
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA review on α-glucosidase inhibitory activity of first row transition metal complexes: a futuristic strategy for treatment of type 2 diabetes
Marzieh Sohrabia,
Mohammad Reza Binaeizadeh b,
Aida Irajicde,
Bagher Larijania,
Mina Saeedi*fg and
Mohammad Mahdavi*a
b,
Aida Irajicde,
Bagher Larijania,
Mina Saeedi*fg and
Mohammad Mahdavi*a
aEndocrinology and Metabolism Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran. E-mail: momahdavi@tums.ac.ir
bSchool of Chemistry, College of Science, University of Tehran, Tehran, Iran
cStem Cells Technology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
dCentral Research Laboratory, Shiraz University of Medical Sciences, Shiraz, Iran
eLiosa Pharmed Parseh Company, Shiraz, Iran
fMedicinal Plants Research Center, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran. E-mail: m-saeedi@tums.ac.ir
gPersian Medicine and Pharmacy Research Center, Tehran University of Medical Sciences, Tehran, Iran
First published on 20th April 2022
Abstract
Type 2 diabetes mellitus (T2DM) is characterized by high blood glucose levels and has emerged as a controversial public health issue worldwide. The increasing number of patients with T2DM on one hand, and serious long-term complications of the disease such as obesity, neuropathy, and vascular disorders on the other hand, have induced a huge economic impact on society globally. In this regard, inhibition of α-glucosidase, the enzyme responsible for the hydrolysis of carbohydrates in the body has been the main therapeutic approach to the treatment of T2DM. As α-glucosidase inhibitors (α-GIs) have occupied a special position in the current research and prescription drugs are generally α-GIs, researchers have been encouraged to design and synthesize novel and efficient inhibitors. Previously, the presence of a sugar moiety seemed to be crucial for designing α-GIs since they can attach to the carbohydrate binding site of the enzyme mimicking the structure of disaccharides or oligosaccharides. However, inhibitors lacking glycosyl structures have also shown potent inhibitory activity and development of non-sugar based inhibitors is accelerating. In this respect, in vitro anti-α-glucosidase activity of metal complexes has attracted lots of attention and this paper has reviewed the inhibitory activity of first-row transition metal complexes toward α-glucosidase and discussed their probable mechanisms of action.
Introduction
Diabetes mellitus (DM) is a chronic metabolic disease, known as one of the important causes of death in the world due to acute side effects.1 The long-term high levels of blood glucose in patients with DM result in various disorders such as microvascular complications or destruction of very small blood vessels in the body causing serious kidney, eye, nerve and heart diseases.2 There are two primary forms of diabetes including type 1 (T1DM) and type 2 (T2DM). T1DM is defined as an insulin-dependent form of the disease and T2DM is described as a non-insulin-dependent disorder.3 The majority of the world's patients with T2DM and the prevalence of the disease have become an increasing public health issue. According to the prediction of the World Health Organization (WHO) in 2006, the number of patients with T2DM may increase to 366 million by 2030 which accounts for 9.9% of the world's adult population.4 Several factors related to lifestyle such as (i) obesity and weight,5,6 (ii) physical inactivity (independent of obesity),7,8 (iii) cigarette smoking,9 (iv) a low-fiber diet with a high glycemic index,10 and (v) depression,11 are involved in increasing the risk of T2DM.Currently, lowering blood glucose levels has been known as the most effective anti-diabetic therapeutic tool.12,13 At this juncture, α-glucosidase inhibitors (α-GIs) have occupied a special position in drug development research since α-glucosidase is a key enzyme converting complex carbohydrates (polysaccharides) into simple sugars (monosaccharides) (Fig. 1a and b).14 α-Glucosidase located in the brush border of the small intestinal can selectively hydrolyze terminal (1 → 4)-linked α-glucosidase residues (starch or disaccharides), leading to the formation of glucose.15,16 α-GIs can reduce the rate of glucose uptake by delaying the digestion of carbohydrates. Thus, reducing the effect of dietary carbohydrates on blood sugar has been revealed to be vital in avoiding the progression of impaired glucose tolerance factor (GTF) to T2DM. Generally, the basic modes of action of α-GIs can be competitive (Fig. 1c), non-competitive (allosteric) (Fig. 1d), and uncompetitive (Fig. 1e).
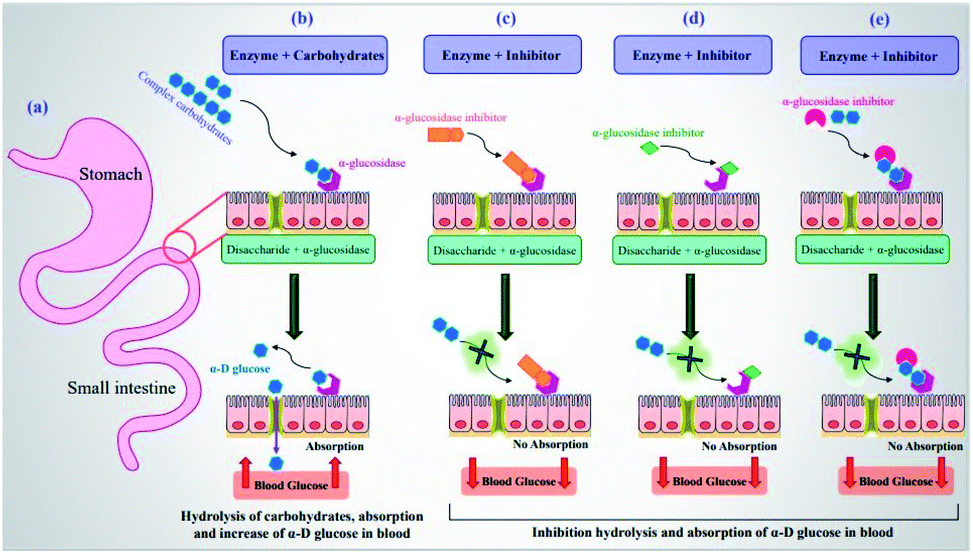 | ||
| Fig. 1 (a) Schematic diagram of small intestine. (b) Hydrolysis of complex carbohydrates by α-glucosidase along the brush border of small intestine to release α-D-glucose. Basic modes of action of α-GIs: (c) competitive, (d) non-competitive, and (e) uncompetitive inhibition of α-glucosidase leading to decrease of postprandial blood glucose levels. | ||
Conventional α-GIs are structurally similar to disaccharides or oligosaccharides, able to attach to the carbohydrate binding site of enzyme. Acarbose (glucobay), voglibose (volix, Basen), and miglitol (Glyset), are three prescription α-GIs available in the market and frequently used for the treatment of T2DM (Fig. 2). However, they suffer from various side effects specially gastrointestinal problems and following that, a wide range of studies have been conducted on the anti-α-glucosidase activity of natural17–19 and synthetic heterocyclic compounds20–27 since the efficacy of α-GIs in the treatment of T2DM has been completely proven. Biological activity of metal ions and their complexes28,29 have made them an important topic in drug discovery research. Recently, they have been investigated for their anti-α-glucosidase activity and it is hoped that metallotherapy can open a new horizon in the field of anti-T2DM agents.
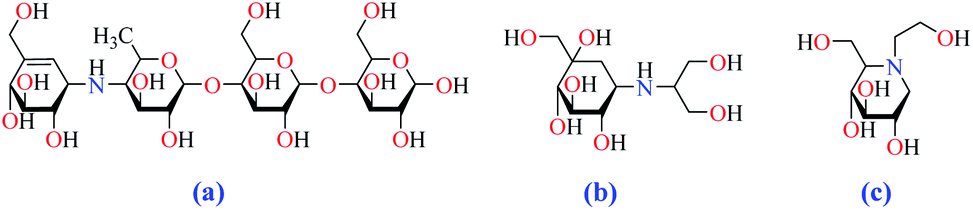 | ||
| Fig. 2 Chemical structures of clinically used α-GIs, (a) acarbose, (b) voglibose, and (c) miglitol. | ||
Biological activity of first-row transition metals
The 10 first-row transition metals (Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, and Zn) profit from both structural and functional roles to induce desired biological properties and diagnostic applications.30,31 Among them, manganese, iron, cobalt, copper, and zinc are five essential elements to human health.31 However, three first-row transition metals including chromium, vanadium, and nickel are generally used as nutritional additives in different diets and possess favorable.30 The structural or catalytic function of these metal ions allows them to bind to the proteins, DNA, RNA or other biological structures to be able to exert a certain role.Oxidation state of the metal center especially that of transition metal complexes plays a significant role in the induction of medicinal properties. The redox environment and the nature of the ligands as well as the group number of metal center lead to different oxidation states of the first-row transition metal complexes.32
The idea of using metal ions to treat diabetes dates back to 1899 and the anti-diabetic properties of the orally active metal complexes such as oxovanadium(IV) (vanadyl) were initially considered in 1990.33,34
In 1979, vanadium salts were reported to have in vitro insulin-mimetic effects35 and their insulin-like activity was revealed in 1899.36 However, vanadium's pharmacological potential was systematically investigated by Heyliger et al.37 Bis(2-ethyl-3-hydroxy-4-pyronato)oxovanadium(IV) (BMOV) which was first synthesized in the late 1990s,38,39 showed desired activity on STZ diabetic rats, possessing higher potency and efficacy than a common inorganic vanadium salt.40 Bis(ethylmaltolato)oxidovanadium(IV) (BEOV) was also found to be anti-diabetic agent derived from vanadium.41 Also, different vanadium, copper and zinc metal complexes42 were found to be effective for the treatment of diabetes under in vivo conditions.33
Mechanistic role of the metal center in the inhibitory activity of the corresponding complexes was completely reviewed by Dyson et al.43 Subsequently, metal complexes are divided into three main classes: (I) metal complexes having biologically active ligands (non-innocent ligands): in this group, metal centers have a structural role and unlike ligands that are biologically active components, they do not play a role in the enzyme inhibition. However, the metal center can indirectly affect the binding of ligand to the enzyme by undergoing a redox process. (II) Metal complexes having biologically active metal center (innocent ligands): in this class of compounds, ligands are usually biologically inactive and their main role is only masking and stabling the metal ion. (III) Metal complexes having biologically active metal and ligands: these metal complexes are predicted to have synergistic effects in inhibiting enzymes due to the biological effects of both metal ion and ligands. However, the mechanism of action of metal complexes in the inhibition of α-glucosidase has not been definitely reported in the literature.
