
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinking metal–organic cages pairwise as a design approach for assembling multivariate crystalline materials†
Adrian W.
Markwell-Heys
 a,
Michael
Roemelt
b,
Ashley D.
Slattery
c,
Oliver M.
Linder-Patton
a and
Witold M.
Bloch
*a
a,
Michael
Roemelt
b,
Ashley D.
Slattery
c,
Oliver M.
Linder-Patton
a and
Witold M.
Bloch
*a
aDepartment of Chemistry, The University of Adelaide, Adelaide, Australia. E-mail: witold.bloch@adelaide.edu.au; Tel: +61 8 8313 5039
bInstitut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor Str. 2, 12489 Berlin, Germany
cAdelaide Microscopy, The University of Adelaide, Adelaide 5005, Australia
First published on 30th November 2021
Abstract
Using metal–organic cages (MOCs) as preformed supermolecular building-blocks (SBBs) is a powerful strategy to design functional metal–organic frameworks (MOFs) with control over the pore architecture and connectivity. However, introducing chemical complexity into the network via this route is limited as most methodologies focus on only one type of MOC as the building-block. Herein we present the pairwise linking of MOCs as a design approach to introduce defined chemical complexity into porous materials. Our methodology exploits preferential Rh-aniline coordination and stoichiometric control to rationally link Cu4L4 and Rh4L4 MOCs into chemically complex, yet extremely well-defined crystalline solids. This strategy is expected to open up significant new possibilities to design bespoke multi-functional materials with atomistic control over the location and ordering of chemical functionalities.
Introduction
The ability to rationally integrate multiple chemical entities within a crystalline porous solid is one of the defining goals in the field of metal–organic frameworks (MOFs).1–3 Such control offers exciting opportunities to design multi-functional materials capable of performing complex and sophisticated operations relevant to applications in gas separation and catalysis.4–8 However, integrating multiple different metal ions and ligands within a MOF structure is difficult to achieve with the typical ‘one-pot’ synthesis.9–11 This relates to the challenge in predicting the assembly outcome of multi-component mixtures and their tendency to crystallise as simple binary phases.1,12,13In contrast, Nature integrates chemical complexity into multi-functional architectures by ordering preorganised subunits in a step-wise manner.14,15 This elegant, step-wise approach has indeed inspired the design of synthetic porous solids. For example, preformed molecular cages have been co-crystallised into multi-functional solids via hydrogen bonding,16 chiral-recognition,17 and ionic interactions.18 However, predicting the crystal structure for solids formed by non-covalent interactions is a long-standing challenge in solid-state chemistry.19,20
In light of this, we propose that linking of metal–organic cages (MOCs) by coordination is a rational approach to introduce multiple functionalities into porous solids. In this regard, MOCs with [M2(COO)4(solvent)2] (M = Cu(II), Rh(II) and Cr(II)) paddlewheel sites represent versatile supermolecular building-blocks (SBBs) owing to their exterior coordination sites and compatible ligand-based functionalities.21,22
The synthesis of MOFs from MOCs was first demonstrated by Zhou who showed that preformed Cu12L12 octahedra could be linked with 4,4′-bipyridine to generate crystals of an interpenetrated MOF (Fig. 1a).23 More recently, Maspoch and co-workers showed that Rh24L24 MOCs can be utilised as SBBs to generate highly-connected MOFs via coordination of metal clusters to the cage exterior (Fig. 1b).24 These methodologies however, are limited to only one type of MOC as the building-block.25,26 The coordinative linking of two different MOCs represents a significant advancement in the design of multi-functional porous solids, but is hitherto unrealized. This may be due to the increased complexity of such a system, in terms of the challenge of directing the selective coordination of one MOC to another.
