DOI:
10.1039/D2RA04485D
(Paper)
RSC Adv., 2022,
12, 26042-26050
A simple and direct ionic chromatography method to monitor galactose oxidase activity†
Received
20th July 2022
, Accepted 6th September 2022
First published on 13th September 2022
Abstract
Galactose oxidase (GalOx, EC.1.1.3.9) is one of the most extensively studied copper radical oxidases (CROs). The reaction catalyzed by GalOx leads to the oxidation of the C-6 hydroxyl group of galactose and galactosides (including galactosylated polysaccharides and glycoproteins) to the corresponding aldehydes, coupled to the reduction of dioxygen to hydrogen peroxide. Despite more than 60 years of research including mechanistic studies, enzyme engineering and application development, GalOx activity remains primarily monitored by indirect measurement of the co-product hydrogen peroxide. Here, we describe a simple direct method to measure GalOx activity through the identification of galactosylated oxidized products using high-performance anion-exchange chromatography coupled to pulsed amperometric detection (HPAEC-PAD). Using galactose and lactose as representative substrates, we were able to separate and detect the C-6 oxidized products, which were confirmed by LC-MS and NMR analyses to exist in their hydrated (geminal-diol) forms. We show that the HPAEC-PAD method is superior to other methods in terms of sensitivity as we could detect down to 0.08 μM of LacOX (eq. 30 μg L−1). We believe the method will prove useful for qualitative detection of galactose oxidase activity in biological samples or for quantitative purposes to analyze enzyme kinetics or to compare enzyme variants in directed evolution programs.
Introduction
Carbohydrate-Active enZymes (CAZymes) are naturally produced by microorganisms to catalyze the assembly, modification, and degradation of complex biomacromolecules, oligo- and polysaccharides.1–3 CAZymes are widely used and essential in numerous biotechnological processes including the production of biomaterials and bioproducts.4 Within the CAZy classification (https://www.cazy.org/),5 the Auxiliary Activity (AA) families comprise a diversity of redox enzymes acting mainly on carbohydrates and/or aromatic compounds.6 Auxiliary Activity family 5 (AA5) contains exclusively copper radical oxidases (CROs) and is subdivided into two subfamilies, viz. AA5_1, containing glyoxal oxidases (GlOx, EC 1.2.3.5),7 and AA5_2 that is now known to include diverse CROs such as galactose oxidases (GalOx, EC 1.1.3.9)8, alcohol oxidases (AlcOx, EC 1.1.3.13),9 and aryl alcohol oxidases (AAO, EC 1.1.3.7).10
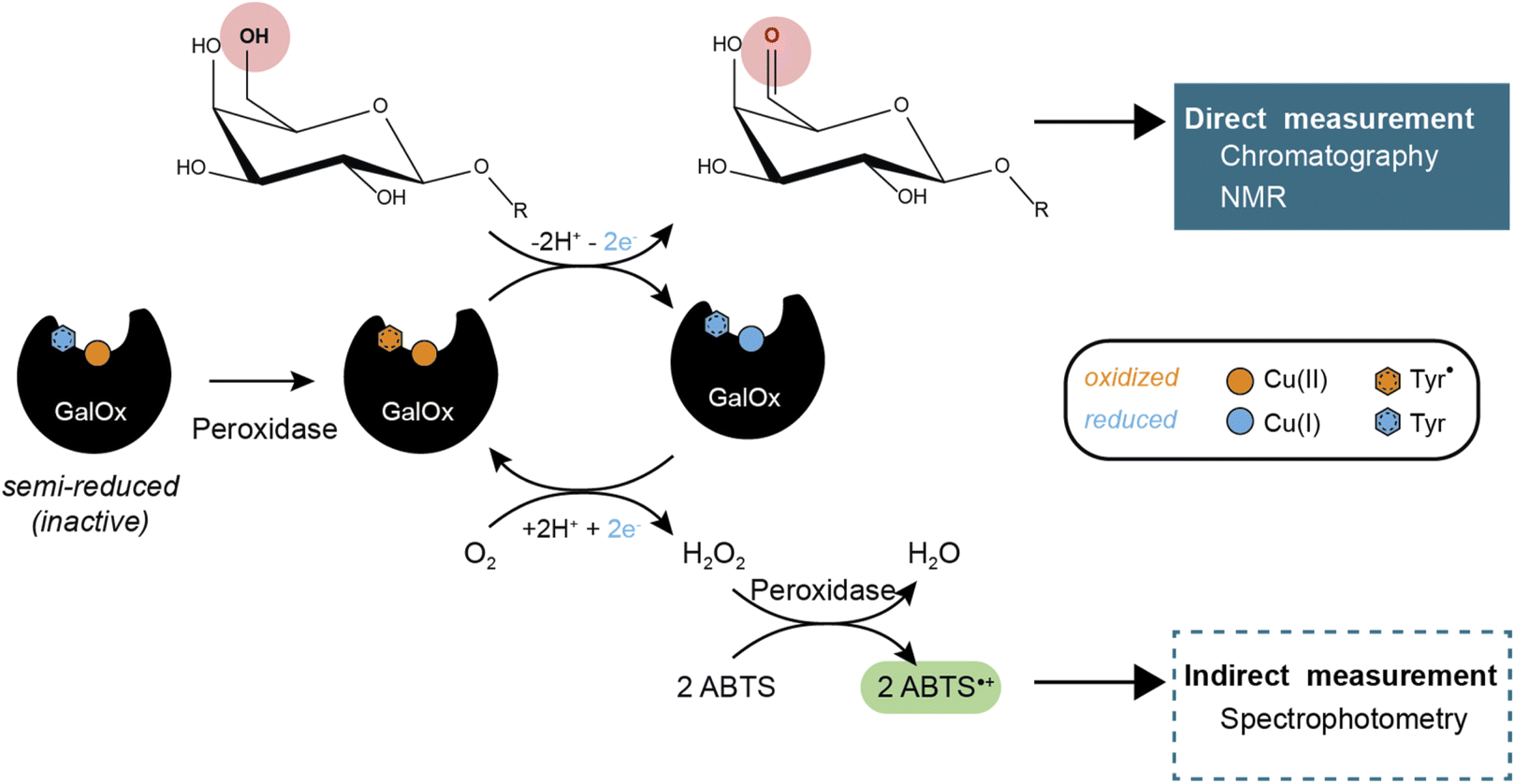
GalOx catalyzes the regioselective oxidation of the 6-hydroxyl group of galactose/galactosylated polysaccharides to the corresponding aldehydes. The two electron oxidation of the carbohydrate is coupled to the two electron reduction of molecular oxygen to H2O2 (Scheme 1).11 AA5 CROs can also exist in an off-cycle, inactive resting state, and therefore require activation by addition of inorganic oxidants12,13 or by accessory redox enzymes (e.g., horseradish peroxidase (HRP)14–16 or fungal peroxidases17) to achieve full activity.
