
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling the optical and catalytic properties of artificial metalloenzyme photocatalysts using chemogenetic engineering†
Yasmine S.
Zubi‡
 a,
Bingqing
Liu‡
a,
Yifan
Gu§
b,
Dipankar
Sahoo¶
a and
Jared C.
Lewis
*a
a,
Bingqing
Liu‡
a,
Yifan
Gu§
b,
Dipankar
Sahoo¶
a and
Jared C.
Lewis
*a
aDepartment of Chemistry, Indiana University, Bloomington, Indiana 47405, USA. E-mail: jcl3@iu.edu
bDepartment of Chemistry, University of Chicago, Chicago, IL 60637, USA
First published on 10th January 2022
Abstract
Visible light photocatalysis enables a broad range of organic transformations that proceed via single electron or energy transfer. Metal polypyridyl complexes are among the most commonly employed visible light photocatalysts. The photophysical properties of these complexes have been extensively studied and can be tuned by modifying the substituents on the pyridine ligands. On the other hand, ligand modifications that enable substrate binding to control reaction selectivity remain rare. Given the exquisite control that enzymes exert over electron and energy transfer processes in nature, we envisioned that artificial metalloenzymes (ArMs) created by incorporating Ru(II) polypyridyl complexes into a suitable protein scaffold could provide a means to control photocatalyst properties. This study describes approaches to create covalent and non-covalent ArMs from a variety of Ru(II) polypyridyl cofactors and a prolyl oligopeptidase scaffold. A panel of ArMs with enhanced photophysical properties were engineered, and the nature of the scaffold/cofactor interactions in these systems was investigated. These ArMs provided higher yields and rates than Ru(Bpy)32+ for the reductive cyclization of dienones and the [2 + 2] photocycloaddition between C-cinnamoyl imidazole and 4-methoxystyrene, suggesting that protein scaffolds could provide a means to improve the efficiency of visible light photocatalysts.
Introduction
Visible light photocatalysts enable a wide range of chemical reactions and provide access to compounds that are difficult to produce using other synthetic methods.1,2 After photoexcitation, these catalysts can react with substrates via single-electron transfer (SET) or energy transfer (ET) to produce high energy intermediates. In the former case, photoredox catalysis is enabled by a second SET event with a sacrificial oxidant or reductant (the order of reaction with the substrate and oxidant/reductant can also be reversed), while in the latter case, the bidirectional nature of ET leads to catalyst turnover. Both organic3,4 and inorganic5 molecules are used as photocatalysts, but metal polypyridyl complexes like Ru(Bpy)32+, Ir(Ppy)3, and derivatives thereof are particularly valuable for synthetic organic methodology.6 Upon absorbing visible light, these complexes efficiently form excited states that are suitably long-lived to enable reaction with organic substrates.7,8 Furthermore, metal polypyridyl complexes exhibit reversible redox properties and are not prone to decomposition or photobleaching.5Structure–activity relationships have provided insight into the effects of polypyridine substituents on the photophysical and electronic properties of metal polypyridine complexes.5,9 Ligand modification has also been used to tune the selectivity of these complexes in reactions involving organic substrates.10 Given that many photocatalysts are only involved in the initial electron or energy transfer to/from a substrate and that the resulting high energy intermediates can undergo rapid subsequent reactions, the substrate must either be in a chiral environment during the excitation step and remain there during the selectivity-determining step or later engage with a chiral co-catalyst during the selectivity-determining step.11 Reactions proceeding via electron transfer can also proceed via radical chain mechanisms that do not involve the catalyst and compete with the desired closed catalytic cycle to erode catalyst controlled selectivity.12,13
Many enzyme-catalyzed reactions,14,15 including recently reported examples of non-native photocatalytic reactions,16 are initiated via SET, and highly efficient ET occurs in photosystems I and II.17 The remarkable selectivity and specificity of these systems results from their ability to bind intermediates and cofactors throughout their respective catalytic cycles. On the other hand, the functional properties of such systems are limited relative to the rapidly expanding repertoire of synthetic photoredox18,19 and energy transfer3 catalysts. A variety of approaches have therefore been taken to explore the photophysical and photocatalytic capabilities of metal polypyridine complexes in the context of protein scaffolds.20 For example, Gray has extensively studied SET in Ru(II)-polypyridyl modified-P450 BM3 enzymes21 and other scaffolds.22–24 Cheruzel later developed related systems to enable light-driven P450 hydroxylation catalysis25 and to modulate the photophysical and catalytic properties of these systems through pyridyl ligand substitution.26 Ward linked Ru(II)-polypyridyl complexes to (strept)avidin via non-covalent biotin binding27 or covalent cysteine bioconjugation28,29 and explored SET in donor-photosensitizer-acceptor triads involving these systems. Importantly, in these cases, the metal polypyridine complexes were solvent exposed, often residing in shallow clefts with limited capacity to interact with the cofactor. While changes in the photophysical or redox properties of the protein-linked complexes were reported in these examples20–29 and others,30–33 none of these systems were used as photocatalysts for direct transformations of organic substrates.
We envisioned that site-specific incorporation of metal polypyridine complexes into a suitable protein scaffold might enable greater control over the photophysical and catalytic properties of these complexes. Herein, we report the synthesis and characterization of a series of artificial metalloenzymes (ArMs) comprising different Ru(Bpy)32+ cofactors linked either covalently (1a–f) or non-covalently (3a–i) to a prolyl oligopeptidase (POP)34 from Pyrococcus furiosus (Pfu)35 (Fig. 1a). Systematic manipulation of both the cofactor and the POP scaffold were conducted to establish how structural changes impacted cofactor binding and photophysical properties. Improved binding, luminescence intensities, and luminescence lifetimes were observed for ArM variants due to hydrophobic and electrostatic interactions with residues in the POP active site. The catalytic activity of a panel of covalent ArMs was then evaluated using a reductive cyclization reaction proceeding via SET and a [2 + 2] cycloaddition enabled by ET. Yields of the desired products and reaction rates were increased by the engineered ArMs in both cases. These results indicate that POP variants can both modulate metal polypyridyl photophysical properties and improve photocatalysis yields, highlighting their potential as scaffolds for the further development of ArM photocatalysts.