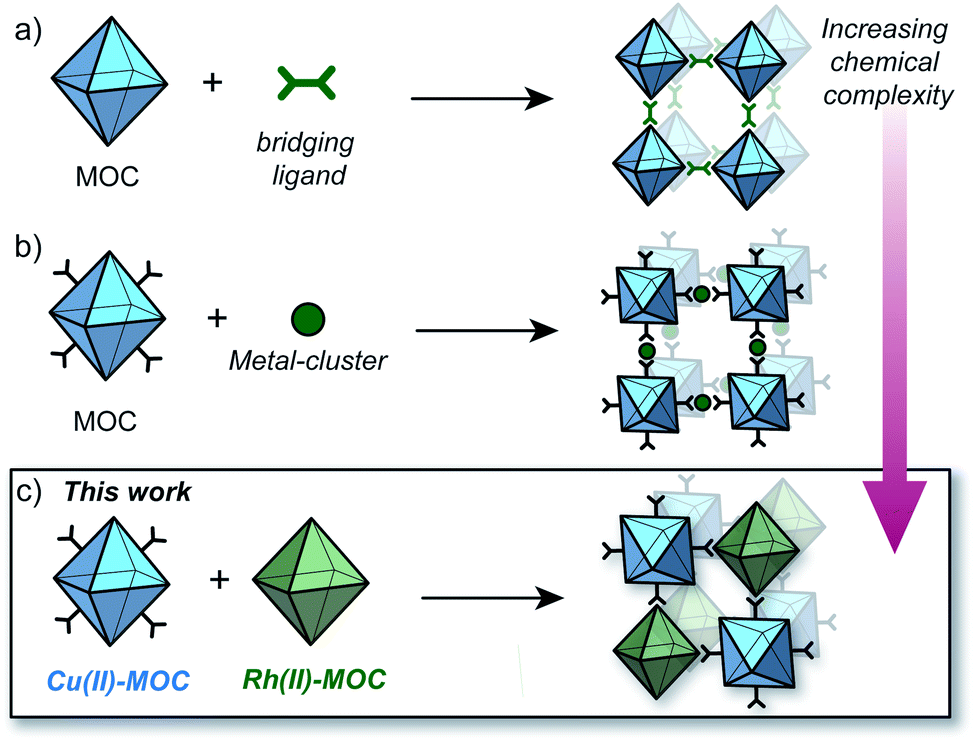 | ||
| Fig. 1 Strategies to synthesise MOFs from preformed MOCs; (a) utilising bridging ligands to link MOCs via the available coordination sites;23 (b) linking surface-functionalised MOCs via a metal cluster;24 (c) this work: linking a surface-functionalised Cu(II) MOC to a Rh(II) MOC via preferential Rh-aniline coordination. | ||
Herein we report the pairwise linking of two different MOCs as a design approach for generating an unprecedented class of multivariate crystalline materials (Fig. 1c). The MOC building-blocks utilised in this study are based on a M4L4 lantern architecture (M = Cu(II) or Rh(II)) and their pairwise linking is achieved by preferential Rh-aniline coordination. The hallmark of this approach is the ability to pre-program the positioning of multiple metal ions and ligand-based functionalities within a structurally well-defined porous material.
Results and discussion
Synthesis of MOC building-blocks
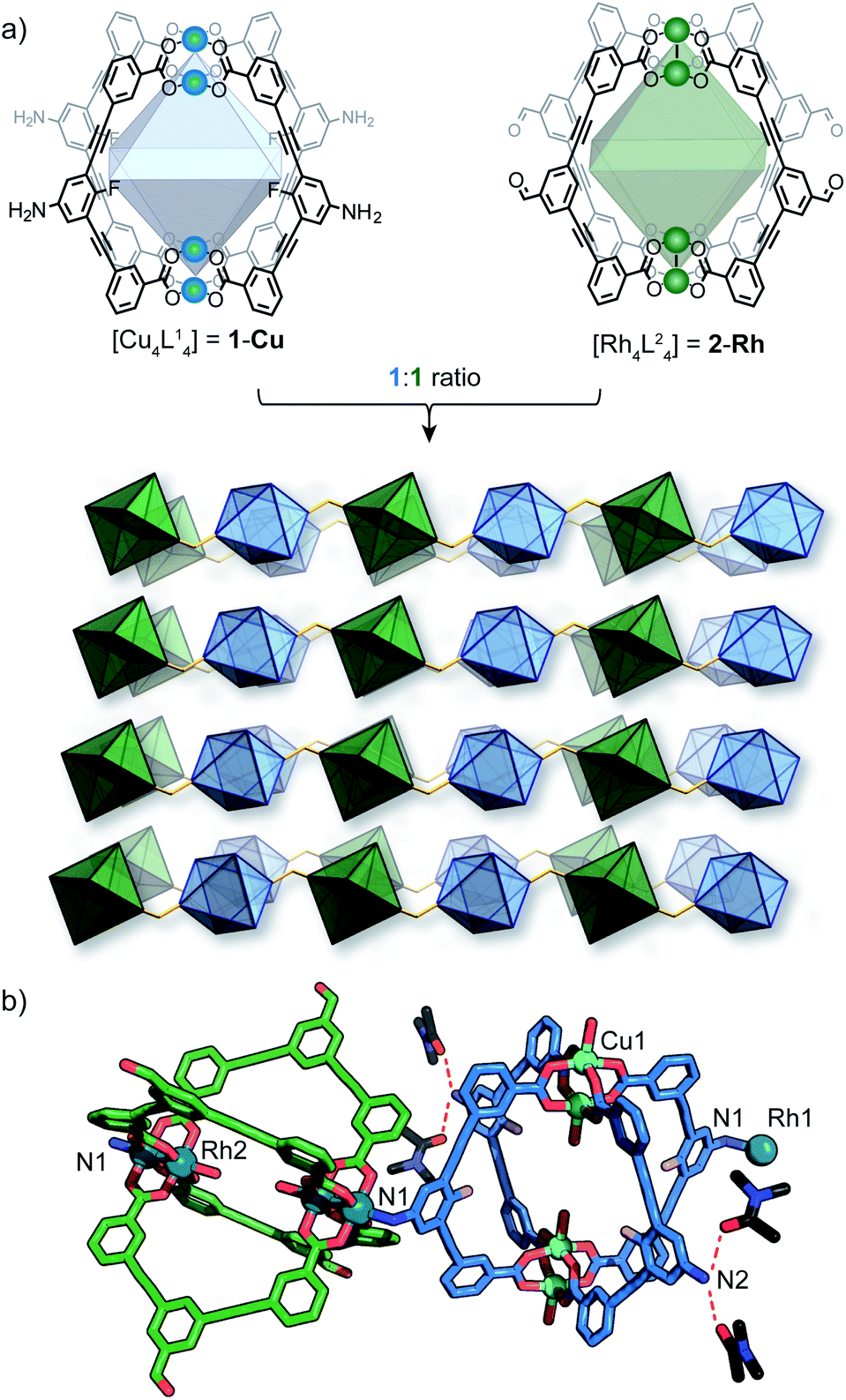
As part of our efforts to design MOCs as SBBs, we recently reported [Cu4L41] – denoted here as 1-Cu.271-Cu is a soluble cage structure composed of two [Cu2(COO)4(solvent)2] paddlewheel nodes and four ligands based on a fluoroaniline backbone (Fig. 2a). 1-Cu is a rare example of a MOC that possesses exterior amine groups and vacant coordination sites yet remains stable in solution.‡28–30 | ||
| Fig. 2 (a) The molecular structure of 1-Cu and 2-Rh and their pairwise linking into a 2-D multivariate MOF {[1-Cu]·[2-Rh]2}n (3); (b) the asymmetric unit of 3; (c) a view of 1-Cu and 2-Rh as four and two connecting nodes, respectively; (d) a perspective view of the 2-D structure of 3, where the two MOCs are represented as polyhedra and their ordered functionalities (Rh(II) and Cu(II), A–D) are annotated. | ||
In order to target a heterogeneous bimetallic system, we prepared a kinetically inert [Rh4L42] cage as the linking partner. We selected the aldehyde-functionalised ligand L2 based on the established chemistry of the [Cu4L42] MOC analogue, as well as the ability to explore covalent linking options.31 Thus, [Rh4L42] (2-Rh, Fig. 2a) was prepared by combining L2, Rh2(OAc)4 and Na2CO3 in DMA and heating the resultant mixture at 85 °C for 32 h. Single-crystals of 2-Rh were grown by slow-vapor diffusion of MeCN into a DMF solution of the cage. Single-crystal X-ray diffraction (SCXRD) confirmed the M4L4 structure and revealed that the axial paddlewheel sites of the MOC are occupied by MeCN ligands (Fig. S26†).
Synthesis and structure of mixed-cage MOF 3
In conceptualizing 1-Cu and 2-Rh as SBBs, both MOCs represent planar four-connecting nodes when considering the positioning and orientation of their covalent functionalities. However, when combining the two MOCs in DMF, we observed a crystallisation phenomenon that was dominant over their covalent reactivity. A microcrystalline precipitate with a Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Rh ratio of 1:2 (as determined by EDX analysis) was isolated from a DMF mixture of 1-Cu and 2-Rh after 2 h at room temperature. This co-crystallisation, however, did not occur in DMA as the solvent, which enabled us to grow larger crystals of the material for SCXRD analysis.