In this review, α-glucosidase inhibitory activity of the first-row transition metal complexes (Zn, Cu, Ni, Co, Fe, Mn, Cr, V) with different oxidation states (II, III, and IV) was in the center of attention as they have shown potent activity and can be considered as novel and efficient surrogates of organic small molecules in the treatment of T2DM. For this purpose, discussed complexes were categorized into three classes based on the oxidation state of the central metal including divalent, trivalent, and tetravalent metal complexes. Moreover, to get better insight into the mechanism of action of metal complexes, docking study was performed for some selected complexes which confirmed the construction of desired interactions with the enzyme.
Divalent metal complexes as α-glucosidase inhibitors
Although some metal(II) ions and their complexes have demonstrated anti-diabetes properties,44–46 their mechanism of action is not completely clear. For example, manganese has played a significant role in the glucose metabolism45 and zinc(II) complexes have shown an insulin-like effect on rat adipocytes.47 In 1980, Coulston and Dandona reported that zinc could stimulate lipogenesis in rat adipocytes similar to the function of insulin.48 Recently, various studies have reported anti-α-glucosidase activity of divalent first row transition metals (Zn(II), Cu(II), Ni(II), Co(II), Fe(II), and Mn(II)) complexes as listed in Tables 1–4 and Fig. 3. The complexes were categorized based on the functional group of ligands such as hydrazides, picolinic acids (PicAs), Schiff bases, and diamines coordinated to the metal(II) ions. These organic ligands generally have not demonstrated α-glucosidase inhibitory activity while the corresponding complexes have shown potent inhibitory activity.| Entry | Compounds | Structure number | IC50 (μM) | Ref. |
|---|---|---|---|---|
| a Acarbose as the reference drug (IC50 = 378.25 μM).b Acarbose as the reference drug (IC50 = 99 μM). | ||||
| 1 |  |
L1 | NA | 53a |
 |
L2 | NA | ||
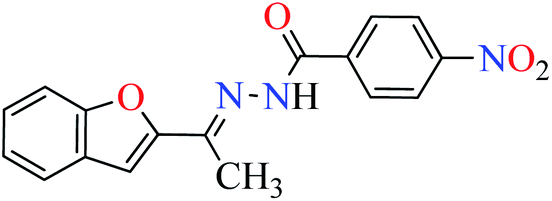 |
L3 | 47.51 | ||
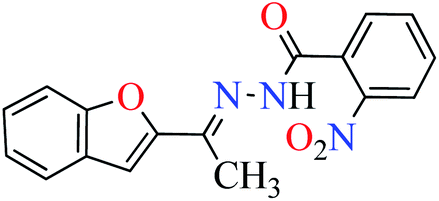 |
L4 | NA | ||
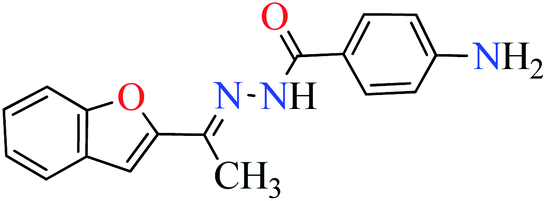 |
L5 | NA | ||
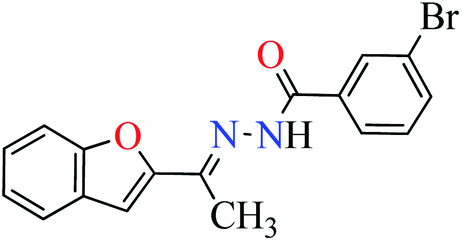 |
L6 | 396.35 | ||
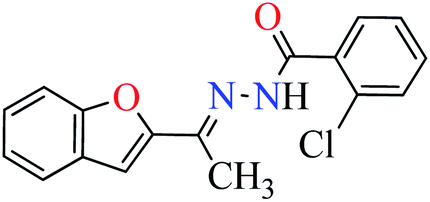 |
L7 | NA | ||
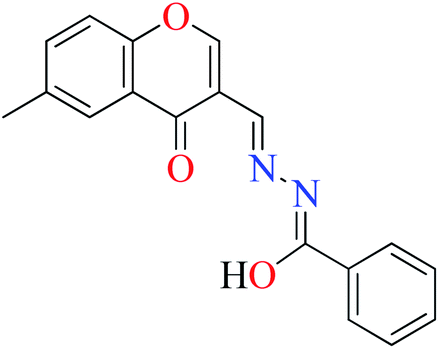 |
L8 | 240 | 54b | |
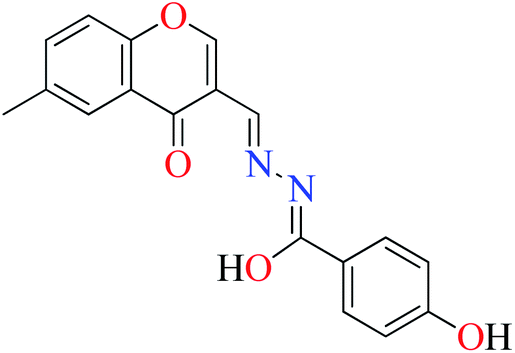 |
L9 | 230 | ||
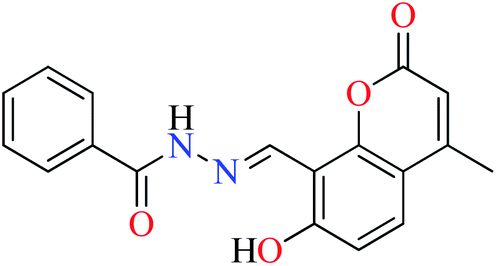 |
L10 | Not reported clearly | 55b | |
| 2 | 53a | |||
| 2a | 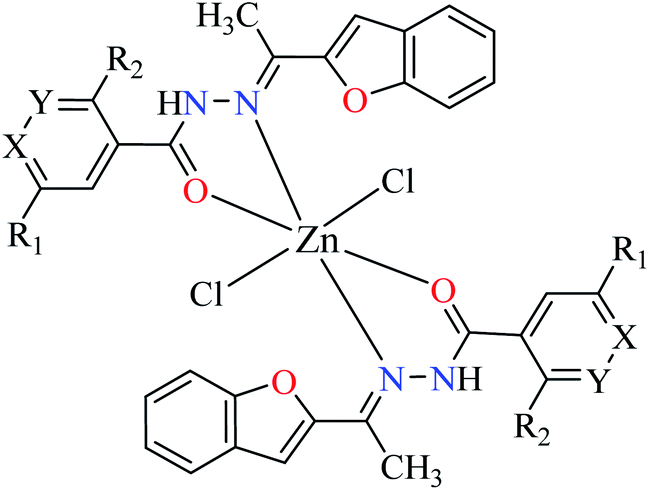 |
|||
| 1 = [Zn(L1)2]Cl2 | 1 X = N; Y = C; R1 = H; R2 = H | 101.29 | ||
| 2 = [Zn(L2)2]Cl2 | 2 X = C; Y = N; R1 = H; R2 = H | 56.27 | ||
| 3 = [Zn(L3)2]Cl2 | 3 X = C–NO2; Y = C; R1 = H; R2 = H | 27.71 | ||
| 4 = [Zn(L4)2]Cl2 | 4 X = C; Y = C; R1 = H; R2 = NO2 | 97.26 | ||
| 5 = [Zn(L5)2]Cl2 | 5 X = C; Y = C–NH2; R1 = H; R2 = H | NA | ||
| 6 = [Zn(L6)2]Cl2 | 6 X = C; Y = C; R1 = Br; R2 = H | 121.19 | ||
| 2b | 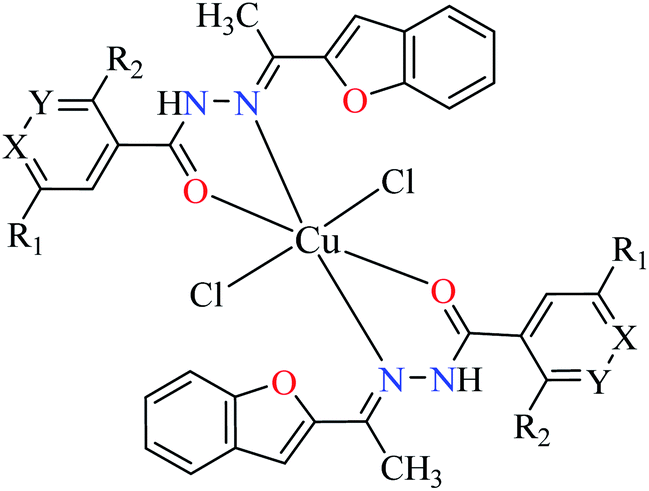 |
|||
| 9 = [Cu(L1)2]Cl2 | 9 X = N; Y = C; R1 = H; R2 = H | >500 | ||
| 10 = [Cu(L2)2]Cl2 | 10 X = C; Y = N; R1 = H; R2 = H | 17.73 | ||
| 11 = [Cu(L3)2]Cl2 | 11 X = C–NO2; Y = C; R1 = H; R2 = H | 1.15 | ||
| 12 = [Cu(L4)2]Cl2 | 12 X = C; Y = C; R1 = H; R2 = NO2 | 18.91 | ||
| 13 = [Cu(L5)2]Cl2 | 13 X = C; Y = C–NH2; R1 = H; R2 = H | 0.15 | ||
| 14 = [Cu(L6)2]Cl2 | 14 X = C; Y = C; R1 = Br; R2 = H | 0.21 | ||
| 15 = [Cu(L7)2]Cl2 | 15 X = C; Y = C; R1 = H; R2 = Cl | NA | ||
| 2c | 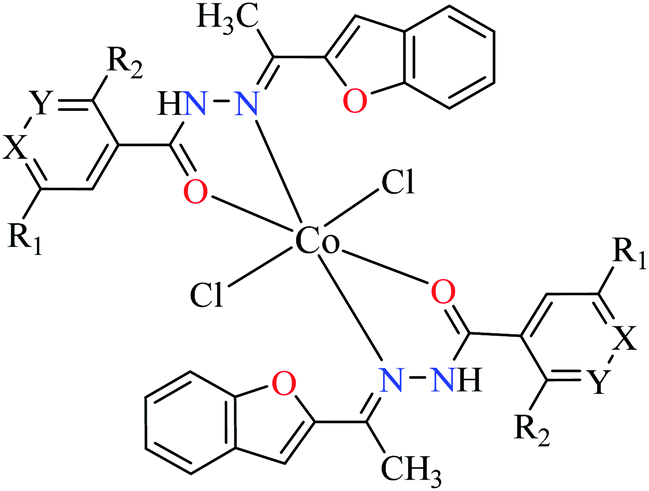 |
|||
| 22 = [Co(L1)2]Cl2 | 22 X = N; Y = C; R1 = H; R2 = H | NA | ||
| 23 = [Co(L2)2]Cl2 | 23 X = C; Y = N; R1 = H; R2 = H | 66.48 | ||
| 24 = [Co(L3)2]Cl2 | 24 X = C–NO2; Y = C; R1 = H; R2 = H | 153.23 | ||
| 25 = [Co(L4)2]Cl2 | 25 X = C; Y = C; R1 = H; R2 = NO2 | 96.95 | ||
| 26 = [Co(L5)2]Cl2 | 26 X = C; Y = C–NH2; R1 = H; R2 = H | NA | ||
| 27 = [Co(L6)2]Cl2 | 27 X = C; Y = C; R1 = Br; R2 = H | 213.30 | ||
| 28 = [Co(L7)2]Cl2 | 28 X = C; Y = C; R1 = H; R2 = Cl | NA | ||
| 2d | 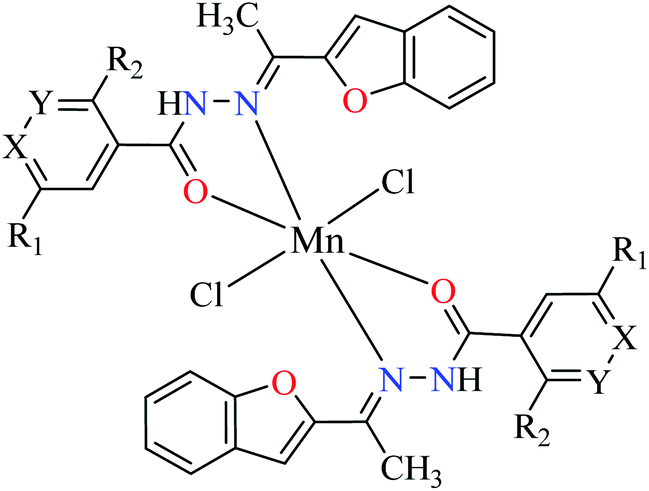 |
|||
| 30 = [Mn(L1)2]Cl2 | 30 X = N; Y = C; R1 = H; R2 = H | 45.63 | ||
| 31 = [Mn(L2)2]Cl2 | 31 X = C; Y = N; R1 = H; R2 = H | 143.21 | ||
| 32 = [Mn(L3)2]Cl2 | 32 X = C–NO2; Y = C; R1 = H; R2 = H | 345.62 | ||
| 33 = [Mn(L4)2]Cl2 | 33 X = C; Y = C; R1 = H; R2 = NO2 | NA | ||
| 34 = [Mn(L5)2]Cl2 | 34 X = C; Y = C–NH2; R1 = H; R2 = H | NA | ||
| 35 = [Mn(L7)2]Cl2 | 35 X = C; Y = C; R1 = H; R2 = Cl | 457.28 | ||
| 3 | 54b | |||
| 3a | 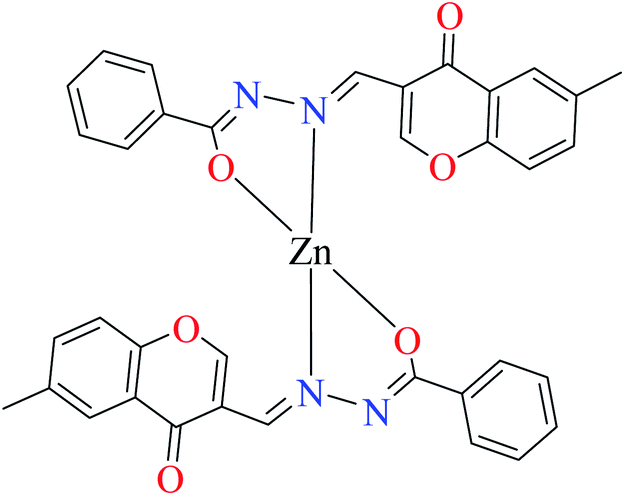 |
|||
| 7 = [Zn(L8)2]·2H2O | 7 | 180 | ||
| 3b | 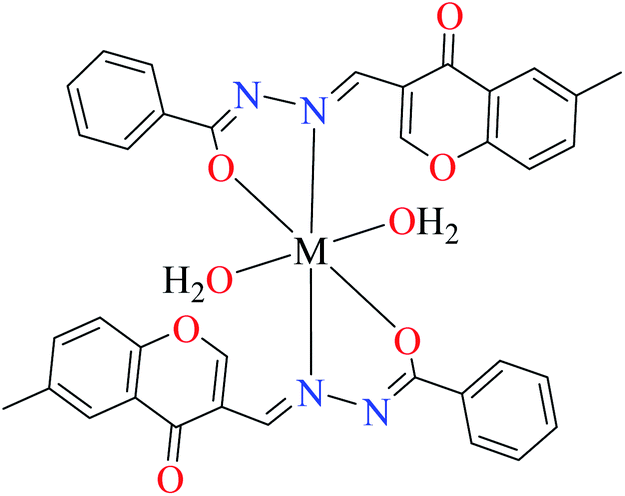 |
|||
| M(II) = Cu, Ni | ||||
| 16 = [Cu(L8)2(OH2)2]·H2O | 16 | 140 | ||
| 19 = [Ni(L8)2(OH2)2]·H2O | 19 | 200 | ||
| 3c | 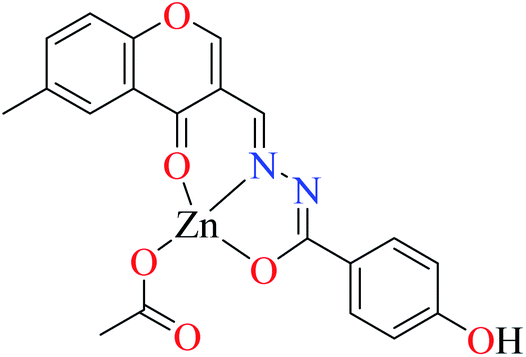 |
|||
| 8 = [Zn(L9)CH3COO]·H2O | 8 | 190 | ||
| 3d | 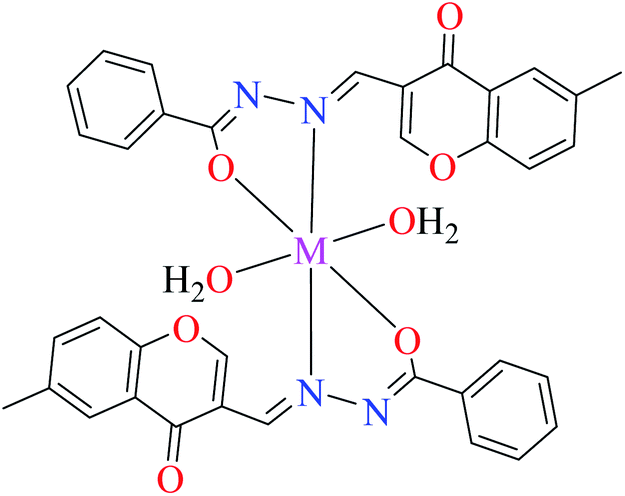 |
|||
| M(II) = Cu, Ni | ||||
| 17 = [Cu(L9)2(OH2)2]·H2O | 17 | 170 | ||
| 20 = [Ni(L9)2(OH2)2]·H2O | 20 | 230 | ||
| 4 | 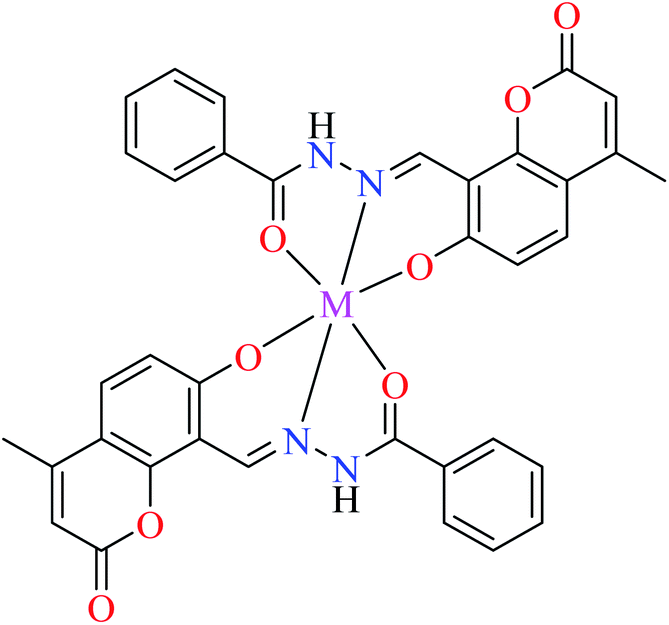 |
55b | ||
| M(II) = Cu, Ni, Co, Mn | ||||
| 18 = [Cu(L10)2] | 18 | Not reported clearly | ||
| 21 = [Ni(L10)2] | 21 | Not reported clearly | ||
| 29 = [Co(L10)2] | 29 | Not reported clearly | ||
| 36 = [Mn(L10)2] | 36 | Not reported clearly | ||
| Entry | Compounds | Structure number | IC50 (μM) | Ref. |
|---|---|---|---|---|
| a Genistein as the reference drug (IC50 = 16.57 ± 0.23 μM).b Genistein as the reference drug (IC50 = 7.85 ± 0.87 μM). | ||||
| 1 | 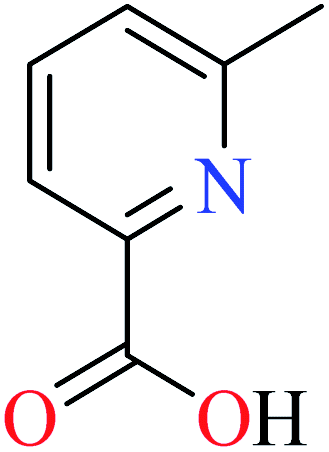 |
L11 | NA | 62a |
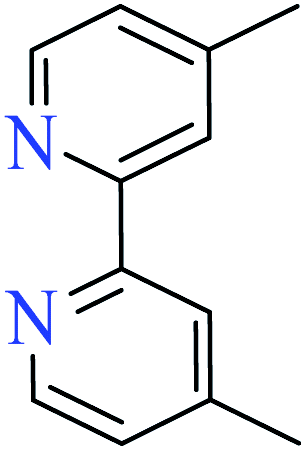 |
L12 | NA | 62a | |
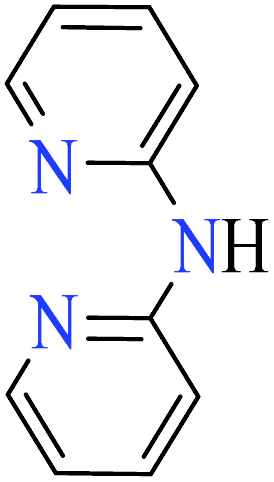 |
L13 | NA | 63a | |
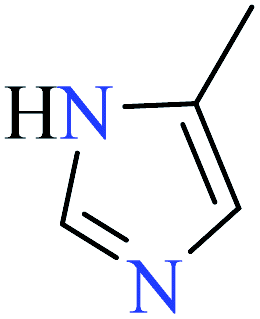 |
L14 | NA | 63a | |
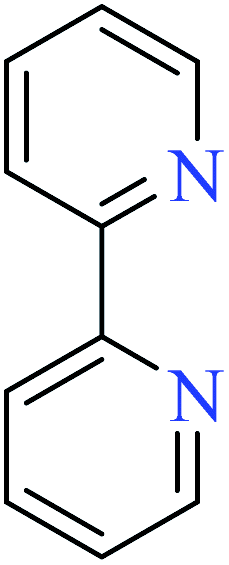 |
L15 | NA | 67a | |
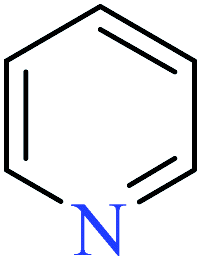 |
L16 | Not reported | 64a | |
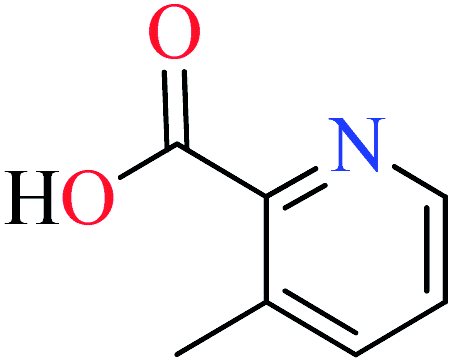 |
L17 | NA | 68a | |
 |
L18 | NA | 68a | |
 |
L19 | NA | 65a | |
 |
L20 | NA | 65a | |
| 2 | 64a | |||
| 2a | 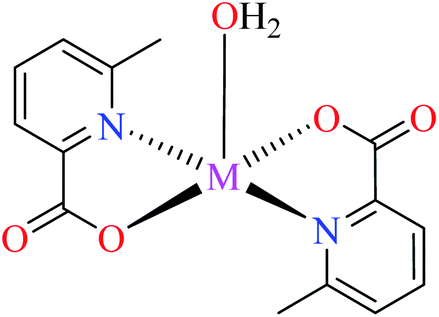 |
|||
| M(II) = Zn, Cu | ||||
| 37 = [Zn(L11)2(H2O)] | 37 | 546.04 | ||
| 42 = [Cu(L11)2(H2O)] | 42 | 2.95 | ||
| 2b | 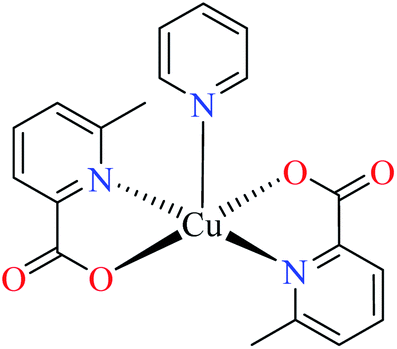 |
|||
| 43 = [Cu(L11)2(L16)] | 43 | 3.49 | ||
| 2c | 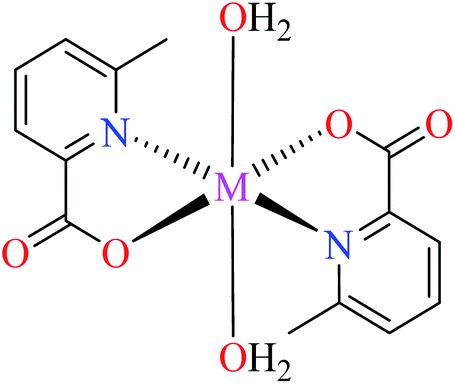 |
|||
| M(II) = Ni, Co, Mn | ||||
| 48 = [Ni(L11)2(H2O)2] | 48 | >600 | ||
| 53 = [Co(L11)2(H2O)2] | 53 | >600 | ||
| 61 = [Mn(L11)2(H2O)2] | 61 | >600 | ||
| 3 | 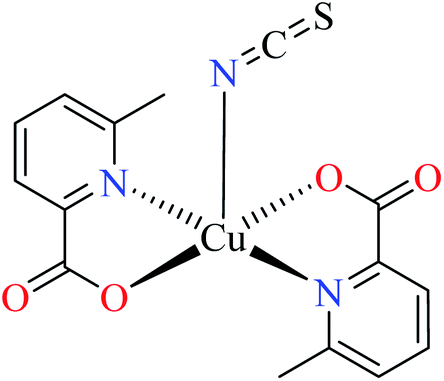 |
|||
| 44 = [Cu(L11)2(NCS)] | 44 | 8.02 | 66b | |
| 4 | 67a | |||
| 4a | 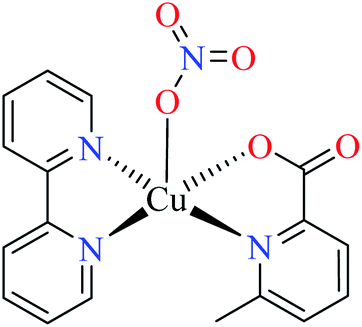 |
|||
| 45 = [Cu(L11)(L15)(NO3)] | 45 | 688.94 | ||
| 4b | 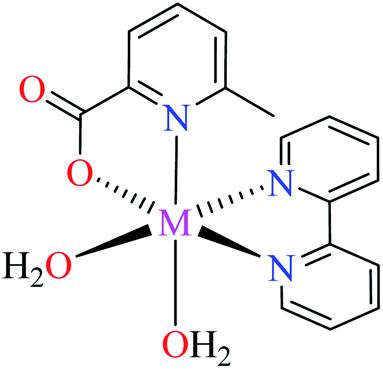 |
|||
| M(II) = Ni, Mn | ||||
| 49 = [Ni(L11)(L15)(H2O)2] | 49 | >600 | ||
| 62 = [Mn(L11)(L15)(H2O)2] | 62 | >600 | ||
| 4c | 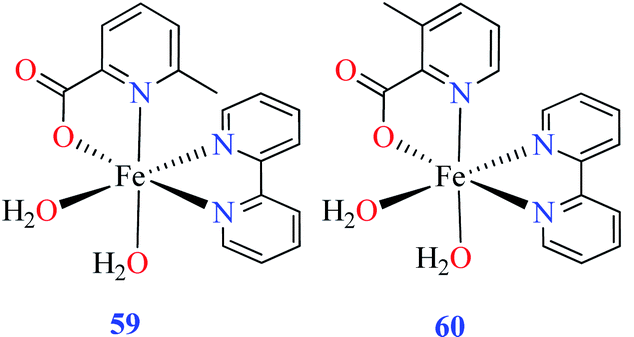 |
|||
| 59 = [Fe(L11)(L15)(H2O)2] | 59 | 97.33 | ||
| 60 = [Fe(L17)(L15)(H2O)2] | 60 | 724.25 | ||
| 4d |  |
|||
| 54 = [Co(L11)2(L15)] | 54 | >600 | ||
| 5 |  |
|||
| M(II) = Zn, Ni | ||||
| 38 = [Zn(L11)(L12)(Cl)2] | 38 | >600 | 62a | |
| 50 = [Ni(L11)(L12)(Cl)2] | 50 | >600 | ||
| 6 | 63a | |||
| 6a | 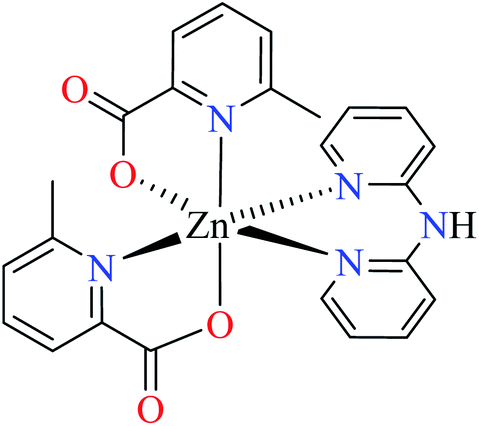 |
|||
| 39 = [Zn(L11)2(L13)] | 39 | >600 | ||
| 6b | 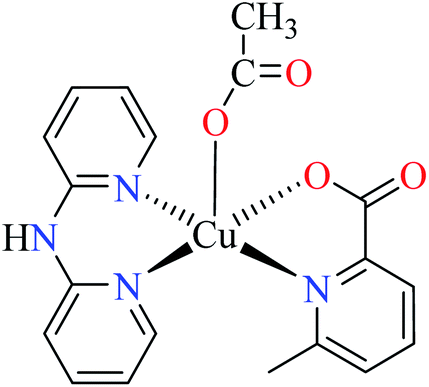 |
|||
| 46 = [Cu(L11)(L13)(CH3COO)] | 46 | 513.10 | ||
| 6c | 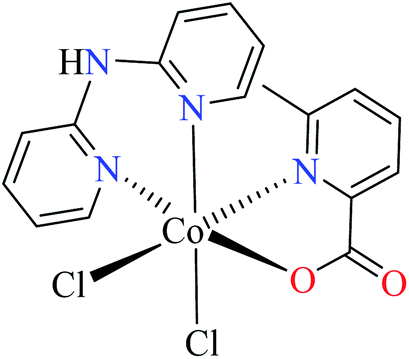 |
|||
| 55 = [Co(L11)(L13)(Cl)2] | 55 | >600 | ||
| 7 | 63a | |||
| 7a | 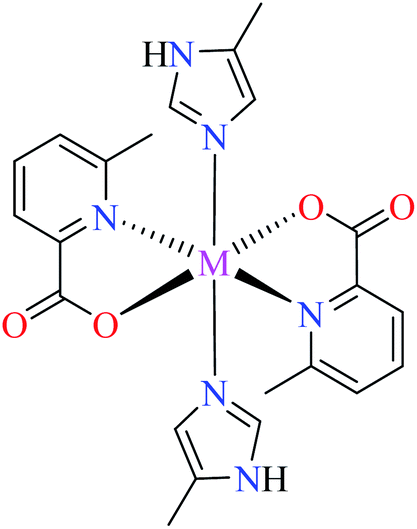 |
|||
| M(II) = Zn, Co, Mn | ||||
| 40 = [Zn(L11)2(L14)2] | 40 | >600 | ||
| 56 = [Co(L11)2(L14)2] | 56 | >600 | ||
| 63 = [Mn(L11)2(L14)2] | 63 | >600 | ||
| 7b | 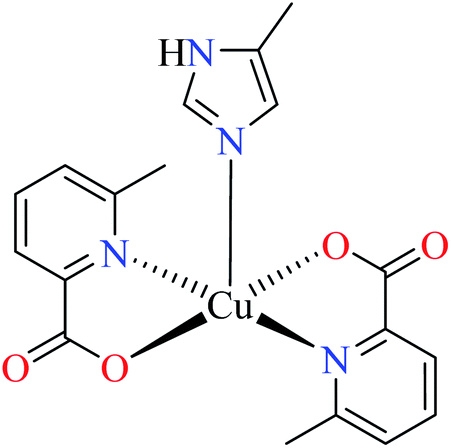 |
|||
| 47 = [Cu(L11)2(L14)] | 47 | 2.91 | ||
| 7c | 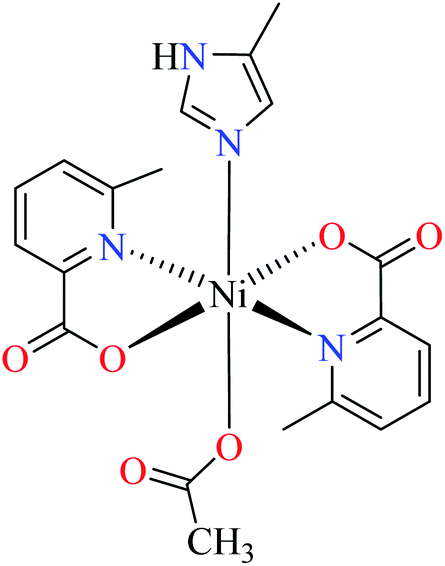 |
|||
| 51 = [Ni(L11)2(L14)(CH3COO)] | 51 | >600 | ||
| 8 | 68a | |||
| 8a |  |
|||
| M(II) = Ni, Co, Mn | ||||
| 52 = [Ni(L11)2(L18)] | 52 | >600 | ||
| 57 = [Co(L11)2(L18)] | 57 | >600 | ||
| 64 = [Mn(L11)2(L18)] | 64 | >600 | ||
| 8b |  |
|||
| 58 = [Co(L17)2(L18)] | 58 | >600 | ||
| 9 | 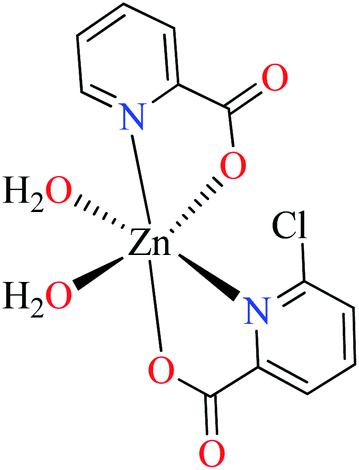 |
|||
| 41 = [Zn(L19)(L20)(H2O)2] | 41 | 440 | 65a | |
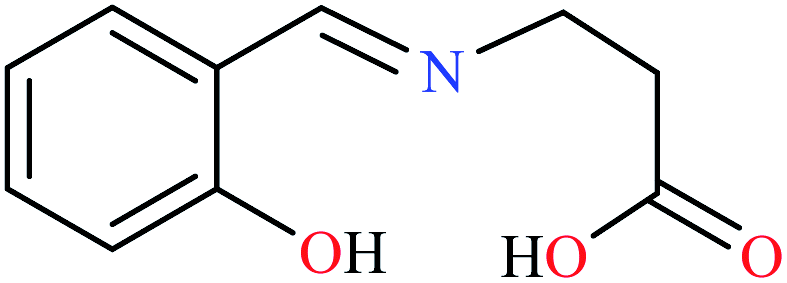
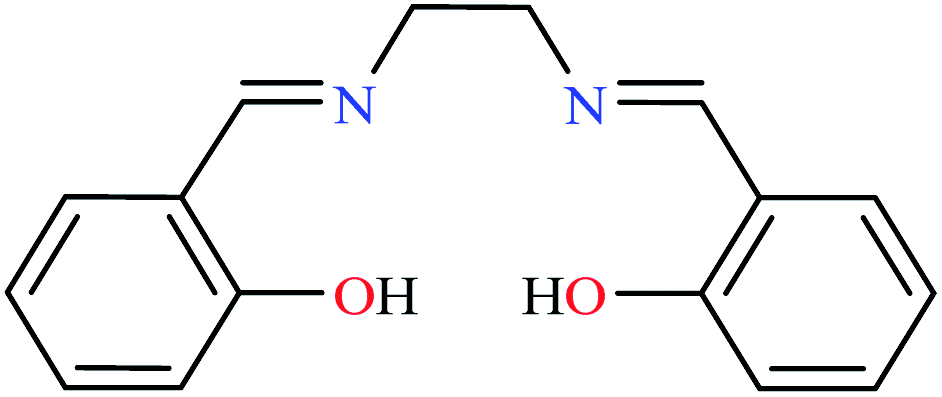
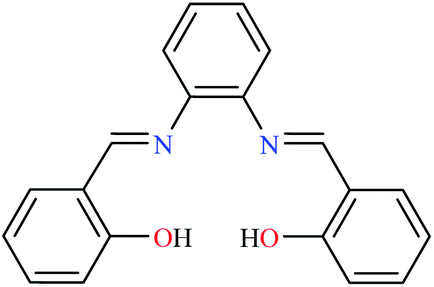
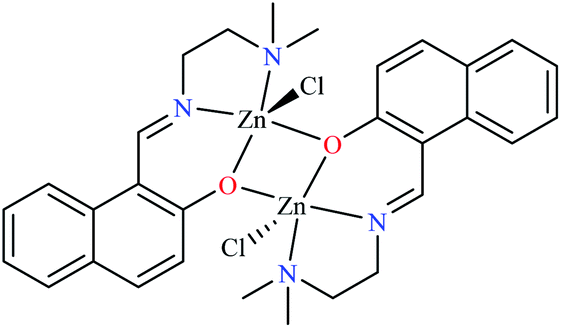
| Entry | Compounds | Structure number | IC50 (mg mL−1) | Ref. |
|---|---|---|---|---|
| a Acarbose as the reference drug (IC50 = 0.140 mg mL−1).b Acarbose as the reference drug (IC50 = 0.143 mg mL−1). | ||||
| 1 | 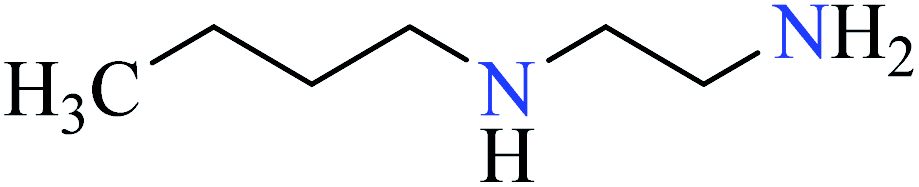 |
L25 | Not reported | 74a |
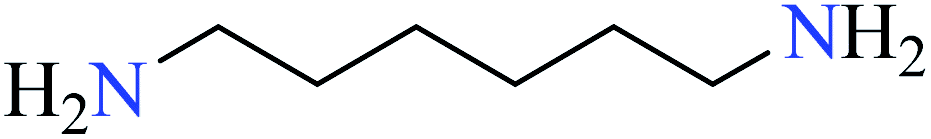 |
L26 | Not reported | 74a | |
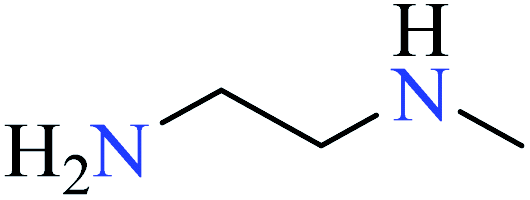 |
L27 | Not reported | 75b | |
 |
L28 | Not reported | 76a | |
| 2 | [Zn(L25)3].2Cl | 69 | 298 | 74a |
| [Zn(L25)3].2NO3 | 70 | 278 | ||
| [Zn(L25)3]·SO4 | 71 | 305 | ||
| [Cu(L25)3]·2Cl | 78 | 266 | ||
| [Cu(L25)3]·2NO3 | 79 | 279 | ||
| [Cu(L25)3]·SO4 | 80 | 256 | ||
| [Ni(L25)3]·2Cl | 87 | 226 | ||
| [Ni(L25)3]·2NO3 | 88 | 198 | ||
| [Ni(L25)3]·SO4 | 89 | 218 | ||
| 3 | [Zn(L26)3]·2Cl | 72 | 1.19 | 74a |
| [Zn(L26)3]·2NO3 | 73 | 1.50 | ||
| [Zn(L26)3]·SO4 | 74 | 1.43 | ||
| [Cu(L26)3]·2Cl | 81 | 1.33 | ||
| [Cu(L26)3]·2NO3 | 82 | 1.26 | ||
| [Cu(L26)3]·SO4 | 83 | 1.76 | ||
| [Ni(L26)3]·2Cl | 90 | 3.63 | ||
| [Ni(L26)3]·2NO3 | 91 | 2.70 | ||
| [Ni(L26)3]·SO4 | 92 | 2.00 | ||
| [Co(L26)3]·Cl2 | 96 | 1348 | ||
| [Co(L26)3]·(NO3)2 | 97 | 1279 | ||
| [Co(L26)3]·SO4 | 98 | 1198 | ||
| 4 | [Zn(L27)3]·2Cl | 75 | 2.13 | 75b |
| [Zn(L27)3]·2NO3 | 76 | 1.05 | ||
| [Zn(L27)3]·SO4 | 77 | 2.0 | ||
| [Ni(L27)3]·2Cl | 93 | 2.53 | ||
| [Ni(L27)3]·2NO3 | 94 | 1.74 | ||
| [Ni(L27)3]·SO4 | 95 | 1.93 | ||
| [Co(L27)3]·Cl2 | 99 | 1.20 | ||
| [Co(L27)3]·(NO3)2 | 100 | 0.90 | ||