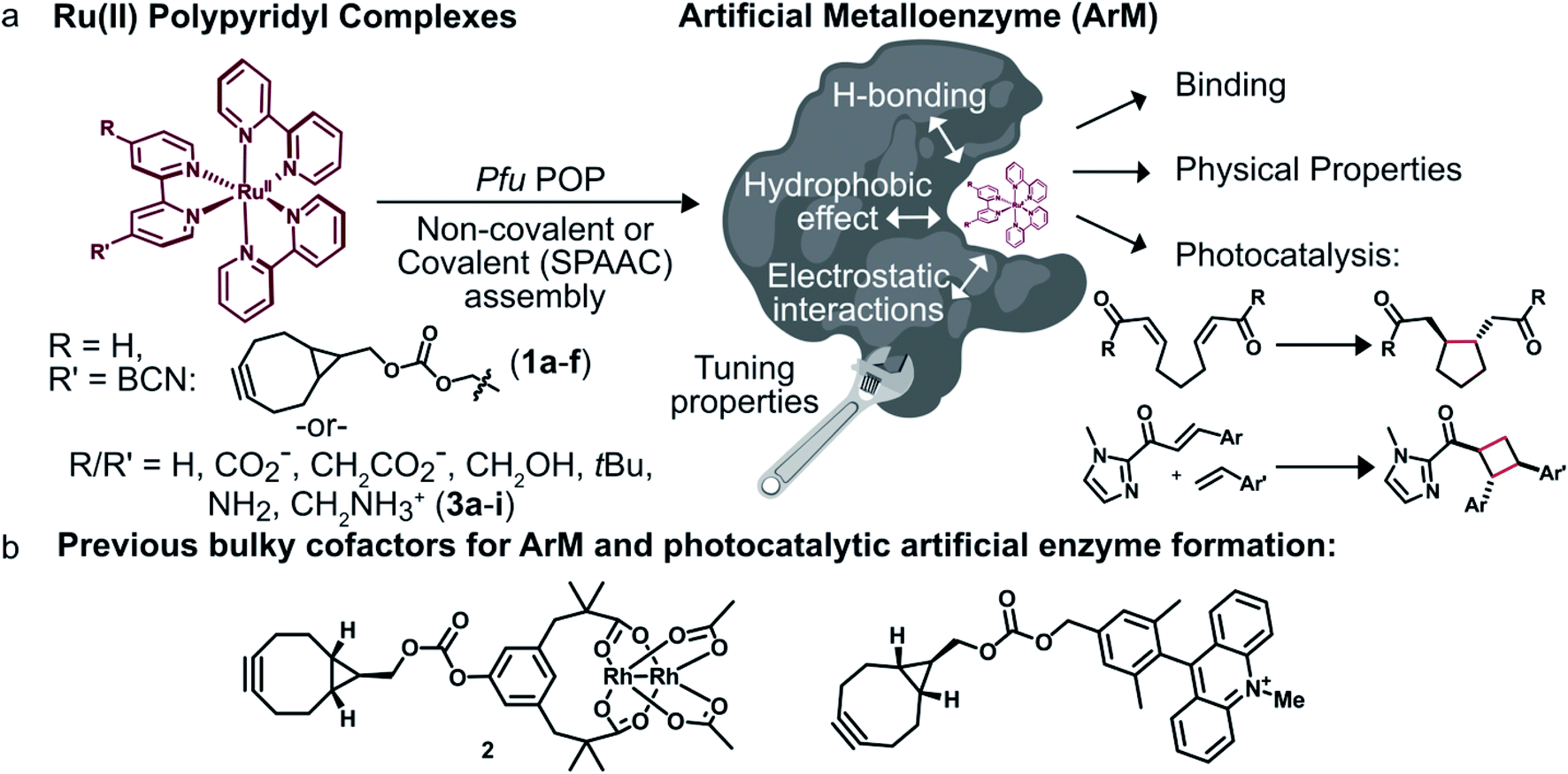 | ||
| Fig. 1 Overview of ArM photocatalyst preparation and characterization. (a) Combining conventional photocatalysts with a protein scaffold enables modulation of the cofactors' properties through interactions with the biomolecule. (b) Previously reported dirhodium38,39 and acridinium37 cofactors. | ||
Results
Scaffold selection
To modulate ArM cofactor properties and catalysis, a protein scaffold must possess sufficient space to accommodate the cofactor(s) of interest while still allowing for beneficial interactions with the cofactor and potential substrates.36 The large size and octahedral geometry of metal polypyridyl complexes places significant constraints on the types of scaffolds that can be used. Our group has used Pfu POP as a scaffold for ArM formation via bioconjugation with bulky acridinium cofactors37 and dirhodium tetracarboxylate (2, Fig. 1b) cofactors.38,39 MD simulations of the latter indicate that POP can fully encapsulate 2 while arraying active site residues proximal to it, and the high selectivity observed for a variety of reactions catalyzed by dirhodium ArMs indicates that these interactions significantly influence cofactor reactivity.38,40 The POP scaffold was therefore selected for construction of ArM photocatalysts.Covalent ArM preparation and characterization
In analogy to the dirhodium and acridinium systems noted above (Fig. 1b), strain-promoted azide–alkyne cycloaddition (SPAAC) was used to covalently link endo-[6.1.0]-bicyclononyne (BCN)-substituted Ru(bpy)32+ cofactor 1a within POP scaffolds containing a genetically encoded azidophenylalanine (Z) residue.41 Cofactor 1a was synthesized by introducing 4-hydroxymethyl-Bpy ii (ref. 42) (prepared from commercially available methyl isonicotinate i) onto Ru(Bpy)2Cl2 (ref. 43) via ligand exchange to generate iii and then reacting this complex with carbonate iv (ref. 44) (Scheme 1). The exo isomer of the cofactor (1b) was also prepared since these isomers could differentially orient the Ru(Bpy)32+ fragment of 1a within the POP active site. Furthermore, because octahedral Ru(Bpy)32+ complexes can adopt Δ and Λ configurations,45 resolved Δ- and Λ-Ru(Bpy)2(Py)2 tartrate salts were used to prepare the endo/Λ, endo/Δ, exo/Λ, and exo/Δ cofactors (1c–f, respectively, ESI Fig. 1†).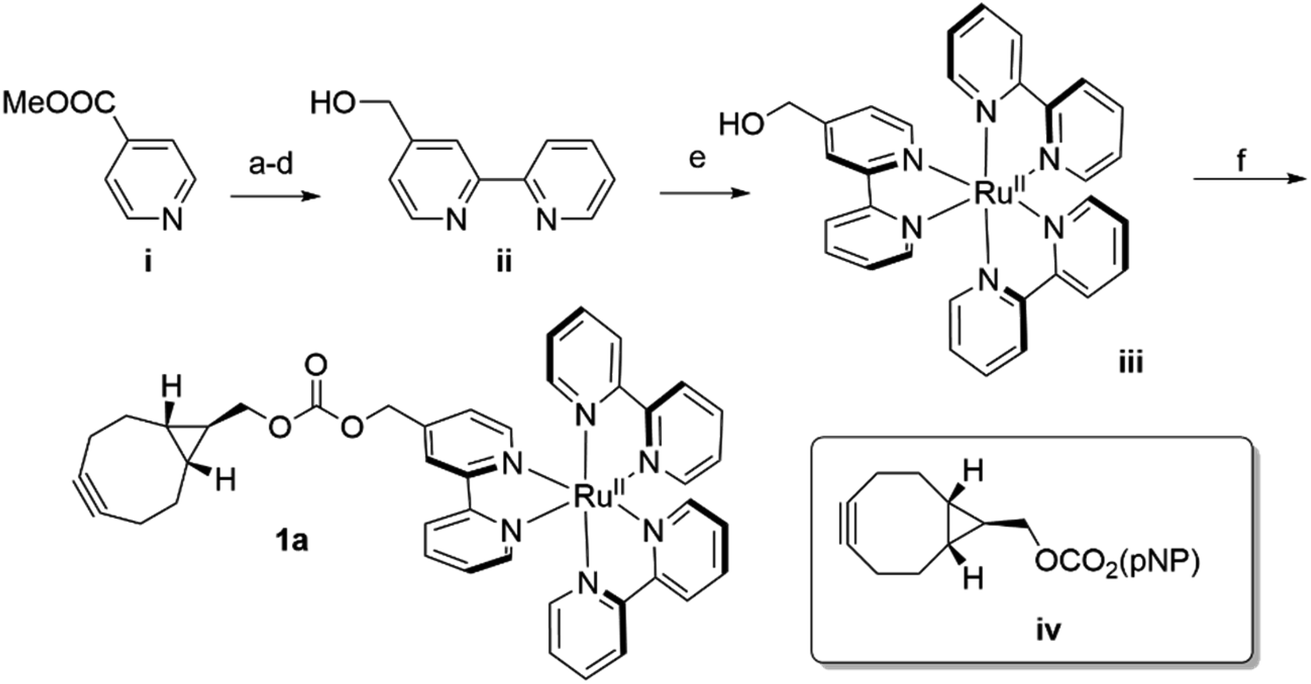 | ||
| Scheme 1 Synthesis of BCN-substituted Ru(Bpy)32+ cofactor 1a (pNP = p-nitrophenyl ester). (a) i, H2O2, acetic acid, 70 °C, O/N (92%); (b) pyridine N-oxide, Pd(OAc)2, K3CO3, 2-bromopyridine, [P(tBu)3H]BF4, toluene, RT → 120 °C, O/N (54%); (c) 4-methoxycarbonyl-2-(pyridin-2-yl)pyridine N-oxide, Pd/C, EtOH, H2, RT, 4 hours (95%); (d) 4-methoxycarbonyl-2-(pyridin-2-yl)pyridine, LiBH4, THF, 0 °C (1 hour) → RT, 12 hours (83%); (e) 2-(pyrid-2-yl)-4-hydroxymethyl-pyridine ii, cis-Ru(Bpy)2Cl2, EtOH, reflux (O/N) → RT, NH4PF6, H2O (54%); (f) iii, NaH, ACN, RT, 5 min → iv, THF, RT, 1 hour → acetone, NH4PF6, H2O (84%). | ||
POP-Z variants were then generated by genetically encoding the Z residue at active site positions 53, 99, 326, 338, and 477 to explore the effects of cofactor linkage site on ArM function. All variants expressed in good yields (∼25–100 mg L−1), and intact ESI-MS confirmed the expected masses of the proteins (ESI Fig. 2†). SPAAC conditions developed for dirhodium ArMs were used to link 1a to these scaffolds,40 and intact ESI-MS again confirmed the expected masses of the ArMs (Fig. 2A and ESI Fig. 2†). Notably, while bioconjugation of 2 is typically complete within 10 min, 6–18 hours of incubation was required to achieve >99% bioconjugation of 1a to all POP-Z variants examined except for POPWT-Z53, which was also complete within 10 min (ESI Fig. 3†).
