligand screening to promote the efficiency of alkyl boronic Suzuki–Miyaura cross-coupling, the development of mechanistically different catalytic protocols to address the problem is highly desirable. A breakthrough in this area has been recently achieved by Molander, who employed nickel/photoredox dual catalysis to streamline the alkylboron transmetalation through a single-electron transfer event. This dual catalytic approach was successfully applied to the cross-coupling of various alkyl trifluoroborates with aryl bromides, offering a diversity of diarylmethane products (Scheme 1a).5 Inspired by their elegant work, we envisioned that the concept of the dual catalytic radical approach might be further explored and modified to enable more types of Suzuki–Miyaura reactions by using easily-available alkylboron reagents.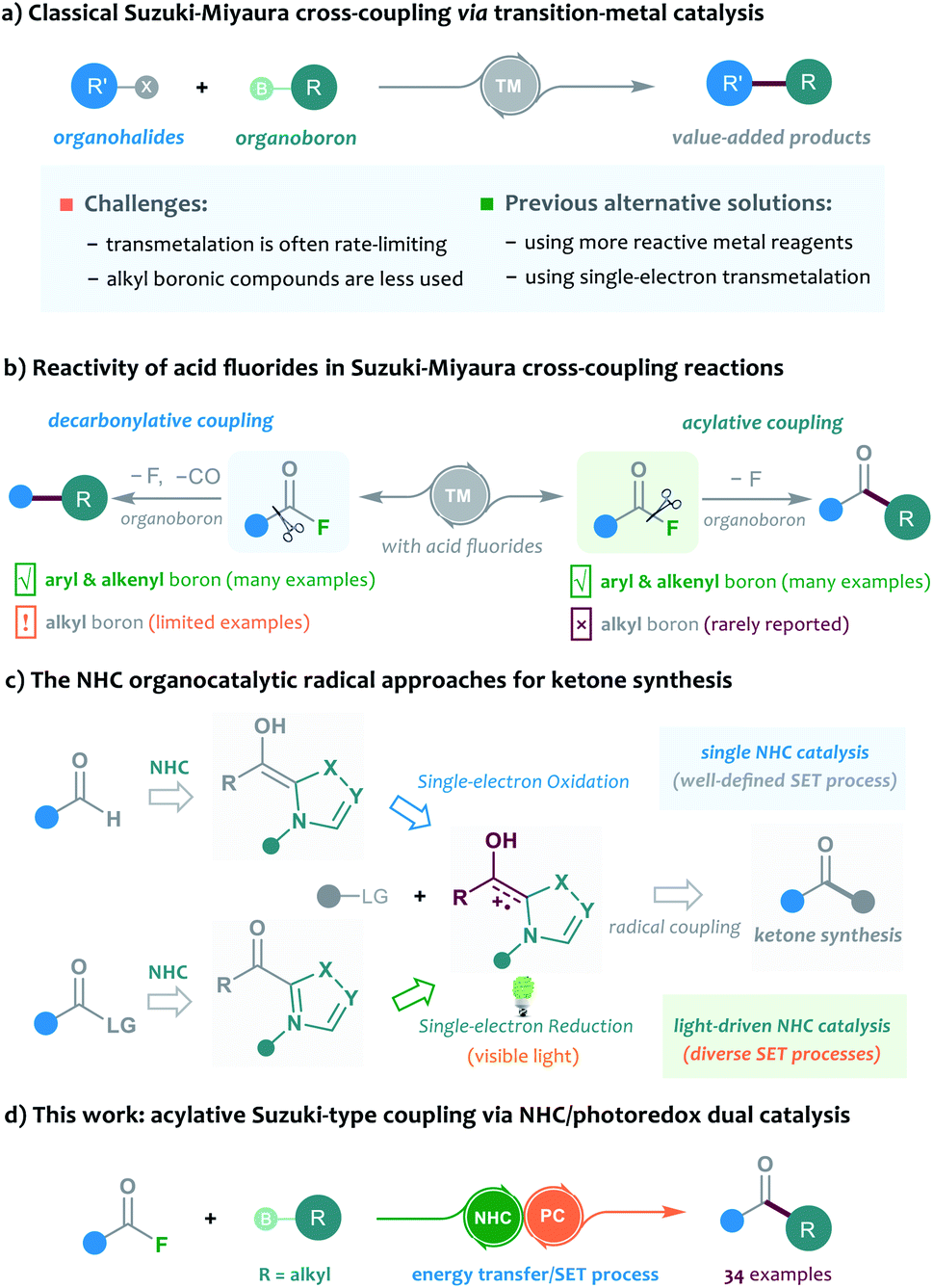 | ||
| Scheme 1 Research motivation for NHC/photoredox dual catalysed Suzuki-type coupling reactions. | ||
The acylative Suzuki reaction, cross-coupling of carboxylic acid derivatives with boron reagents, represents a powerful tool for the synthesis of ketones.6 As an important kind of carboxylic substrate, acid fluorides are regarded as easy-to-handle and versatile building blocks for cross-coupling chemistry, probably because they display a great balance between stability and reactivity.7 Rovis and colleagues for the first time reported the catalytic cross-coupling of acid fluorides in the presence of organozinc reagents in 2004.8 However, the boron-based Suzuki-type coupling of acid fluorides remained unexplored until the recent independent studies of Sakai and Sanford's group.9 In 2017, Sakai et al.9a enriched the acylative Suzuki reaction to include acid fluoride substrates, but the method was still limited to the classical C(sp2)-hybridised boron reagents. Later, Sanford and colleagues disclosed an elegant base-free decarbonylative Suzuki-coupling of acid fluorides with various aryl and alkenyl boronic acids. Intriguingly, a few examples of alkyl boron substrates were found to be productive in the reaction and gave the alkyl substituted aromatic rings; however, the related acylative Suzuki-coupling that leads to selective synthesis of ketones could still not be realised in their catalytic system9b (Scheme 1b). To address the challenge, the direct acylative coupling of acid fluorides with alkyl boronic compounds may require a mechanistically different approach that not only allows the catalytic activation of acid fluorides but also bypasses the traditional boron-transmetalation process.