| [Co(L27)3]·SO4 | 101 | 1.09 | ||
| 5 | [Cu(L28)3]·2Cl | 84 | 0.80 | 76a |
| [Cu(L28)3]·2NO3 | 85 | 0.47 | ||
| [Cu(L28)3]·SO4 | 86 | 0.61 | ||
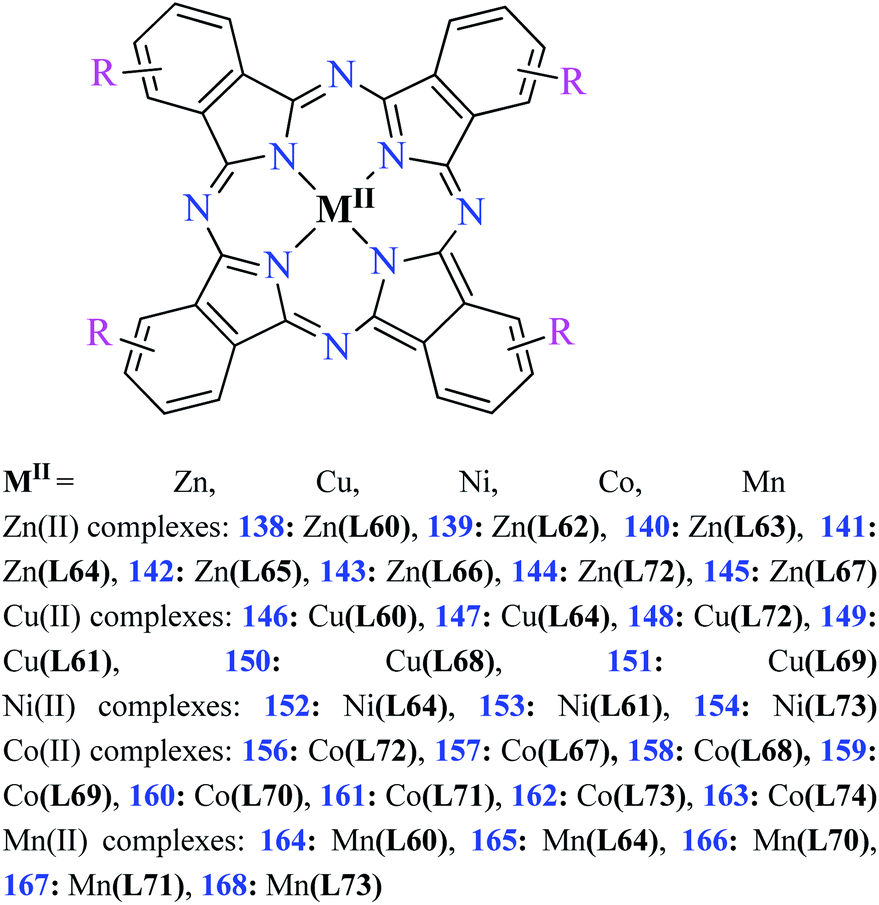 | ||
| Fig. 3 The structure of phthalocyanine complexes with Zn(II), Cu(II), Ni(II), Co(II), and Mn(II) ions. Phthalocyanine ligands L60–L74 were demonstrated in Table 7. | ||
Hydrazide metal(II) complexes
Bioactive compounds containing hydrazide moiety have shown extensive biological properties such as anti-cancer, anti-viral, analgesic and anti-inflammatory, anti-platelet, vasodilator, antioxidant, and hypoglycemic activities.49 They are popular in coordination chemistry due to their ease of synthesis and electronic properties, inducing versatile enzymes inhibitory activity50 through proper binding with the active sites. Hydrazide derivatives have been documented as a potent moiety for the inhibition of α-glucosidase.51,52In this section, various metal(II) complexes containing different hydrazide moieties as a ligand were discussed53–55 (Table 1). It should be noted that the results reported by Naik et al.55 (Table 1, entry 4) had ambiguous values of IC50 or percent inhibition which led us to exclude those data and prevented authors from having a discussion based on the SAR. However, in this study, all complexes showed more potent activity than the free ligand.
α-Glucosidase inhibitory activity of Zn(II) complexes 7 and 8 (Table 1, entries 3a and 3c) were also evaluated (IC50 = 180 and 190 μM, respectively) by Philip et al.,54 and compared with acarbose (IC50 = 99 μM). Complexes were prepared via the coordination of chromone hydrazones ligands (L8 and L9, Table 1, entry 1) and Zn(II). Inhibitory activity of L8 and L9 was approximately similar to each other (IC50 = 240 and 230 μM, respectively), however, weaker than the corresponding complexes. It should be mentioned that L9 possessing phenolic OH group showed no higher activity than L8 lacking hydroxyl group, a potent group for the construction of H-bonding interaction. However, coordination of the Zn(II) had no significant effect on the inhibitory effect. It seems that the formation of complex did not afford either structural or functional role.
Using L8 and L9 containing a chromenone hydrazone moiety in the synthesis of complexes 16 and 17 (IC50s = 140 and 170 μM, respectively, Table 1, entries 3b and 3d) as reported by Philip et al.,54 led to the reduction of anti-α-glucosidase activity comparing with complex 13 (IC50 = 0.15 μM).
Similar to Zn(II) complexes (7 and 8), the inhibitory activity of their counterparts (16 and 17) was independent of phenolic OH group of the ligand. In addition, the narrow IC50 range of complexes 7, 8, 16, and 17 suggested that the structural properties of complexes do not play a role in the inhibitory activity as 7 and 8 are disordered tetrahedral, 16 and 17 are disordered octahedral.
In the same series of ligands, Ni(II) complexes were found to be weaker than those of Zn(II) and Cu(II).
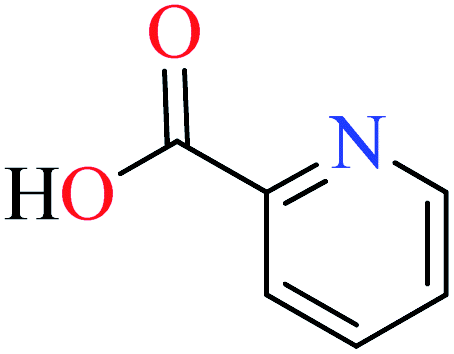
Picolinic acids metal(II) complexes
Picolinic acid (PicA) known as the tryptophan metabolite has a low toxicity to mammals. Oral administration of PicA helps the absorption of several metal ions through the small intestine.56–58 This compound which is an isomer of nicotinic acid and isonicotinic acid is a chelating agent for metals such as chromium, zinc, manganese, copper, iron and molybdenum in the human body.59 Most of its complexes are lipophilic, so PicA can play an important role in the absorption of metals such as zinc in the human body.60 Because different applications of picolinic acid derivatives (PicAs) such as catalysis, ion exchange, nonlinear optics and physicochemical properties are used for the synthesis of different structures containing coordination complexes. Therefore, this compound is very widely used in the field of bio-inorganic chemistry.PicA complex of copper(II) has demonstrated very potent in vitro anti-α-glucosidase activity (IC50 = 1.28 μM) comparing with acarbose (IC50 = 747 μM). It was also evaluated in vivo and could inhibit the α-glucosidase action in ddy mice after oral administration of sucrose and the blood glucose levels were remarkably suppressed. However, it could not change the blood glucose levels in mice administered with glucose.61
6-Methylpicolinic acid (L11) and 3-methylpicolinic acid (L17) was found to be inactive toward α-glucosidase, however, the coordinated products showed proper activity (Table 2) to confirm desired role of metal(II) ions in the enzyme inhibitory activity.
Complex 3764 possessing a square pyramidal geometry, contained two bidentate L11 ligands and depicted low inhibitory activity (IC50 > 546 μM) compared with genistein (IC50 = 16.57 μM). However, replacement of one of L11 by 4,4′-dimethyl-2,2′-bipyridyl (L12),62 2,2′-dipyridylamine (L13),63 and 4(5) methylimidazole (L14)63 to form complexes 38–40 having distorted octahedral geometry (Table 2, entries 5, 6a, and 7a), could not improve the IC50 values (IC50 > 600 μM). Moreover, Dege et al. have synthesized a novel Zn(II) complex having 2-picolinic acid (L19) and 6-chloropicolinic acid (L20) ligands with distorted octahedral geometry (Table 2, entry 1) and examined the inhibitory activity of the complex (41, Table 2, entry 9),65 comparing with genistein as a reference drug (IC50 = 8 μM). Comparing of the inhibitory activity of complex 37 (IC50 = 546.04 μM) with 41 (IC50 = 440 μM) revealed that the replacement of methyl group with Cl did not lead to significant inhibitory activity. In general, the low activity of Zn(II) complexes containing PicA moiety can be associated with the full d orbitals of metal center ion. It should be mentioned that all ligands (L11–L20) were also inactive toward α-glucosidase.
In complex 43,64 the third ligand, water was replaced by the pyridine (L16, Table 2, entry 1) which led to the reduction of activity from IC50 = 2.95 μM to 3.49 μM compared with genistein (IC50 = 16.57 μM). That was associated with the π-conjugation effect of L16. Comparison of the activity of complex 42 with its counterpart, complex 37, indicated the essential role of Cu2+ ions as the metal center in the α-glucosidase inhibitory activity.
Although the introduction of isothiocyanate into the Cu(II) complex 44 afforded the inhibitory potency (IC50 = 8.02 μM) approximately as potent as the genistein (IC50 = 7.85 μM), it was not as efficient as water and pyridine (42 and 43, respectively).66
The efficacy of other complexes containing mixed-ligands 4567 (IC50 = 688.94 μM) and 46 (IC50 = 513.10 μM)63 has been also investigated. The presence of 2,2′-bipyridyl (L15) and nitrate in complex 45 and 2,2′-dipyridylamine (L13) and acetate in complex 46, reduced the inhibitory activity compared with 42, 43, 44, and 47 in spite of the same geometry.
Distinguished α-glucosidase inhibitory activity of Cu(II) complexes of PicAs 42–47 compared with non-active free ligands can be explained from various points of view. The structural and electronic role of Cu(II) ion in providing desired binding affinity toward the enzyme, is significant.
Comparison of Fe(II) complexes with their counterparts 57 and 58 demonstrated that the position of methyl group on PicA moiety played an important role in inhibitory activity, however, it was not found to be important for 57 and 58. It may be related to the more significant electronic property of Fe(II) than that of Co(II) in spite of same geometry of distorted octahedral.
Schiff bases metal(II) complexes
Schiff bases are the popular organic ligands in the coordination chemistry possessing significant role in the induction of different biological activities. It was found that Schiff base complexes especially those having tetrahedral or octahedral geometry, have demonstrated DNA-binding properties and DNA-cleavage activities,69 antifungal, antibacterial, antimalarial, anti-proliferative and anticancer, anti-inflammatory, antiviral, and antipyretic activity.70,71 The imine nitrogen is basic and exhibits pi-acceptor properties. Most of the biological properties of these compounds are due to their pi-acceptor properties. Schiff bases are commonly bi- or tri-dentate ligands, able to form stable complexes with transition metals. Schiff base reactions are important for the formation of nitrogen–carbon double bonds in organic chemistry. Also, they are used as liquid crystals.70Diamines metal(II) complexes
Diamines are organic compounds with two groups of amino groups. The term diamine refers to type-I diamines because of the high reactivity of the first type of amines. They are used as monomers to produce polyimides and polyureas. 1,2-Diamine derivatives are the most practical moieties in the structure of biological compounds and coordination chemistry.73 Many natural products that have significant biological properties contain at least one 1,2-diamino component in their structure. Also, complexes of 1,2-diamino groups have been widely investigated for their biological properties. For example, 1,2-diamino complexes of Pt(II) demonstrated more antitumor activity than cisplatin due to lower toxicity and drug resistance.73Four types of diamines including N-(n-butyl) ethylenediamine (L25),74 1,6-diaminohexane (L26),74 N-methylethylenediamine (L27),75 and ethylenediamine (L28)76 were used and the effect of the counterion (chloride, nitrate, and sulphate) was also considered in this series. As can be seen in Table 4, Cu(II) complexes of ethylenediamine (L28) possessed much higher activity than Zn(II), Ni(II), and Co(II).