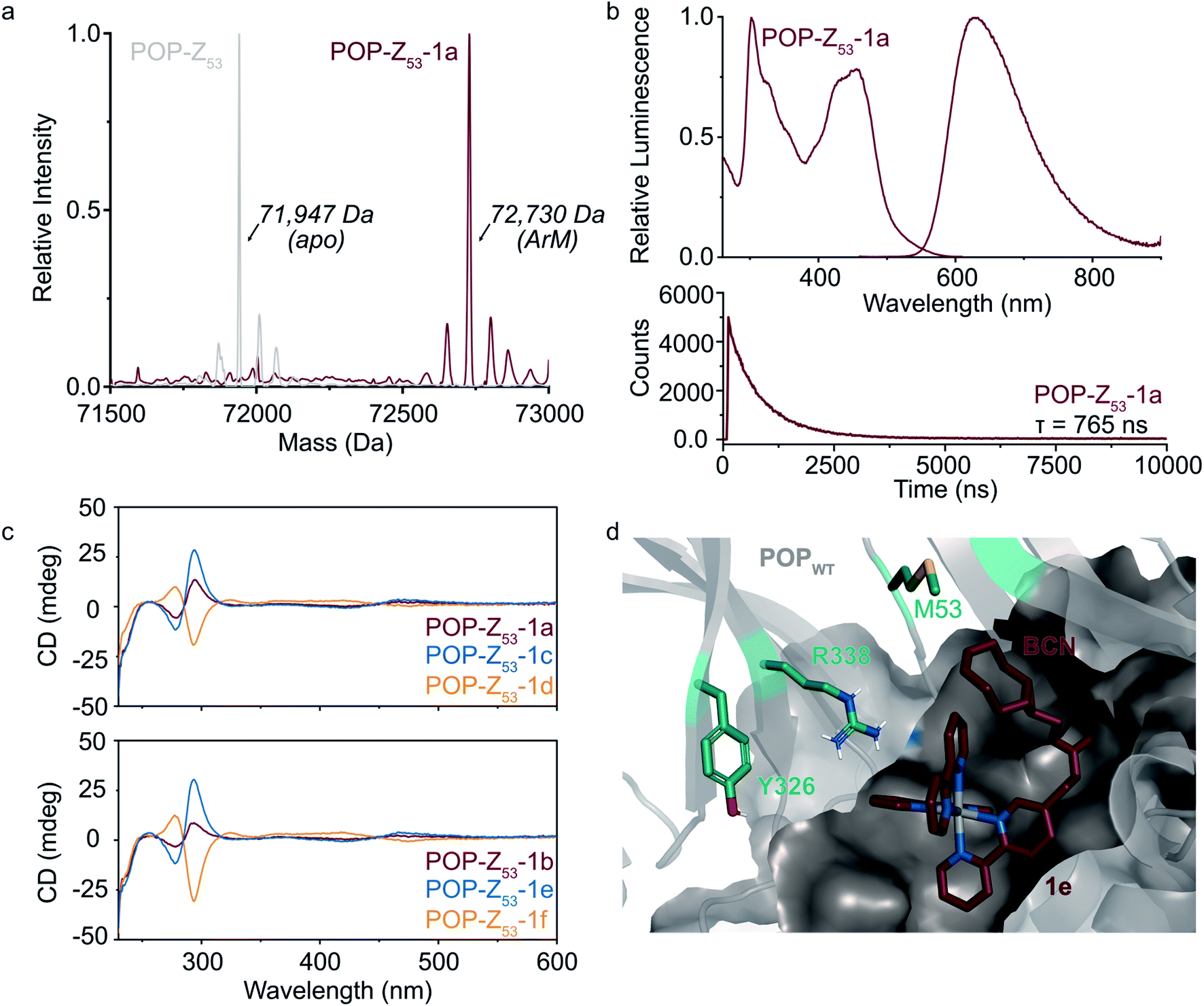 | ||
Fig. 2 Covalent ArM characterization. (a) Intact ESI-MS characterization of POPWT-Z53 before (theoretical: 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 947 Da) and after SPAAC reaction (theoretical: 72723 Da) with cofactor 1a (endo/racemic). (b) Excitation (λexc = 260–610 nm, λem = 620 nm) and emission (λexc = 450 nm, λem = 460–900 nm) spectra (upper) for POPWT-Z53-1a ArM (50 μM). Luminescence lifetime (λexc = 450 nm, λem = 620 nm) decay (lower) obtained for POPWT-Z53-1a ArM (50 μM). (c) Circular dichroism spectra of various POPWT-Z53 ArMs (20 μM). (d) Docking of cofactor 1e (exo/Λ; crimson) with POPWT (grey) scaffold using AutoDock Vina.51 The alkyne functional group on the BCN moiety is oriented towards residue M53, and not towards other residues (e.g. Y326 and R338) targeted for incorporation of azidophenylalanine. 947 Da) and after SPAAC reaction (theoretical: 72723 Da) with cofactor 1a (endo/racemic). (b) Excitation (λexc = 260–610 nm, λem = 620 nm) and emission (λexc = 450 nm, λem = 460–900 nm) spectra (upper) for POPWT-Z53-1a ArM (50 μM). Luminescence lifetime (λexc = 450 nm, λem = 620 nm) decay (lower) obtained for POPWT-Z53-1a ArM (50 μM). (c) Circular dichroism spectra of various POPWT-Z53 ArMs (20 μM). (d) Docking of cofactor 1e (exo/Λ; crimson) with POPWT (grey) scaffold using AutoDock Vina.51 The alkyne functional group on the BCN moiety is oriented towards residue M53, and not towards other residues (e.g. Y326 and R338) targeted for incorporation of azidophenylalanine. | ||
A variety of spectroscopic methods were next used to provide insight into binding of 1a–f within the different POP-Z variants. The λmax values in the absorbance and emission spectra of the cofactors did not change significantly upon bioconjugation (ESI Fig. 4 and 5†), consistent with the fact that the orbital energies of Ru(Bpy)32+ and related complexes are generally not sensitive to changes in solvent/environment.46 The luminescence lifetimes of the ArMs were also evaluated since it is known that the lifetime of luminophores like Ru(Bpy)32+ increases in more hydrophobic environments,30,47,48 such as that expected within the POP active site, and substantial increases compared to the free complexes were observed (Table 1).