Recently, the radical-mediated N-heterocyclic carbene (NHC) organocatalysis has emerged as a fertile platform for the discovery of novel acylation reactions.10 Of particular interest are the diverse newly-developed approaches that allow radical synthesis of ketones from various feedstock materials. For example, Ohmiya and co-workers for the first time combined aldehydes with carboxylic acid-derived redox esters to synthesise ketones under NHC radical catalysis.11 Schiedt and Chi's group independently employed acyl imidazoles or carboxylic esters to react with Hantzsch ester for ketone synthesis by using light-driven NHC catalysis.12 Moreover, the NHC-catalysed three-component reactions were developed for the preparation of structurally complex ketones by Ohmiya, Studer, our group and other research teams.13 In these NHC catalysed reactions, the radical Breslow intermediate, generated through either the single-electron oxidation or single-electron reduction process, played a key role in the catalytic process (Scheme 1c). Encouraged by these previous achievements and our continuing interest in this area,14 the NHC radical catalysis, showing great potential in ketonisation chemistry,15 was envisaged to be a feasible alternative solution to the aforementioned acylative Suzuki-type couplings. Compared to the traditional preparation of ketones, such as Weinreb ketone synthesis,16 the radical organocatalytic methods could avoid the use of strong organometallic reagents at a low reaction temperature, thereby improving the synthetic generality and practicability. Herein, we report our recent progress in the development of cross-coupling of acid fluorides with alkyl trifluoroborates by using an NHC/photoredox dual catalysis strategy17 (Scheme 1d). Preliminary mechanistic studies, including control reactions, photophysical experimentation, determination of redox potential through cyclic voltammetry,18 and DFT calculations, suggested a unique sequential energy transfer and single-electron transfer activation pathway.
Results and discussion
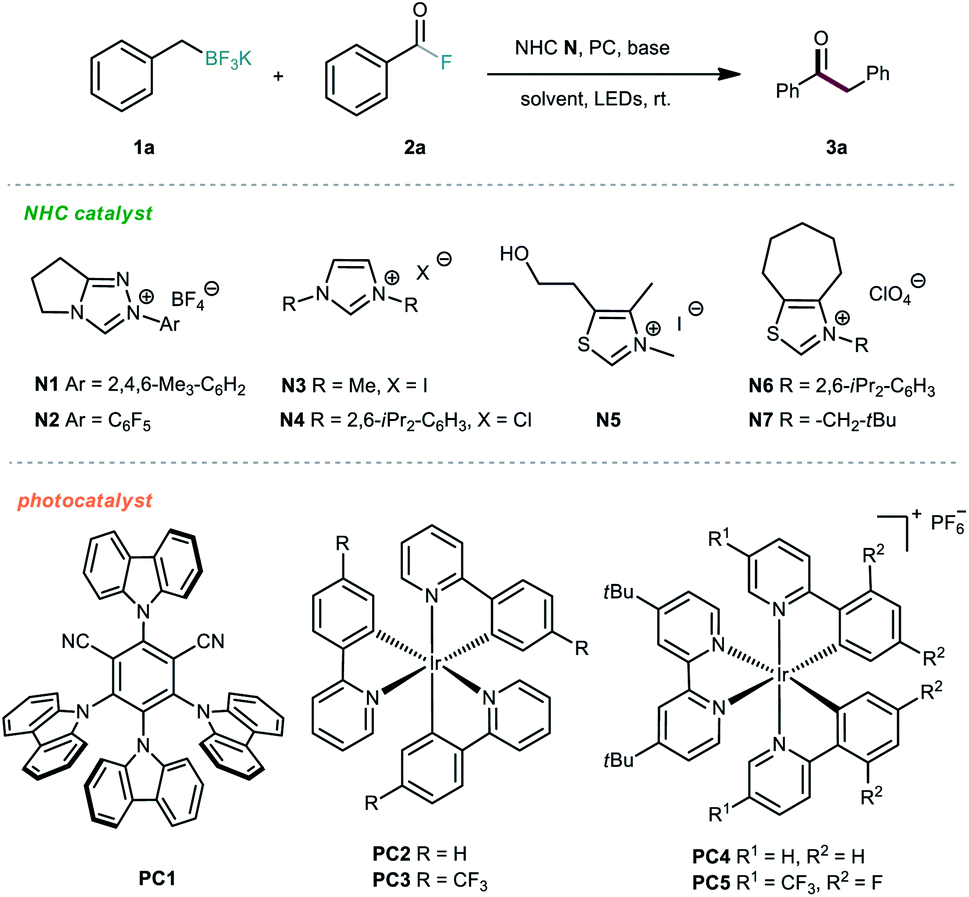
We commenced the investigations by selecting alkyl trifluoroborate 1a and benzoyl fluoride 2a as model substrates, and a blue light-emitting diode (LED) was used as the visible-light source. First, several photocatalysts were screened in the presence of NHC organocatalyst N1 and Cs2CO3 in acetone at room temperature. As shown in Table 1, the iridium-based photocatalyst PC4 could promote the desired reaction with the highest efficiency, providing the ketone product 3a in 61% yield (Table 1, entries 1–5). Then, various NHC catalysts (N2–N7), including triazolium, imidazolium