Trivalent metal complexes as α-glucosidase inhibitors
The activity of trivalent transition metal complexes as α-glucosidase inhibitors is not well understood and a few studies have been reported in this field. All known modes of life need iron. Among iron compounds, iron(III) ion is usually the most stable species in the air, as demonstrated by the pervasiveness of rust, an unsolved iron(III)-containing material. Iron(III) is a d5 center, indicating that the iron(III) has five valence electrons in the 3d orbital shell. These d-orbitals can accept a large variety of ligands to form coordination complexes. Normally, ferric ions are enclosed by six ligands arranged in octahedral; but sometimes three and sometimes as many as seven ligands are seen.77 Many proteins in existing beings contain fixed iron(III) ions which are important subclass of the metalloproteins including oxyhemoglobin, ferredoxin, and cytochromes.78Chromium especially Cr(III) is an important bioelement for the mammalian organism.79 It plays an essential role in the maintenance of a normal glucose tolerance to maintain regular carbohydrate and lipid metabolism.80,81 The direct relation between chromium deficiency and glucose intolerance has been clinically studied.82 Cr(III) glycinate complex has also been reported to improve blood glucose levels and reduces the copper to zinc ratio in tissues of rats with mild hyperglycaemia.83
Metals(III) are able to form complexes possessing unique properties due to strong interactions with nitrogen-giving and oxygenating ligands resulting in versatile electronic structure and electron density of the complex. For this purpose, study of their biological activities such as α-glucosidase inhibitory activity was considered.
Considering the fact that all ligands L11, L12, L17 (Table 2, entry 1) and L29–L32 (Table 5, entry 1) used for the preparation of trivalent metal complexes are not active toward α-glucosidase, inhibitory activity of resulted complexes is valuable.
| Entry | Compounds | Structure number | IC50 (μM) | Ref. |
|---|---|---|---|---|
| a Acarbose as the reference drug (IC50 = 418 ± 0.55 μM).b Genistein as the reference drug (IC50 = 16.57 ± 0.23 μM).c Genistein as the reference drug (IC50 = 7.85 ± 0.87 μM). | ||||
| 1 |  |
L29 | NA | 85a |
 |
L30 | NA | ||
 |
L31 | NA | ||
 |
L32 | NA | ||
| 2 | 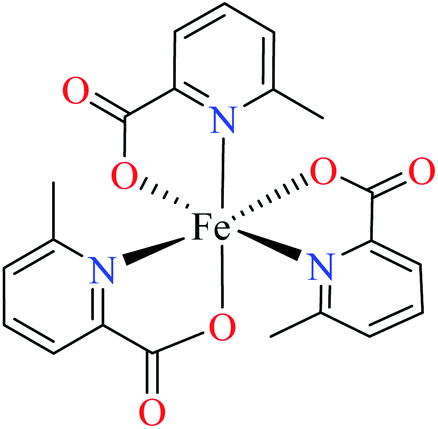 |
64b | ||
| 102 = [Fe(L11)3] | 102 | 446.2 | ||
| 3 | 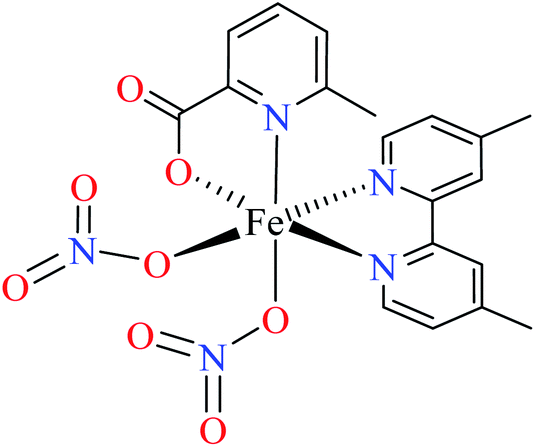 |
|||
| 103 = [Fe(L11)(L12)(NO3)2] | 103 | 492.3 | 62b | |
| 4 | 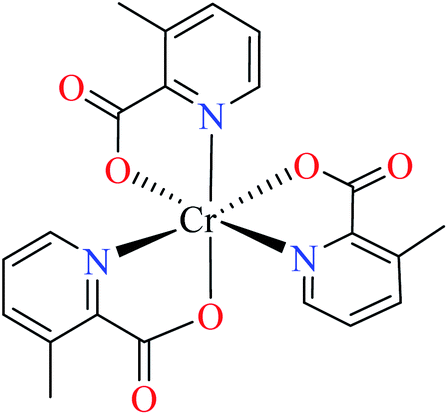 |
84b | ||
| 104 = [Cr(L17)3] | 104 | >600 | ||
| 5 | 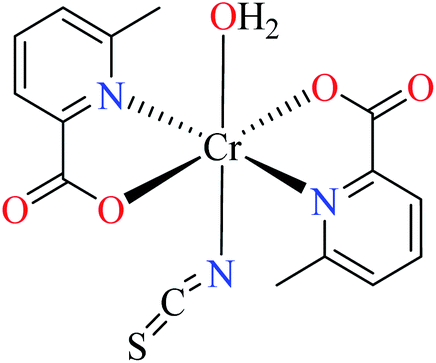 |
66c | ||
| 105 = [Cr(L11)2(NCS)(H2O)] | 105 | >600 | ||
| 6 | 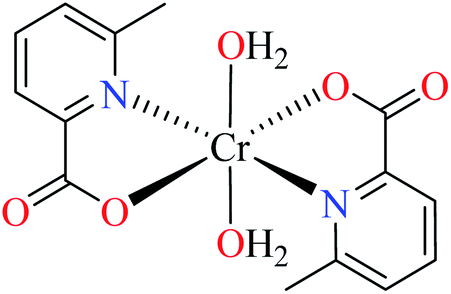 |
64b | ||
| 106 = [Cr(L11)2(H2O)2] | 106 | 164.87 | ||
| 7 | 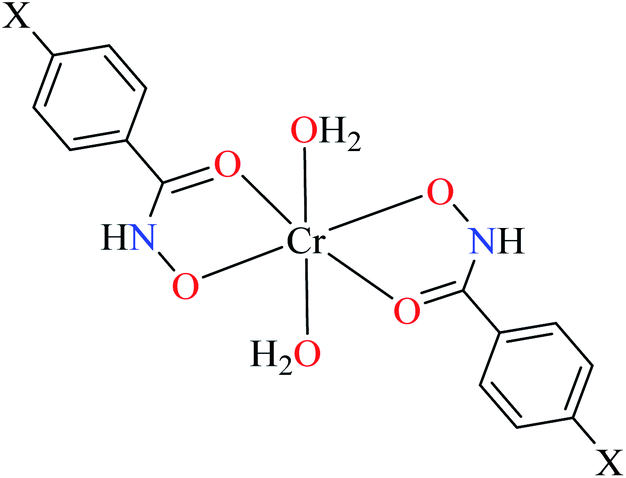 |
85a | ||
| 107 = [Cr(L29)2(H2O)2] | 107 | 28.7 | ||
| 108 = [Cr(L30)2(H2O)2] | 108 | 69.28 | ||
| 109 = [Cr(L31)2(H2O)2] | 109 | 355.9 | ||
| 110 = [Cr(L32)2(H2O)2] | 110 | 169.5 | ||
Picolinic acid metal(III) complexes
In this part, Fe(III) and Cr(III) complexes containing picolinic acid (PicA) were considered.Hydroxamic acid chromium(III) complexes
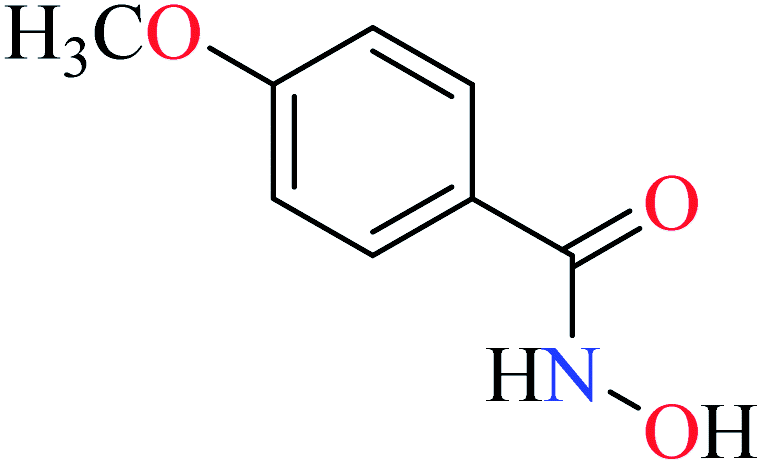
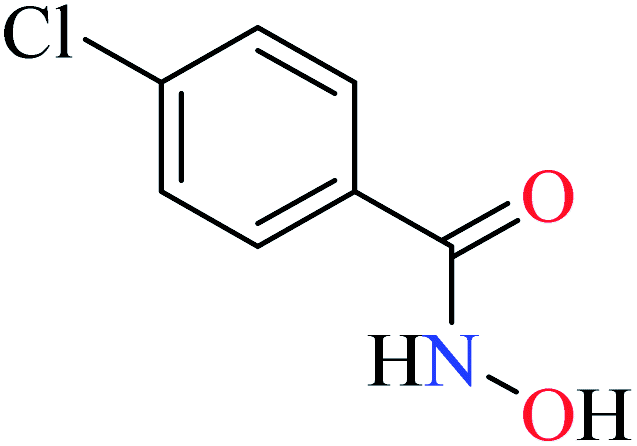
Hassan et al. reported α-glucosidase inhibitory activity of Cr(III) complexes 107–110 (Table 5, entry 7) of hydroxamic acid derivatives L29–L32 (Table 5, entry 1) comparing with acarbose as the standard drug (IC50 = 418 μM).85 The efficacy of hydroxamic acid derivatives (L29–L32) were found to be more effective than PicAs (L11, L12, and L17), in spite the fact that all ligands were not inhibitor of α-glucosidase. Also, the presence of various substituents on hydroxamic acid moiety played an important role in the inhibitory activity. Among 107–110, the best activity was reported by complex 107 (IC50 = 28.7 μM) with no substituent on the aryl moiety. Introduction of other substituents including CH3 (108), Cl (110), and CH3O (109) into the 4-position of the aryl group deteriorated anti-α-glucosidase activity. However, the order of activity was found as CH3 (IC50 = 69.28 μM) > Cl (IC50 = 169.5 μM) > OCH3 (IC50 = 355.9 μM).Tetravalent metal complexes as α-glucosidase inhibitors
Considering the fact that the stability of central metal of the complexes in the high oxidation state is difficult, they have been generally found in polyoxo materials. Among the first row transition metals, vanadium and oxo-vanadium complexes have been frequently synthesized and investigated for their biological properties such as anti-α-glucosidase activity.Vanadium in the oxidation states III, IV, and V readily forms V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and can coordinate to N and S of small molecules to form stable complexes. The redox potential of V(V) to V(IV) or V(IV) to V(III) have made vanadium a versatile metal in the biological and medicinal applications.86–90 Under in vitro conditions, vanadium compounds stimulate glucose uptake and inhibit lipid breakdown, in a way remarkably suggestive of insulin's effects. Under in vivo conditions, vanadium improves insulin's plasma glucose and lipid-lowering properties, leading to normalization of diabetic symptoms in the presence of only minimal endogenous insulin.87