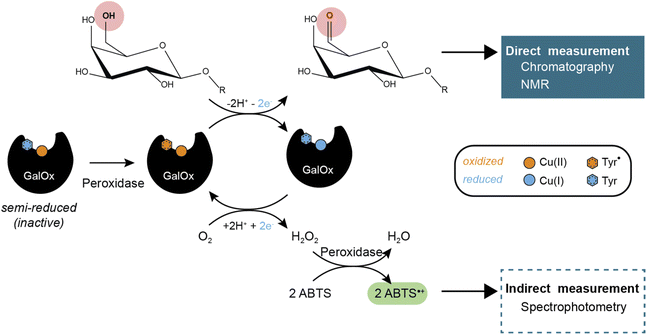 |
| | Scheme 1 GalOx reaction mechanism and products detection methods. | |
Since its discovery in 1959, GalOx from the cereal head blight ascomycete Fusarium graminearum (FgrGalOx, EC.1.1.3.9)18 has been the most extensively studied CRO and is therefore the archetype and benchmark AA5 CRO. For instance, protein engineering has been used extensively to increase FgrGalOx catalytic efficiency19–21 and extended its substrate scope to include diverse carbohydrates22,23 and/or alcohols.24,25 Hence, both wild-type and engineered FgrGalOx variants have been used in different biotechnological applications, such as for the labeling of glycoproteins,26 the chemo-enzymatic modification of galactose and galactosides,27 the engineering of lactose biosensors,28 the modification of polysaccharide for the development of functional materials,29–31 or, more recently, in a cascade reaction to produce the anti-HIV-1 drug Islatravir.32 Taking advantage of recombinant enzyme production pipelines, a large diversity of AA5_2 enzymes has been recently characterized.9,10,33–36
GalOx activity has historically been measured by indirect, spectrophotometric methods that rely on quantifying the co-product H2O2 using a coupled horseradish peroxidase (HRP)/2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid (ABTS) assay.37 Despite widespread use, such assays have certain limitations due to the cross-reactivity of H2O2 or the instability of the reporter molecule (ABTS cation radical). Yet, some studies have investigated the use of alternative methods that require specific equipment and expertise, such as electrochemical detection of oxygen consumption38 or H2O2 production,39 or NMR analysis of carbohydrate products.40 Therefore, there is considerable interest in the development of easily accessible methods for the direct measurement of the oxidized carbohydrate product, especially for mechanistic, protein engineering, and biotechnological studies.
High-performance anion-exchange chromatography coupled to pulsed amperometric detection (HPAEC-PAD) is a powerful, widely used analytical technique enabling the high-resolution separation and sensitive quantification of monosaccharides and oligosaccharides.41 Hence, HPAEC-PAD is widely used in analytical glycobiology, including for quantitative assays of carbohydrate-active enzymes such as glycoside hydrolases,42 lytic polysaccharide monooxygenases,43 and oligosaccharide oxidases/dehydrogenases.44 Here, we present a simple method to quantify GalOx activity through the direct measurement of oxidized galactosylated products using HPAEC-PAD. This method can be readily and widely deployed to monitor GalOx enzyme kinetics, given the prevalence of such HPLC systems in research laboratories.
Experimental
General information
Most chemicals were purchased from Sigma-Aldrich (Germany). HRP type II and catalase from bovine liver were purchased from Sigma-Aldrich. Molar concentrations of HRP (MW: 33.89 kDa) and catalase (MW of monomer: 62.5 kDa) were estimated by Bradford assay.
FgrGalOx production and purification
Bioreactor production. Building on an Invitrogen protocol (Pichia Fermentation Process Guidelines), FgrGalOx production was carried out in a 1.3 L bioreactor (New Brunswick BioFlo 115 fermentor, Eppendorf, Germany) as follows: preculture was prepared as described previously15 and was used to inoculate at 0.2% (v/v) 100 mL of BMGY medium, in a 500 mL flask, incubated (30 °C, 200 rpm) until the OD600nm reached 4–6. 400 mL of basal salt medium containing 4.5 mL L−1 PTM1 trace salts (both made according to the P. pastoris fermentation process guidelines – Invitrogen) were inoculated with 10% (v/v) of the BMGY culture. Temperature was set to 30 °C. 100 μL of Pluronic E8100 (BASF, Germany) were added after 6 h of culture to prevent foam. After full consumption of glycerol (as indicated by a return of dissolve oxygen (DO) level at 100%), sorbitol–methanol transition phase was initiated by addition of 80 mL sorbitol (250 g L−1 stock solution), 1.6 mL PTM1 traces salts and 2 mL methanol. After full consumption of carbon sources, the temperature was lowered to 20 °C and a methanol fed-batch was initiated with a feeding rate of 3.9 mL h−1 L−1 (mL per hour per liter of initial fermentation volume) of a methanol and PTM1 trace salts (12 mL L−1). New additions of 100 μL Pluronic E8100 were made after 30 h and 54 h of fermentation. Methanol feeding rate was increased to 7.8 mL h−1 L−1 after 52 h of fermentation. Throughout the fermentation, pH was maintained at 5 by automated adjustment with NH3 base. Air flow was maintained at 0.5 slpm (standard liter per minute). A cascade with a set point of 20% dissolved oxygen is maintained through agitation between 400 to 900 rpm and the percentage of pure oxygen addition between 0 to 50%. Fermentation was ceased after 122 hours. The culture was centrifuged (10 min, 5500×g, 4 °C). The supernatant harvested and filtered through a 0.45 μm membrane (Millipore), aliquoted and flash-frozen in liquid nitrogen and stored at −80 °C. We verified that flash-freezing did not cause any enzyme activity loss for FgrGalOx. One aliquot of frozen supernatant was thawed at 4 °C overnight before purification (vide infra).
Protein purification. The pH of the culture supernatant was adjusted to 7.8 before purification, filtered on 0.22 μm filters (Merck-Millipore, Germany), and loaded onto a 5 mL HiTrap HP column (GE Healthcare, Buc, France) equilibrated with buffer A (50 mM Tris–HCl, pH 7.8, 150 mM NaCl and 10 mM imidazole) that was connected to an Äkta purifier 100 (GE Healthcare). (His)6-tagged recombinant enzyme was eluted with buffer B (50 mM Tris–HCl, pH 7.8, 150 mM NaCl and 500 mM imidazole). Fractions containing the recombinant enzyme were pooled, concentrated with a 10-kD Vivaspin concentrator (Sartorius, Palaiseau, France) to remove imidazole and buffer exchanged in sodium phosphate buffer (50 mM, pH 6.0).
Protein analysis. The protein concentration was determined by UV absorption at 280 nm using a Nanodrop ND-200 device (Thermo Fisher Scientific, USA) with calculated molecular mass (73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 247 Da) and molar extinction coefficients (124135 M−1 cm−1) derived from the sequence using the ProtParam online tool (Expasy.org). The protein was loaded onto 10% SDS-PAGE gels (Thermo Fisher Scientific, IL, USA), which was stained with Imperial Protein Stain (Thermo Fisher Scientific, IL, USA). The molecular mass under denaturating conditions was determined with reference standard proteins (Page Ruler Prestained Protein Ladder, Thermo Fisher Scientific).
247 Da) and molar extinction coefficients (124135 M−1 cm−1) derived from the sequence using the ProtParam online tool (Expasy.org). The protein was loaded onto 10% SDS-PAGE gels (Thermo Fisher Scientific, IL, USA), which was stained with Imperial Protein Stain (Thermo Fisher Scientific, IL, USA). The molecular mass under denaturating conditions was determined with reference standard proteins (Page Ruler Prestained Protein Ladder, Thermo Fisher Scientific).
Enzymatic reactions
Spectrophotometric assay. FgrGalOx initial rates were determined by monitoring H2O2 released during FgrGalOx-catalyzed oxidation of galactose or lactose, using the coupled ABTS/HRP assay.45 Routine assays were performed in 96-wells transparent microtiter plates (flat bottom, polystyrene – Greiner Bio One, Austria) in 100 μL final volume containing ABTS powder (0.25 mg mL−1), HRP powder (as provided by the supplier; 0.1 mg mL−1), substrate (0.1–7 mM) and 5 nM FgrGalOx (unless indicated otherwise) in sodium phosphate buffer (50 mM, pH 7.0), at 23 °C. Reactions were initiated by the addition of substrate to a premix containing all other reagents. Evolution of the absorbance at 414 nm (ABTS˙+ cation radical) was measured over time with a Tecan Infinite M200 (Tecan, Switzerland) plate reader. Oxidation of 1 mole of substrate by FgrGalOx consumes 1 equivalent of O2 and generates 1 equivalent of H2O2 which is in turn used by HRP as co-substrate to oxidize 2 equivalents of ABTS. Standard curves of know concentrations of H2O2 (1 to 10 μM) were made and used to quantify H2O2 production. One unit of FgrGalOx activity was defined as the amount of enzyme necessary to produce 1 μmole of H2O2 per minute.