and thiazolium salts, were tested in this dual catalytic system, but poor conversions were generally observed (Table 1, entries 6–11). The effect of the solvent was subsequently investigated, but no better results were achieved (Table 1, entries 12–15). Further screening of various inorganic and organic bases did not improve the reaction outcome (Table 1, entries 16–19). Finally, the solvent amount was screened, and we were delighted to find that the isolated yield could be improved to 71% by using 0.05 M reaction concentration (Table 1, entries 20–22).|
|
|||||
|---|---|---|---|---|---|
| Entry | NHC | PC | Solvent | Base | Yieldb (%) |
| a Unless noted otherwise, reactions were performed with 0.10 mmol of 1a, 0.20 mmol of 2a, 0.02 mmol of NHC catalyst N, 0.10 mmol of base and 2 mol% of photocatalyst PC in 1 mL solvent at room temperature for 3 h. b Isolated yields. c 0.5 mL solvent. d 2.0 mL solvent. e 3.0 mL solvent. | |||||
| 1 | N1 | PC1 | Acetone | Cs2CO3 | 25 |
| 2 | N1 | PC2 | Acetone | Cs2CO3 | <5 |
| 3 | N1 | PC3 | Acetone | Cs2CO3 | 15 |
| 4 | N1 | PC4 | Acetone | Cs2CO3 | 61 |
| 5 | N1 | PC5 | Acetone | Cs2CO3 | 51 |
| 6 | N2 | PC4 | Acetone | Cs2CO3 | 8 |
| 7 | N3 | PC4 | Acetone | Cs2CO3 | <5 |
| 8 | N4 | PC4 | Acetone | Cs2CO3 | 22 |
| 9 | N5 | PC4 | Acetone | Cs2CO3 | <5 |
| 10 | N6 | PC4 | Acetone | Cs2CO3 | 5 |
| 11 | N7 | PC4 | Acetone | Cs2CO3 | <5 |
| 12 | N1 | PC4 | Toluene | Cs2CO3 | 44 |
| 13 | N1 | PC4 | MeCN | Cs2CO3 | 42 |
| 14 | N1 | PC4 | DCM | Cs2CO3 | 42 |
| 15 | N1 | PC4 | DMF | Cs2CO3 | 27 |
| 16 | N1 | PC4 | Acetone | K2CO3 | 43 |
| 17 | N1 | PC4 | Acetone | NaOAc | 15 |
| 18 | N1 | PC4 | Acetone | Et3N | <5 |
| 29 | N1 | PC4 | Acetone | DBU | 33 |
| 20c | N1 | PC4 | Acetone | Cs2CO3 | 36 |
| 21 | N1 | PC4 | Acetone | Cs 2 CO 3 | 71 |
| 22e | N1 | PC4 | Acetone | Cs2CO3 | 64 |
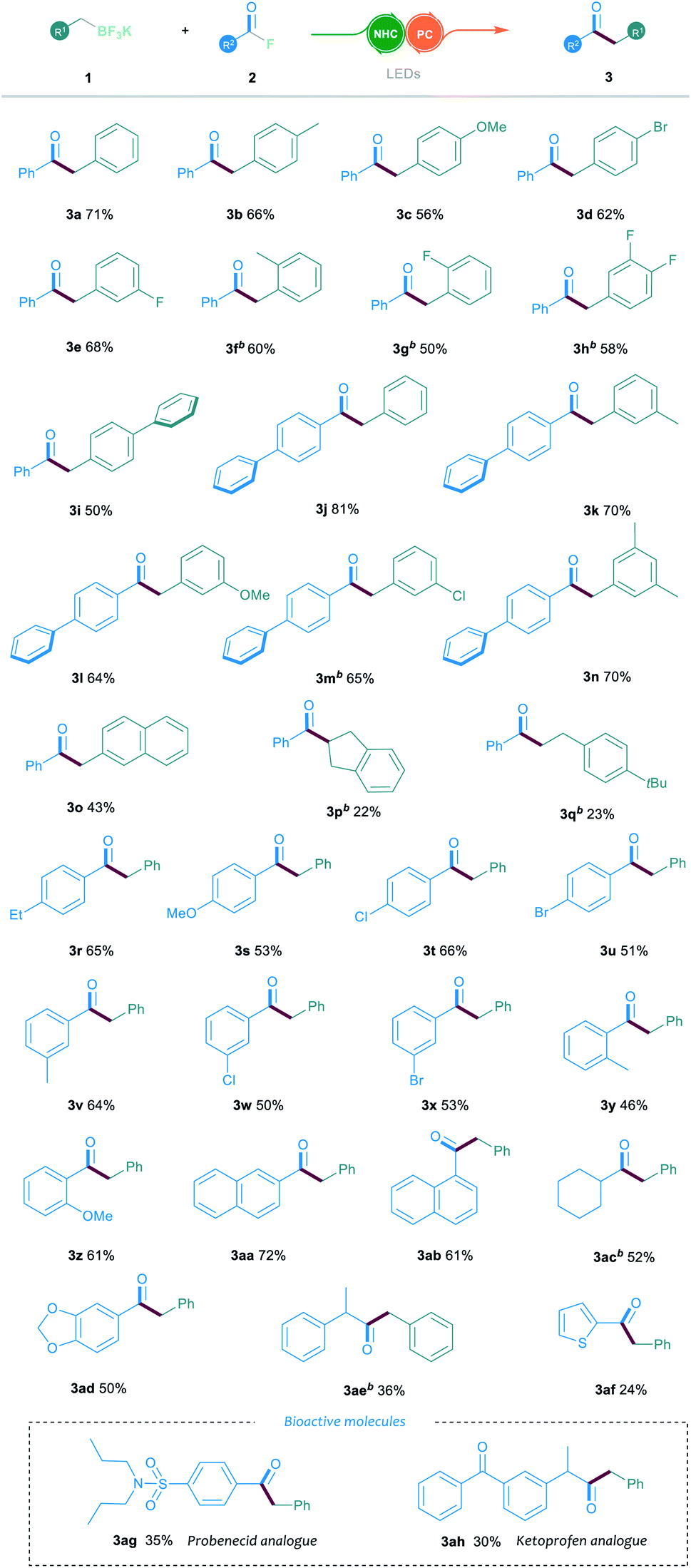
With the optimal conditions in hand, we then explored the generality and limitations of the NHC/photoredox dual catalysed cross-coupling reaction by testing various substituted alkyl trifluoroborates 1 and acyl fluorides 2. In general, the electronic variation of the aromatic ring on trifluoroborate 1 had limited effect on the reaction outcome. As shown in Table 2, the substituted trifluoroborates bearing either an electron-withdrawing or electron-donating group, such as the halogen, methyl or methoxyl group, were well tolerated to give the corresponding ketone products 3a–3e in 56–71% yields. It is noteworthy that the ortho-substituted trifluoroborate could smoothly participate in this reaction, affording the desired products 3f and 3g in 60% and 50% yields, respectively.