| Entry | Scaffold | Cofactor | Stereochemistry | Lifetime (ns) |
|---|---|---|---|---|
| a Measurements performed in 0.5% ACN. All other measurements were performed in MQ H2O. | ||||
| 1 | N/A | 3a | Racemic/n/a | 573 |
| 2 | N/A | 1a | Racemic/endo | 530 |
| 3 | POPWT-Z53 | 1a | Racemic/endo | 765 |
| 4 | POPWT-Z99 | 1a | Racemic/endo | 791 |
| 5 | POPWT-Z326 | 1a | Racemic/endo | 761 |
| 6 | POPWT-Z338 | 1a | Racemic/endo | 773 |
| 7 | POPWT-Z477 | 1a | Racemic/endo | 837 |
| 8 | POPWT-Z53 | 1b | Racemic/exo | 725 |
| 9 | POPWT-Z53 | 1c | Λ/endo | 737 |
| 10 | POPWT-Z53 | 1d | Δ/endo | 715 |
| 11 | POPWT-Z53 | 1e | Λ/exo | 726 |
| 12 | POPWT-Z53 | 1f | Δ/exo | 715 |
Finally, CD spectroscopy was used to more directly interrogate possible interactions between 1a–f and the different protein scaffolds (Fig. 2C and ESI Fig. 6†). Optically-active chiral molecules like Ru(Bpy)32+ differentially absorb left- and right-circularly polarized light,49 and the resulting Cotton effects (i.e. CD signals near absorbance bands) can be used to analyze optical activity of metal complexes bound to a protein host.50 While the apo proteins were spectroscopically silent above 250 nm, positive and negative Cotton effects near 300 nm were observed for ArMs prepared with enantiopure cofactors (Fig. 2C). Similar Cotton effects were observed for POPWT-Z53-1a (endo/racemic) and POP-Z53-1b (exo/racemic). These observations indicate that POPWT-Z53 possesses sufficient affinity for the Ru(Bpy)32+ fragment of 1c/1e (Λ isomers) to enable partial resolution of racemic 1a during bioconjugation with an excess of this cofactor (2.75 equivalents). This apparent affinity explains the increased rate of bioconjugation for POPWT-Z53. Consistent with these observations, docking simulations of 1e revealed that the Ru(Bpy)32+ fragment of 1e preferred to bind in a pocket that projected the BCN fragment toward residue 53 but not any of the other residues in the POP-Z variants examined (Fig. 2D).
Non-covalent ArM preparation and characterization
To further explore the apparent binding of the Ru(Bpy)32+ fragments of 1a–f within POP, the affinity of POPWT toward a variety of substituted Ru(Bpy)32+ complexes was examined. Luminescence lifetime titrations52–54 established that Ru(Bpy)32+ (3a) binds to POPWT with a Kd of 28 ± 9 μM. Control experiments with BSA indicate that 3a possesses minimal non-specific binding affinity towards a similarly-sized globular protein (ESI Fig. 7†). The unexpected observation of 3a binding to POP thus provides a means to readily characterize the effects of changes in both protein and cofactor structure on binding affinity and photophysical properties.We first aimed to increase the affinity of POP for 3avia targeted mutations within the POP active site (Fig. 3a). Several active site arginine and lysine residues (R55, R198, K255, and R338) that could have repulsive interactions with positively charged Ru(Bpy)32+ derivatives were mutated to alanine in POP G99A. Active site tyrosine and tryptophan residues (W142 and Y326) were also mutated to alanine to avoid potential oxidation by Ru(Bpy)33+ derivatives, which could be accessed via oxidative quenching with an exogenous oxidant,55,56 giving POPNeut. Residues 99, 142, and 326, all projecting into the active site, were also mutated to aspartic acid in the POPNeut scaffold to generate variant POPNeg. With Kd values of 1.7 ± 0.7 μM and 1.1 ± 1.7 μM for POPNeut and POPNeg, respectively, both variants had improved affinities for 3a (Fig. 3). The apparent dissociation constant for POPNeg is only an upper limit due to experimental restrictions arising from the high affinity of the interaction (see ESI† for details).57
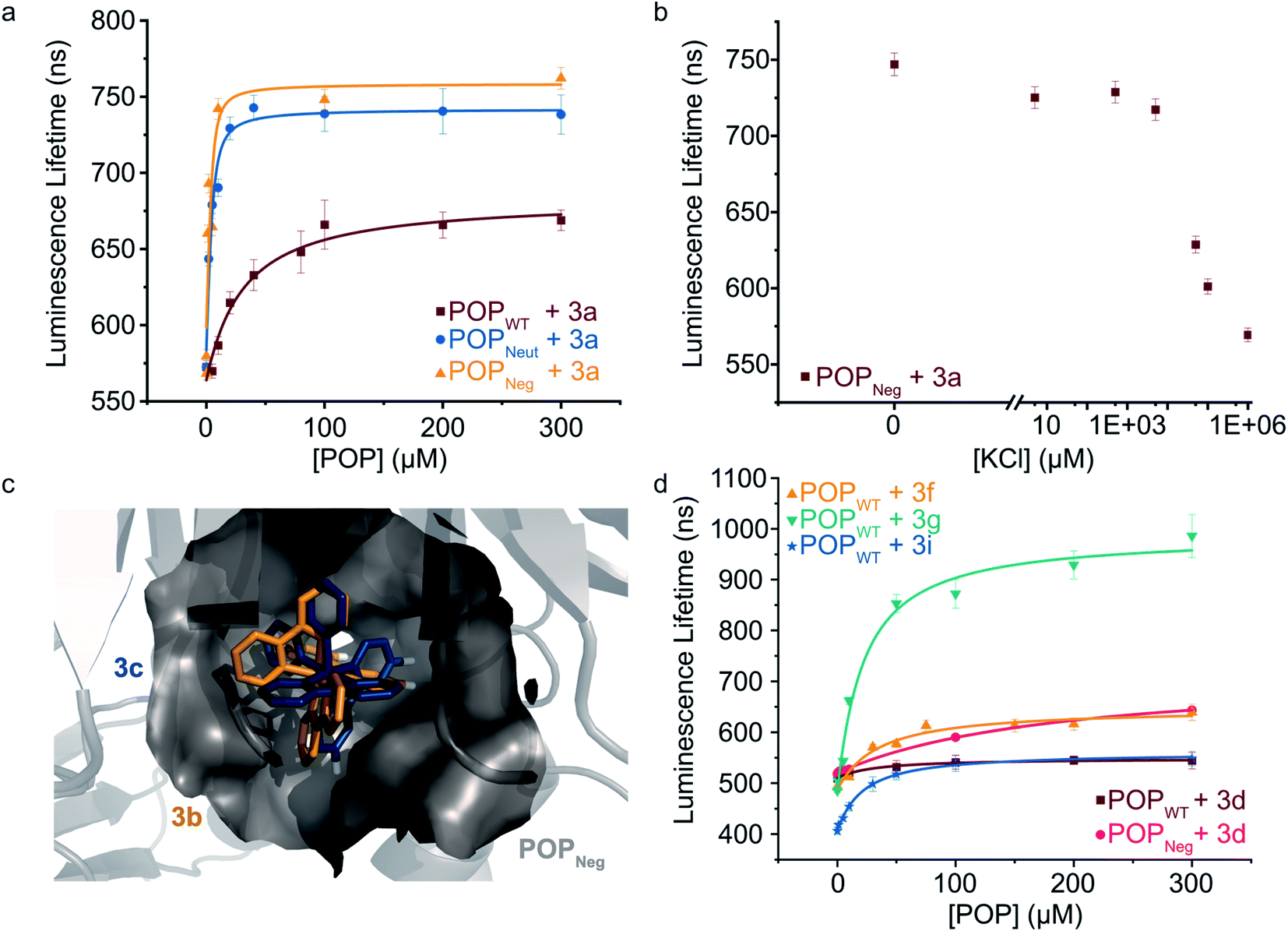 | ||
| Fig. 3 Modulating non-covalent ArM formation through protein and cofactor engineering. (a) Luminescence lifetime titrations (λexc = 450 nm, λem = 620 nm) demonstrate binding of POP variants to 3a. (b) 3a binding to POPNeg is disrupted by increasing concentrations of KCl. (c) Docking simulations between 3b (gold) and 3c (blue) with POPNeg (grey) reveal potential binding in the enzyme's active site. The 4,4′-hydrogens (white) of one ligand project into the hydrophobic pocket. (d) Luminescence lifetime titrations (λexc = 450 nm, λem = 620–650 nm) demonstrate binding of POPWT and POPNeg to various 4,4′-substituted ruthenium complexes. Plotted data points represent single measurements and error bars represent standard deviations resulting from single exponential tail fitting of decay data. Data were fit with a quadratic binding equation to give curves shown (eqn (1) in the ESI†). | ||
To confirm that 3a was binding in the active site of POP, a competition experiment was conducted using this complex and the known covalent POP inhibitor, Z-Pro-Prolinal (ZPP).58 Increasing inhibitor concentrations resulted in lower lifetime values, though the values were still significantly greater than the free cofactor, consistent with reduced affinity of the covalently modified active site for 3a (ESI Fig. 8†). In a series of complementary experiments inspired by the improved binding of 3a to POPNeut and POPNeg relative to POPWT, the ionic strength of the medium was increased to better solvate dicationic 3a and thus disfavor binding in the POP active site (Fig. 3b and ESI Fig. 9, 10†). Increased concentrations of KCl and various other salts/buffers significantly perturbed binding of 3a to POPNeg. Furthermore, non-covalent ArMs treated with different aqueous salts during spin filtration retained no 3a while those treated only with water retained a significant amount of cofactor.
We next explored the extent to which the chirality of Ru(Bpy)32+ derivatives and substitution on the Bpy ligands affects cofactor binding. As noted above, docking simulations suggest that BCN-substituted cofactor 1c can bind to a pocket in the hinge region of the POP active site. Analogous simulations using Ru(Bpy)32+ complexes 3b and 3c (Λ and Δ configurations, respectively) indicate that this pocket could accommodate both cofactor enantiomers. These simulations also indicate that the 4,4′-hydrogens of one Bpy ligand extended into this pocket with enough room to accommodate modifications at these positions (Fig. 3c). Consistent with the docking simulations, comparable binding of enantiopure Ru(Bpy)32+ complexes 3b and 3c to POPWT (Kd values of 30 μM and 37 μM, respectively) was observed (Table 2 and ESI Fig. 11†). Racemic complexes displaying 4,4′-CO2−, –CH2CO2−, –CH2OH, –tBu, –NH2, and –CH2NH3+ substituents (3d–i) were prepared, and luminescence lifetime titrations (Fig. 3d and ESI Fig. 12†) revealed minor differences in affinity towards POPWT, except for 3g and 3i, which bind 1.4- and 1.5-fold tighter than 3a, respectively (Table 2).
| Entry | Variant | Cofactor | R | K d (μM) | Δ lifetime (ns) |
|---|---|---|---|---|---|
| a This value is only an estimated upper bound of affinity. The binding data exhibited characteristics consistent with being in the “titration” regime.57 Refer to the ESI for more details. b Low luminescence intensities precluded accurate determination of the lifetime of this complex at reasonable concentrations (i.e. ≪[POP]). | |||||
| 1 | POPWT | 3a | H | 28.3 ± 8.8 | 96 |
| 2 | POPNeut | 3a | H | 1.65 ± 0.71 | 166 |
| 3 | POPNeg | 3a | H | 1.08 ± 1.70a | 183 |
| 4 | POPWT | 3b | H | 30.1 ± 8.2 | 110 |
| 5 | POPWT | 3c | H | 36.8 ± 11.1 | 86 |
| 6 | POPWT | 3d | –CO2− | 29.5 ± 9.8 | 35 |
| 7 | POPNeg | 3d | –CO2− | 185 ± 20 | 125 |
| 8 | POPWT | 3e | –CH2CO2− | 30.8 ± 10.1 | 60 |
| 9 | POPWT | 3f | –CH2OH | 30.8 ± 10.4 | 147 |
| 10 | POPWT | 3g | –tBu | 19.6 ± 5.7 | 500 |
| 11 | POPWT | 3h | –NH2 | NAb | NA |
| 12 | POPWT | 3i | –CH2NH3+ | 18.7 ± 2.0 | 142 |
We also observed different changes in the luminescence lifetime (Table 2) of the free vs. bound cofactors (Δlifetime), and in many cases, significant increases in the luminescence intensity of the cofactor (ESI Fig. 13†). Analogous binding experiments were also performed using POPNeg (ESI Fig. 14 and 15†). While similar trends were observed, there were notable differences between acid-substituted complex binding to the protein variants, as 3d had a moderately lower affinity for the POPNeg scaffold (Kd = 185 μM) compared to POPWT (Kd = 30 μM), perhaps due to electrostatic repulsion between the anionic cofactor and the aspartate residues (i.e. 99D, 142D, and 326D) in the active site of POPNeg.
POP ArMs for photocatalysis
Having established two approaches to link different photocatalysts to POP scaffolds, we next sought to evaluate the catalytic properties of these systems. Yoon has reported two mechanistically distinct reactions involving catalytic quantities of Ru(Bpy)32+ and a Brønsted acid co-catalyst. One of these involves 5-exo-trig reductive cyclization of dienones to generate 5-membered rings via electron transfer (Scheme 2a);59 the second involves [2 + 2] cycloaddition between C-cinnamoyl imidazoles and different alkenes to give cyclobutanes via energy transfer (Scheme 2b).60,61 Both reactions worked well in 10% aqueous ACN. Brønsted acid co-catalysts were not required in this medium, though a slightly acidic pH was optimal (ESI Fig. 16†) for the cycloaddition (25 mM MES pH 6.0; pKa of related N-methyl imidazole ∼7.0), and ascorbate served as a water-soluble reductant for the reductive cyclization reaction (ESI Tables 1 and 2†).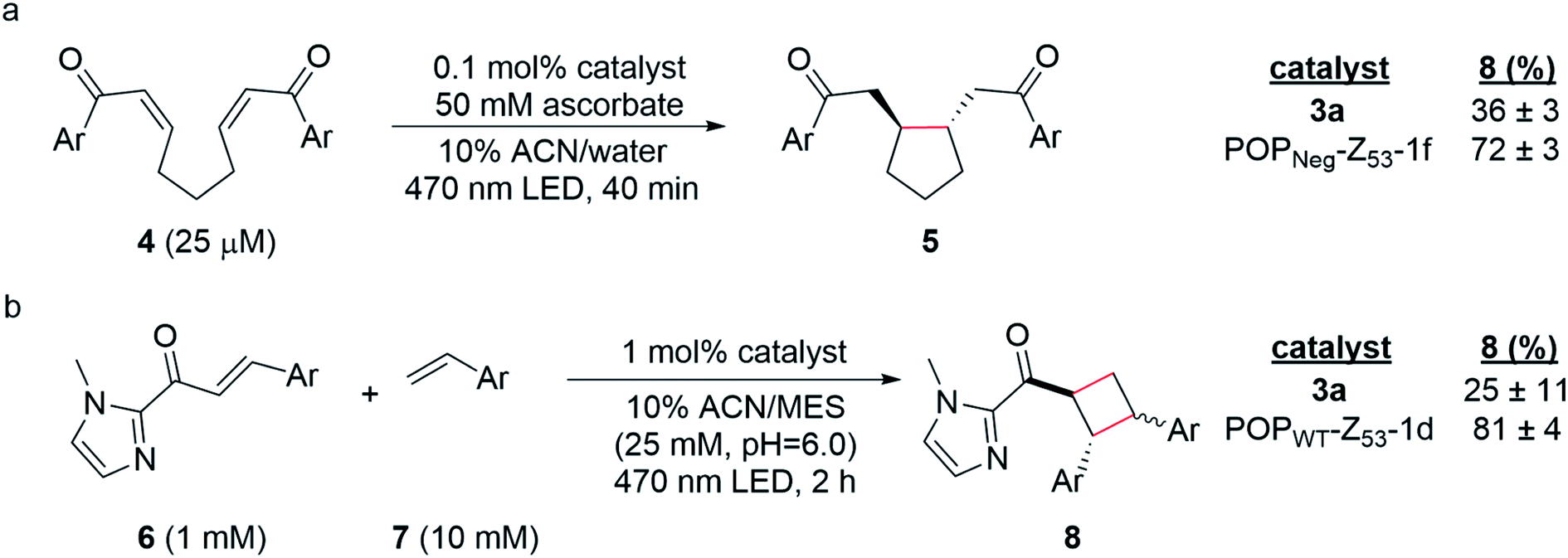 | ||
| Scheme 2 (a) Photoreductive cyclization reaction of dienone 4 and (b) [2 + 2] photocycloaddition of cinnamoyl imidazole 6 and 4-methoxystyrene 7. Yields were determined by UHPLC relative to an internal standard, 1,3,5-trimethoxybenzene (TMB). Reactions were performed in triplicate (n = 3) and are reported as averages with standard deviations. | ||
The optimized conditions for both reactions were used to screen the reactivity and selectivity of a small panel of ArMs generated from different POP scaffolds. Only covalent ArMs were evaluated since ascorbate and MES, like other salts and buffers (vide supra), displaced the complex from non-covalent ArMs (ESI Fig. 17†). Reactions were conducted in an inert atmosphere wetbox containing a custom-built LED photoreactor compatible with 96-well plates to enable consistent irradiation samples (ESI Fig. 18†). The desired products, 5 and 8, were observed in all cases, and higher conversions were generally observed for ArM-catalyzed reactions relative to the free cofactor (Scheme 2, ESI Tables 3–5†). Furthermore, increased rates of reaction were observed relative to Ru(Bpy)32+ with a 2-fold increase for reaction with 4 (0.21 μM min−1vs. 0.41 μM min−1 for 3a and POPNeg-Z53-1f, respectively; ESI Fig. 19†) and a 2.6-fold increase for reaction with 6 (4.7 μM min−1vs. 12.0 μM min−1 for 3a and POPWT-Z53-1d, respectively; ESI Fig. 20†). In the case of the reductive cyclization reaction, similar yields were observed for the ArM and free cofactor 3a if reactions were irradiated for longer periods of time (ESI Table 6†).
Discussion
This study describes the formation of ArMs via covalent and non-covalent incorporation of Ru(II) polypyridine visible light photocatalysts into engineered POP variants. While our selection of metal polypyridine photocatalysts was motivated by their stability and the diversity of reactions that they catalyze,5 their large size presents a challenge for ArM design. Privileged scaffolds like myoglobin62 or LmrR63 that bind a variety of planar complexes, for example, would likely not accommodate bulky cofactors like 1a–f. More open scaffolds like streptavidin do not fully encapsulate anchored catalysts,64 so their ability to modulate cofactor properties and control reactivity is limited (though fused lid structures such as that recently reported for streptavidin could correct this issue).65 We previously established that Pfu POP is a versatile scaffold for ArM formation66 and that directed evolution can be used to engineer selective ArMs for a variety of dirhodium-catalyzed carbene addition and insertion reactions.38,40 We have also used this scaffold to develop an artificial enzyme containing an acridinium cofactor (Fig. 1b) for visible light photoredox catalysis.37 This system catalyzes sulfide oxidation via SET, but no selectivity was obtained, presumably due to poor substrate binding within the active site. Rapid photobleaching of the acridinium cofactor limits the potential of this system even if selectivity could be achieved.Preliminary efforts to develop POP ArM photocatalysts focused on cofactor 1a (Fig. 1a), which contains a [6.1.0]-bicyclononyne anchor for SPAAC to proteins containing a genetically encoded azidophenylalanine residue. SPAAC proceeded efficiently, giving >99% bioconjugation in all cases. Interestingly, the linkage site strongly influenced the rate of SPAAC, and POPWT-Z53 was even able to partially resolve racemic cofactor 1a to form an ArM enriched in the Λ cofactor enantiomer. Similar stereoselective bioconjugation of ruthenium complexes to proteins has been described previously.30–33 For example, Salmain showed that Michael addition of the active site cysteine of papain to maleimide-substituted Ru(II) complexes proceeded stereoselectively and that substituents on these complexes affect bioconjugation rates.30 Tiede later showed that bioconjugation of PpcA via cysteine alkylation also led to cofactor resolution, and differences in stereoselectivity based on bioconjugation site were noted.31 Together, these studies provide precedent for the site-dependent bioconjugation rate acceleration observed in the current study, but both involved relatively small scaffolds that left their cofactors solvent exposed. POP is large enough to accommodate both the cofactor and potential substrate(s) in its hydrophobic active site, and secondary Ru(II) polypyridine binding provides a means to reduce cofactor movement, which has been found to improve the activity and selectivity of ArMs generated from other scaffolds.67,68
Although covalent cofactor bioconjugation allows for complete removal of free cofactor and associated background spectroscopic signals or catalysis,40 it necessitates the incorporation of bioorthogonal linkage sites into both cofactor and scaffold.41 High affinity non-covalent cofactor binding eliminates these issues and inherently provides a direct interface between active site residues and the cofactor to modulate cofactor properties.69 Consistent with the apparent binding of the Ru(II) fragment of cofactor 1a, POP exhibited Ru(Bpy)32+ binding affinities in the μM range. Reduced luminescence lifetimes for this system in the presence of a known covalent inhibitor of POP, Z-Pro-Prolinal,58 and negligible binding to BSA highlighted the unique affinity of the POP active site for Ru(Bpy)32+. This fortuitous discovery provides a starting point to develop non-covalent ArM photocatalysts in which both cofactor and scaffold can be tuned to modulate cofactor binding, photophysical properties, and reactivity.
Initial non-covalent ArM engineering efforts focused on further improving cofactor binding. Given that Ru(Bpy)32+ is dicationic, several positively charged residues in the POP active site were mutated to Ala or Asp to create POPNeut and POPNeg, respectively, with the goal of improving electrostatic scaffold/cofactor complementarity. This approach mirrors analogous charge complementation in both natural proteins (e.g. cytochrome f in photosystem I)70–72 and synthetic systems (e.g. methyl viologen in streptavidin).29 Non-covalent binding in POPNeut and POPNeg increases substantially, and Kd values approaching the nM regime are observed for Ru(Bpy)32+ binding. The electrostatic nature of cofactor binding in these systems is supported by control experiments in which added KCl and other salt solutions that could better solvate the cofactor significantly reduce binding of 3a even to POPNeg. Cofactor substitution was also found to modulate binding. Docking simulations were used to guide the installation of substituents on the 4 and 4′ positions of one Bpy ligand that could interact with the protein via different mechanisms (e.g. hydrophobic effect, hydrogen bonding, ion pairing, etc.). tBu-substituted complex 3g was found to bind better than the homoleptic complex 3a, highlighting the importance of the hydrophobic effect for cofactor binding.
The Ru(II) polypyridine ArMs developed in this study also provide a unique platform to tune and study the photophysical properties and reactivity of visible light photocatalysts. Protein scaffolds have been previously demonstrated to enhance the photophysical properties of metal cofactors in a variety of systems.30–33,73–75 In the context of the POP scaffold, both covalent and non-covalent ArMs exhibit increased luminescence lifetimes and intensities relative to free cofactors. These changes were dependent on the identities of the cofactor and scaffold. For example, while the average increase in the luminescence lifetime upon non-covalent binding to POPWT was around 100 ns for most cofactors, the lifetime of 3g was increased by 500 ns (from 486 ns to 986 ns), similar to the lifetime of Ru(Bpy)32+ in deoxygenated acetonitrile (930 ns).5,8 Similar enhancements in different Ru(II) polypyridine–protein conjugates have been ascribed to Ru(II) polypyridine binding in hydrophobic sites that can shield the complexes from luminophore-quenching oxygen.30 Both of these mechanisms could be operative for POP ArMs given the large, hydrophobic Ru(II) polypyridine binding pocket within the POP active site (Fig. 3c). The site of cofactor attachment and electrostatic complementation were also observed to modulate the luminescence of covalent cofactors. For instance, ArMs constructed by bioconjugating 1a at different active site residues had lifetimes ranging from 761 ns to 837 ns (Table 1), and the addition of an anionic carboxylate group (3d) limited the increase in lifetime to only 35 ns upon non-covalent association with POPWTvs. 125 ns for POPNeg, highlighting the dependence of cofactor properties on the local protein environment. The importance of linker design/placement in synthetic systems was recently demonstrated in a systematic study focused on the effects of ligand modification (i.e. length and chiral configuration) and chromophore placement on the photophysics of sulforhodamine B.76 While we only investigated one linker in this study, the rigidity, length, and stereochemistry of the BCN moiety could be tuned to further control the nature of protein-cofactor interactions.
Finally, covalent ArM photocatalysts exhibited promising activity towards two mechanistically distinct transformations. In the first, 5-exo-trig reductive cyclization of a dienone (e.g.4) generates a cyclopentane (e.g.5) via SET using a sacrificial reductant.59 In the other model reaction, [2 + 2] cycloaddition between a C-cinnamoyl imidazole (e.g.6) and an electron-rich styrene like 4-methoxystyrene yields a cyclobutane (e.g.8).60,61 In both cases, enhanced yields and rate acceleration compared to the free cofactor were observed under optimized aqueous conditions. These improvements could result from increased local concentration of both substrate and photocatalyst within the ArM active site relative to solution. The increased luminescence lifetime of ArM photocatalysts could also improve yields since longer-lived excited states increase the probability of the catalyst encountering substrates in a bimolecular reaction. Small differences in the diastereomeric ratios (d.r.) for ArM- and Ru(Bpy)32+-catalyzed [2 + 2] cycloaddition were observed (1.2 and 1.5, respectively; ESI Fig. 21†), though no significant enantioselectivity was measured. These results suggest that while the protein scaffold can influence the rate and stereochemistry of the reactions studied, the naïve active site pocket is not suitable for imparting high selectivity. Ongoing efforts are focused on engineering the POP scaffold to better accommodate and orient the cofactors and substrates explored in this study and related systems to enable selective visible light photocatalysis.
Conclusion
In this study, we developed a series of covalent and non-covalent ArMs using metal polypyridyl cofactors and a prolyl oligopeptidase scaffold. Modifications to both the scaffold and the cofactor were used to increase cofactor binding affinity and extend its luminescence lifetime, an important metric when assessing the potential of a photocatalyst. The ArM photocatalysts exhibited good activity toward reactions that proceed via SET (reductive cyclization) and ET ([2 + 2] photocycloaddition) under optimized aqueous reaction conditions. Moreover, the yields of the desired products and rates of reactions were enhanced for ArM-catalyzed reactions compared to Ru(Bpy)32+ under aqueous conditions. The ability of POP to bind and modulate the properties of different Ru(II) cofactors suggests that ArM photocatalysts could serve as a general platform for the further development of diverse catalysts with tunable photophysical properties and reactivity.Data availability
All data needed to evaluate the conclusions in the paper are present in the main text and/or the ESI.† Additional data related to this study may be requested from the authors.Author contributions
J. C. L.: conceptualization; writing – original draft; writing – review and editing; visualization; project administration; funding acquisition. Y. S. Z.: conceptualization; methodology; validation; formal analysis; investigation; resources; writing – original draft; writing – review & editing; and visualization. B. L.: methodology; validation; formal analysis; investigation; resources; visualization; and writing – review & editing. Y. G.: conceptualization; methodology; and resources. D. S.: resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the U.S. Army Research Laboratory and the U.S. Army Research Office under Contract/Grant W911NF-19-1-0074 and by an NSF CAREER Award to J. C. L. (CHE-1351991). Y. S. Z. gratefully acknowledges receipt of a predoctoral fellowship from the Graduate Training Program in Quantitative and Chemical Biology at Indiana University (T32 GM131994). NMR data were acquired on a spectrometer funded by the NSF (MRI CHE-1920026) using a Prodigy probe that was partially funded by the Indiana Clinical and Translational Sciences Institute. We thank Dr Jonathan Trinidad for assistance with intact protein ESI-MS; Dr Giovanni Gonzalez-Gutierrez for assistance with various instrumentation in the IU Physical Biochemistry Instrumentation Facility and for allowing us to borrow a low-volume cuvette for luminescence measurements; and Prof. Amar Flood for access to a UV-Vis spectrophotometer and a fluorometer. We thank IU Mechanical Instrument Services and IU Electronic Instrument Services, including Mr John Poehlman and Ms Rose Burchfield in particular, for assistance with the design and fabrication of the photoreactor used in these studies.References
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- D. van der Westhuizen, K. G. von Eschwege and J. Conradie, Electrochim. Acta, 2019, 320, 134540 CrossRef CAS.
- J. Zheng, W. B. Swords, H. Jung, K. L. Skubi, J. B. Kidd, G. J. Meyer, M. H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 13625–13634 CrossRef CAS PubMed.
- M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chem. Rev., 2021 DOI:10.1021/acs.chemrev.1c00467.
- M. P. Sibi, S. Manyem and J. Zimmerman, Chem. Rev., 2003, 103, 3263–3295 CrossRef CAS PubMed.
- T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef CAS PubMed.
- P. A. Frey, Annu. Rev. Biochem., 2003, 70, 121–148 CrossRef PubMed.
- H. R. Williamson, B. A. Dow and V. L. Davidson, Bioorg. Chem., 2014, 57, 213–221 CrossRef CAS PubMed.
- T. K. Hyster, Synlett, 2020, 31, 248–254 CrossRef CAS.
- N. Nelson and W. Junge, Annu. Rev. Biochem., 2015, 84, 659–683 CrossRef CAS PubMed.
- R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- Q. Lam, M. Kato and L. Cheruzel, Biochim. Biophys. Acta, 2016, 1857, 589–597 CrossRef CAS PubMed.
- M. E. Ener, Y. T. Lee, J. R. Winkler, H. B. Gray and L. Cheruzel, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18783–18786 CrossRef CAS PubMed.
- J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2014, 136, 2930–2939 CrossRef CAS PubMed.
- H. B. Gray and J. R. Winkler, Annu. Rev. Biochem., 2003, 65, 537–561 CrossRef PubMed.
- J. R. Winkler and H. B. Gray, Chem. Rev., 2013, 114, 3369–3380 CrossRef PubMed.
- N. H. Tran, N. Huynh, T. Bui, Y. Nguyen, P. Huynh, M. E. Cooper and L. E. Cheruzel, Chem. Commun., 2011, 47, 11936–11938 RSC.
- H. Shalan, A. Colbert, T. T. Nguyen, M. Kato and L. Cheruzel, Inorg. Chem., 2017, 56, 6558–6564 CrossRef CAS PubMed.
- A. Loosli, U. E. Rusbandi, J. Gradinaru, K. Bernauer, C. W. Schlaepfer, M. Meyer, S. Mazurek, M. Novic and T. R. Ward, Inorg. Chem., 2005, 45, 660–668 CrossRef PubMed.
- S. G. Keller, A. Pannwitz, F. Schwizer, J. Klehr, O. S. Wenger and T. R. Ward, Org. Biomol. Chem., 2016, 14, 7197–7201 RSC.
- S. G. Keller, A. Pannwitz, H. Mallin, O. S. Wenger and T. R. Ward, Chem.–Eur. J., 2017, 23, 18019–18024 CrossRef CAS PubMed.
- P. Haquette, J. Jacques, S. Dagorne, C. Fosse and M. Salmain, Eur. J. Inorg. Chem., 2010, 2010, 5087–5095 CrossRef.
- N. S. Ponomarenko, O. Kokhan, P. R. Pokkuluri, K. L. Mulfort and D. M. Tiede, Photosynth. Res., 2020, 143, 99–113 CrossRef CAS PubMed.
- J. Luo, J. F. Wishart and S. S. Isied, J. Am. Chem. Soc., 1998, 120, 12970–12971 CrossRef CAS.
- I. J. Dmochowski, J. R. Winkler and H. B. Gray, J. Inorg. Biochem., 2000, 81, 221–228 CrossRef CAS PubMed.