GalOx reactions for chromatographic analyses. Routine assays were performed in 2 mL-Eppendorf tubes in 1 mL final volume containing FgrGalOx (50 nM), galactose (3 mM final concentration) or lactose (3 mM), and HRP (0.05 mg mL−1), in sodium phosphate buffer (50 mM, pH 7.0) and incubated in a Thermomixer (30 °C, 1000 rpm, 2 min to 24 h). 200 μL of the reaction were sampled and heated (100 °C, 5 min) in a dry bath incubator to stop the reaction. The mixture was centrifuged (5 min, 12000×g, 4 °C) and the supernatant collected and diluted in milliQ water before injection on HPAEC column (vide infra). For LC-MS analyses, reactions were carried out as detailed above with the following variations: 1 mM substrate was used and reactions were prepared in milliQ water only instead of buffer. Regarding the experiment comparing the HPAEC-PAD and spectrophotometric methods, the reaction (2 mL final volume) contained FgrGalOx (10 nM), lactose (3 mM), HRP (0.1 mg mL−1) and ABTS (0.25 mg mL−1) in sodium phosphate buffer (50 mM, pH 7.0) and incubated at 23 °C in a cuvette, under magnetic stirring. At regular intervals, two samples of 100 μL were taken and either (i) boiled (100 °C, 5 min) and mixed with 900 μL of milliQ water (for HPAEC-PAD analysis), or (ii) mixed with 900 μL of acetic acid (0.1% final) (for spectrophotometric monitoring of ABTS˙+ cation radical formation). Of note, we used the acid treatment to stop the reaction as this method did not affect the signal of the ABTS˙+ cation radical whereas boiling and NaOH did.
HPAEC-PAD analyses
The detection method is performed using a high-performance anion-exchange chromatography (HPAEC) coupled with pulsed amperometric detection (PAD) (DIONEX ICS6000 system, Thermo Fisher Scientific, Waltham, MA, USA). The system is equipped with a CarboPac-PA1 guard column (2 × 50 mm) and a CarboPac-PA1 column (2 × 250 mm) kept at 30 °C. Elution was carried out at a flow rate of 0.25 mL min−1 and 25 μL of sample was injected. The solvents used were 100 mM NaOH (eluent A) and NaOAc (1 M) in 100 mM NaOH (eluent B). The initial conditions were set to 100% eluent A, and the following gradient was applied: 0–10 min, 0–10% B; 10–20 min, 10–18% B; 20–26 min, 18–100% B (non-linear gradient profile called “curve 6”); 26–27 min, 100–0% B; 27–36 min, 100% A. Integration was performed using the Chromeleon 7.2.10 chromatography data software.
LC-MS analyses
LC-MS analysis was performed on a UHPLC Ultimate 3000RS (Thermo Scientific) coupled to Charged Aerosol Detector (CAD Corona, Thermo Scientific) and a ISQ-EM mass spectrometer with Heated ESI-interface (Thermo Scientific). The eluent was splitted 1:1 and the resulting flow from the LC to the MS was in all cases 0.125 mL min−1. The heated ESI was operated at 75 °C in negative mode at −2 kV spray current, with a sheath gas flow of 23.5 and an auxiliary gas flow of 2.6 (arbitrary units). The capillary temperature was 250 °C. UHPLC-ESI-MS data were acquired and analyzed with Chromeleon software v7.2.10 (Thermo Scientific). An Acquity UHPLC BEH Amide column (2.1 mm × 150 mm, 1.7 mm, Waters, Milford, USA) was used for chromatographic separation of analytes. Enzyme assay aliquots were diluted 5-fold in acetonitrile (20 μL assay aliquot + 80 μL acetonitrile) and 2 μL of diluted samples were injected. The column temperature was maintained at 30 °C. The isocratic elution method uses ammonium formate 12 mM-acetonitrile 35/65% (v/v) at a flow rate of 0.25 mL min−1. Masses from 50 to 1500 m/z were monitored.
NMR spectroscopy analyses
Reactions containing 50 mM of substrate (galactose and lactose) and 0.7 mg mL−1 of both catalase and HRP were initiated by the addition of purified FgrGalOx (10 μM) in a final volume of 1 mL, in sodium phosphate buffer (50 mM, pH 7.0). Reactions were incubated in a Thermomixer (30 °C, 1000 rpm, 24 h). Reaction mixtures were frozen in liquid nitrogen and lyophilized. The resulting powders were resuspended in D2O for NMR analysis. Control reactions without enzyme were performed under the same conditions. NMR spectra were acquired on a Bruker AV III HD 400 MHz spectrometer equipped with a BBFO smart probe and on the AVANCE 600 MHz spectrometer. NMR spectra were calibrated using an internal standard of acetone (25 mM and 50 mM for the galactose and lactose samples, respectively). The 1H and 13C{1H} spectra were referenced at 2.22 ppm and 30.89 ppm, respectively. Peak integration values were used to determine the extent of substrate conversion to product(s).
Results and discussion
Lactose is a good model substrate for monitoring galactose oxidase activity
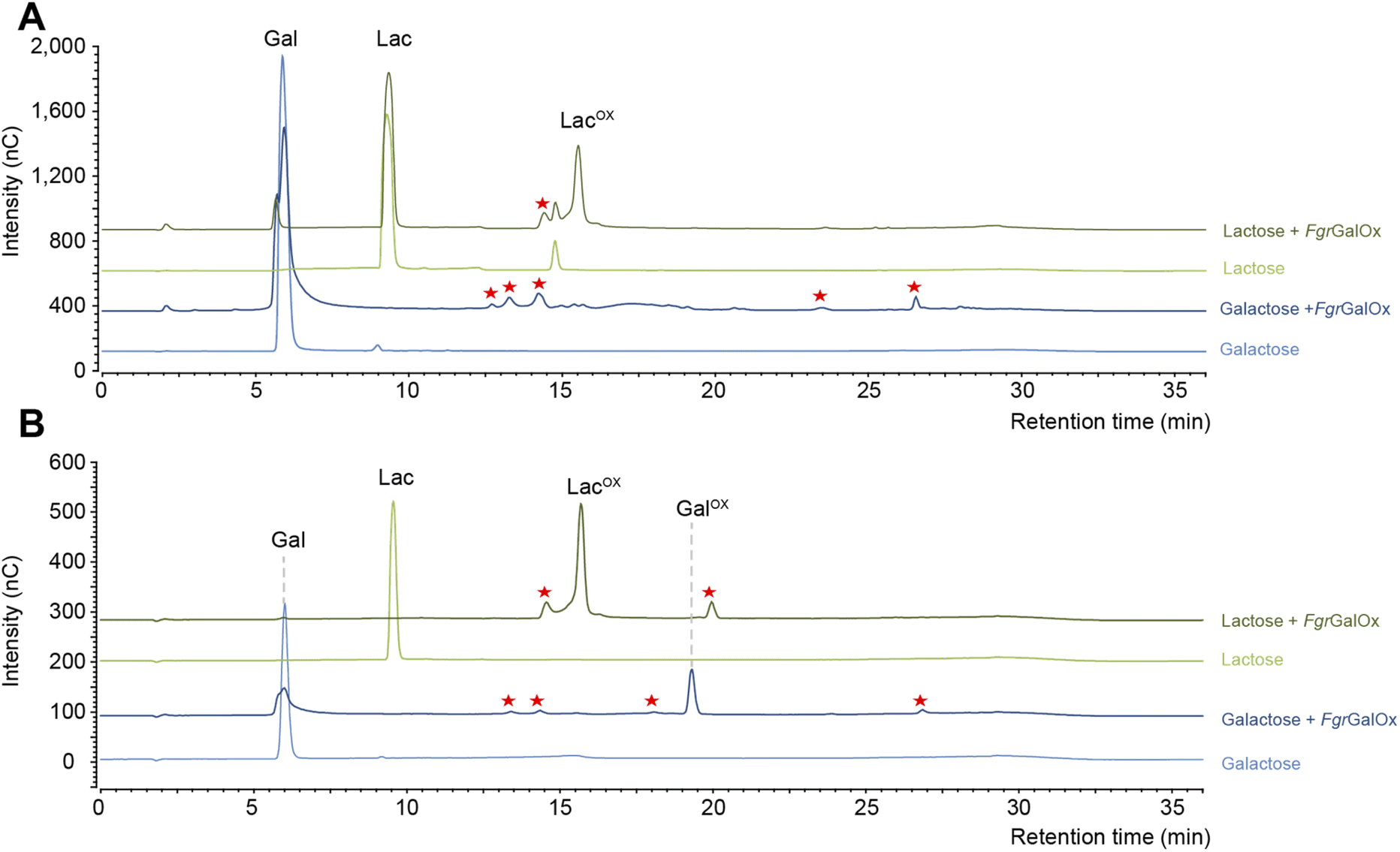
Early NMR studies have shown that FgrGalOx-catalyzed oxidation of galactose can yield a very complex mixture of secondary products derived from the aldehyde (i.e., hydrated geminal-diol, dehydrated form, overoxidized carboxylic form, disaccharide formation).46 Accordingly, we also observed a complex product profile using the HPAEC-PAD method developed in the frame of this study (Fig. 1A). Interestingly, using a galactosylated substrate (such as lactose) in which the C1 position of the galactose unit is engaged in a glycosidic bond (with D-glucose here), yielded a simpler product profile (Fig. 1A). Of note, the complexity of the product profile could be resolved by playing on reaction conditions, as shown in Fig. 1B, where conditions aiming at optimizing the conversion yield (i.e. high enzyme dosage, presence of catalase) were employed. Thus, depending on the reaction set-up, the extent of complexity of the product profile can be tuned. Nevertheless, in the absence of prior knowledge on optimal conditions, the use of D-lactose seems more appropriate as it yields a much simpler product profile, regardless of reaction conditions.