| a Unless noted otherwise, reactions were performed with 0.10 mmol of 1, 0.20 mmol of 2, 0.02 mmol of the N1 catalyst, 0.10 mmol of Cs2CO3 and 2% mmol of PC4 in 2 mL acetone at room temperature for 3 h; the yield refers to isolated yield of the reactions. b Using Ir[dF(CF3)ppy]2(Phen)PF6 instead of PC4. |
|---|
|
In addition, the reaction proceeded smoothly when trifluoroborate bearing a biphenyl group was used (3i). Furthermore, the reactions between various substituted trifluoroborates and the biphenyl benzoylfluoride were also efficient, which offered the ketone products 3j–3n in 64–81% yields. The 2-naphthyl-substituted 1 was compatible in this reaction, delivering the corresponding product 3o in 43% yield. However, when the indenyl or benzyl-ethyl trifluoroborates were tested, a decrease in the reaction yield was observed (3p and 3q). Then, the reactions with various aroyl fluorides 2 were examined under the established NHC/photoredox dual catalytic system. A broad range of acyl fluorides 2 with diverse electronic and steric properties were tested. As shown in Table 2, the aroyl fluorides bearing electron-withdrawing or electron-donating substituents at the para-position could all participate in this reaction and offer the products 3r–3u in 51–66% yields. The reaction could also proceed smoothly when testing various substituents in the meta-position of the aroyl fluorides (3v–3x). Notably, the ortho-substituted aroyl fluorides could successfully afford the desired products 3y and 3z in 46% and 61% yields, respectively. Aroyl fluorides bearing a 1-naphthyl or 2-naphthyl group reacted well to afford ketones 3aa and 3ab in 72% and 61% yields, respectively. Importantly, the reaction proceeded smoothly when the aliphatic acyl fluorides were employed as the substrates (3ac and 3ae). To our gratification, this catalytic protocol could also be suitable for the late-stage functionalisation of bioactive molecules. For example, the acyl fluorides, derived from probenecid and ketoprofen, could readily be converted into the corresponding ketone derivatives 3ag and 3ah through the established cross-coupling reaction.
To further illustrate the generality and expandability of this protocol, this catalytic method was subsequently tested for the acylation reaction of α-silyl carbazole 4. In general, the reactions proceeded efficiently with various substituted α-silyl carbazoles 4 and acyl fluorides 2. As shown in Table 3, the aroyl fluorides bearing either electron-withdrawing or electron-donating substituents at the para-, meta- or ortho-position could all participate in this reaction and offered the products 5a–5j in 50–90% yields. In addition, heteroaryl fluoride was also compatible in the reaction albeit with lower yield (5k). Notably, the acyl fluoride featuring a cycloheptyl substituent could also be suitable for the catalytic transformation to provide the corresponding product 5l with good yield. Furthermore, the reactions between various substituted α-silyl carbazoles and the benzoyl fluorides also occurred efficiently, offering the ketone products 5m–5v in 56–90% yields.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| a Reactions were performed with 0.10 mmol of 1, 0.20 mmol of 2, 0.02 mmol of N1, 0.10 mmol of Cs2CO3 and 2% mmol of PC4 in 1 mL MeCN at room temperature for 1 h, isolated yields. |
|---|

|
To gain insights into the dual catalytic system, we set up control experiments to shed light on the reaction mechanism (Scheme 2). First, in the presence of alkyl trifluoroborate 1a, the direct use of acyl azolium ion 6 under photoredox catalysis provided ketone 3a in 74% yield.19 This result indicated that the acyl azolium intermediate, in situ generated from acyl fluorides and the NHC catalyst, should be involved in this catalytic reaction. Moreover, the catalytic process could be completely suppressed by adding TEMPO in the reaction, suggesting a radical nature of this transformation (Scheme 2a). Next, a light on–off experiment was carried out with alkyl trifluoroborate 1a and benzoyl fluoride 2a (Scheme 2b). We observed that product formation only occurred during the periods of constant irradiation. In addition, we also measured the quantum yield of the reaction, and determined that φ was 0.69 (see the ESI† for details).20 The results indicated that the reaction underwent a catalytic radical process rather than a radical chain pathway.