:Rh ratio of 1:2 (as determined by EDX analysis) was isolated from a DMF mixture of 1-Cu and 2-Rh after 2 h at room temperature. This co-crystallisation, however, did not occur in DMA as the solvent, which enabled us to grow larger crystals of the material for SCXRD analysis.
Slow-vapor diffusion of ethyl acetate into the 1:2 mixture of 1-Cu and 2-Rh in DMA resulted in the formation of ∼200 μm-sized plate-like crystals of {[1-Cu]·[2-Rh]2}n, (herein denoted as 3) after 5 days. Synchrotron SCXRD revealed that 3 crystallizes in the tetragonal space group I4/m. The asymmetric unit contains 1/8 of the structure 1-Cu and 1/4 of the structure of 2-Rh (Fig. 2b), which is in agreement with the 1:2 Cu:Rh stoichiometry observed by EDX. The X-ray structure reveals that 1-Cu and 2-Rh crystallise through coordinative linking; the exterior coordination sites of 2-Rh are both occupied by an aniline donor from a separate molecule of 1-Cu (DN1–Rh1: 2.23 Å; C14–N1–Rh1: 109.0° – Fig. 2a). Their connectivity is both highly directional and rational; 1-Cu behaves as a four-connecting node through its exterior aniline donors, whilst 2-Rh behaves as a two-connecting node through the two exterior paddlewheel sites (Fig. 2c). This combination produces a (4,4)-net, where the 2-D layers pack in a staggered ABAB fashion (Fig. 3c). The eclipsed orientation of 2-Rh along the c axis creates channels approximating the dimensions of internal cavity of the MOC (∼7 × 8 Å).
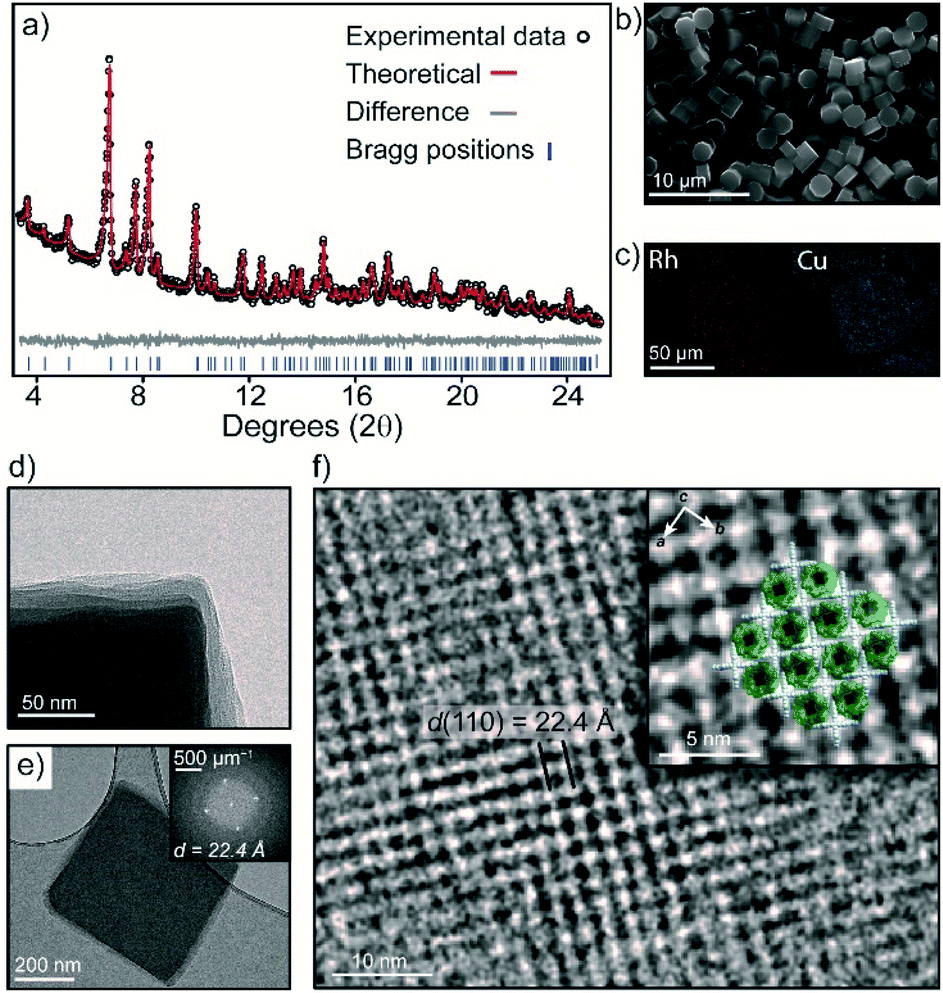 | ||
| Fig. 3 (a) Rietveld refinement of the PXRD data of 3; (b) SEM image of 3 obtained from a DMF recrystallization; (c) EDX elemental mapping of Cu and Rh within crystals of 3; TEM images of 3 (d) the edge of a crystal, showing misalignment of layers; (e) the FFT pattern of a selected crystal; (f) an enlarged image of the same crystal showing the superimposition of the crystal structure of 3 along the c axis. | ||
The bulk purity of 3 was confirmed by Powder X-ray Diffraction (PXRD). A Rietveld refinement of the data yielded Rwp and GoF (goodness of fit) parameters of 3.89 and 1.15 respectively, indicating that the bulk sample is phase pure and in excellent agreement with the SCXRD structure (Fig. 3a and S10†). This is remarkable given the propensity of 1-Cu and 2-Rh to crystallize as discrete structures under similar conditions.27,31 The 1:2 stoichiometry of 1-Cu and 2-Rh as observed by SEM/EDX analysis could also be validated by 1H NMR spectroscopy. A sample of 3 was dissolved in DMF-d7 by heating which produced a soluble mixture of the pure discrete MOCs in a 1:2 ratio, respectively (Fig. S5†). Allowing the solution to stand at 25 °C for 5 days resulted in the re-crystallization of 3 (Fig. 3b and S13†).
Whilst multiple MOC entities are often isolated within MOFs through in situ assembly,32,33 this is the first example where two preformed MOCs have been linked into a multivariate MOF material. Here, the aniline-metal interaction has been precisely tuned to facilitate the pairwise linking of two soluble MOCs upon crystallisation.‡ In doing so, the location and coordination environment of the Cu(II) and Rh(II) metal ions is both predictable and extremely well-defined. In addition, the checkerboard arrangement of 1-Cu and 2-Rh results in the ordering of two alternating pore environments, and therefore, multiple ligand-based functionalities (A–D, Fig. 2d).
Activation and porosity of 3
Activation of 3 was carried out by super-critical CO2 exchange of an acetone-solvated sample. Whilst PXRD revealed a reduction in long-range order (Fig. S13†), transmission electron microscopy (TEM) revealed that the MOC ordering within the layers of 3 is largely maintained. An image focused at the edge of a ∼400 nm sized crystal clearly shows an irregular alignment of the layers in the activated sample (Fig. 3d). Nevertheless, the Fast Fourier Transform (FFT) pattern in Fig. 3e displays prominent peaks that correspond to a d spacing of 22.4 Å. This d spacing is in good agreement with the hkl 110 plane of 3 (hkl 110 = 22.9 Å) which represents the distance between 1-Cu along the crystallographic c axis. In Fig. 3f, the bright regions in the TEM image represent the interconnected 1-Cu and 2-Rh MOCs, where 1-Cu is oriented vertically to the direction of the electron beam. The dark areas correspond to the pores of 2-Rh, which is oriented perpendicular to the electron beam.The crystal packing and pore-structure of 3 is clearly dictated by the coordinative linking of 1-Cu and 2-Rh. This is in stark contrast to most molecular cage solids, where weak non-covalent interactions are responsible for their solid-state ordering and resulting pore-structure.34,35 As such, we sought to evaluate the porosity of 3 by N2 and CO2 adsorption measurements at 77 K and 195 K, respectively. The N2 isotherm of 3 displays a typical type-I adsorption profile with a Brunauer–Emmett–Teller surface area (SABET) of 422 m2 g−1 (Langmuir = 722 m2 g−1). The pore-size distribution (PSD) derived from the low-pressure region shows a maximum at 8.0 Å (Fig. S18†) which agrees well with the expected dimensions of the internal cavity of 2-Rh. In contrast, the two discrete MOC solids are non-porous to N2 at 77 K (Fig. S18†), presumably because of the random structural aggregation that accompanies their activation (both solids are amorphous – Fig. S10† and ref. 28). The CO2 isotherms measured 195 K revealed that 1-Cu, 2-Rh, and 3 display type I profiles (Fig. S19†). A marked increase in surface area is evident for 3 when comparing to the discrete MOCs; SABET of 3 = 359 m2 g−1 (ca. 95 and 282 m2 g−1 for 1-Cu and 2-Rh, respectively – Table S1†).
Stoichiometric control over MOC connectivity and network structure
Given the molecular nature of 1-Cu and 2-Rh, we sought to examine whether their connectivity can be dictated by their relative stoichiometry in solution. Owing to their directional linking, we predicted that a change in MOC stoichiometry should result in a change in network structure. Indeed, whilst a 1:2 stoichiometry of 1-Cu and 2-Rh gave crystals of the 2D MOF 3, a completely different mixed-cage solid (denoted here as 4) was obtained from the crystallisation of a 1:1 MOC mixture.
Synchrotron SCXRD revealed that 4 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with a half of each M4L4 structure in the asymmetric unit. Again, both exterior axial sites of 2-Rh are coordinated by an aniline donor from a separate molecule of 1-Cu (DN–Rh: 2.22 Å; C–N–Rh: 113.2° – Fig. 4b). Due to the change in stoichiometry, only two of the four aniline donors of 1-Cu participate in coordination, and the remaining two donors hydrogen bond with co-crystallized DMA solvent; DNH⋯O = 2.02 Å and 2.46 Å, 151.1° and 166.2°, respectively. Thus, both MOCs act as linear two-connecting nodes to give rise to a 1-D coordination polymer with a formula of {[1-Cu]·[2-Rh]}n (Fig. 4a).
, with a half of each M4L4 structure in the asymmetric unit. Again, both exterior axial sites of 2-Rh are coordinated by an aniline donor from a separate molecule of 1-Cu (DN–Rh: 2.22 Å; C–N–Rh: 113.2° – Fig. 4b). Due to the change in stoichiometry, only two of the four aniline donors of 1-Cu participate in coordination, and the remaining two donors hydrogen bond with co-crystallized DMA solvent; DNH⋯O = 2.02 Å and 2.46 Å, 151.1° and 166.2°, respectively. Thus, both MOCs act as linear two-connecting nodes to give rise to a 1-D coordination polymer with a formula of {[1-Cu]·[2-Rh]}n (Fig. 4a).
 | ||
| Fig. 4 (a) assembly of {[1-Cu]·[2-Rh]}n (4) from a 1:1 mixture of 1-Cu (left) and 2-Rh (right) showing a perspective view of the crystal packing (MOCs represented as polyhedra); (b) a view of the coordination of 1-Cu to 2-Rh. The bulk characterisation of 4 is described in the ESI.† | ||
Elucidating the preference for Rh-aniline coordination by DFT calculations
To rationalize the observed preference for aniline-Rh(II) coordination, we carried out DFT calculations on representative paddlewheel complexes (full details in ESI†). The calculated energy for the ligand exchange reaction shown in Fig. 5a amounts to ΔE = −7.6 kcal mol−1 which supports a thermodynamic argument in favour of preferential coordination of aniline to the Rh(II) paddlewheel complex (as compared to the Cu(II) counterpart). For the optimized complexes, the Cu–N bond is ∼0.7 Å shorter compared to the Rh–N bond, presumably due to the smaller ionic radius of Cu(II) and the trans effect of the Rh(II) complex.36 This agrees well with the crystallographic results where the Rh–N aniline bond of the linked MOCs is longer by ∼0.4 Å compared to Cu–N.27 Furthermore, the optimized geometry of [Cu2(PhCOO)4(PhNH2)2] exhibits Cu–O–O–Cu dihedral angles between 14.7° and 15.9° while the corresponding the Rh–O–O–Rh dihedral angles are below 1.3° (Fig. 5b and S32†). The shorter Cu–N bond length and distortion of [Cu2(PhCOO)4(PhNH2)2] compared to the Rh analogue therefore suggests that the observed preference is associated with a reduced steric hindrance for Rh-aniline coordination. It is noteworthy that Rh24L24 MOCs have recently been used to separate regioisomers of methyl pyridine based on their steric hindrance.37 | ||
| Fig. 5 (a) The ligand exchange reaction between Cu(II) and Rh(II) paddle-wheel complexes as investigated by DFT calculations; (b) a comparison of the optimized structures of [Cu2(PhCOO)4(PhNH2)2] and [Rh2(PhCOO)4(PhNH2)2]. | ||
Conclusions
In summary, we have reported the pairwise linking of MOCs as an approach to precisely control the coordination environment and distribution of multiple metal sites and ligands within crystalline porous materials. In doing so, we reported the synthesis and structure of two mixed-cage crystalline solids which can be selectively obtained from the same Cu4L4 and Rh4L4 MOC mixture through stoichiometric control. In the case of the 2-D MOF (3), the benefit of linking 1-Cu and 2-Rh into a mixed-cage network extends also to an improved porosity compared to the discrete cage counterparts. It is worth noting that the selective coordination of 1-Cu to 2-Rh also addresses the major challenge of synthesizing coordination polymers from kinetically inert Rh(II) metal ions.38 Assuming that the interaction energy and coordination preference are optimised, we expect that this method will extend to a broad range of paddlewheel MOCs22,39,40 and enable access to a wide range of multi-functional MOFs with control over the connectivity and dimesionality.41 Indeed, efforts in this direction are currently underway in our laboratory.Data availability
We have no extra experimental or computational data associated with this article to deposite.Author contributions
WMB: conceptualisation, synthesis, analysis, characterisation (SCXRD, PXRD, TGA, IR, adsorption analysis), writing and editing. AM-H: ligand synthesis and analysis. MR: DFT calculations. ADS: TEM measurements. OL-P: SEM measurements. All authors provided comments and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. M. B gratefully acknowledges the Australian Research Council for financially supporting this project (DE190100327). MR thanks the Deutsche Forschungsge-meinschaft (DFG) for funding through Emmy-Noether grant RO 5688/1-1. Jorge Albalad is thanked for advice and helpful discussions. Aspects of this research were undertaken on the MX1 (ref. 42) and MX2 (ref. 43) beamlines at the Australian Synchrotron, Victoria, Australia. The authors acknowledge the instruments and scientific and technical assistance of Microscopy Australia at Adelaide Microscopy, The University of Adelaide, a facility that is funded by the University, and State and Federal Governments.Notes and references
- W. Xu, B. Tu, Q. Liu, Y. Shu, C.-C. Liang, C. S. Diercks, O. M. Yaghi, Y.-B. Zhang, H. Deng and Q. Li, Nat. Rev. Mater., 2020, 5, 764–779 CrossRef CAS.
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC.
- Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- R.-B. Lin, S. Xiang, W. Zhou and B. Chen, Chem, 2020, 6, 337–363 CAS.
- W. Fan, S. Yuan, W. Wang, L. Feng, X. Liu, X. Zhang, X. Wang, Z. Kang, F. Dai, D. Yuan, D. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2020, 142, 8728–8737 CrossRef PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- H. Furukawa, U. Müller and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 3417–3430 CrossRef CAS PubMed.
- M. Y. Masoomi, A. Morsali, A. Dhakshinamoorthy and H. Garcia, Angew. Chem., Int. Ed., 2019, 58, 15188–15205 CrossRef CAS PubMed.
- R.-B. Lin, S. Xiang, B. Li, Y. Cui, G. Qian, W. Zhou and B. Chen, Coord. Chem. Rev., 2019, 384, 21–36 CrossRef CAS.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS.
- A. K. Cheetham, G. Kieslich and H. H.-M. Yeung, Acc. Chem. Res., 2018, 51, 659–667 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619–1636 RSC.
- Y.-P. He, G.-H. Chen, D.-J. Li, Q.-H. Li, L. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2920–2923 CrossRef CAS PubMed.
- J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed.
- A. J. Gosselin, A. M. Antonio, K. J. Korman, M. M. Deegan, G. P. A. Yap and E. D. Bloch, J. Am. Chem. Soc., 2021, 143, 14956–14961 CrossRef CAS PubMed.
- G. R. Desiraju, Nat. Mater., 2002, 1, 77–79 CrossRef CAS.
- A. Pulido, L. Chen, T. Kaczorowski, D. Holden, M. A. Little, S. Y. Chong, B. J. Slater, D. P. McMahon, B. Bonillo, C. J. Stackhouse, A. Stephenson, C. M. Kane, R. Clowes, T. Hasell, A. I. Cooper and G. M. Day, Nature, 2017, 543, 657–664 CrossRef CAS.
- D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS PubMed.
- S. Lee, H. Jeong, D. Nam, M. S. Lah and W. Choe, Chem. Soc. Rev., 2021, 50, 528–555 RSC.
- J. R. Li, D. J. Timmons and H. C. Zhou, J. Am. Chem. Soc., 2009, 131, 6368–6369 CrossRef CAS.
- T. Grancha, A. Carné-Sánchez, F. Zarekarizi, L. Hernández-López, J. Albalad, A. Khobotov, V. Guillerm, A. Morsali, J. Juanhuix, F. Gándara, I. Imaz and D. Maspoch, Angew. Chem., Int. Ed., 2021, 60, 5729–5733 CrossRef CAS.
- H.-N. Wang, X. Meng, G.-S. Yang, X.-L. Wang, K.-Z. Shao, Z.-M. Su and C.-G. Wang, Chem. Commun., 2011, 47, 7128–7130 RSC.
- H.-N. Wang, F.-H. Liu, X.-L. Wang, K.-Z. Shao and Z.-M. Su, J. Mater. Chem. A, 2013, 1, 13060–13063 RSC.
- M. L. Schneider, A. W. Markwell-Heys, O. M. Linder-Patton and W. M. Bloch, Front. Chem., 2021, 9, 382 Search PubMed.
- M. L. Schneider, O. M. Linder-Patton and W. M. Bloch, Chem. Commun., 2020, 56, 12969–12972 RSC.
- G. A. Taggart, A. M. Antonio, G. R. Lorzing, G. P. A. Yap and E. D. Bloch, ACS Appl. Mater. Interfaces, 2020, 12, 24913–24919 CrossRef CAS.
- H. Furukawa, J. Kim, N. W. Ockwig, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11650–11661 CrossRef CAS.
- W. M. Bloch, R. Babarao and M. L. Schneider, Chem. Sci., 2020, 11, 3664–3671 RSC.
- J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS.
- E. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120, 8987–9014 CrossRef PubMed.
- Y. B. Koh and G. G. Christoph, Inorg. Chem., 1978, 17, 2590–2596 CrossRef CAS.
- L. Hernández-López, J. Martínez-Esaín, A. Carné-Sánchez, T. Grancha, J. Faraudo and D. Maspoch, Angew. Chem., Int. Ed., 2021, 60, 11406–11413 CrossRef.
- W.-Y. Gao, A. Sur, C.-H. Wang, G. R. Lorzing, A. M. Antonio, G. A. Taggart, A. A. Ezazi, N. Bhuvanesh, E. D. Bloch and D. C. Powers, Angew. Chem., Int. Ed., 2020, 59, 10878–10883 CrossRef CAS PubMed.
- S. Furukawa, N. Horike, M. Kondo, Y. Hijikata, A. Carné-Sánchez, P. Larpent, N. Louvain, S. Diring, H. Sato, R. Matsuda, R. Kawano and S. Kitagawa, Inorg. Chem., 2016, 55, 10843–10846 CrossRef CAS PubMed.
- E.-S. M. El-Sayed and D. Yuan, Chem. Lett., 2019, 49, 28–53 CrossRef.
- V. Guillerm and M. Eddaoudi, Acc. Chem. Res., 2021, 54, 3298–3312 CrossRef CAS.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS PubMed.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and supporting analysis. CCDC 2106776–2106778. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05663h |
| ‡ The fluoroaniline backbone of L1 is electronically tuned to reduce the possibility of Cu(II)-aniline coordination; phenylene diamine derived ligands were previously observed to interfere with Cu4L4 MOC assembly.28 We also attempted to assemble Cu4L4 MOCs from a pyridyl-functionalised dicarboxylate ligand (ESI†). However, this type of SBB could not be accessed due to the propensity of pyridine to coordinate to Cu(II) during MOC assembly (Fig. S3†). This further demonstrates the importance of tuning the metal–ligand electronics of the linking donor group. |
| This journal is © The Royal Society of Chemistry 2022 |