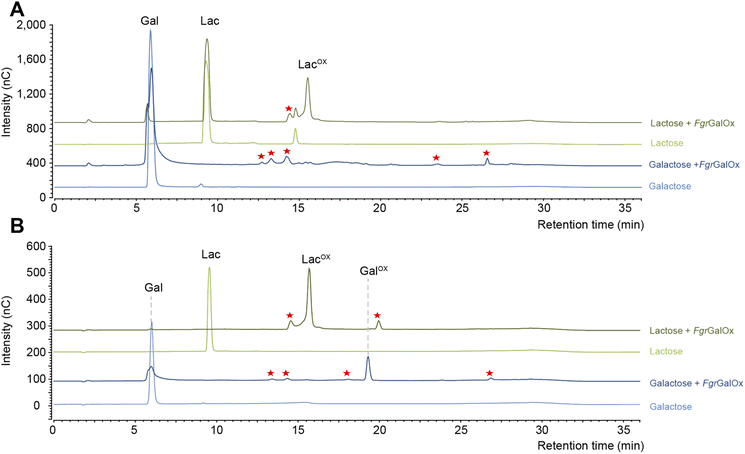 |
| | Fig. 1 Comparison of FgrGalOx product profile on galactose and lactose. The graphs show HPAEC-PAD chromatograms of products generated under (A) short reaction conditions and (B) optimized conditions for NMR analysis. In panel (A), FgrGalOx (50 nM) was incubated with D-galactose (3 mM final concentration) or D-lactose (3 mM), and HRP (0.05 mg mL−1). Reactions were prepared in sodium phosphate buffer (50 mM, pH 7.0) and incubated 1 h at 30 °C, 1000 rpm. In panel (B), FgrGalOx (10 μM) was incubated with D-galactose (50 mM final concentration) or lactose (50 mM), HRP and catalase (0.7 mg mL−1 each). Enzymatic reactions were prepared in sodium phosphate buffer (50 mM, pH 7.0) and incubated 24 h at 30 °C, 1000 rpm. Experiments were carried out in triplicate (n = 3 independent replicates). For the sake of clarity, only one replicate is shown. Red stars indicate secondary products. | |
The main product detected is a geminal-diol on the C6 position of the galactose unit
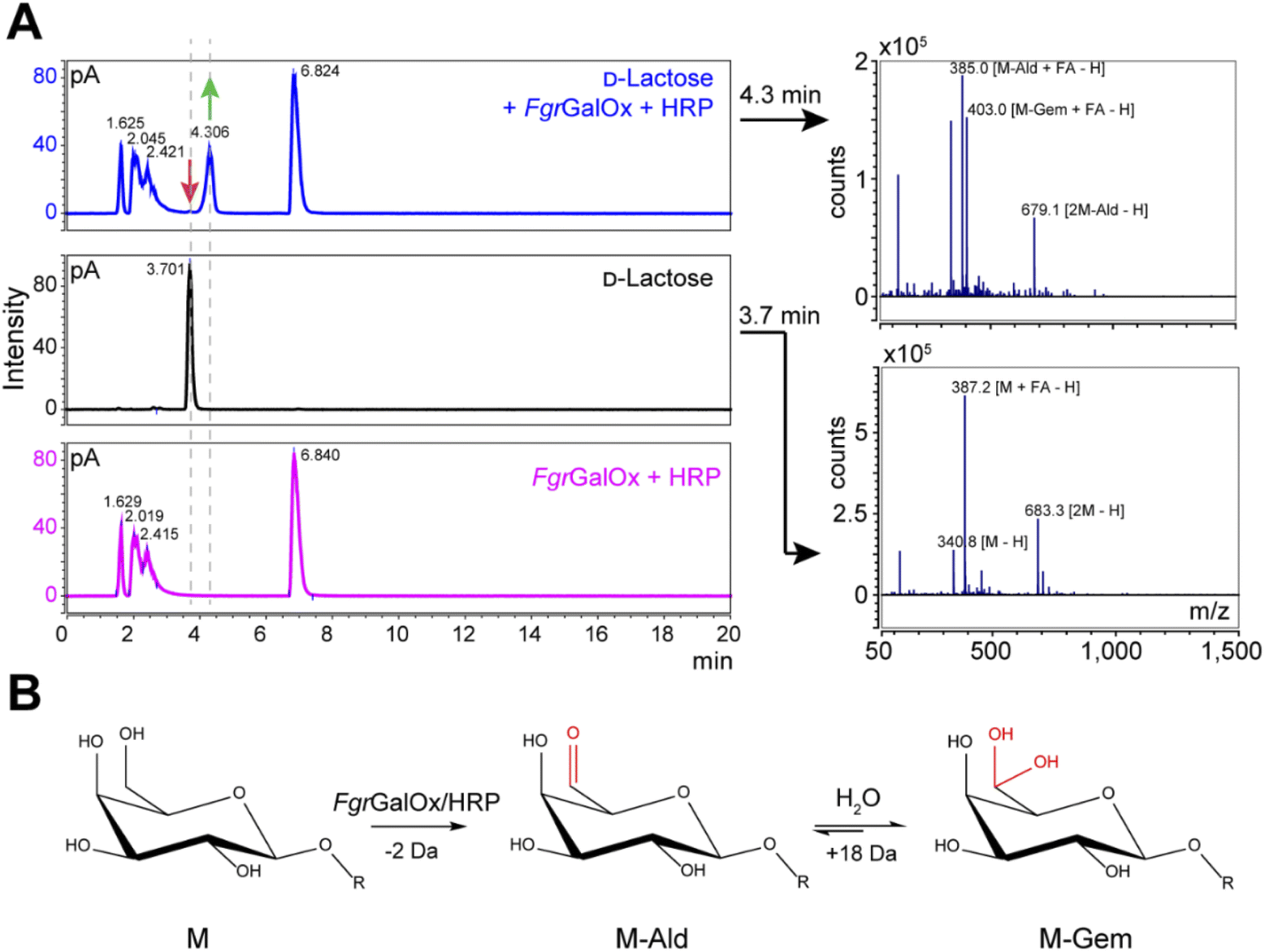
Analyses using liquid chromatography (LC) coupled to mass spectrometry (MS) allowed to separate the initial substrate (lactose, with a mass M) from its oxidized form. The latter was observed to be a mixture of aldehyde (M-2 Da; abbreviated M-Ald) and geminal-diol forms (M + 16 Da; abbreviated M-Gem) (Fig. 2A). We note that the M-2 form could also be a dehydrated form of the geminal-diol [M + 16–18 (H2O)]. Similar results were obtained when using D-galactose as substrate (Fig. S1†).