- Z. Szeltner and L. Polgár, Curr. Protein Pept. Sci., 2008, 9, 96–107 CrossRef CAS PubMed.
- K. Ellis-Guardiola, H. Rui, R. L. Beckner, P. Srivastava, N. Sukumar, B. Roux and J. C. Lewis, Biochemistry, 2019, 58, 1616–1626 CrossRef CAS PubMed.
- S. Abe, T. Ueno and Y. Watanabe, Top. Organomet. Chem., 2009, 25, 25–43 CrossRef CAS.
- Y. Gu, K. Ellis-Guardiola, P. Srivastava and J. C. Lewis, ChemBioChem, 2015, 16, 1880–1883 CrossRef CAS PubMed.
- D. M. Upp, R. Huang, Y. Li, M. J. Bultman, B. Roux and J. C. Lewis, Angew. Chem., Int. Ed., 2021, 60(44), 23672–23677 CrossRef CAS PubMed.
- J. C. Lewis, Acc. Chem. Res., 2019, 52, 576–584 CrossRef CAS PubMed.
- H. Yang, A. M. Swartz, H. June Park, P. Srivastava, K. Ellis-Guardiola, D. M. Upp, G. Lee, K. Belsare, Y. Gu, C. Zhang, R. E. Moellering and J. C. Lewis, Nat. Chem., 2018, 10, 318–324 CrossRef CAS PubMed.
- H. Yang, P. Srivastava, C. Zhang and J. C. Lewis, ChemBioChem, 2014, 15, 223–227 CrossRef CAS PubMed.
- Y. Kobayashi, M. Hoshino, T. Kameda, K. Kobayashi, K. Akaji, S. Inuki, H. Ohno and S. Oishi, Inorg. Chem., 2018, 57, 5475–5485 CrossRef CAS PubMed.
- L. Wang, H. Yin, M. A. Jabed, M. Hetu, C. Wang, S. Monro, X. Zhu, S. Kilina, S. A. McFarland and W. Sun, Inorg. Chem., 2017, 56, 3245–3259 CrossRef CAS PubMed.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. vanHest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed.
- X. Hua and A. von Zelewsky, Inorg. Chem., 1995, 34, 5791–5797 CrossRef CAS.
- N. R. M. Simpson, M. D. Ward, A. F. Morales and F. Barigelletti, J. Chem. Soc., Dalton Trans., 2002, 2449–2454 RSC.
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641 CrossRef CAS PubMed.
- S. H. Mejías, G. Roelfes and W. R. Browne, Phys. Chem. Chem. Phys., 2020, 22, 12228–12238 RSC.
- R. D. Gillard, in Progress in Inorganic Chemistry, ed. F. A. Cotton, John Wiley & Sons Inc., New York, 1st edn, 1966, ch. 4, vol. 7, pp. 215–276 Search PubMed.
- L. Quintanar and L. Rivillas-Acevedo, Methods Mol. Biol., 2013, 1008, 267–297 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- H. Szmacinski, E. Terpetschnig and J. R. Lakowicz, Biophys. Chem., 1996, 62, 109–120 CrossRef CAS PubMed.
- S. P. Bernhard, C. K. Goodman, E. G. Norton, D. G. Alme, C. M. Lawrence and M. J. Cloninger, ACS Omega, 2020, 5, 29017–29024 CrossRef CAS PubMed.
- J. N. Lampe and W. M. Atkins, Biochemistry, 2006, 45, 12204–12215 CrossRef CAS PubMed.
- M. Sjödin, S. Styring, B. Åkermark, L. Sun and L. Hammarström, J. Am. Chem. Soc., 2000, 122, 3932–3936 CrossRef.
- M. T. Zhang and L. Hammarström, J. Am. Chem. Soc., 2011, 133, 8806–8809 CrossRef CAS PubMed.
- I. Jarmoskaite, I. Alsadhan, P. P. Vaidyanathan and D. Herschlag, Elife, 2020, 9, 1–34 CrossRef PubMed.
- M. N. Harris, J. D. Madura, L. J. Ming and V. J. Harwood, J. Biol. Chem., 2001, 276, 19310–19317 CrossRef CAS PubMed.
- J. Du, L. R. Espelt, I. A. Guzei and T. P. Yoon, Chem. Sci., 2011, 2, 2115–2119 RSC.
- E. M. Sherbrook, H. Jung, D. Cho, M. H. Baik and T. P. Yoon, Chem. Sci., 2020, 11, 856–861 RSC.
- E. M. Sherbrook, M. J. Genzink, B. Park, I. A. Guzei, M. H. Baik and T. P. Yoon, Nat. Commun., 2021, 12, 5735 CrossRef CAS PubMed.
- K. Oohora and T. Hayashi, Dalton Trans., 2021, 50, 1940–1949 RSC.
- G. Roelfes, Acc. Chem. Res., 2019, 52, 545–556 CrossRef CAS PubMed.
- T. Heinisch and T. R. Ward, Acc. Chem. Res., 2016, 49, 1711–1721 CrossRef CAS PubMed.
- F. Christoffel, N. V. Igareta, M. M. Pellizzoni, L. Tiessler-Sala, B. Lozhkin, D. C. Spiess, A. Lledós, J. D. Maréchal, R. L. Peterson and T. R. Ward, Nat. Catal., 2021, 4, 643–653 CrossRef CAS.
- P. Srivastava, H. Yang, K. Ellis-Guardiola and J. C. Lewis, Nat. Commun., 2015, 6, 7789 CrossRef CAS PubMed.
- H. J. Davis and T. R. Ward, ACS Cent. Sci., 2019, 5, 1120–1136 CrossRef CAS PubMed.
- J. R. Carey, S. K. Ma, T. D. Pfister, D. K. Garner, H. K. Kim, J. A. Abramite, Z. Wang, Z. Guo and Y. Lu, J. Am. Chem. Soc., 2004, 126, 10812–10813 CrossRef CAS PubMed.
- L. Fruk, C. H. Kuo, E. Torres and C. M. Niemeyer, Angew. Chem., Int. Ed., 2009, 48, 1550–1574 CrossRef CAS PubMed.
- E. L. Gross, Photosynth. Res., 1993, 37, 103–116 CrossRef CAS PubMed.
- E. J. Haddadian and E. L. Gross, Biophys. J., 2005, 88, 2323–2339 CrossRef CAS PubMed.
- M. R. Redinbo, T. O. Yeates and S. Merchant, J. Bioenerg. Biomembr., 1994, 26, 49–66 CrossRef CAS PubMed.
- (a) M. Delor, J. Dai, T. D. Roberts, J. R. Rogers, S. M. Hamed, J. B. Neaton, P. L. Geissler, M. B. Francis and N. S. Ginsberg, J. Am. Chem. Soc., 2018, 140, 6278–6287 CrossRef CAS PubMed; (b) K. K. W. Lo, T. K. M. Lee, J. S. Y. Lau, W. L. Poon and S. H. Cheng, Inorg. Chem., 2007, 47, 200–208 CrossRef PubMed.
- K. K. W. Lo, T. K. M. Lee and K. Y. Zhang, Inorg. Chim. Acta, 2006, 359, 1845–1854 CrossRef CAS.
- K. K. W. Lo and T. K. M. Lee, Inorg. Chem., 2004, 43, 5275–5282 CrossRef CAS PubMed.
- M. Delor, J. Dai, T. D. Roberts, J. R. Rogers, S. M. Hamed, J. B. Neaton, P. L. Geissler, M. B. Francis and N. S. Ginsberg, J. Am. Chem. Soc., 2018, 140, 6278–6287 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplemental figures, detailed procedures and results for all reported experiments, and data for compound characterization. See DOI: 10.1039/d1sc05792h |
| ‡ These authors contributed equally to this study. |
| § Current address: 5F Lab Building, No. 100 Chongwen Road, Suzhou Industrial Park, Suzhou, Jiangsu Province, P. R. China. |
| ¶ Current address: Department of Chemistry, Virginia Tech, Blacksburg, VA 24061, USA. |
| This journal is © The Royal Society of Chemistry 2022 |