O bond and can coordinate to N and S of small molecules to form stable complexes. The redox potential of V(V) to V(IV) or V(IV) to V(III) have made vanadium a versatile metal in the biological and medicinal applications.86–90 Under in vitro conditions, vanadium compounds stimulate glucose uptake and inhibit lipid breakdown, in a way remarkably suggestive of insulin's effects. Under in vivo conditions, vanadium improves insulin's plasma glucose and lipid-lowering properties, leading to normalization of diabetic symptoms in the presence of only minimal endogenous insulin.87
Discovery of the insulin-mimetic effect of vanadium ions and their complexes as potential agents for the treatment or opposition of DM has been the most remarkable improvement in the treatment of the disease in the last decade of the 20th century.39 Subsequently, the effect of vanadyl sulfate (VOSO4) was clinically tested to treat T2DM in the USA since 1995.91,92
Vanadium complexes containing O-coordinating atoms are known for their high hydrolytic stability; however, they have shown low redox stability. In this respect, vanadium complexes coordinated to both O- and N-atoms are known for their high hydrolytic stability.93 Accordingly, complexes of hydrazides, PicAs, and Schiff bases can be versatile candidates for desired α-glucosidase inhibitory activity (Table 6).
| Entry | Compounds | Structure number | IC50 (μM) | Ref. |
|---|---|---|---|---|
| a Acarbose as the reference drug (IC50 = 18.59 μM).b Acarbose as the reference drug (IC50 = 780 ± 0.28 μM).c Not reported.d Genistein as the reference drug (IC50 = 16.75 ± 0.23 μM). | ||||
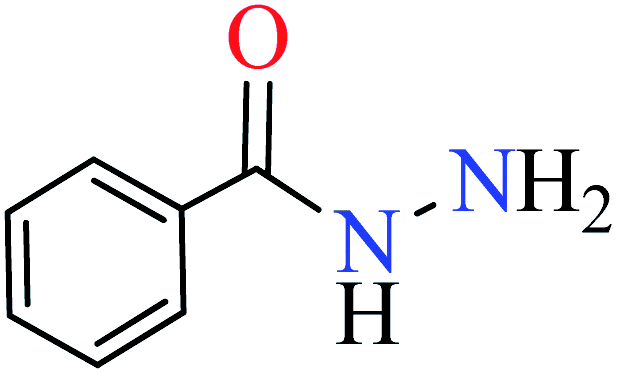
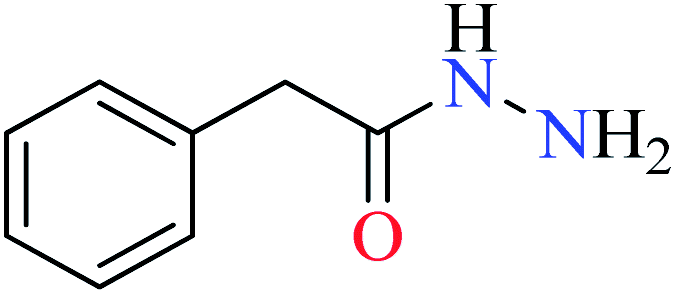
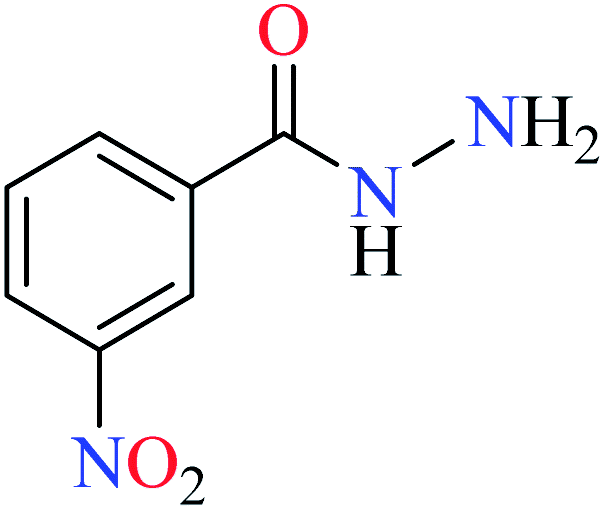
| 1 | 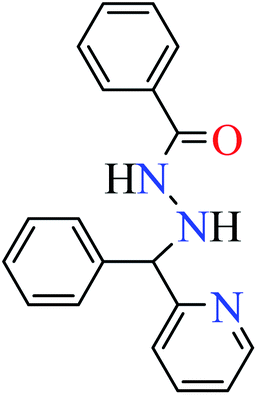 |
L33 | Not reported | 94a |
 |
L34 | Not reported | 94a | |
 |
L35 | NA | 93b | |
 |
L36 | NA | 93b | |
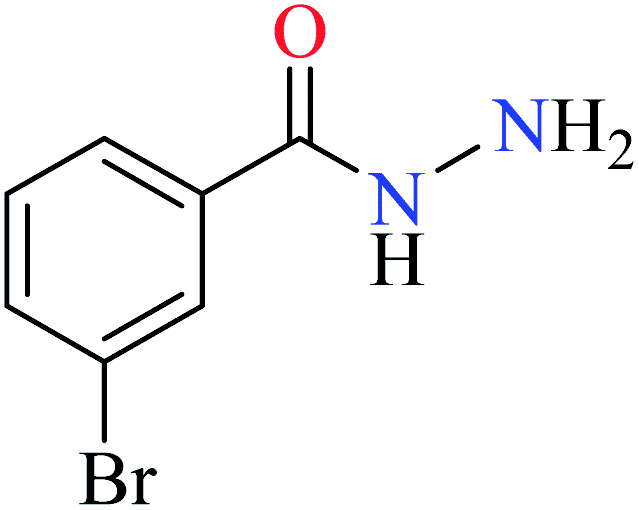 |
L37 | NA | 93b | |
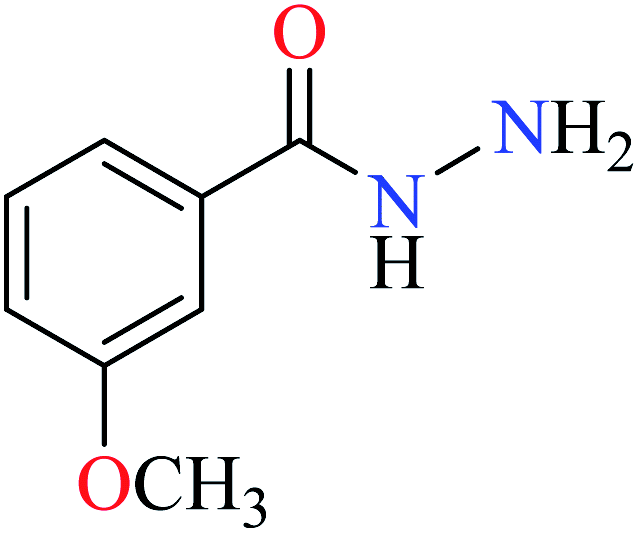 |
L38 | NA | 93b | |
 |
L39 | NA | 93b | |
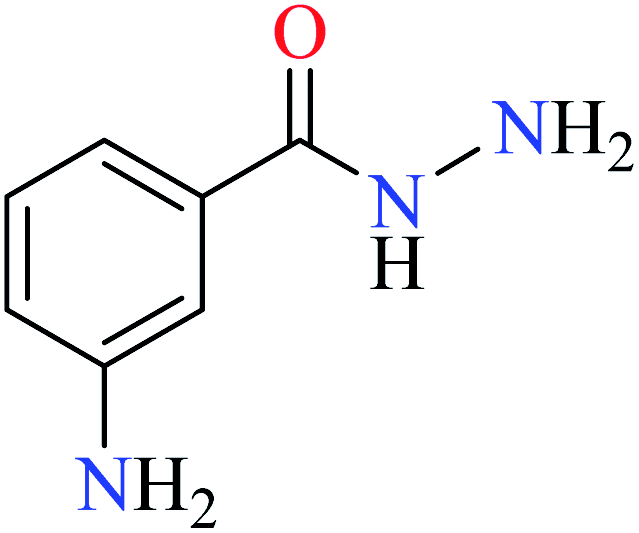
 |
L40 | NA | 93b | |
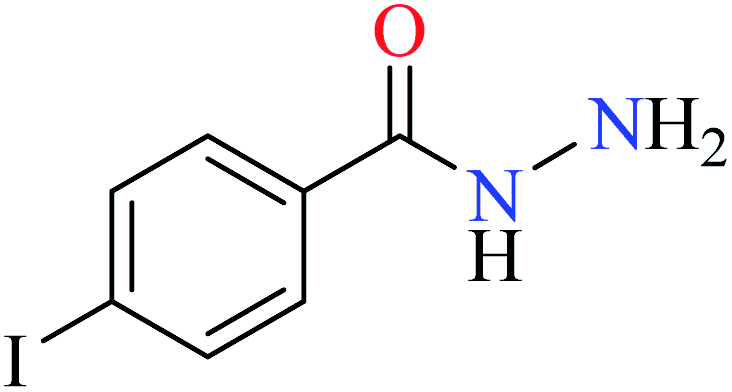 |
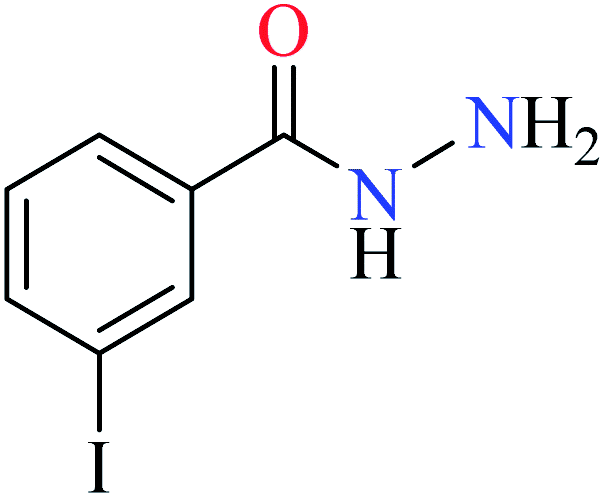
L41 | NA | 93b | |
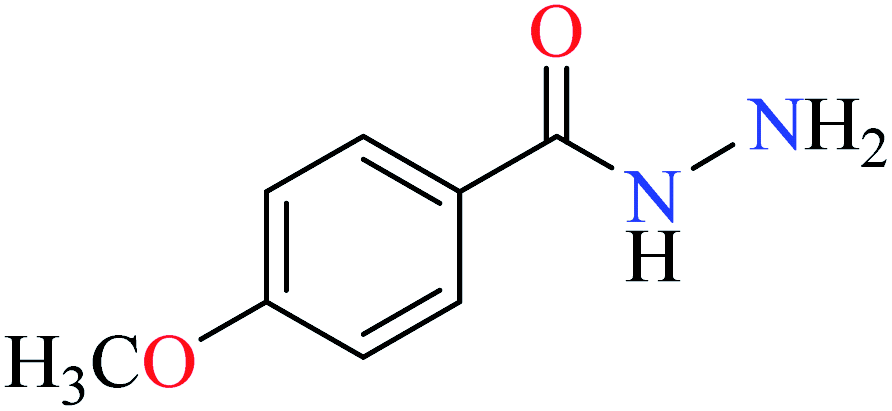 |
L42 | NA | 93b | |
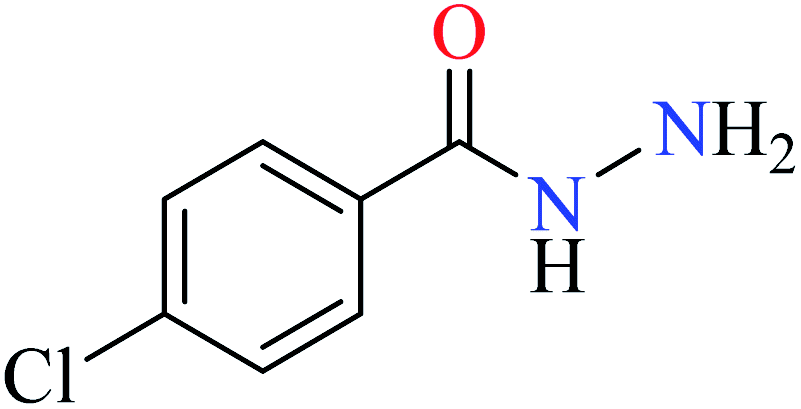 |
L43 | NA | 93b | |
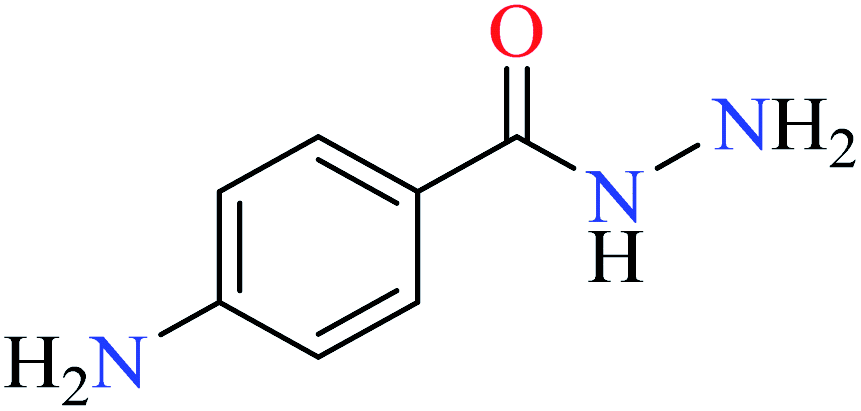 |
L44 | NA | 93b | |
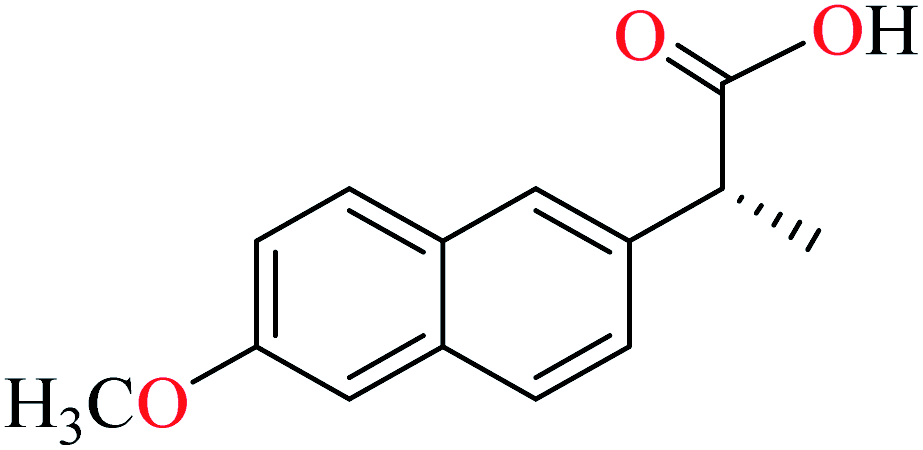 |
L45 | Not reported | 95c | |
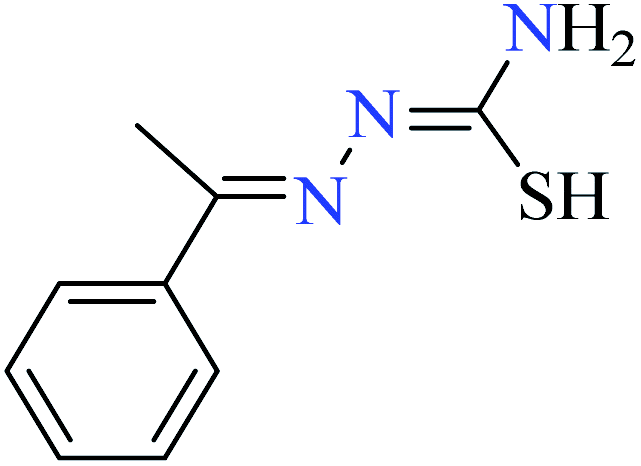 |
L46 | Not reported | 95c | |
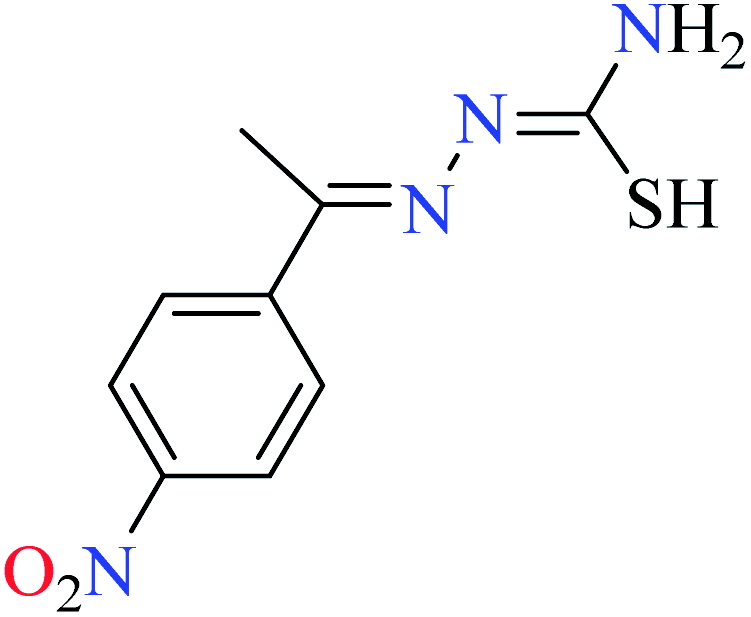 |
L47 | Not reported | 95c | |
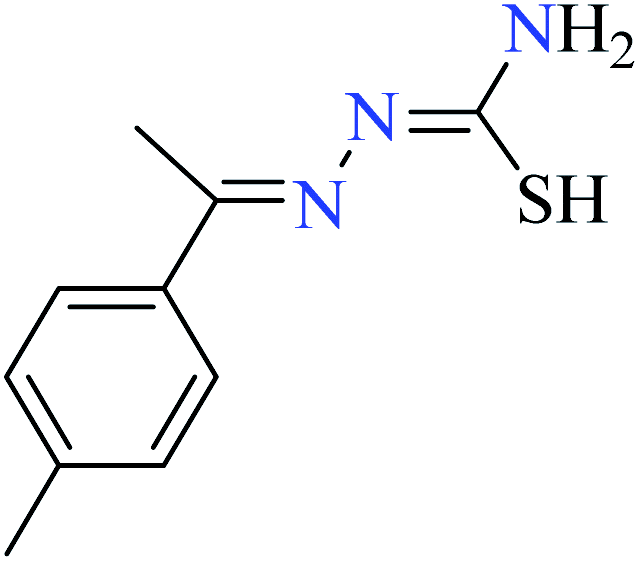 |
L48 | Not reported | 95c | |
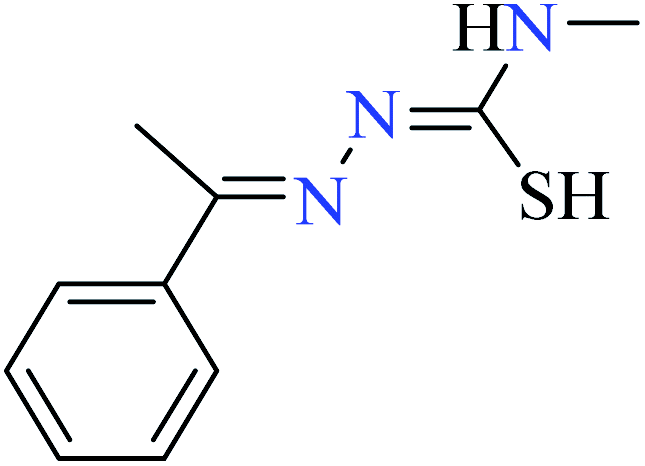 |
L49 | Not reported | 95c | |
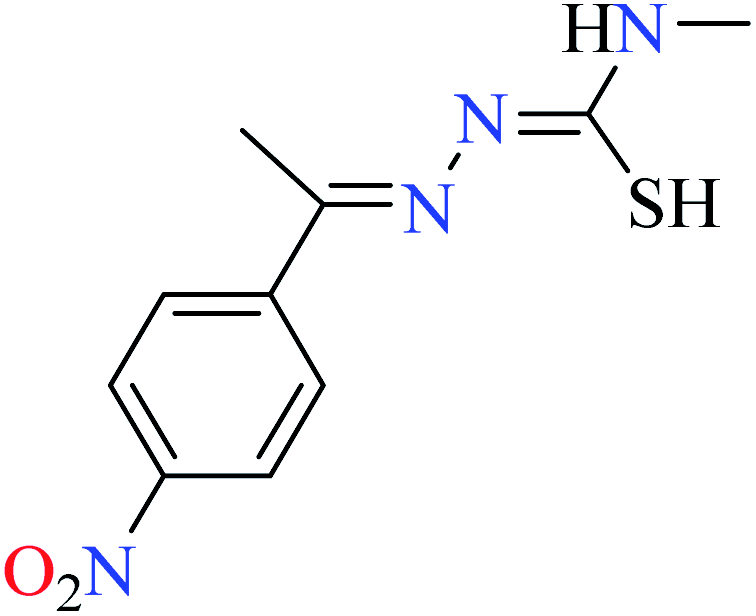 |
L50 | Not reported | 95c | |
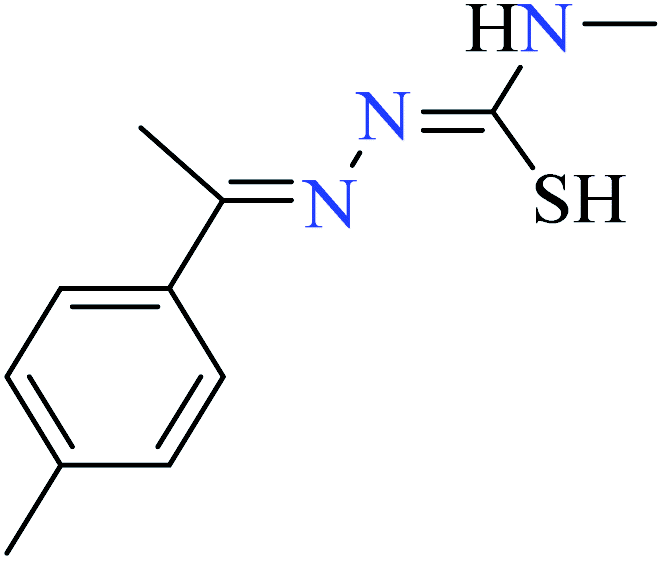 |
L51 | Not reported | 95c | |
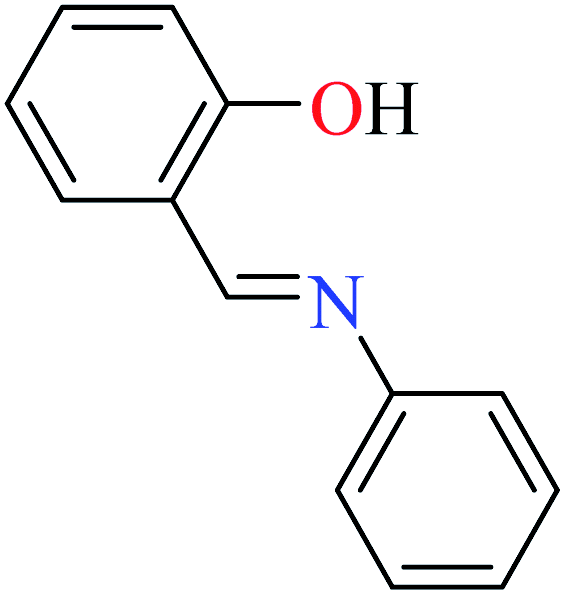 |
L52 | Not reported | 96a | |
 |
L53 | Not reported | 96a | |
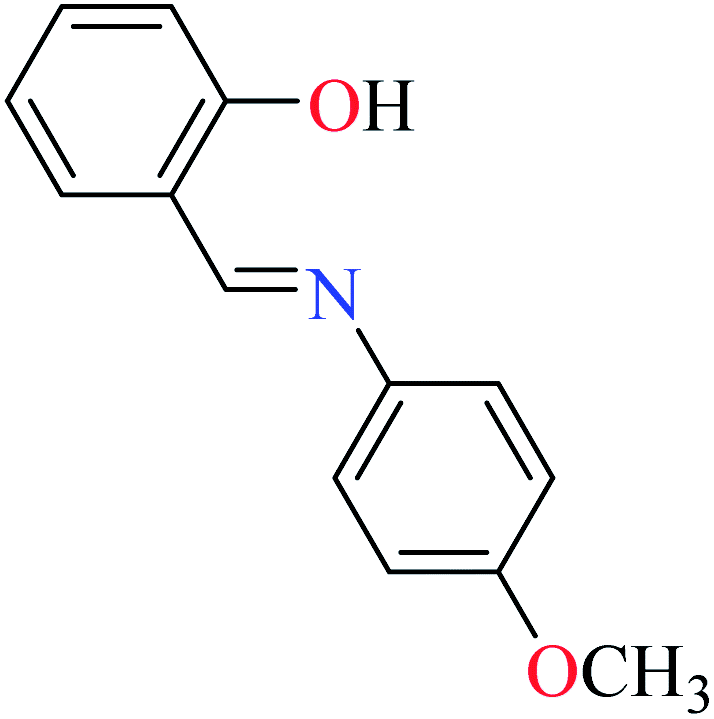 |
L54 | Not reported | 96a | |
 |
L55 | 22.72 | 96a | |
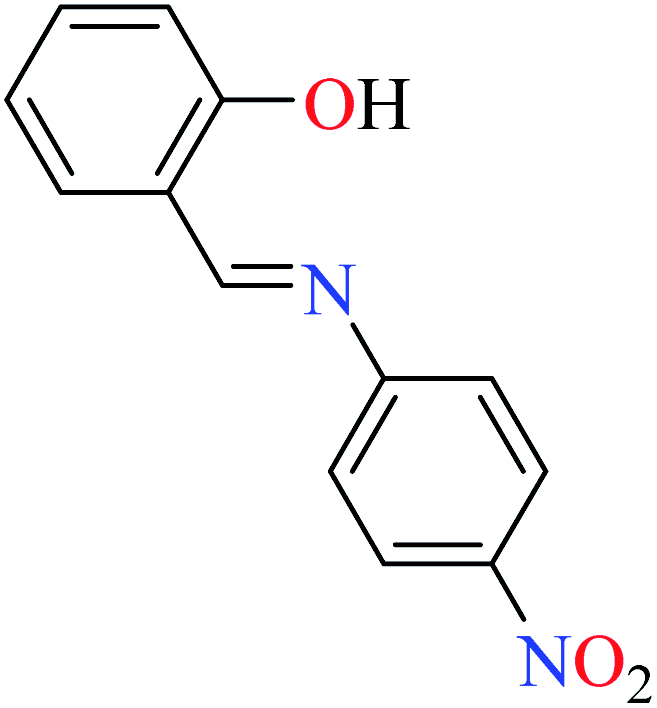
 |
L56 | Not reported | 96a | |
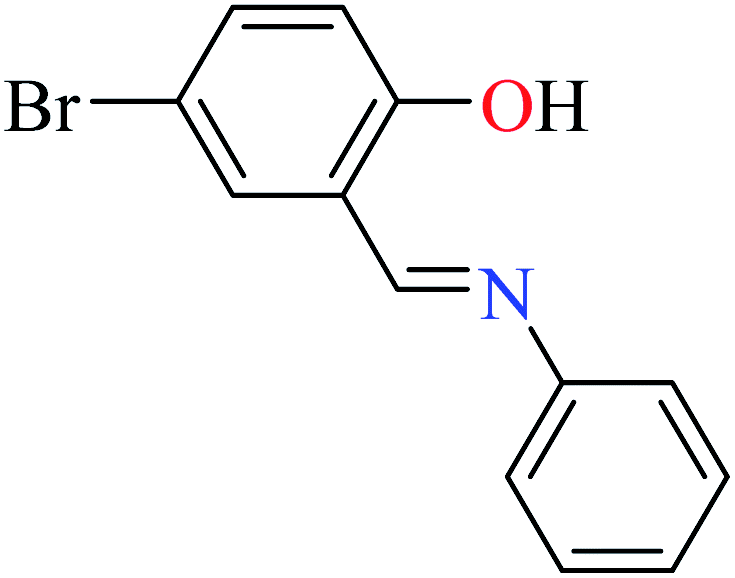
 |
L57 | Not reported | 96a | |
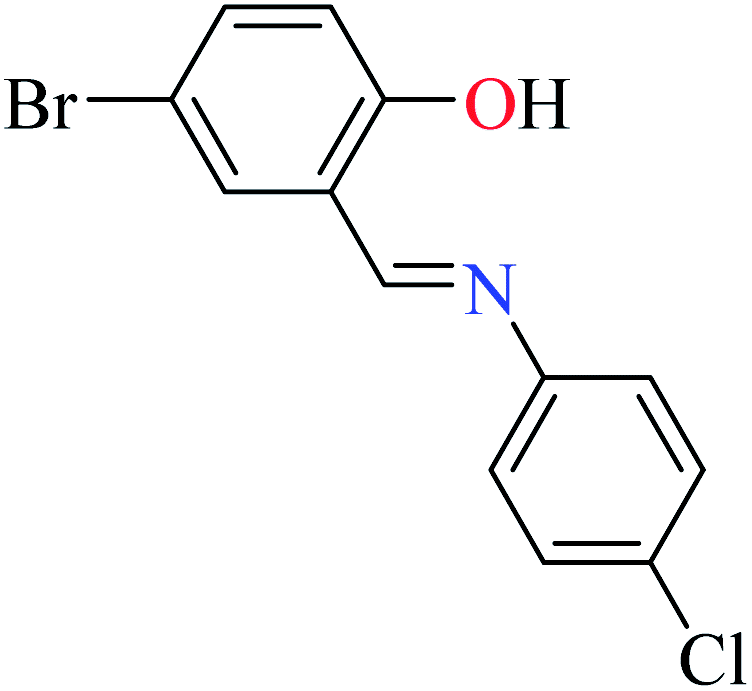
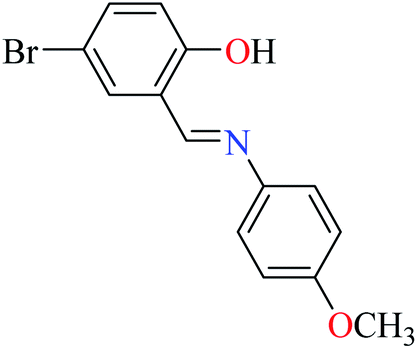 |
L58 | 74.11 | 96a | |
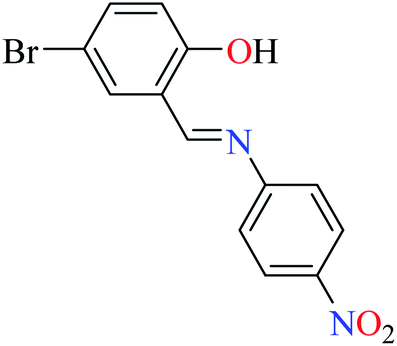 |
L59 | 245 | 96a | |
| 2 |  |
94a | ||
| 111 = [VO(L33)(L34)] | 111 | 14.75 | ||
| 3 | 93b | |||
| 3a |  |
|||
| 112 = [(VO)2(L35)2(H2O)2]·SO42− | 112 | 10.70 | ||
| 3b | 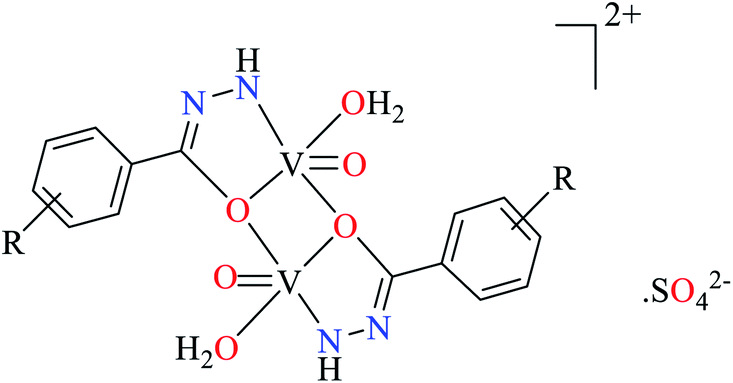 |
|||
| R = Br, OCH3, I, NH2, NO2 | ||||
| 113 = [(VO)2(L36)2(H2O)2] | 113 | 67.00 | ||
| 114 = [(VO)2(L37)2(H2O)2] | 114 | 20.10 | ||
| 115 = [(VO)2(L38)2(H2O)2] | 115 | 0.68 | ||
| 116 = [(VO)2(L39)2(H2O)2] | 116 | 12.50 | ||
| 117 = [(VO)2(L40)2(H2O)2] | 117 | 800 | ||
| 118 = [(VO)2(L41)2(H2O)2] | 118 | 758 | ||
| 119 = [(VO)2(L42)2(H2O)2] | 119 | 38.00 | ||
| 120 = [(VO)2(L43)2(H2O)2] | 120 | 13.50 | ||
| 121 = [(VO)2(L44)2(H2O)2] | 121 | 16.40 | ||
| 4 | 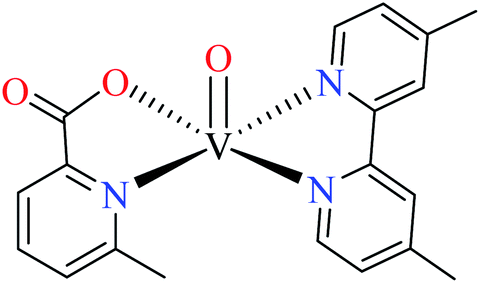 |
62d | ||
| 122 = [VO(L11)(L12)] | 122 | >600 | ||
| 5 | 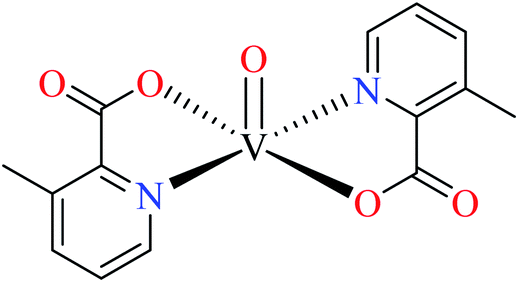 |
84d | ||
| 123 = [VO(L17)2] | 123 | >600 | ||
| 6 |  |
95c | ||
| R1 = H, NO2, CH3 | ||||
| R2 = H, CH3 | ||||
| 124 = [VO(L45)(L46)] | 124 | Not reported clearly | ||
| 125 = [VO(L45)(L47)] | 125 | Not reported clearly | ||
| 126 = [VO(L45)(L48)] | 126 | Not reported clearly | ||
| 127 = [VO(L45)(L49)] | 127 | Not reported clearly | ||
| 128 = [VO(L45)(L50)] | 128 | Not reported clearly | ||
| 129 = [VO(L45)(L51)] | 129 | Not reported clearly | ||
| 7 | 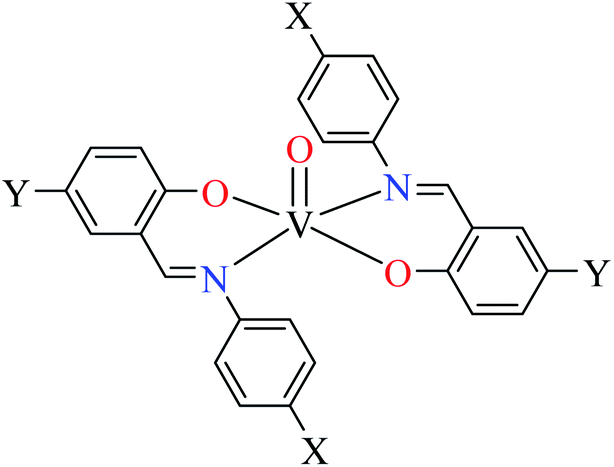 |
96a | ||
| X = H, Cl, OCH3, NO2 | ||||
| Y = H, Br | ||||
| 130 = [VO(L52)2] | 130 | 2.11 | ||
| 131 = [VO(L53)2] | 131 | 13.12 | ||
| 132 = [VO(L54)2] | 132 | 1.26 | ||
| 133 = [VO(L55)2] | 133 | 1.26 | ||
| 134 = [VO(L56)2] | 134 | 16.92 | ||
| 135 = [VO(L57)2] | 135 | Not reported | ||
| 136 = [VO(L58)2] | 136 | 92.43 | ||
| 137 = [VO(L59)2] | 137 | 31.10 | ||
Patel et al. synthesized mononuclear vanadium(IV) complex (111, Table 6, entry 2) of N′-[(E)-phenyl(pyridin-2-yl)methylidene]benzohydrazide (L33) and benzhydrazide (L34) (Table 6, entry 1)94 to evaluate its anti-α-glucosidase activity comparing with acarbose (IC50 = 18.59 μM). Complex 111 showed good inhibitory activity with IC50 value of 14.75 μM.