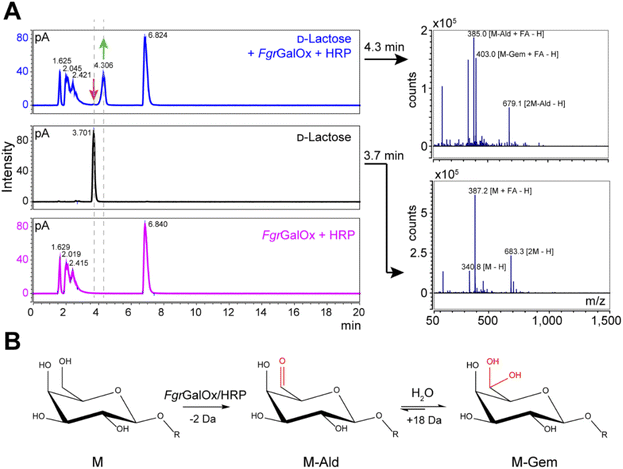 |
| | Fig. 2 LC-MS analysis of FgrGalOx reaction products. (A) LC-MS analysis showing the detection of adducts (in negative mode; FA, formate) of D-lactose, m/z (g mol−1) = 387.1 [M + FA − H] and 683.3 [2M−H]); and derived oxidized forms: aldehyde, m/z = 385.1 [M-Ald + FA − H]); and geminal-diol, m/z = 403.1 [M-Gem + FA − H]), 679.1 [2M-Gem − H]). We note that the −2 Da form (m/z = 385.1) can also be a dehydrated form of the geminal-diol [M-Gem − H2O − H]. Reaction mixtures contained D-lactose (1 mM final concentration), FgrGalOx (50 nM) and HRP (0.05 mg mL−1) in water and were incubated in a Thermomixer (23 °C, 1000 rpm, 24 h). (B) Reaction scheme of FgrGalOx-catalyzed production of aldehyde (M-Ald) and derived geminal-diol (M-Gem). For D-lactose R = (1,4)-D-Glcp; for D-galactose, R = H (see Fig. S1†). | |
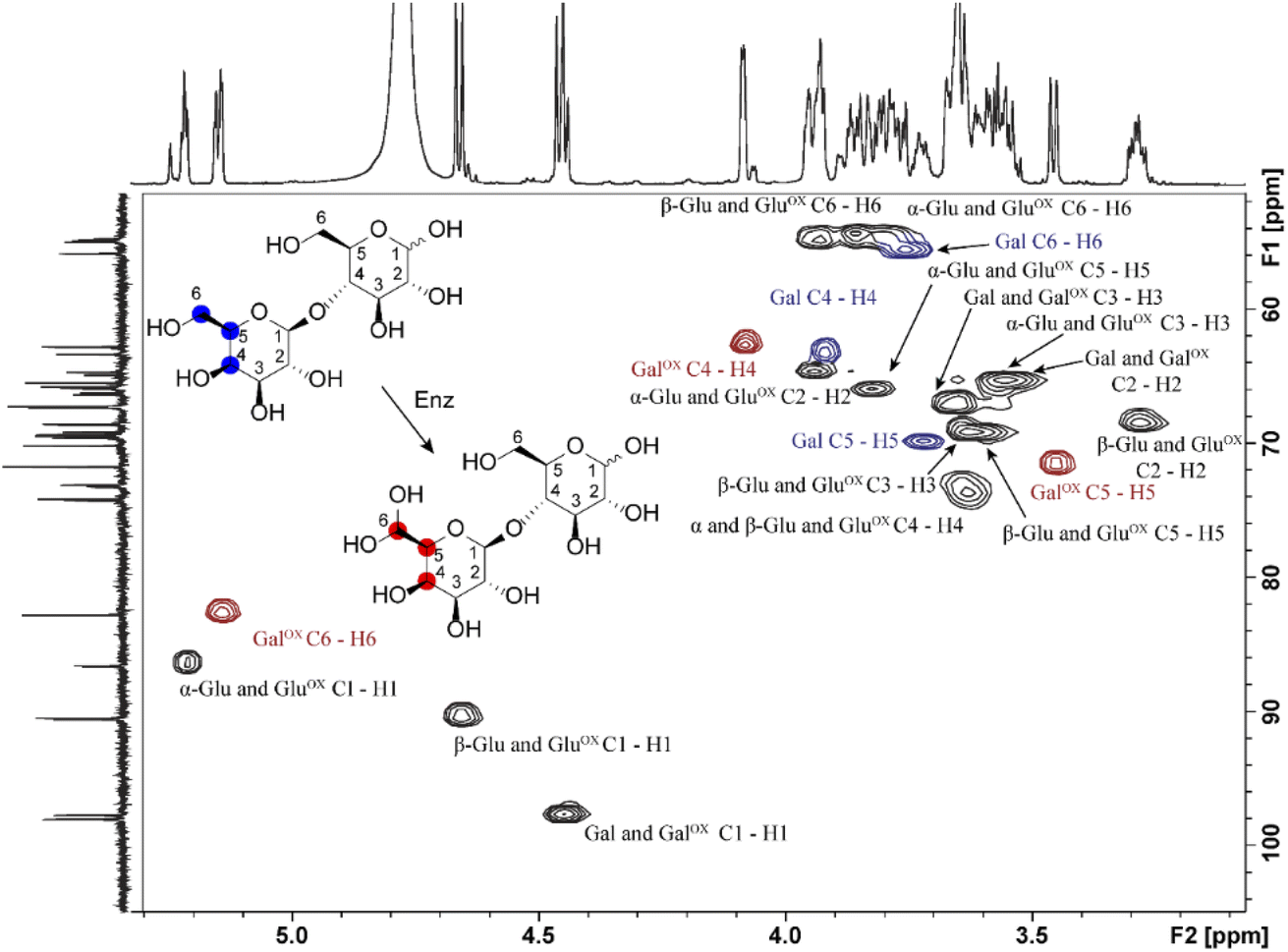
Samples generated under optimized conditions and analyzed by HPAEC-PAD (Fig. 1B) were also analyzed by NMR spectroscopy (Fig. 3). The data indicate that both galactose (Fig. S2–S5, Table S1†) and lactose (Fig. 3, S6–S8, Table S2†) were oxidized at the C6 position of the galactose unit to the corresponding aldehyde, which spontaneously hydrated to the gem-diol (Fig. 2B), as expected.46 This was notably shown by the downfield shift of the H6 hydrogen of the galactose unit: for galactose, from 3.74 ppm in the substrate to 5.09/5.12 ppm (α-/β-anomers) in the product; for lactose, from 3.76 to 5.15 ppm, respectively (Tables S1 and S2†). A downfield shift was also observed for the C6 carbon: for galactose, from 61.61/61.81 to 88.82/89.07 ppm (β-/α-anomers); for lactose, from 61.70 to 88.64 ppm (Tables S1 and S2†). In the case of galactose, full assignment of all peaks, using HSQC and HMBC NMR (Table S1†), revealed the presence of both alpha and beta anomers of the C6-oxidized product (Fig. S4–S5†).
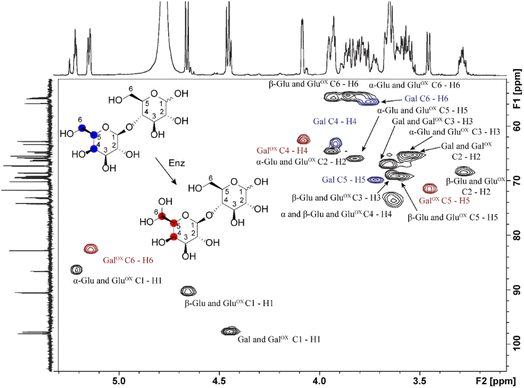 |
| | Fig. 3 1H–13C HSQC NMR analysis of the oxidation of D-lactose by FgrGalOx. Each crosspeak corresponds to a 1JC,H-coupling interaction from the HSQC experiment. Red crosspeaks correspond to peaks unique to the oxidized product, blue crosspeaks correspond to unique peaks found in the unoxidized substrate and black crosspeaks correspond to signals overlapping from substrate and product. The notation states the sugar ring and its corresponding atoms(s). Abbreviations used: Gal, galactose; Glu, glucose; ox, Oxidized. | |
In agreement with HPAEC-PAD analyses (Fig. 1), NMR analyses showed that galactose conversion was not fully complete (Fig. S3†). Peak integration values indicate 77–94% conversion. Regarding lactose oxidation, calculation of the conversion yield by NMR is more complicated. Indeed, the HPAEC-PAD shows full consumption of lactose and formation of minor side products (Fig. 1B), while NMR shows only one product containing a C6-oxidized galactose unit and some apparent remaining lactose (we underscore that both analyses were done on the same samples) (Fig. S6–S8, Table S2†). One plausible explanation for this apparent contradiction is that the side products that form during the reaction with lactose are in low abundance and possess signals that likely overlap with other signals previously identified as lactose.
Characteristics of the HPAEC-PAD detection method
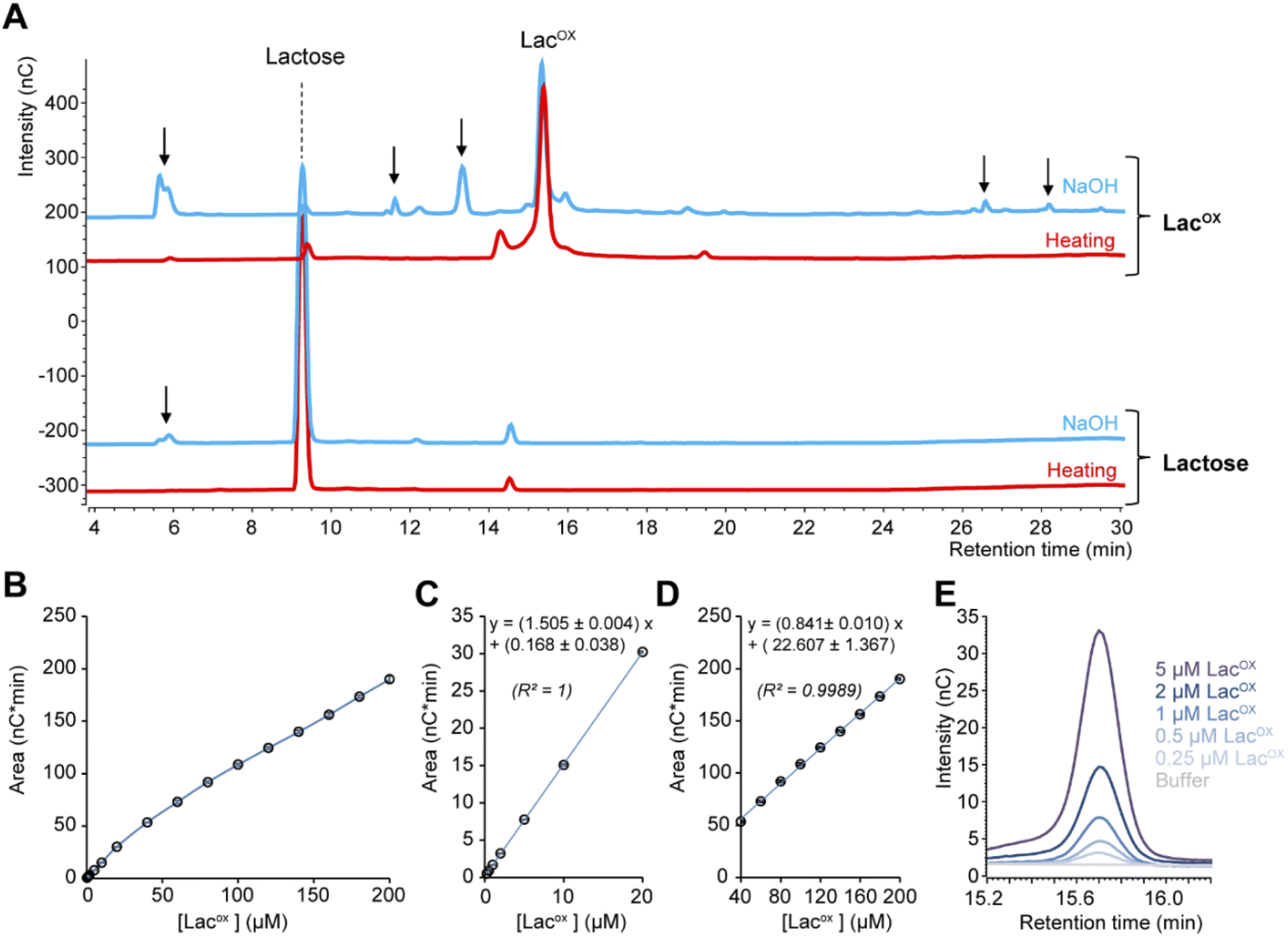
When monitoring enzymatic reactions by HPAEC-PAD, a common practice is to add NaOH to the reaction mixture to stop the reaction before injection on the column, especially when the reaction has to be monitored over time. However, we noticed that NaOH addition to lactose and oxidized lactose (LacOX) solutions yielded degradation products (Fig. 4A). Therefore, we tested heating (100 °C, 5 min) as an alternative method to stop the reaction, which proved more suitable, as evidenced by the significant decrease in side products (Fig. 4A). This method is thus recommended and was used in all experiments shown in this manuscript (including Fig. 1). We also tested the linearity range of the measured amperometric signal of LacOX (Fig. 4B–D). We observed two linear regions (Fig. 4B), a first one in the 0–20 μM range (Fig. 4C) and a second one in the 40–200 μM range (Fig. 4D). For best reliability of signal conversion into concentration values, we thus recommend to dilute samples accordingly. Regarding the sensitivity of the method, we calculated the limit of blank (LOB), the limit of detection (LOD) and the limit of quantification (LOQ).
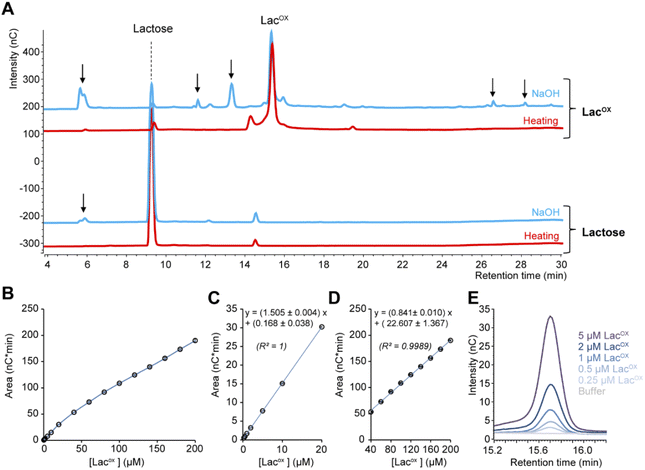 |
| | Fig. 4 Features of the HPAEC-PAD LacOX detection method. (A) HPAEC-PAD chromatograms showing the product profile of D-lactose (250 μM final) and LacOX (250 μM final) after treatment with NaOH (0.15 M final; blue lines) or heat (100 °C, 5 min; red lines) (note that the samples were further diluted in water before injection). (B–D) Standard curve of oxidized lactose (LacOX). Panels (C) and (D) show zoom-in views on the [0–20 μM] and [40–200 μM] ranges, respectively. Data points show average values and error bars show standard deviations (n = 3 independent technical replicates; error bars are <3% of each corresponding average value). (E) Zoom-in view of the HPAEC-PAD chromatogram in the LacOX region showing the detection of very low concentrations. | |
We underscore that the LOB (eqn (1)), which represents the highest apparent analyte concentration expected to be found when replicates of a blank sample containing no analyte are tested, was found to be null (i.e. no signal measured at retention time corresponding to LacOX when only buffer is injected). The LOD (eqn (2)), which is the lowest analyte concentration likely to be reliably distinguished from the LOB and at which detection is feasible, was found to be equal to 0.08 μM (eq. 30 μg L−1). The LOQ (eqn (3)), i.e. the lowest value of a signal that can be quantified with acceptable accuracy and precision, was found to be equal to 0.25 μM (eq. 90 μg L−1) (see Fig. 4E for an illustration). As a comparison, the LOQ of the HRP/ABTS assay rather lie in the low micromolar range,47 i.e. one order of magnitude less sensitive than the HPAEC-PAD method presented here.