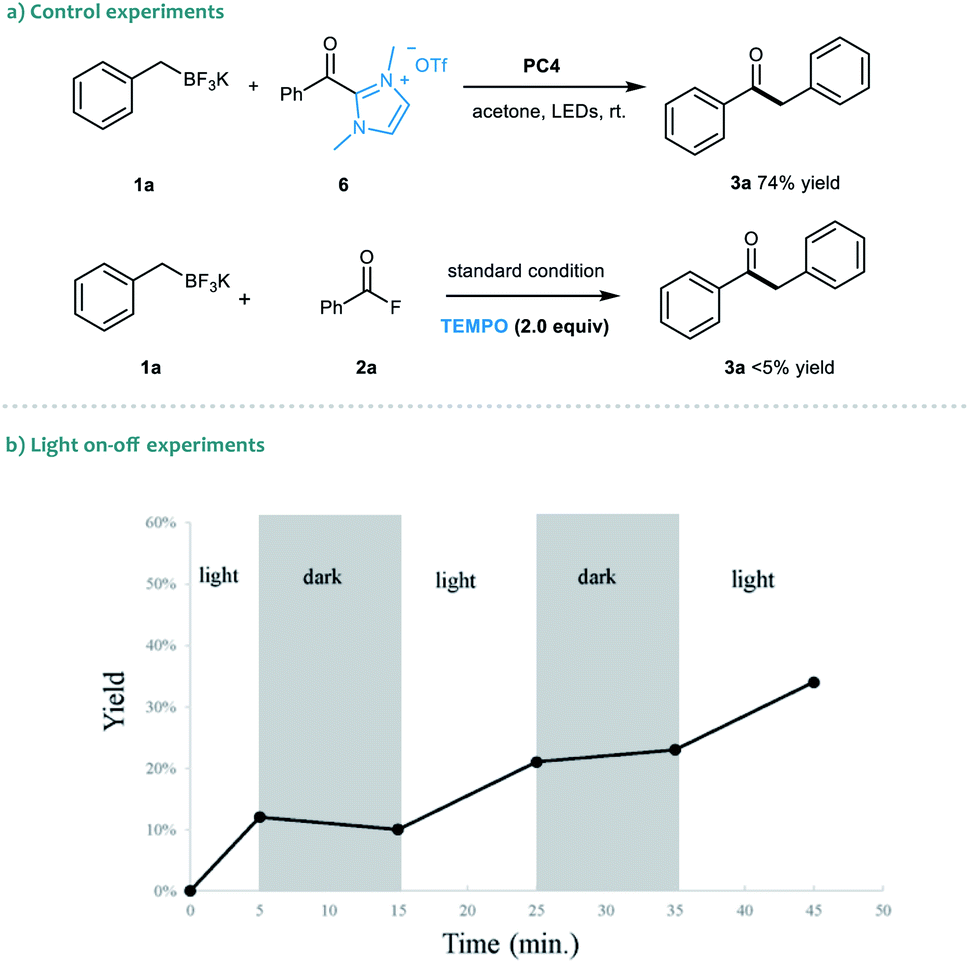 | ||
| Scheme 2 Control experiments and the light on–off experiment. | ||
Moreover, the UV-vis absorption spectrum of the photocatalyst [Ir(ppy)2(dtbbpy)]PF6 revealed significant absorption of visible light, and the tail wavelength reached over 500 nm (Scheme 3a, I). Without a photocatalyst, no absorption was observed in the spectrum of visible light for the substrates, the NHC catalyst or their combinations (Scheme 3a, II–V). These results indicated that the photocatalytic process is initiated by the visible light excitation of the photocatalyst. Besides, we also performed Stern–Volmer fluorescence quenching experiments. As illustrated in Scheme 3b, the excited state of photocatalyst PC4 could be readily quenched by acyl azolium, rather than trifluoroborates. Furthermore, the reduction potential of acyl azolium 6 (Ered = −1.31 V vs. SCE) was measured by cyclic voltammetry analysis (Scheme 3c). Compared with the oxidation potential of the excited [Ir(ppy)2(dtbbpy)]PF6 (E1/2IV/III* = −0.96 V vs. SCE),21 a single-electron transfer between the excited photocatalyst and acyl acylium is unlikely. Then, we performed density functional theory (DFT) calculations of the triplet energy of acyl azolium intermediates and photocatalyst using the (U)B3LYP functional (for computational details, see the ESI†).22 The calculated triplet–singlet energy gaps [ΔG(T1–S0)] of the key intermediates 6 and 7 are 39.5 and 27.1 kcal mol−1, respectively, which are below the calculated [ΔG(T1–S0)] (49.7 kcal mol−1) and experimentally determined result (49.2 kcal mol−1)23 of PC4. Thus, these experimental and computational results indicated that an energy transfer (EnT) process might be feasible under the current circumstance.24
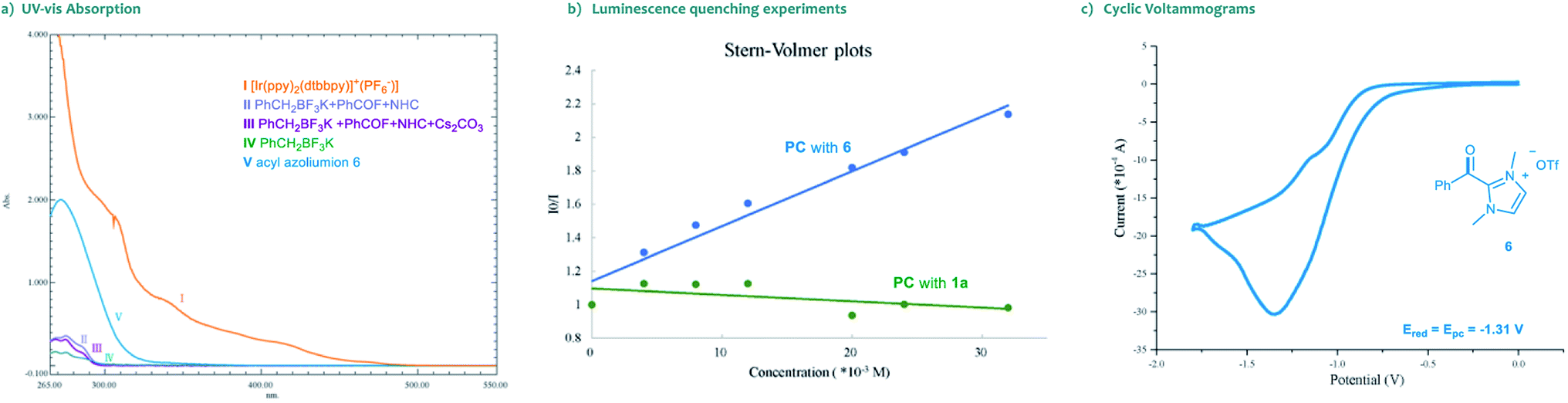 | ||
| Scheme 3 Mechanistic investigations of the reaction. | ||
On the basis of the above observations, the mechanism of the present reaction is proposed in Scheme 4. Upon visible light irradiation, the Ir-based photocatalyst was excited to its excited state. Then, the acyl azolium intermediate, generated from the NHC catalyst and acyl fluoride, was activated by the triple state photocatalyst through an EnT process. Subsequently, the single electron transfer between the excited acyl azolium and benzylic trifluoroborate generated a ketyl radical and a benzyl radical species. Finally, the cross-coupling of these two radical intermediates afforded a ketone product and released the free carbene catalyst.