Ashiq et al. prepared various hydrazide vanadium(IV) complexes 112 (Table 6, entry 3a) using 2-phenylacetohydrazide (L35) and complexes 113–121 (Table 6, entry 3b) using benzylidenehydrazine derivatives (L36–L44).93 The complex 112 showed very good inhibitory activity (IC50 = 10.7 μM) comparing with acarbose (IC50 = 780 μM) and deoxynojirimycin (IC50 = 426 μM) in spite of lacking activity of L35. The inhibitory activity of complexes 113–121 (IC50 = 0.68–800 μM) was completely depended on the electronic property of substituents on the aryl ring, as compared with acarbose (IC50 = 780 μM) and deoxynojirimycin (IC50 = 426 μM). Complex 115 possessing strong electron-releasing methoxy group at 3-position of aryl ring, showed the best activity. Changing the position of methoxy group (complex 119) led to the reduction of activity (IC50 = 38 μM). However, similar trend was observed in the case of complexes 116 and 121 having a strong electron-releasing group (NH2). Complex 116 having the group at meta position of aryl ring showed better activity (IC50 = 12.5 μM) than complex 121 possessing the amino group at para position (IC50 = 16.4 μM). However, the efficacy of amino group at 3-position was lower than the methoxy group by comparing the IC50 values of complexes 115 and 116. However, comparing those of complexes 119 and 121 gave inverse result.
Introduction of a strong electron-withdrawing group (NO2) into the meta opposition of aryl moiety 113 led to much lower activity (IC50 = 67.0 μM) than those complexes having a strong electron-releasing group (115, 116, 119, and 121). Moreover, insertion of halogens (Cl, Br, and I) into the meat and para position of aryl group (complexes 120, 114, 117, and 118) gave interesting results. Among halogenated derivatives, 120 having Cl at para position of aryl ring was found to be the most active compound (IC50 = 13.5 μM). It also was more potent than its counterparts, complexes 119 and 121. The presence of Br at meta position of aryl ring (complex 114) led to relatively good activity (IC50 = 20.1 μM). Complexes 117 (IC50 = 800 μM) and 118 (IC50 = 758 μM) having iodine at 3- or 4-position of the aryl ring demonstrated very low activity which may be related to the steric hindrance of iodine. In the case of complex 120, electron-donating property of Cl via resonance may be important to increase the electron density at central metal inducing desired inhibitory potential.
Picolinic acid vanadium(IV) complexes
Avcı et al. synthesized vanadyl complexes 122 (Table 6, entry 4)62 and 123 (Table 6, entry 5)84 for their α-glucosidase inhibitory activity comparing with genistein (IC50 = 16.57 μM). Complex 122 coordinated to 6-methylpicolinic acid (L11, Table 2, entry 1) and 4,4-dimethyl-2,2-bipyridyl (L12, Table 2, entry 1). In complex 123, two ligands were replaced by two 3-methylpicolinic acid ligands (L17). Both complexes have distorted square pyramidal structure and showed no activity. It worth mentioning that theoretical calculations from frontier molecular orbitals (FMO) revealed high reactivity and polarizability of complexes 122 and 123. In addition, the molecular electrostatic potential (MEP) surface indicated high electrophilic and nucleophilic reactivity of both complexes.Schiff base vanadium(IV) complexes
Rahiman et al. synthesized various oxovanadium(IV) complexes (124–129, Table 6, entry 6) using 2-(6-methoxynaphthalen-2-yl)propanoic acid (L45) and 2-(1-ethylidene)hydrazinecarbothioamide derivatives (L46–51).95 However, ambiguous results in IC50 values which were calculated as (%) prevented us to discuss in details. Misra et al. synthesized and evaluated oxovanadium(IV) complexes of salicylaldehyde Schiff bases L52–59 (Table 6, entry 1, complexes 130–137, Table 6, entry 7) toward rat intestinal α-glucosidase.96 L56–59 possessed Br on the aryl ring of salicylaldehyde. L55, L58, and L59 showed inhibitory activity with IC50 values of 22.72, 74.11, 245.0 μM, respectively, comparing with acarbose (IC50 = 18.59 μM). All complexes showed very good anti-α-glucosidase activity, however, those lacking Br on the salicylaldehyde moiety (130–133) showed better activity (IC50 = 1.26–13.12 μM) than brominated derivatives (134–137, IC50 = 16.92–62.43 μM). The best activity was obtained by complexes 132 and 133 (IC50 = 1.26 μM) which possessed strong electron-donating and electron-withdrawing groups, OMe and NO2, respectively. The presence of Cl (131) at the same position reduced activity and the absence of substituent on the aryl moiety (130) was found to be 6 times more potent than 131. In the case of brominated compounds (134–137), complex 136 as the counterpart of 132, was the weakest inhibitor. Totally, each brominated derivative showed lower activity than counterpart lacking Br, which may be related to the halogen size interrupting the complex from appropriate interactions with the enzyme. In this study,96 complexe 132 was candidate for the in vivo evaluation and it could decrease the blood glucose levels by approximately 12%. However, the corresponding value for acarbose was reported as 35%. The lower activity of 132 in in vivo conditions may be related to dissociation of the complex due to low pH inside the intestine.Phthalocyanine metal complexes
In this review, anti-α-glucosidase activity of complexes of the first-row transition metals including Zn2+, Cu2+, Ni2+, Co2+, Fe2+, Mn2+, Fe3+, Cr3+, and V4+ complexes with small molecules; hydrazide, PicAs, Schiff bases, diamines, and hydroxamic acid was discussed. However, macromolecules such as phthalocyanines have been found as the versatile ligands for the formation of desired complexes with first-row transition metals possessing significant α-glucosidase inhibitory properties.Phthalocyanines and their metal complexes which are structurally related to porphyrin metal complexes, have been widely investigated as chemical sensors,97 liquid crystals,98 Langmuir Blodgett films,99 photovoltaic cells,100,101 electro-chromic materials,101 optical and electrical materials, catalysts,102 and photosensitizers in the photodynamic cancer therapy.103 Also, their enzyme inhibitory activity have been in the center of attention.104,105 Herein, α-glucosidase inhibitory activity of several metal(II) complexes 138–168 (Fig. 3) bearing different phthalocyanine derivatives L60–L74 (Table 7) was discussed. Although all of them possessed square planar geometry; however, the electronic properties of metal ions and ligands were found to play a significant role in the inhibitory activity.
| Entry | Compounds | Structure number | IC50 (μM) | Ref. |
|---|---|---|---|---|
| a Acarbose as the reference drug (IC50 = 0.38 μM).b Acarbose as the reference drug (IC50 = 60.55 μM).c Acarbose as the reference drug (IC50 = 63.03 μM).d Acarbose as the reference drug (IC50 = 15.92 μM).e Acarbose as the reference drug (IC50 = 51.45 μM).f Acarbose as the reference drug (IC50 = 189.20 μM).g Acarbose as the reference drug (IC50 = 22.80 μM).h Acarbose as the reference drug (IC50 = 12.60 μM).i Acarbose as the reference drug (IC50 = 58.47 μM). | ||||
| 1 | 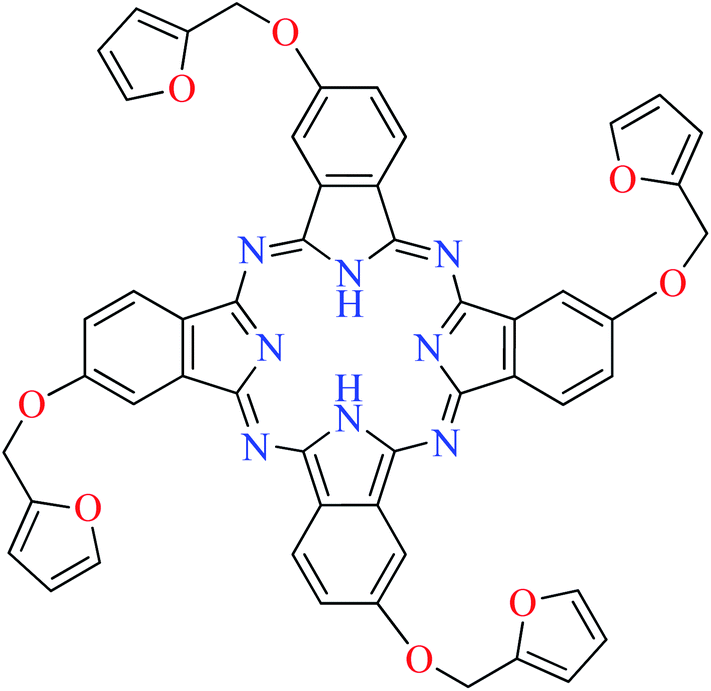 |
L60 | 1104.04 | 106a |
| 2 |  |
L61 | 158.66 | 105b |
| 3 |  |
L62 | Not reported | 107c |
| 4 |  |
L63 | Not reported | 109d |
| 5 |  |
L64 | 11.65 | 108e |
| 6 |  |
L65 | Not reported | 110f |
| 7 |  |
L66 | Not reported | 110g |
| 8 |  |
L67 | Not reported | 111g |
| 9 |  |
L68 | Not reported | 112h |
| 10 |  |
L69 | Not reported | 109f |
| 11 |  |
L70 | Not reported | 113i |
| 12 |  |
L71 | Not reported | 113i |
| 13 |  |
L72 | Not reported | 110g |
| 14 |  |
L73 | Not reported | 114g |
| 15 |  |
L74 | Not reported | 111g |
Prospects and challenges
Evaluation of α-glucosidase inhibitory activity of metal complexes has recently absorbed the attention of researchers working on the field of treatment of T2DM. In this paper, the inhibitory activity of the first-row transition complexes toward α-glucosidase was reviewed.α-Glucosidase inhibitory activity of divalent transition metal complexes 1–101(Tables 1–4)
Discussed metal centers coordinated to hydrazide, picolinic acid, Schiff base, and diamine moieties. Among them, Cu(II) complexes were usually found to be significant inhibitors, however, the complex of hydrazide moiety attracted more attention.In the case of divalent transition metal complexes bearing hydrazide ligands 1–36 (Table 1):
• Complex 13 bearing L5 was the most potent compound.
• Complexes of L1: complex 30 Mn(L1) showed the best inhibitory activity (IC50 = 45.63 μM) and replacement of Zn(II) afforded more than 2-fold reduction in the activity (IC50 = 101.29 μM). Cu(II) and Co(II) complexes of the same ligand were not active toward α-glucosidase.
• Complexes of L2: complex 10 Cu(L2) depicted the best anti-α-glucosidase activity (IC50 = 17.73 μM) and replacement of metal ion by Zn(II), Co(II), and Mn(II) afforded lower activity (IC50s = 101.29, 66.48, and 143.21 μM, respectively).
• Complexes of L3: complex 11 Cu(L3) with IC50 value of 1.15 μM was found to the most active. Replacement of the metal ion by Zn(II) gave the lower activity (IC50 = 27.71 μM) and the order of activity in Co(II) and Mn(II) complexes followed a declining trend (IC50s = 153.23 and 354.62 μM, respectively).
• Complexes of L4: complex 12 Cu(L4) was the most potent anti-α-glucosidase compound (IC50 = 18.91 μM). Complexes 4 Zn(L4) and 25 Co(L4) showed similar activity (IC50s = 97.26 and 96.95 μM, respectively), however, lower than 12 Cu(L4). 33 Mn(L4) was completely inactive toward α-glucosidase.
• Complexes of L5: complex 13 Cu(L5) was the most potent inhibitor (IC50 = 0.15 μM) and the other ML5 complexes were inactive toward α-glucosidase (Table 1, entry 2).
• Complexes of L6: complex 14 Cu(L6) was the most potent inhibitor (IC50 = 0.21 μM) and 6 Zn(L6) and 27 Co(L6) showed at least a 500 and 1000-fold reduction of activity with IC50 values of 121.19 and 213.30 μM, respectively.
• Complexes of L7: most complexes were inactive, only 35 Mn(L7) showed moderate activity (IC50 = 457.28 μM).
• Complexes of L8: 16 Cu(L8) represented the higher inhibitory effect (IC50 = 140 μM) than 7 Zn(L8) and 19 Ni(L8) with IC50 values of 180 and 200 μM, respectively.
• Complexes of L9: with the replacement of L9, no noteworthy change in the inhibitory activity of Cu(II), Zn(II), and Ni(II) complexes was observed. However, 17 Cu(L9) showed the best inhibition (IC50 = 170 μM). In fact, the presence of the hydroxyl group in L9 did not have a significant effect on the inhibitory activity of the complexes.
• The inhibitory activity of L10 as well as related Cu(II), Ni(II), Co(II), and Mn(II) complexes were not reported clearly (Table 1, entry 4).
In the case of divalent transition metal complexes bearing PicA ligands 37–64 (Table 2).
• Among the complexes 37–64, complex 47 including Cu(II) metal center had the best inhibitory effect (IC50 = 2.91 μM) which was 5.7-fold more potent than genistein (IC50 = 16.57 μM), as the reference compound.
• Interestingly, three complexes 38, 37, and 35 with same ion center (Cu(II)) and structure showed different inhibitory effects. They differed only in one of the substituents.
• The order of inhibitory effect for 42–44, complexes based on L11 having water, pyridine, and thiocyanate ligands, respectively, with trigonal bipyramidal geometry was 42 (IC50 = 2.95 μM) > 43 (IC50 = 3.49 μM) > 44 (IC50 = 8.02 μM). It is apparent that the replacement of thiocyanate moiety in 44 and pyridine in 43 with the water ligand in 42 reduced the inhibition of α-glucosidase by approximately 2.71 and 1.18-fold, respectively. The difference for complexes 42 and 43 can be associated with the effect of the π-conjugation of the pyridine ligand. In addition, H-bonding interactions in 42 including a water ligand could increase the inhibitory activity.
• Furthermore, by replacing Zn(II) metal with Cu(II) in complex 42, the inhibitory properties of 37 (IC50 = 546.04 μM) reduced almost 185-fold (Table 2, entry 2a). These results suggested that metal ions can play a significant role in enzyme inhibition due to their electronic properties. It seems that Cu(II) with d9 electronic configuration is able to be more stable via changing its coordination geometry leading to the better interactions with enzyme. In the case of Zn(II) complexes in which the metal center possesses d10 electronic configuration, no structural changes are observed due to lacking crystal field stabilization energy (CFSE).
• All complexes which have the distorted octahedral geometry coordination (38–41, 48, 49, 51–55, and 57–64) were found to be weak inhibitors (IC50 > 440 μM), except Fe(II) complex 59 depicting moderate activity (IC50 = 97.33 μM). It can be drawn from the obtained results that the presence of metal ion and the coordination environment of complexes can play a significant role in the inhibitory activity of the complex.
• There are two molecules of water in complexes 48, 49, 53, 61, and 62 (Table 2) which are expected to increase the number of H-bonding interactions and improve the inhibitory activity of the complexes. However, this was not observed and the same IC50 values were obtained as reported for other distorted octahedral tris chelates complexes.