| | |
LOB = mean value of 10 blank reactions + 1.645 × SD (blank reactions)
| (1) |
where SD is the standard deviation of the measure of blank reactions,
S is the slope of the 1st linear region (see
Fig. 4C) and SE the standard error of the
y-intercept (
Fig. 4C).
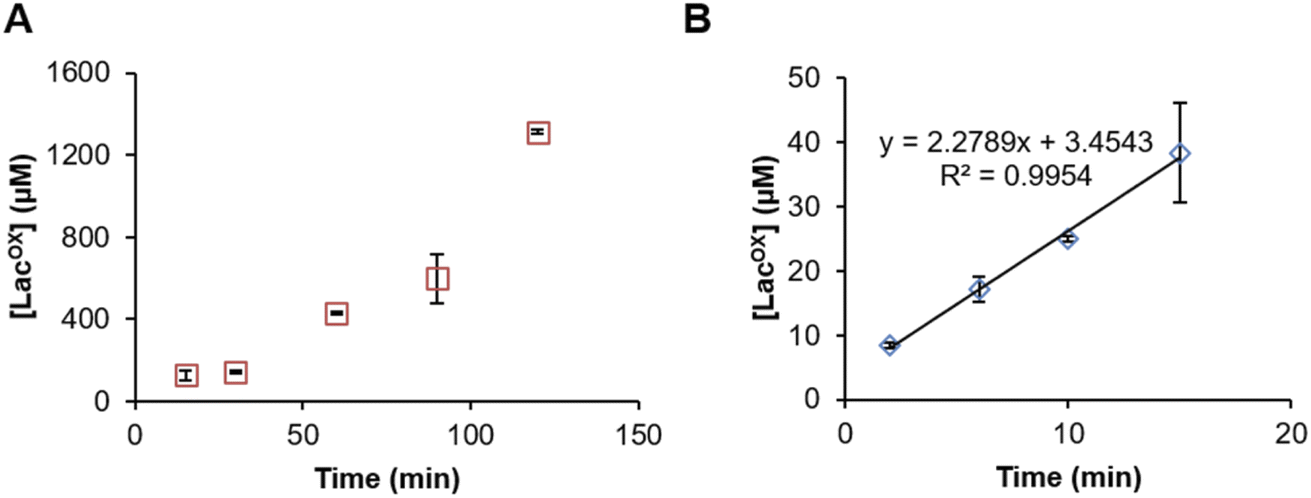
Example of GalOx activity monitoring over time
In Fig. 5, we show that the LacOX detection method presented here can be harnessed to monitor the GalOx activity over time. The method can be used for long incubation times (Fig. 5A) and also for short kinetics to determine initial rates (Fig. 5B; Vi/E = 3.8 s−1). In the latter case, we wanted to compare the initial rate with that obtained by indirect spectrophotometric measurement. To do so, at each time point of the reaction, the sample was split in two aliquots and analyzed in parallel by HPAEC-PAD and indirect spectrophotometric method (see Materials and methods for details on reaction setup). We observed that the spectrophotometric method yielded higher initial rates (Vi/E = 17.9 s−1). The latter rate reflects a total activity (as it is the co-product H2O2 that is indirectly measured) and may also account for other uncontrolled side redox reactions promoting ABTS cation radical formation. The HPAEC-PAD assay allows to reflect the exact turnover of lactose oxidation into LacOX.
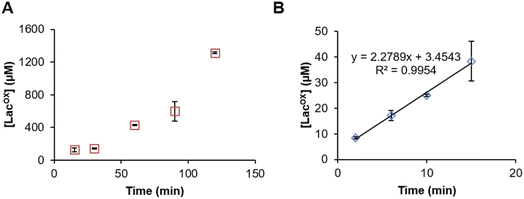 |
| | Fig. 5 Time-course monitoring of FgrGalOx-catalyzed D-lactose oxidation. (A) Long and (B) short kinetic experiments. In panel (A), the FgrGalOx (50 nM) was activated with HRP (0.05 mg mL−1). In panel (B), we used conditions (10 nM FgrGalOx; 0.1 mg mL−1 HRP, 0.25 mg mL−1 ABTS) compatible with the determination of an initial rate in order to compare it with rates determined by spectrophotometric assay. In all experiments (panels (A) and (B)), reactions contained D-lactose (3 mM) in sodium phosphate buffer (50 mM, pH 7.0). | |
Conclusions
In this study, we presented a simple ionic chromatographic method for the direct detection of oxidized galactosylated products resulting from GalOx activity. The method can be used for qualitative approaches to detect very low enzyme activities, as low as 30 μg L−1 of LacOX. Such approach could be used to detect galactose oxidase activity in diluted biological samples (e.g., in fungal secretomes) or to screen for enzyme mutants. Indeed, in the latter case, the study of the effect of enzyme mutations requires the accurate detection of the reaction products (main and secondary carbohydrate products) whereas indirect methods relying on the measurement of the co-product H2O2 only provide a partial understanding of the impact of mutations. For quantitation purposes (e.g., sample titration or enzyme kinetics), we have also established a protocol to prepare the samples to avoid product degradation and determined the concentration range in which quantitative analyses can be carried out in a reliable way. We hope this method will prove useful in the field of copper radical oxidases, and more broadly in the field of carbohydrate research.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by INRAE through the “EvoFun” project (PAF_02), the SATT-Sud Est through the “PANDORA” project, and by the French National Agency for Research (“Agence Nationale de la Recherche”) through the ANR-NSERC “FUNTASTIC” project (ANR-17-CE07-0047).
Notes and references
- J. D. Marth, Nat. Cell Biol., 2008, 10, 1015 CrossRef CAS PubMed.
- C. M. Payne, B. C. Knott, H. B. Mayes, H. Hansson, M. E. Himmel, M. Sandgren, J. Ståhlberg and G. T. Beckham, Chem. Rev., 2015, 115, 1308–1448 CrossRef CAS PubMed.
- L. L. Lairson, B. Henrissat, G. J. Davies and S. G. Withers, Annu. Rev. Biochem., 2008, 77, 521–555 CrossRef CAS PubMed.
- V. K. Gupta, C. P. Kubicek, J. G. Berrin, D. W. Wilson, M. Couturier, A. Berlin, E. X. F. Filho and T. Ezeji, Trends Biochem. Sci., 2016, 41, 633–645 CrossRef CAS PubMed.
- E. Drula, M. L. Garron, S. Dogan, V. Lombard, B. Henrissat and N. Terrapon, Nucleic Acids Res., 2022, 50, D571–D577 CrossRef CAS PubMed.
- A. Levasseur, E. Drula, V. Lombard, P. M. Coutinho and B. Henrissat, Biotechnol. Biofuels, 2013, 6, 41 CrossRef CAS PubMed.
- P. J. Kersten and T. K. Kirk, J. Bacteriol., 1987, 169, 2195–2201 CrossRef CAS.
- J. W. Whittaker, Chem. Rev., 2003, 103, 2347–2363 CrossRef CAS PubMed.