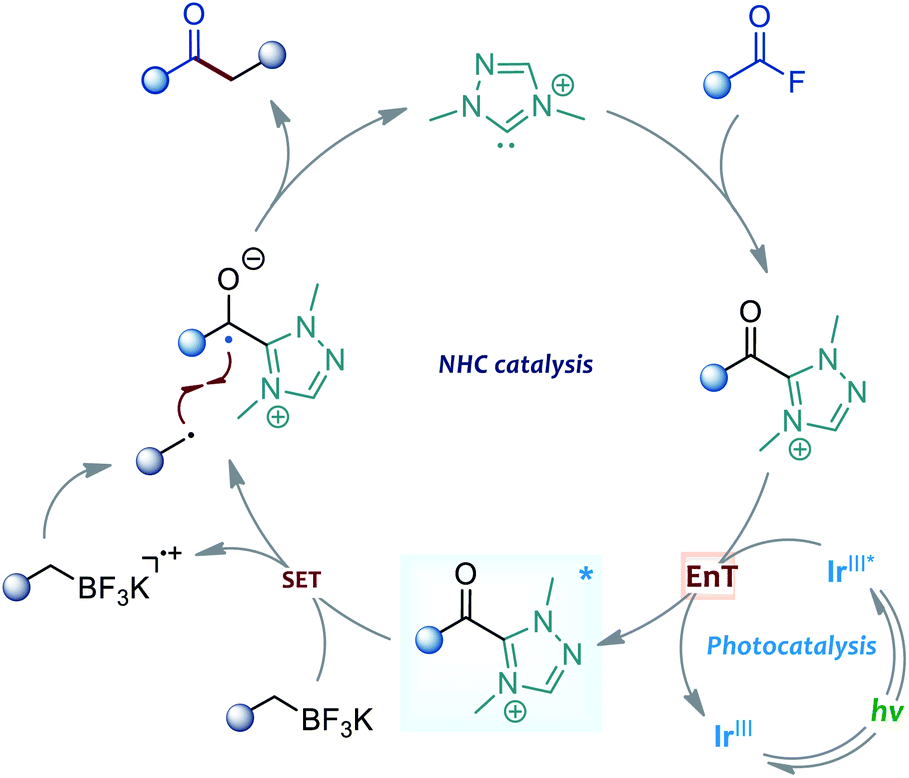 | ||
| Scheme 4 Plausible reaction pathway. | ||
Conclusions
In summary, we have developed an alternative strategy to realise the acylative Suzuki-type cross-coupling of alkyl trifluoroborates and acid fluorides by merging NHC organocatalysis with photoredox catalysis. With the developed dual catalytic system, a broad spectrum of ketones could be facilely synthesised from easily accessible substrates under mild reaction conditions. To further demonstrate the generality of this protocol, the catalytic acylation of α-silyl carbazoles was also disclosed. In addition, we have performed several mechanistic investigations, such as control reactions, photophysical experimentation, cyclic voltammetry analysis, and computational stimulations, to shed light on the unique sequential energy transfer and single-electron transfer activation pathway. Further detailed studies on the reaction mechanism and extension of this dual catalytic system to activate more challenging substrates are currently underway in our laboratory, and the results will be reported in due course.Data availability
All experimental data is available in the ESI.†Author contributions
J.-L. L. and H. H. conceived and designed the study. H. H., Q.-S. D., S.-L. Y., and Y.-M. T. performed the synthetic experiments with input from J.-L. L. The mechanistic investigations were performed by H. H., H.-J. L., and Q.-Z. L. The DFT calculations were performed by X. Z. and T. Q. The manuscript was prepared by J.-L. L. and H. H. All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from NSFC (21871031 and 22071011), the Science & Technology Department of Sichuan Province (2021YJ0404), and Longquan Talents Program, and the start-up founding from Chengdu University is gratefully acknowledged.Notes and references
- (a) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6723–6737 Search PubMed; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- N. Schneider, D. M. Lowe, R. A. Sayle, M. A. Tarselli and G. A. Landrum, J. Med. Chem., 2016, 59, 4385–4402 CrossRef CAS PubMed.
- (a) J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010 Search PubMed; (b) L. S. Hegedus and B. C. G. Soderberg, Transition Metals in the Synthesis of Complex Organic Molecules, University Science Books, Sausalito, CA, 2010 Search PubMed.
- For related reviews, see: (a) A. Sekiya and N. Ishikawa, J. Org. Chem., 1977, 125, 281–290 CrossRef CAS; (b) M. J. Zacuto, C. S. Shultz and M. Journet, Org. Process Res. Dev., 2011, 15, 158–161 CrossRef CAS; (c) D. Haas, J. M. Hammann, R. Greiner and P. Knochel, ACS Catal., 2016, 6, 1540–1552 CrossRef CAS; (d) C. Cordovilla, C. Bartolomé, J. M. Martínez-Ilarduya and P. Espinet, ACS Catal., 2015, 5, 3040–3053 CrossRef CAS; (e) B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed; (f) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed; (g) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed.