• According to Table 2, entry 4c, complexes 59 and 60, including Fe(II) metal ion, with the same coordination geometry showed different inhibitory effects. It should be noted that complex 59 (IC50 = 97.33 μM) having a methyl substitution group at 6-position, displayed almost 7.5-fold increase in the activity comparing with 60 (IC50 = 724.25 μM) which has the same substituent at 3-position of the pyridine ring. Therefore, it can be concluded that in addition to the change of the central metal, the presence of a substituent at different positions can change the α-glucosidase inhibitory effect of a complex.
• Zn(II) 38 and Ni(II) 50 complexes (Table 2, entry 5) containing L11 and L12 (Table 2, entry 1) mixed ligands and two methyl groups at 4-position of 2,2′-bipyridyl ligand (L12), showed no inhibitory activity (IC50 = >600 μM). They were similar to 59 from structure point of view.
• Comparing the inhibitory activity of complexes 39, 46, and 55 (Table 2, entry 6) having L11 and L13 mixed-ligand revealed that there is no a significant difference between IC50 values of these complexes. Zn(II) and Co(II) complexes with a distorted octahedral geometry showed weak activity (IC50 > 600 μM) and Cu(II) complex that had a distorted trigonal bipyramidal geometry revealed better anti-α-glucosidase activity (IC50 = 513.10 μM). It should be noted that the synthesized ligands were not active toward α-glucosidase.
• The IC50 values of the synthesized complexes of Zn(II), Cu(II), Ni(II), Mn(II), and Co(II) metals with mixed-ligands including L11 and L14 showed that except Cu(II) complex 47 (IC50 = 2.91 μM), the variation of metals in coordination did not affect the inhibition of α-glucosidase enzyme (Table 2, entry 7). The IC50 value of Cu(II) complex was approximately 6-fold higher than genistein as the reference drug (IC50 = 16.575 μM). Also, it was >206-fold more potent than other complexes (IC50 > 600 μM).
• The activity of complexes 52, 57, 58, and 64 (IC50 > 600 μM) bearing L11 and L18 showed that distorted octahedral geometry had no effect on the inhibition of α-glucosidase.
• Among metal(II) complexes having PicA derivatives (37–64), only Cu(II) complexes 42 (IC50 = 2.95 μM) and 47 (IC50 = 2.91 μM) depicted strong inhibitory properties.
• Comparison of the inhibitory activity of Cu(II) coordinated to L11 with same geometry of distorted trigonal bipyramidal (42–47) demonstrated that those having two L11 ligands (42–44 and 47, IC50 ≤ 8 μM) were much more potent than complexes containing one L11 ligand (45 and 46, IC50 > 500 μM).
In the case of divalent transition metal complexes bearing Schiff base ligands 65–68 (Table 3):
• These complexes have not been widely investigated and only Zn(II) complexes were reported. Complex 68 bearing naphthalene moiety depicted the highest activity toward α-glucosidase both yeast and rat intestinal enzymes.
In the case divalent transition metal complexes bearing diamine ligands 69–101 (Table 4):
• Diamine complexes with the central metal Zn(II), Cu(II), Ni(II), and Co(II) having distorted octahedral structures did not demonstrate significant inhibitory potency. Among these complexes, only Cu(II) complex 85 possessing L28 ligand revealed better IC50 value (IC50 = 0.47 mg mL−1) than other metal compounds.
α-Glucosidase inhibitory activity of trivalent transition metal complexes 102–110 (Table 5)
A limited number of studies have investigated the α-glucosidase inhibitory activity of trivalent metal transition complexes in comparison to divalent complexes.• Cr(III) complexes were generally found to be more potent than those Fe(III). The best inhibitory effect was shown by complex 107 with an IC50 value of 28.7 μM, which is approximately 14.5-fold higher than acarbose (IC50 = 418.0 μM), used as a reference compound.
• The hydroxamic acid ligand induced more important activity than PicA ligand. However, the electronic property of substituents on the hydroxamic acid moiety was significant. Complex 107 lacking substituents was more potent than 108 (IC50 = 69.28 μM), 109 (IC50 = 355.90 μM) and 110 (IC50 = 169.50 μM), possessing CH3, OCH3, and Cl, respectively. It seemed that electron-donating via a strong electron donating group (OMe) or inductive effect (Cl) deteriorated inhibitory activity.
• In one case, the complex of PicA ligand (106) showed relatively good activity which found to be more active than 109 and 110. It seems that the presence of water as the ligand had a supportive role. Replacement of water by other ligands reduced or deleted the inhibitory activity.
α-Glucosidase inhibitory activity of tetravalent transition metal complexes 111–137 (Table 6)
• Complex 115 (IC50 = 0.68 μM) having L38 with meta-methoxy substitution showed the most potent inhibitory activity. While complex 119 (IC50 = 38 μM) bearing L42 in which OCH3 group was at para position displayed a reduction by approximately 56-fold in the inhibitory activity.• Among hydrazide vanadyl complexes (111–121), the lowest inhibitory activity belonged to the complexes 117 (IC50 = 800 μM) and 118 (IC50 = 758 μM) containing L36 and L37 ligands with iodine substitution at meta and para positions, respectively. This may be related to the steric hindrance of iodine substituent.
• PicA vanadyl complexes (122 and 123) with distorted square pyramidal geometry showed no activity against α-glucosidase.
• Among Schiff base vanadyl complexes (124–137) possessing square pyramidal geometry, 132 and 133 including L54 and L55, having strong electron-donating (OMe) and electron-withdrawing (NO2) groups, respectively, showed the best inhibitory effect (IC50 = 1.26 μM).
α-Glucosidase inhibitory activity of phthalocyanine transition metal complexes 138–168 (Table 7)
• Considering the most potent complexes of the first row transition metals with phthalocyanines revealed that the best inhibitory activity was related to the complexes 168, 162, and 154 possessing 5-amino-4-aryl-3-methyl-1-oxylethyl pyrazole substituted phthalocyanine (L73, Table 7, entry 14). It should be noted that the inhibitory activity of complex 168 (IC50 = 0.002 μM, acarbose = 22.80 μM) was much more significant than other phthalocyanine complexes (138–168, Fig. 3).• Complexes 138, 146, and 164 possessing oxy methyl furan group (L60, Table 7, entry 1) demonstrated the lowest inhibitory activity.
• No comparable inhibitory activity was found between L64-based (phenyl-1,2,3-triazole substituted phthalocyanine) complexes 141, 147, 152, 155 and 165 of Zn(II), Cu(II), Ni(II), Co(II), and Mn(II), respectively.
• The inhibitory activity of metallo(Zn(II), Cu(II), and Co(II))phthalocyanine complexes (144, 148, and 156) having L72 (3,4-dimethoxyphenethoxy substituted phthalocyanine) was approximately 9-fold, 28-fold, and 25-fold more potent than acarbose, respectively.
• Among the copper(II) (149) and nickel(II) (153) complexes containing L61 5-chloro-2-(2,4-dichlorophenoxy)phenol substituted phthalocyanine, 149 showed relatively good activity, which was more potent than 153. It confirms that changing the metal ion affect the inhibitory activity of the complexes.
• Complexes of Cu(II) and Co(II), 151 and 159, having L69 (methylated amino thio pyrimidine substituted phthalocyanine) showed no noteworthy changes for the anti-α-glucosidase activity. However, 159 Co(L69) exhibited approximately 2-fold higher activity than acarbose.
• Among complexes 160 Co(L70) and 161 Co(L71) as well as complexes 166 Mn(L70) and 167 Mn(L71), 161 showed very good inhibitory activity (IC50 = 3.05 μM), however, the others more potent than acarbose.
Conclusions and future perspectives
Survey of literature on novel α-glucosidase inhibitors led us to metal complexes, which have shown versatile properties from both structural and electronic points of view to induce desired activity. Hence, they have occupied a particular attention in the field of enzyme inhibition comparing with small organic molecules. Herein, we focused on the anti-α-glucosidase activity of complexes as a novel and efficient tool in the treatment of T2DM. The efficacy of metal complexes comes back to the fact that free ligands usually have not been active or had a low activity, whereas the corresponding complexes depicted high inhibitory activity. In this respect, the first-row transition metal complexes were in the center of attention. However, their exact mechanism of action toward enzyme is not definitely clear. In this respect, the interaction mode of some potent compounds 5, 13, 26, 34, 42, 47, 85, 115, 132, 133, and 168 were investigated (Fig. 4). | ||
| Fig. 4 Binding conformations of top rank docked compounds inside the active site of α-glucosidase. | ||
The inhibitory activity of discussed complexes in this review and probable mechanism of action can be explained from different perspectives of electronic and structural properties.
• The electronic property of the metal complexes plays a significant role in the anti-α-glucosidase activity. The electronic property is depended on the oxidation state of central metal ions and monodentate ligands, which have been coordinated to the central metal. According to the study of Gu et al.,115 metal ions can be considered as the versatile candidates for the inhibition of α-glucosidase. Various metal ions such as Cu2+, Ni2+, Mg2+, Fe2+, Fe3+, Hg2+, Zn2+, Ca2+, Pb2+, Ag+, V4+, V5+, Al3+, B3+, and Mn2+ were evaluated for their in vitro α-glucosidase inhibitory activity.
(i) Transitions state metals such as copper, vanadium, nickel, and zinc were found to be much more active that main group metals.
(ii) Among divalent cations, Cu2+ was the most potent ion (IC50 = 2.28 μM). However, it depicted the highest activity in the studied series of ions. Trivalent cations were not found to be important inhibitors as Fe3+ and Al3+ were completely inactive and B3+ could weakly inhibit α-glucosidase (IC50 = 2291 μM). In the case of tetravalent and higher metal ions (V4+ and V5+), good activity was observed. V4+ also showed much more inhibition potency (IC50 = 44.8 μM) than that of V5+. Our survey on the inhibitory activity of the first-row transition metal complexes confirmed the high efficacy of Cu2+ complexes among divalent complexes. Trivalent complexes were also low active and V4+ complexes possessed moderate to good potency in the series of tetravalent and higher complexes. It seems that the role of electronic property of metal complexes is important in α-glucosidase inhibition and also is in good agreement with those of free metal ions property.115
• The structural property of the metal complexes is also an important factor, which should be taken in account for anti-α-glucosidase activity.
(i) The literature review indicated that the α-glucosidase inhibitory activity of metal(II) complexes was completely depended on geometry of the complex. For example, in the series of M(II) complexes 40, 47, 51, 56, and 63 reported by Avcı et al. complex 47 with TBP geometry showed the best inhibitory activity (IC50 = 2.91 μM) when compared with other complexes possessing octahedral geometry (40, 51, 56, and 63) (IC50 > 600 μM).63
(ii) The spatial hindrance of the ligand, the presence of electron-donating or electron-withdrawing substituents as well as their position on the ligand moiety were crucial factors which affected the structural property of the metal complexes. For instance, two Fe(II) complexes 59 and 60 having 3- and 6-methylpicolinic acid (L11 and L17), respectively, with the same geometry revealed different inhibitory activity. Changing the position of methyl group from 6- of PicA moiety in complex 60 (IC50 = 724.25 μM) to 3- in complex 59 (IC50 = 97.33 μM) led to a 7-fold increase of activity.67
• The mechanism of α-glucosidase inhibitory activity of the first row transition metal complexes can be investigated from different aspects.
(i) According to Gu et al. report,115 metal ions are enable to inhibit the enzyme through impact on the secondary structure of the enzyme by reduction of α-helix to β-sheet ratio which is occurred by weakening the hydrophobic property.
(ii) Molecular docking of some complexes (5, 13, 26, 34, 42, 47, 85, 115, 132, and 133, and 168) were performed to predict binding conformations and interactions between ligands and the binding site of α-glucosidase.
As far as we know no study was performed to fully evaluate and compare different software to apply in docking of organometallic complexes. All of the complexes were drawn using GaussView 6.0 and were converted to the energetically most stable structure using energy minimization with Hyperchem and Gaussian program. The ligands and protein were converted to their proper readable file format based on the software applied. Different molecular docking programs were applied, to detect similar trends between biological results and in silico outputs. In this context, different docking software named Autodock tools (version 4.2), Gold (Hermes v1.3.1), IGEM (version, 2.1) and Molegro Virtual Docker dock (version, 6.0) software was conducted. Also, various fitness functions, scoring functions, and algorithms were applied. According to Table 8, Molegro Virtual Docker dock software demonstrated more power to properly predict the bio-results in comparison with the rest of the docking programs used in this study. Molegro Virtual Docker software with Max iterations 1500, population size 200, energy threshold 100, and the number of run 100 with MolDock scoring function recorded the best results. The final visualization of the docked structure was performed using Discovery Studio Visualizer 2.5 (Accelrys Software Inc., San Diego, CA, USA). As can be seen, the potent complex was 13 with Molegro Score of −156.14 and IC50 value of 0.15 μM followed by 85 (Molegro Score = −133.73 and IC50 = 0.47 μM) and 115 (Molegro Score = −129.37 and IC50 = 0.68 μM). Our preliminary evaluations showed that the most active derivatives participated in the interactions with Asp404 and Asp282. Also, it was exhibited that binding energy below ≥−100 can be considered as not active while −110≥ categorized as potent inhibitors. The deviation to predict α-glucosidase inhibitory activity can be seen in compound 26. The predicted binding pose of top-ranked docked complexes was presented in Fig. 4. All the residues involved in molecular interaction are shown in line form and colored by atom types in which carbon is depicted in grey and oxygen in red.
| Complex | Molegro score | IC50 (μM) | Residues | Interaction type |
|---|---|---|---|---|
| 5 | −90.0604 | Not active | Trp616 | Pi–donor hydrogen bond |
| Ser676 | Pi–donor hydrogen bond | |||
| Leu678 | Hydrogen bond | |||
| Trp618 | Pi–pi stack | |||
| Trp376 | Pi–pi stack | |||
| Trp376 | Pi–pi stack | |||
| Asp616 | Pii–anion | |||
| Leu677 | Unfavorable bump | |||
| Leu678 | Unfavorable bump | |||
| Leu650 | Unfavorable bump | |||
| Ser676 | Unfavorable bump | |||
| 13 | −156.14 | 0.15 | Trp376 | Pi–pi T-shaped |
| Phe525 | Pi–pi stacked | |||
| Phe525 | Pi–pi stacked | |||
| Trp481 | Pi–pi stacked | |||
| Leu650 | Pi-alkyl | |||
| Asp282 | Pi–anion | |||
| Leu650 | Pi–sigma | |||
| Leu678 | Pi–sigma | |||
| Leu678 | Pi–sigma | |||
| Asp404 | Hydrogen bond | |||
| Asp616 | Hydrogen bond | |||
| Asp649 | Hydrogen bond | |||
| 26 | −109.165 | NA | Trp481 | Pi–pi T-shaped |
| His674 | Pi–pi stack | |||
| Phe649 | Pi–pi stack | |||
| Trp376 | Pi–pi stack | |||
| Arg375 | Pi-alkyl | |||
| Leu650 | Pi-alkyl | |||
| Leu677 | Pi-alkyl | |||
| Asp616 | Hydrogen bond | |||
| Ser679 | Hydrogen bond | |||
| Leu678 | Pi–sigma | |||
| Asn675 | Unfavorable bump | |||
| Ser676 | Unfavorable bump | |||
| Leu677 | Unfavorable bump | |||
| Leu678 | Unfavorable bump | |||
| 34 | −74.292 | NA | Trp376 | Pi–pi T-shaped |
| Trp376 | Pi–pi stack | |||
| Trp376 | Pi–pi stack | |||
| Leu678 | Pi-alkyl | |||
| Asp616 | Pi–anion | |||
| Ser676 | Unfavorable bump | |||
| Leu677 | Unfavorable bump | |||
| Leu677 | Unfavorable bump | |||
| 42 | −110.0231 | 2.95 | Trp481 | Pi-alkyl |
| Trp516 | Pi-alkyl | |||
| Trp519 | Pi-alkyl | |||
| Trp613 | Pi-alkyl | |||
| Phe649 | Pi-alkyl | |||
| His674 | Pi-alkyl | |||
| Asp404 | Hydrogen bond | |||
| Trp481 | Hydrogen bond | |||
| Asp518 | Unfavorable bump | |||
| Asp616 | Unfavorable bump | |||
| 47 | −136.527 | 2.91 | Trp376 | Pi-alkyl |
| Trp376 | Pi-alkyl | |||
| Trp481 | Pi-alkyl | |||
| Phe649 | Pi-alkyl | |||
| Phe649 | Pi-alkyl | |||
| Phe650 | Pi-alkyl | |||
| Asp282 | van der Waals | |||
| Asp616 | van der Waals | |||
| Asp282 | Hydrogen bond | |||
| Met519 | Hydrogen bond | |||
| Ala284 | Unfavorable bump | |||
| Asp616 | Unfavorable bump | |||
| 85 | −133.7351 | 0.47 | Asp404 | van der Waals |
| Asp404 | van der Waals | |||
| Asp616 | van der Waals | |||
| Asp616 | van der Waals | |||
| His674 | van der Waals | |||
| Asp404 | Hydrogen bond | |||
| Asp518 | Hydrogen bond | |||
| Asp518 | Hydrogen bond | |||
| Asp518 | Hydrogen bond | |||
| Trp481 | Unfavorable bump | |||
| 115 | −129.3691 | 0.68 | Ser523 | Carbon hydrogen bond |
| Ser523 | Carbon hydrogen bond | |||
| Phe649 | Pi–pi stack | |||
| Asp282 | Pi–anion | |||
| Asp518 | Hydrogen bond | |||
| Asp616 | Hydrogen bond | |||
| Asp616 | Hydrogen bond | |||
| Trp481 | Unfavorable bump | |||
| Met519 | Unfavorable bump | |||
| Arg600 | Unfavorable bump | |||
| 132 | −126.219 | 1.26 | Phe525 | Pi–pi T-shaped |
| Asp282 | Pi–anion | |||
| Asp518 | Pi–anion | |||
| Met519 | Pi–sulfur | |||
| Asp282 | Carbon hydrogen bond | |||
| Phe525 | Carbon hydrogen bond | |||
| His674 | Carbon hydrogen bond | |||
| 133 | −139.723 | 1.26 | Phe525 | Pi–pi T-shaped |
| Asp282 | Pi–anion | |||
| Asp518 | Pi–anion | |||
| Met519 | Pi–sulfur | |||
| Phe525 | Pi–sigma | |||
| Asp404 | Hydrogen bond | |||
| Asp404 | Hydrogen bond | |||
| His674 | Hydrogen bond | |||
| 168 | −237.455 | Glu346 | Pi–anion | |
| Ser349 | H-bond | |||
| Ser349 | H-bond | |||
| His708 | H-bond | |||
| Leu712 | Alkyl | |||
| Glu730 | Pi–anion | |||
| Glu730 | Pi–anion | |||
| Phe731 | Pi–pi T shaped | |||
| Phe731 | Pi–pi T shaped | |||
| Lys733 | Pi-alkyl | |||
| Lys733 | Pi–sigma | |||
| Glu748 | Pi–anion | |||
| Glu748 | Pi–anion | |||
| His799 | Pi–pi T shaped | |||
| His799 | Pi-alkyl | |||
| Glu801 | Pi–anion | |||
| Arg819 | Pi–cation | |||
| Lys849 | Alkyl | |||
| Lys849 | Pi-alkyl | |||
| Arg854 | H-bound | |||
| Arg854 | Pi-alkyl | |||
| Arg854 | Pi–cation | |||
| Glu856 | Pi–anion |
The main point is that complex 168 behaved differently against α-glucosidase. The enzyme kinetic study showed that it was a non-competitive inhibitor and it binds to the enzyme at a location other than the active site. As a result, the maestro sitemap tool was applied to find the possible cavity of the enzyme. Five possible binding sites were detected on the surface of the enzyme which can be suitable for non-competitive inhibition. Next, 168 as the potent inhibitor was docked on all of the potential binding sites of the enzyme. The cavity with Ser349 His708, Leu712, Glu730, Glu801, and Arg819 residue (reported in Table 8) was selected as an allosteric site. Considering the Moledock score results, 168 participated in several interactions with the allosteric binding site which justifies its potency.
Even with all the significant improvements in the computational study in drug discovery, proper prediction of how ligand binds to its target is an extremely challenging task. It is important to note that no specific docking software or scoring function exists, that can be successfully applied to all molecular docking studies, due to the variability in nature of the ligands and its type of inhibition. However, according to our preliminary study, Molegro Virtual Docker software had more power to properly predict binding energy of organic metallic compounds against α-glucosidase.
Author contributions
Marzieh Sohrabi and Mohammad Reza Binaeizadeh searched databases and prepared the original draft. Aida Iraji performed docking study. Bagher Larijani and Mohammad Mahdavi analysed the results. Mina Saeedi edited the manuscript and supervised all steps.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledged partial support from Tehran University of Medical Sciences.References
- A. Bastaki, Int. J. Diabetes Mellitus, 2005, 13, 111 CrossRef.
- W. Sami, T. Ansari, N. S. Butt and M. R. Ab Hamid, Int. J. Health Sci., 2017, 11, 65 Search PubMed.