- D. (Tyler) Yin, S. Urresti, M. Lafond, E. M. Johnston, F. Derikvand, L. Ciano, J.-G. Berrin, B. Henrissat, P. H. Walton, G. J. Davies and H. Brumer, Nat. Commun., 2015, 6, 10197 CrossRef PubMed.
- Y. Mathieu, W. A. Offen, S. M. Forget, L. Ciano, A. H. Viborg, E. Blagova, B. Henrissat, P. H. Walton, G. J. Davies and H. Brumer, ACS Catal., 2020, 10, 3042–3058 CrossRef CAS.
- N. Ito, S. E. V Phillips, C. Stevens, Z. B. Ogel, M. J. McPherson, J. N. Keen, K. D. S. Yadav and P. F. Knowles, Nature, 1991, 350, 87–90 CrossRef CAS.
- A. Toftgaard Pedersen, W. R. Birmingham, G. Rehn, S. J. Charnock, N. J. Turner and J. M. Woodley, Org. Process Res. Dev., 2015, 19, 1580–1589 CrossRef CAS.
- H. C. Johnson, S. Zhang, A. Fryszkowska, S. Ruccolo, S. A. Robaire, A. Klapars, N. R. Patel, A. M. Whittaker, M. A. Huffman and N. A. Strotman, Org. Biomol. Chem., 2021, 19, 1620–1625 RSC.
- J. W. Whittaker, Arch. Biochem. Biophys., 2005, 433, 227–239 CrossRef CAS PubMed.
- D. Ribeaucourt, B. Bissaro, M. Yemloul, V. Guallar, H. Brumer, F. Lambert, J.-G. Berrin and M. Lafond, ACS Sustainable Chem. Eng., 2021, 9, 4411–4421 CrossRef CAS.
- S. M. Forget, F. R. Xia, J. E. Hein and H. Brumer, Org. Biomol. Chem., 2020, 18, 2076–2084 RSC.
- B. Bissaro, S. Kodama, H. Hage, D. Ribeaucourt, M. Haon, S. Grisel, A. J. Simaan, S. M. Forget, H. Brumer, M.-N. Rosso, R. O'Connell, M. Lafond, Y. Kubo and J.-G. Berrin, DOI:10.21203/rs.3.rs-493001/v1.
- J. A. D. Cooper, W. Smith, M. Bacila and H. Medina, J. Biol. Chem., 1959, 234, 445–448 CrossRef CAS PubMed.
- L. Sun, I. P. Petrounia, M. Yagasaki, G. Bandara and F. H. Arnold, Protein Eng., 2001, 14, 699–704 CrossRef CAS PubMed.
- S. E. Deacon and M. J. McPherson, ChemBioChem, 2011, 12, 593–601 CrossRef CAS PubMed.
- S. Delagrave, D. J. Murphy, J. L. R. Pruss, A. M. Maffia, B. L. Marrs, E. J. Bylina, W. J. Coleman, C. L. Grek, M. R. Dilworth, M. M. Yang and D. C. Youvan, Protein Eng., 2001, 14, 261–267 CrossRef CAS.
- L. Sun, T. Bulter, M. Alcalde, I. P. Petrounia and F. H. Arnold, ChemBioChem, 2002, 3, 781–783 CrossRef CAS PubMed.
- S. E. Deacon, K. Mahmoud, R. K. Spooner, S. J. Firbank, P. F. Knowles, S. E. V. Phillips and M. J. McPherson, Chembiochem, 2004, 5, 972–979 CrossRef CAS PubMed.
- W. R. Birmingham and N. J. Turner, ACS Catal., 2018, 8, 4025–4032 CrossRef CAS.
- F. Escalettes and N. J. Turner, ChemBioChem, 2008, 9, 857–860 CrossRef CAS PubMed.
- G. P. Roberts and S. K. Gupta, Nature, 1965, 207, 425–426 CrossRef CAS PubMed.
- R. Schoevaart and T. Kieboom, Top. Catal., 2004, 27(1), 3–9 CrossRef CAS.
- R. Monosik, M. Stredansky, J. Tkac and E. Sturdik, Food Anal. Methods, 2011, 5(1), 40–53 CrossRef.
- K. S. Mikkonen, K. Parikka, J. P. Suuronen, A. Ghafar, R. Serimaa and M. Tenkanen, RSC Adv., 2014, 4, 11884–11892 RSC.
- C. Xu, O. Spadiut, A. C. Araújo, A. Nakhai and H. Brumer, ChemSusChem, 2012, 5, 661–665 CrossRef CAS PubMed.
- A. S. Leppänen, C. Xu, K. Parikka, P. Eklund, R. Sjöholm, H. Brumer, M. Tenkanen and S. Willför, Carbohydr. Polym., 2014, 100, 46–54 CrossRef.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- M. Andberg, F. Mollerup, K. Parikka, S. Koutaniemi, H. Boer, M. Juvonen, E. Master, M. Tenkanen and K. Kruus, Appl. Environ. Microbiol., 2017, 83, e01383 CrossRef CAS PubMed.
- M. E. Cleveland, Y. Mathieu, D. Ribeaucourt, M. Haon, P. Mulyk, J. E. Hein, M. Lafond, J. G. Berrin and H. Brumer, Cell. Mol. Life Sci., 2021, 78, 8187–8208 CrossRef CAS PubMed.
- D. Ribeaucourt, S. Saker, D. Navarro, B. Bissaro, E. Drula, L. O. Correia, M. Haon, S. Grisel, N. Lapalu, B. Henrissat, R. J. O'Connell, F. Lambert, M. Lafond and J. G. Berrin, Appl. Environ. Microbiol., 2021, 87, e01526–21 CrossRef CAS PubMed.
- M. Cleveland, M. Lafond, F. R. Xia, R. Chung, P. Mulyk, J. E. Hein and H. Brumer, Biotechnol. Biofuels, 2021, 14, 1–19 CrossRef.
- A. J. Baron, C. Stevens, C. Wilmot, K. D. Seneviratne, V. Blakeley, D. M. Dooley, S. E. V. Phillips, P. F. Knowles and M. J. McPherson, J. Biol. Chem., 1994, 269, 25095–25105 CrossRef CAS PubMed.
- F. S. Cheng and G. D. Christain, Anal. Chim. Acta, 1979, 104, 47–53 CrossRef CAS.
- C. Figueiredo, A. L. De Lacey and M. Pita, Electrochem. Sci. Adv., 2021, e2100171 Search PubMed.
- V. Bonnet, R. Duval and C. Rabiller, J. Mol. Catal. B: Enzym., 2003, 24–25, 9–16 CrossRef CAS.
- S. Roseman, R. H. Abeles and A. Dorfman, Arch. Biochem. Biophys., 1952, 36, 232–233 CrossRef CAS.
- N. McGregor, G. Arnal and H. Brumer, Methods in Molecular Biology, Humana Press Inc., 2017, 1588, pp. 15–25 Search PubMed.
- B. Westereng, M. Ø. Arntzen, F. L. Aachmann, A. Várnai, V. G. Eijsink and J. W. Agger, J. Chromatogr. A, 2016, 46–54 CrossRef CAS PubMed.
- M. Haddad Momeni, F. Fredslund, B. Bissaro, O. Raji, T. V. Vuong, S. Meier, T. S. Nielsen, V. Lombard, B. Guigliarelli, F. Biaso, M. Haon, S. Grisel, B. Henrissat, D. H. Welner, E. R. Master, J. G. Berrin and M. Abou Hachem, Nat. Commun., 2021, 12, 2132 CrossRef CAS.
- O. Spadiut, L. Olsson and H. Brumer, Microb. Cell Fact., 2010, 9, 1–13 CrossRef PubMed.
- K. Parikka and M. Tenkanen, Carbohydr. Res., 2009, 344, 14–20 CrossRef CAS PubMed.
- R. Ravani Ananda, M. Kempegowda and M. Guin, Anal. Chem. Lett., 2017, 7, 779–791 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
David Navarro
b,
David Navarro