- For selected examples, see: (a) C. S. Cho, K. Itotani and S. Uemura, J. Org. Chem., 1993, 443, 253–259 CrossRef CAS; (b) L. J. Goossen, D. Koley, H. L. Hermann and W. Thiel, J. Am. Chem. Soc., 2005, 127, 11102–11114 CrossRef CAS PubMed; (c) R. Kakino, H. Narahashi, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2002, 75, 1333–1345 CrossRef CAS; (d) B. W. Xin, Y. H. Zhang and K. Cheng, J. Org. Chem., 2006, 71, 5725–5731 CrossRef CAS PubMed; (e) C. G. Frost and K. J. Wadsworth, Chem. Commun., 2001, 2316–2317 RSC; (f) S. F. Si, C. Wang, N. Zhang and G. Zou, J. Org. Chem., 2016, 81, 4364–4370 CrossRef CAS PubMed; (g) L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260–11261 CrossRef CAS; (h) Y. Yu and L. S. Liebeskind, J. Org. Chem., 2004, 69, 3554–3557 CrossRef CAS PubMed; (i) H. Yang, H. Li, R. Wittenberg, M. Egi, W. Huang and L. S. Liebeskind, J. Am. Chem. Soc., 2007, 129, 1132–1140 CrossRef CAS PubMed; (j) L. J. Gooßen and K. Ghosh, Chem. Commun., 2001, 2084–2085 Search PubMed; (k) H. Wu, B. Xu, Y. Li, F. Hong, D. Zhu, J. Jian, X. Pu and Z. Zeng, J. Org. Chem., 2016, 81, 2987–2992 CrossRef CAS PubMed; (l) H. Wu, B. Xu, Y. Li, F. Hong, D. Zhu, J. Jian, X. Pu and Z. Zeng, J. Org. Chem., 2016, 81, 2987–2992 CrossRef CAS PubMed; (m) H. Tatamidani, F. Kakiuchi and N. Chatani, Org. Lett., 2004, 6, 3597–3599 CrossRef CAS PubMed; (n) J. Wang, S. Zuo, W. Chen, X. Zhang, K. Tan, Y. Tian and J. Wang, J. Org. Chem., 2013, 78, 8217–8231 CrossRef CAS PubMed; (o) T. B. Halima, W. Zhang, I. Yalaoui, X. Hong, Y. F. Yang, K. N. Houk and S. G. Newman, J. Am. Chem. Soc., 2017, 139, 1311–1318 CrossRef PubMed , for a related review, see:; (p) H. Prokopcová and C. O. Kappe, Angew. Chem., Int. Ed., 2009, 48, 2276–2286 CrossRef PubMed.
- Y. Ogiwara and N. Sakai, Angew. Chem., Int. Ed., 2020, 59, 574–594 CrossRef CAS PubMed.
- Y. Zhang and T. Rovis, J. Am. Chem. Soc., 2004, 126, 15964–15965 CrossRef CAS PubMed.
- (a) Y. Ogiwara, D. Sakino, Y. Sakurai and N. Sakai, Eur. J. Org. Chem., 2017, 4324–4327 CrossRef CAS; (b) C. A. Malapit, J. R. Bour, C. E. Brigham and M. S. Sanford, Nature, 2018, 563, 100–104 CrossRef CAS PubMed.
- (a) T. Ishii, K. Nagao and H. Ohmiya, Chem. Sci., 2020, 11, 5630–5636 RSC; (b) Q. Z. Li, R. Zeng, B. Han and J. L. Li, Chem.–Eur. J., 2021, 27, 3238–3250 CrossRef CAS PubMed; (c) Q. Liu and X. Y. Chen, Org. Chem. Front., 2020, 7, 2082–2087 RSC; (d) A. Mavroskoufis, M. Jakob and M. N. Hopkinson, ChemPhotoChem, 2020, 4, 5147–5153 CrossRef CAS; (e) L. Dai and S. Ye, Chin. Chem. Lett., 2021, 32, 660–667 CrossRef CAS; (f) H. Ohmiya, ACS Catal., 2020, 10, 6862–6869 CrossRef CAS; (g) J. Liu, X.-N. Xing, J.-H. Huang, L.-Q. Lu and W.-J. Xiao, Chem. Sci., 2020, 11, 10605–10613 RSC.
- (a) T. Ishii, K. Ota, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 14073–14077 CrossRef CAS PubMed; (b) T. Ishii, Y. Kakeno, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 3854–3858 CrossRef CAS PubMed.
- (a) A. V. Bay, K. P. Fitzpatrick, R. C. Betori and K. A. Scheidt, Angew. Chem., Int. Ed., 2020, 59, 9143–9148 CrossRef CAS PubMed; (b) S.-C. Ren, W.-X. Lv, X. Yang, J.-L. Yan, J. Xu, F.-X. Wang, L. Hao, H. Chai, Z. Jin and Y. R. Chi, ACS Catal., 2021, 11, 2925–2934 CrossRef CAS; (c) A. V. Bay, K. P. Fitzpatrick, G. A. González-Montiel, A. O. Farah, P. H. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2021, 60, 17925–17931 CrossRef CAS PubMed; (d) A. A. Bayly, B. R. McDonald, M. Mrksich and K. A. Scheidt, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 13261–13266 CrossRef CAS PubMedfor other elegant examples, see: (e) K. Liu and A. Studer, J. Am. Chem. Soc., 2021, 143, 4903–4909 CrossRef CAS PubMed; (f) Q.-Y. Meng, N. Döben and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 19956–119960 CrossRef CAS PubMed; (g) Q.-Y. Meng, L. Lezius and A. Studer, Nat. Commun., 2021, 12, 2068–2075 CrossRef CAS PubMed; (h) Z. Zuo, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 25252–25257 CrossRef CAS PubMed; (i) M.-S. Liu, L. Min, B.-H. Chen and W. Shu, ACS Catal., 2021, 11, 9715–9721 CrossRef CAS; (j) H. Sheng, Q. Liu, X.-D. Su, Y. Lu, Z.-X. Wang and X.-Y. Chen, Org. Lett., 2020, 22, 7187–7192 CrossRef CAS PubMed; (k) D. Du, K. Zhang, R. Ma, L. Chen, J. Gao, T. Lu, Z. Shi and J. Feng, Org. Lett., 2020, 22, 6370–6375 CrossRef CAS PubMed; (l) K.-Q. Chen, Z.-X. Wang and X.-Y. Chen, Org. Lett., 2020, 22, 8059–8064 CrossRef CAS PubMed; (m) A. Mavroskoufis, K. Rajes, P. Golz, A. Agrawal, V. Ruß, J. P. Götze and M. N. Hopkinson, Angew. Chem., Int. Ed., 2020, 59, 3190–3194 CrossRef CAS PubMed; (n) L. Dai, Z.-H. Xia, Y.-Y. Gao, Z.-H. Gao and S. Ye, Angew. Chem., Int. Ed., 2019, 58, 18124–18130 CrossRef CAS PubMed.
- (a) Y. Matsuki, N. Ohnishi, Y. Kakeno, S. Takemoto, T. Ishii, K. Nagao and H. Ohmiya, Nat. Commun., 2021, 12, 3848 CrossRef CAS PubMed; (b) Y. Kakeno, M. Kusakabe, K. Nagao and H. Ohmiya, ACS Catal., 2020, 10, 8524–8529 CrossRef CAS; (c) W. Liu, A. Vianna, Z. Zhang, S. Huang, L. Huang, M. Melaimi, G. Bertrand and X. Yan, Chem. Catal., 2021, 1, 196–206 CrossRef; (d) Z. Li, M. Huang, X. Zhang, J. Chen and Y. Huang, ACS Catal., 2021, 11, 10123–10130 CrossRef CAS; (e) K. OTa, K. Nagao and H. Ohmiya, Org. Lett., 2020, 22, 3922–3925 CrossRef CAS PubMed; (f) I. Kim, H. Im, H. Lee and S. Hong, Chem. Sci., 2020, 11, 3192–3197 RSC; (g) Y. Gao, Y. Quan, Z. Li, L. Gao, Z. Zhang, X. Zou, R. Yan, Y. Qu and K. Guo, Org. Lett., 2021, 23, 183–189 CrossRef CAS PubMed; (h) J. L. Li, Y. Q. Liu, W. L. Zou, R. Zeng, X. Zhang, Y. Liu, B. Han, Y. He, H. J. Leng and Q. Z. Li, Angew. Chem., Int. Ed., 2020, 59, 1863–1870 CrossRef CAS PubMed; (i) B. Zhang, Q. Peng, D. Guo and J. Wang, Org. Lett., 2020, 22, 443–447 CrossRef CAS PubMed; (j) H. B. Yang, Z. H. Wang, J. M. Li and C. Wu, Chem. Commun., 2020, 56, 3801–3804 RSC.
- (a) J. L. Li, L. Fu, J. Wu, K. C. Yang, Q. Z. Li, X. J. Gou, C. Peng, B. Han and X. D. Shen, Chem. Commun., 2017, 53, 6875–6878 RSC; (b) K. C. Yang, Q. Z. Li, Y. Liu, Q. Q. He, Y. Liu, H. J. Leng, A. Q. Jia, S. Ramachandran and J. L. Li, Org. Lett., 2018, 20, 7518–7521 CrossRef CAS PubMed; (c) Q. Li, L. Zhou, X.-D. Shen, K.-C. Yang, X. Zhang, Q.-D. Dai, H.-J. Leng, Q.-Z. Li and J.-L. Li, Angew. Chem., Int. Ed., 2018, 57, 1913–1917 CrossRef CAS PubMed; (d) H. Huang, Q.-Z. Li, Y.-Q. Liu, H.-J. Leng, P. Xiang, Q.-S. Dai, X.-H. He, W. Huang and J.-L. Li, Org. Chem. Front., 2020, 7, 3862–3867 RSC.
- (a) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed; (b) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (c) X. Y. Chen, Z. H. Gao and S. Ye, Acc. Chem. Res., 2020, 53, 690–702 CrossRef CAS PubMed.
- For related reviews, see: (a) W. Zhao and W. Liu, Chin. J. Org. Chem., 2015, 35, 55–69 CrossRef CAS; (b) N. Chalotra, S. Sultan and B. A. Shah, Asian J. Org. Chem., 2020, 9, 863–881 CrossRef CAS; (c) S. G. Davies, A. M. Fletcher and J. E. Thomson, Chem. Commun., 2013, 49, 8586–8598 RSC; (d) M. Nowak, Synlett, 2015, 26, 561–562 CrossRef CAS , for selected examples, see:; (e) S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS; (f) V. Malathong and S. D. Rychnovsky, Org. Lett., 2009, 11, 4220–4223 CrossRef CAS PubMed; (g) C. O. Kangani, D. E. Kelley and B. W. Day, Tetrahedron Lett., 2006, 47, 6289–6292 CrossRef CAS.
- (a) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (b) S. Witzel, A. S. K. Hashmi and J. Xie, Chem. Rev., 2021, 121, 8868–8925 CrossRef CAS PubMed; (c) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- The triazole-derived acyl azoliums are unstable and are difficult to isolate and handle. Therefore, the imidazole-derived acyl azoliums are used for the mechanistic studies, and the imidazolium species also proved to be the productive intermediate which was confirmed by the result of control experiments. Studer and co-workers also used a similar mechanistic investigation strategy; for details, see ref. 12f.
- (a) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019–8030 CrossRef CAS PubMed; (b) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- (a) K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS PubMed; (b) K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Chem. Rev., 2021, 122, 2353–2428 CrossRef PubMed.
- (a) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzera and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data for new compounds and crystallographic data. See DOI: 10.1039/d1sc06102j |
| This journal is © The Royal Society of Chemistry 2022 |