- Y.-G. Chen, P. Li, P. Li, R. Yan, X.-Q. Zhang, Y. Wang, X.-T. Zhang, W.-C. Ye and Q.-W. Zhang, Molecules, 2013, 18, 4221–4232 CrossRef CAS PubMed.
- R. K. Campbell, J. Am. Pharmaceut. Assoc., 2009, 49, S3–S9 CrossRef PubMed.
- G. A. Colditz, W. C. Willett, M. J. Stampfer, J. E. Manson, C. H. Hennekens, R. A. Arky and F. E. Speizer, Am. J. Epidemiol., 1990, 132, 501–513 CrossRef CAS PubMed.
- G. A. Colditz, W. C. Willett, A. Rotnitzky and J. E. Manson, Ann. Intern. Med., 1995, 122, 481–486 CrossRef CAS PubMed.
- S. P. Helmrich, D. R. Ragland, R. W. Leung and R. S. Paffenbarger Jr, N. Engl. J. Med., 1991, 325, 147–152 CrossRef CAS PubMed.
- F. B. Hu, R. J. Sigal, J. W. Rich-Edwards, G. A. Colditz, C. G. Solomon, W. C. Willett, F. E. Speizer and J. E. Manson, JAMA, 1999, 282, 1433–1439 CrossRef CAS PubMed.
- J. E. Manson, U. A. Ajani, S. Liu, D. M. Nathan and C. H. Hennekens, Am. J. Med., 2000, 109, 538–542 CrossRef CAS PubMed.
- J. Salmeron, J. E. Manson, M. J. Stampfer, G. A. Colditz, A. L. Wing and W. C. Willett, Jama, 1997, 277, 472–477 CrossRef CAS PubMed.
- M. J. Knol, J. W. Twisk, A. T. Beekman, R. J. Heine, F. J. Snoek and F. Pouwer, Diabetologia, 2006, 49, 837–845 CrossRef CAS PubMed.
- C.-Y. Cai, L. Rao, Y. Rao, J.-X. Guo, Z.-Z. Xiao, J.-Y. Cao, Z.-S. Huang and B. Wang, Eur. J. Med. Chem., 2017, 130, 51–59 CrossRef CAS PubMed.
- J. Zhen, Y. Dai, T. Villani, D. Giurleo, J. E. Simon and Q. Wu, Bioorg. Med. Chem., 2017, 25, 5355–5364 CrossRef CAS PubMed.
- S. T. Assefa, E.-Y. Yang, S.-Y. Chae, M. Song, J. Lee, M.-C. Cho and S. Jang, Plants, 2020, 9, 2 CrossRef CAS PubMed.
- H. B. B. Ag, Eur. J. Clin. Invest., 1994, 24, 3–10 CrossRef.
- S. Chiba, Biosci., Biotechnol., Biochem., 1997, 61, 1233–1239 CrossRef CAS PubMed.
- M. Saeedi, A. Hadjiakhondi, S. Mohammad Nabavi and A. Manayi, Curr. Top. Med. Chem., 2017, 17, 428–440 CrossRef CAS PubMed.
- K. Papoutsis, J. Zhang, M. C. Bowyer, N. Brunton, E. R. Gibney and J. Lyng, Food Chem., 2020, 128119 Search PubMed.
- S. Fattaheian-Dehkordi, R. Hojjatifard, M. Saeedi and M. Khanavi, Evidence-Based Complementary Altern. Med., 2021, 2021 Search PubMed.
- D. Shareghi-Boroujeni, A. Iraji, S. Mojtabavi, M. A. Faramarzi, T. Akbarzadeh and M. Saeedi, Bioorg. Chem., 2021, 111, 104869 CrossRef CAS PubMed.
- M. Saeedi, M. Raeisi-Nafchi, S. Sobhani, S. S. Mirfazli, M. Zardkanlou, S. Mojtabavi, M. A. Faramarzi and T. Akbarzadeh, Mol. Diversity, 2020, 1–11 CAS.
- M. Saeedi, M. Mohammadi-Khanaposhtani, M. S. Asgari, N. Eghbalnejad, S. Imanparast, M. A. Faramarzi, B. Larijani, M. Mahdavi and T. Akbarzadeh, Bioorg. Med. Chem., 2019, 27, 115148 CrossRef CAS PubMed.
- M. Saeedi, M. Mohammadi-Khanaposhtani, P. Pourrabia, N. Razzaghi, R. Ghadimi, S. Imanparast, M. A. Faramarzi, F. Bandarian, E. N. Esfahani and M. Safavi, Bioorg. Chem., 2019, 83, 161–169 CrossRef CAS PubMed.
- Z. Liu and S. Ma, ChemMedChem, 2017, 12, 819–829 CrossRef CAS PubMed.
- K. Pedrood, M. Sherafati, M. Mohammadi-Khanaposhtani, M. S. Asgari, S. Hosseini, H. Rastegar, B. Larijani, M. Mahdavi, P. Taslimi and Y. Erden, Int. J. Biol. Macromol., 2021, 170, 1–12 CrossRef CAS PubMed.
- M. S. Asgari, M. Mohammadi-Khanaposhtani, Z. Sharafi, M. A. Faramarzi, H. Rastegar, E. N. Esfahani, F. Bandarian, P. R. Rashidi, R. Rahimi and M. Biglar, Mol. Diversity, 2021, 25, 877–888 CrossRef CAS PubMed.
- M. Sherafati, R. Mirzazadeh, E. Barzegari, M. Mohammadi-Khanaposhtani, H. Azizian, M. S. Asgari, S. Hosseini, E. Zabihi, S. Mojtabavi and M. A. Faramarzi, Bioorg. Chem., 2021, 109, 104703 CrossRef CAS PubMed.
- M. Sohrabi, M. Saeedi, B. Larijani and M. Mahdavi, Eur. J. Med. Chem., 2021, 113308 CrossRef CAS PubMed.
- S. Maikoo, D. Makayane, I. N. Booysen, P. Ngubane and A. Khathi, Eur. J. Med. Chem., 2020, 113064 Search PubMed.
- D. C. Crans and K. Kostenkova, Commun. Chem., 2020, 3, 1–4 CrossRef.
- C. Van Cleave and D. C. Crans, Inorganics, 2019, 7, 111 CrossRef CAS.
- G. Parkin, J. Chem. Educ., 2006, 83, 791 CrossRef CAS.
- H. Sakurai, A. Katoh, T. Kiss, T. Jakusch and M. Hattori, Metallomics, 2010, 2, 670–682 CrossRef CAS PubMed.
- K. Thompson, V. Yuen, J. McNeill and C. Orvig, ACS Symp. Ser., 1998, 711, 329–343 CrossRef CAS.
- E. L. Tolman, E. Barris, M. Burns, A. Pansini and R. Partridge, Life Sci., 1979, 25, 1159–1164 CrossRef CAS PubMed.
- B. Lyonnet, Presse Med., 1899, 1, 191–192 Search PubMed.
- C. E. Heyliger, A. G. Tahiliani and J. H. McNeill, Science, 1985, 227, 1474–1477 CrossRef CAS PubMed.
- K. H. Thompson and C. Orvig, Met. Ions Biol. Syst., 2004, 41, 221–252 CAS.
- K. H. Thompson, J. H. McNeill and C. Orvig, Chem. Rev., 1999, 99, 2561–2572 CrossRef CAS PubMed.
- S. Dai, K. Thompson and J. McNeill, J. Pharmacol. Toxicol., 1994, 74, 101–109 CrossRef CAS PubMed.
- K. H. Thompson, B. D. Liboiron, Y. Sun, K. D. Bellman, I. A. Setyawati, B. O. Patrick, V. Karunaratne, G. Rawji, J. Wheeler and K. Sutton, J. Biol. Inorg Chem., 2003, 8, 66–74 CrossRef CAS PubMed.
- S. Fujimoto, H. Yasui and Y. Yoshikawa, J. Inorg. Biochem., 2013, 121, 10–15 CrossRef CAS PubMed.
- K. J. Kilpin and P. J. Dyson, Chem. Sci., 2013, 4, 1410–1419 RSC.
- Y. Yoshikawa, E. Ueda, K. Kawabe, H. Miyake, H. Sakurai and Y. Kojima, Chem. Lett., 2000, 29, 874–875 CrossRef.
- A. Rubenstein, N. Levin and G. Elliott, Nature, 1962, 194, 188–189 CrossRef CAS PubMed.
- Y. Yoshikawa, E. Ueda, K. Kawabe, H. Miyake, T. Takino, H. Sakurai and Y. Kojima, J. Biol. Inorg Chem., 2002, 7, 68–73 CrossRef CAS PubMed.
- Y. Yoshikawa, E. Ueda, Y. Kojima and H. Sakurai, Life Sci., 2004, 75, 741–751 CrossRef CAS PubMed.
- L. Coulston and P. Dandona, Diabetes, 1980, 29, 665–667 CrossRef CAS PubMed.
- R. Narang, B. Narasimhan and S. Sharma, Curr. Med. Chem., 2012, 19, 569–612 CrossRef CAS PubMed.
- F. Guo, T. Xia, P. Xiao, Q. Wang, Z. Deng, W. Zhang and G. Diao, Bioorg. Chem., 2021, 110, 104764 CrossRef CAS PubMed.
- T.-H. Duong, A. P. Devi, N.-V. Huynh, J. Sichaem, H.-D. Tran, M. Alam, T.-P. Nguyen, H.-H. Nguyen, W. Chavasiri and T.-C. Nguyen, Bioorg. Med. Chem. Lett., 2020, 30, 127359 CrossRef CAS PubMed.
- P. Akhter, U. Ashiq, R. A. Jamal, Z. Shaikh, M. Mahroof-Tahir, M. Lateef and R. Badar, Med. Chem., 2019, 15, 923–936 CrossRef CAS PubMed.
- S. Khan, M. Tariq, M. Ashraf, S. Abdullah, M. Al-Rashida, M. Khalid, P. Taslimi, M. Fatima, R. Zafar and Z. Shafiq, Bioorg. Chem., 2020, 102, 104082 CrossRef CAS PubMed.
- J. E. Philip, M. Shahid, M. P. Kurup and M. P. Velayudhan, J. Photochem. Photobiol., B, 2017, 175, 178–191 CrossRef CAS PubMed.
- K. K. Naik, S. Selvaraj and N. Naik, Spectrochim. Acta, Part A, 2014, 131, 599–605 CrossRef PubMed.
- S. Barrie, J. Wright, J. Pizzorno, E. Kutter and P. Barron, Agents Actions, 1987, 21, 223–228 CrossRef CAS PubMed.
- P. J. Aggett, P. K. Fenwick and H. Kirk, J. Nutr., 1989, 119, 1432–1437 CrossRef CAS PubMed.
- I. Krieger, R. Cash and G. W. Evans, J. Pediatr. Gastroenterol. Nutr., 1984, 3, 62–68 CrossRef CAS PubMed.
- R. Grant, S. Coggan and G. Smythe, Int. J. Tryptophan Res., 2009, 2, S2469 CrossRef PubMed.
- L. Tan, J.-T. Yu and L. Tan, J. Neurol. Sci., 2012, 323, 1–8 CrossRef CAS PubMed.
- Y. Yoshikawa, R. Hirata, H. Yasui and H. Sakurai, Biochimie, 2009, 91, 1339–1341 CrossRef CAS PubMed.
- D. Avcı, S. Altürk, F. Sönmez, Ö. Tamer, A. Başoğlu, Y. Atalay and B. Z. Kurt, J. Mol. Struct., 2020, 1205, 127655 CrossRef.
- D. Avcı, S. Altürk, F. Sönmez, Ö. Tamer, A. Başoğlu, Y. Atalay, B. Z. Kurt and N. Dege, J. Mol. Struct., 2019, 1197, 645–655 CrossRef.
- D. Avcı, S. Altürk, F. Sönmez, Ö. Tamer, A. Başoğlu, Y. Atalay, B. Z. Kurt and N. Dege, Mol. Diversity, 2020, 1–19 Search PubMed.
- N. Dege, Ö. Özge, D. Avcı, A. Başoğlu, F. Sönmez, M. Yaman, Ö. Tamer, Y. Atalay and B. Z. Kurt, Spectrochim. Acta, Part A, 2021, 262, 120072 CrossRef CAS PubMed.
- D. Avcı, S. Altürk, F. Sönmez, Ö. Tamer, A. Başoğlu, Y. Atalay, B. Z. Kurt and N. Dege, Tetrahedron, 2018, 74, 7198–7208 CrossRef.
- D. Avcı, S. Altürk, F. Sönmez, Ö. Tamer, A. Başoğlu, Y. Atalay, B. Z. Kurt and N. Dege, J. Biol. Inorg Chem., 2019, 24, 747–764 CrossRef PubMed.
- D. Avcı, S. Altürk, F. Sönmez, Ö. Tamer, A. Başoğlu, Y. Atalay, B. Z. Kurt and N. Dege, Appl. Organomet. Chem., 2020, 34, e5412 CrossRef.
- L. Li, Q. Guo, J. Dong, T. Xu and J. Li, J. Photochem. Photobiol., B, 2013, 125, 56–62 CrossRef CAS PubMed.
- S. Arulmurugan, H. P. Kavitha and B. Venkatraman, Rasayan J. Chem., 2010, 3, 385–410 Search PubMed.
- E. A. Bajema, K. Roberts and T. Meade, Met. Ions Life Sci., 2019, 19, 267–301 CAS.
- R. Miyazaki, H. Yasui and Y. Yoshikawa, Open J. Inorg. Chem., 2016, 6, 114–124 CrossRef CAS.
- D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580–2627 CrossRef CAS PubMed.
- I. P. Tripathi, A. Dwivedi and M. Mishra, Asian J. Med. Health, 2017, 2, 1–14 CrossRef.
- I. Tripathi and A. Dwivedi, J. Adv. Med. Med. Res., 2016, 1–11 Search PubMed.
- I. P. Tripathi, M. M. Kumar, K. Arti, M. Chinmayi, T. Ruchita, S. L. Kant and P. K. Bihari, Res. J. Chem. Sci., 2013, 2231, 606X Search PubMed.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier, 2012 Search PubMed.
- H. Marschner and V. Römheld, Plant Soil, 1994, 165, 261–274 CrossRef CAS.
- W. T. Cefalu and F. B. Hu, Diabetes Care, 2004, 27, 2741–2751 CrossRef CAS PubMed.
- R. A. Anderson, J. Am. Coll. Nutr., 1998, 17, 548–555 CrossRef CAS PubMed.
- K. Schwarz and W. Mertz, Arch. Biochem. Biophys., 1959, 85, 292–295 CrossRef CAS PubMed.
- K. N. Jeejeebhoy, R. Chu, E. Marliss, G. R. Greenberg and A. Bruce-Robertson, Am. J. Clin. Nutr., 1977, 30, 531–538 CrossRef CAS PubMed.
- E. Król, Z. Krejpcio, M. Okulicz and H. Śmigielska, Biol. Trace Elem. Res., 2020, 193, 185–194 CrossRef PubMed.
- D. Avcı, S. Altürk, F. Sönmez, Ö. Tamer, A. Başoğlu, Y. Atalay, B. Z. Kurt and N. Dege, J. Mol. Struct., 2020, 1220, 128761 CrossRef.
- L. R. Hassan, H. Bahron, F. Abdullah and A. M. Tajuddin, J. Biol. Inorg Chem., 2020, 25, 239–252 CrossRef CAS PubMed.
- N. D. Chasteen, J. K. Grady and C. E. Holloway, Inorg. Chem., 1986, 25, 2754–2760 CrossRef CAS.
- K. H. Thompson and C. Orvig, J. Chem. Soc., Dalton Trans., 2000, 2885–2892 RSC.
- C. Amante, D. Sousa-Coelho, A. Luísa and M. Aureliano, Metals, 2021, 11, 828 CrossRef CAS.
- S. Selvaraj and U. M. Krishnan, J. Med. Chem., 2021, 64, 12435–12452 CrossRef CAS PubMed.
- A. Ścibior and J. Kurus, Curr. Med. Chem., 2019, 26, 5456–5500 CrossRef PubMed.
- K. Cusi, S. Cukier, R. DeFronzo, M. Torres, F. Puchulu and J. P. Redondo, J. Clin. Endocrinol. Metab., 2001, 86, 1410–1417 CAS.
- A. B. Goldfine, M.-E. Patti, L. Zuberi, B. J. Goldstein, R. LeBlanc, E. J. Landaker, Z. Y. Jiang, G. R. Willsky and C. R. Kahn, Metabolism, 2000, 49, 400–410 CrossRef CAS PubMed.
- U. Ashiq, R. Ara, M. Mahroof-Tahir, Z. T. Maqsood, K. M. Khan, S. N. Khan, H. Siddiqui and M. I. Choudhary, Chem. Biodiversity, 2008, 5, 82–92 CrossRef CAS PubMed.
- R. Patel and Y. P. Singh, J. Mol. Struct., 2018, 1153, 162–169 CrossRef CAS.
- S. Bharathi, D. Mahendiran, R. Senthil Kumar and A. Kalilur Rahiman, ChemistrySelect, 2020, 5, 6245–6254 CrossRef CAS.
- S. Misra, K. B. Pandeya, A. K. Tiwari, A. Z. Ali, T. Saradamani, S. B. Agawane and K. Madhusudana, Int. J. Nutr. Metab., 2012, 4, 11–18 CAS.
- T. V. Basova, N. S. Mikhaleva, A. K. Hassan and V. G. Kiselev, Sens. Actuators, B, 2016, 227, 634–642 CrossRef CAS.
- T. Basova, E. Kol'tsov, A. Ray, A. Hassan, A. Gürek and V. Ahsen, Sens. Actuators, B, 2006, 113, 127–134 CrossRef CAS.
- R. J. G. Rubira, P. H. B. Aoki, C. J. L. Constantino and P. Alessio, Appl. Surf. Sci., 2017, 416, 482–491 CrossRef CAS.
- T. Keleş, Z. Biyiklioglu, E. Güzel, M. Nebioğlu and İ. Şişman, Appl. Organomet. Chem., 2021, 35, e6076 Search PubMed.
- B. Yıldız, E. Güzel, D. Akyüz, B. S. Arslan, A. Koca and M. K. Şener, Sol. Energy, 2019, 191, 654–662 CrossRef.
- A. B. Sorokin, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS PubMed.
- P.-C. Lo, M. S. Rodríguez-Morgade, R. K. Pandey, D. K. Ng, T. Torres and F. Dumoulin, Chem. Soc. Rev., 2020, 49, 1041–1056 RSC.
- F. Özen, A. Günel and A. Baran, Bioorg. Chem., 2018, 81, 71–78 CrossRef PubMed.
- B. Barut and Ü. Demirbaş, J. Organomet. Chem., 2020, 923, 121423 CrossRef CAS.
- E. Güzel, Ü. M. Koçyiğit, P. Taslimi, S. Erkan and O. S. Taskin, J. Biochem. Mol. Toxicol., 2021, 35, 1–9 CrossRef PubMed.
- A. Günsel, F. Kalkan, G. Y. Atmaca, B. Barut, A. T. Bilgiçli, H. Pişkin, A. Özel, A. Erdoğmuş and M. N. Yarasir, Appl. Organomet. Chem., 2021, 35, e6202 CrossRef.
- Ü. M. Koçyiğit, P. Taslimi, B. Tüzün, H. Yakan, H. Muğlu and E. Güzel, J. Biomol. Struct. Dyn., 2020, 1–11 CrossRef PubMed.
- A. Günsel, P. Taslimi, G. Y. Atmaca, A. T. Bilgiçli, H. Pişkin, Y. Ceylan, A. Erdoğmuş, M. N. Yarasir and İ. Gülçin, J. Mol. Struct., 2021, 1237, 130402 CrossRef.
- A. Günsel, A. T. Bilgicli, B. Barut, P. Taslimi, A. Özel, İ. Gülçin, Z. Biyiklioglu and M. N. Yarasir, J. Mol. Struct., 2020, 1214, 128210 CrossRef.
- F. Türkan, P. Taslimi, B. Cabir, M. S. Ağırtaş, Y. Erden, H. U. Celebioglu, B. Tuzun, E. Bursal and I. Gulcin, Polycyclic Aromat. Compd., 2021, 1–13 Search PubMed.
- A. Günsel, A. Yıldırım, P. Taslimi, Y. Erden, T. Taskin-Tok, H. Pişkin, A. T. Bilgiçli, İ. Gülçin and M. N. Yarasir, Inorg. Chem. Commun., 2022, 138, 109263 CrossRef.
- B. Barut, T. Keleş, Z. Biyiklioglu and C. Ö. Yalçın, Appl. Organomet. Chem., 2021, 35, e6021 CrossRef CAS.
- E. Güzel, Ü. M. Koçyiğit, B. S. Arslan, M. Ataş, P. Taslimi, F. Gökalp, M. Nebioğlu, İ. Şişman and İ. Gulçin, Arch. Pharm., 2019, 352, 1800292 CrossRef PubMed.
- Y. Wang, L. Ma, Z. Li, Z. Du, Z. Liu, J. Qin, X. Wang, Z. Huang, L. Gu and A. S. Chen, FEBS Lett., 2004, 576, 46–50 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |